Back to Journals » International Journal of Nanomedicine » Volume 14
Local and systemic delivery of mRNA encoding survivin-T34A by lipoplex for efficient colon cancer gene therapy
Authors Zhang X, Men K ![]() , Zhang Y, Zhang R, Yang L, Duan X
, Zhang Y, Zhang R, Yang L, Duan X
Received 19 December 2018
Accepted for publication 11 March 2019
Published 23 April 2019 Volume 2019:14 Pages 2733—2751
DOI https://doi.org/10.2147/IJN.S198747
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
Xueyan Zhang,1 Ke Men,1 Yuanfa Zhang,1 Rui Zhang,1 Li Yang,1 Xingmei Duan2
1State Key Laboratory of Biotherapy and Cancer Center/Collaborative Innovation Center for Biotherapy, West China Hospital, Sichuan University, Chengdu 610041, People’s Republic of China; 2Department of Pharmacy and Personalized Drug Therapy Key Laboratory of Sichuan Province, Sichuan Academy of Medical Sciences & Sichuan Provincial People’s Hospital, Chengdu 610072, People’s Republic of China
Background: In vitro transcribed (IVT) mRNA has been applied as an alternative therapeutic molecule to plasmid DNA in the field of cancer therapy and biomedical research studies. mRNA-based therapy has demonstrated several advantages over its DNA counterparts. However, its further therapeutic application is largely restricted by delivery method.
Methods: In this work, a liposome-protamine lipoplex (CLPP) was prepared to deliver IVT mRNA encoding survivin-T34A gene, forming a novel core-shell structured nanoparticle formulation (CLPP/mSur-T34A).
Results: The prepared CLPP/mSur-T34A particle had an average size of 186.1±3.1 nm, displaying high mRNA transfecting and expression efficiency on C26 tumor cells through lipid rafts-mediated endocytosis. CLPP/mSur-T34A mRNA formulation demonstrated obvious therapeutic effects on various models of C26 colon cancer both in vitro and in vivo. Particularly, local and systemic administration of CLPP/mSur-T34A particle exhibited superior antitumor effect regarding its DNA plasmid counterpart with high safety.
Conclusion: Our results indicated the high delivery capacity of liposome-protamine lipoplex and further suggested CLPP/mSur-T34A mRNA formulation to be a potential candidate for colon cancer therapy.
Keywords: mRNA gene therapy, colon cancer, nonviral vector, systemic delivery
Introduction
Cancer is one of the leading causes of death in both economically developed and developing countries. Colon carcinoma holds the second most common cause of death among cancers.1 The wide catalog of biological therapeutics such as gene therapy, immunotherapy and cell therapy has been intensively developed for cancer treatment.2–4 Among them, benefited from the progress of non-viral and viral vectors, gene therapy has demonstrated outstanding potential in cancer treatment owing to its high specificity and safety.5 Recently, several products have been approved for clinical application worldwide.6–8 Despite much progress being achieved, developing novel therapeutic targets as well as nucleic acid agents is still in high demand.
As a natural product of gene, messenger RNA (mRNA) is a transient entity that mediates the translation of genetic information from DNA to proteins in cells. Previously, mRNA has been successfully applied as an alternative genetic molecule in the field of cancer immunotherapy and zygote-based biomedical research.9–12 However, innate mRNA was previously considered to be unstable in both structural and expression level which somehow hinders its use as a therapeutic agent. The introduction of chemical modifications to mRNA, such as 5′ cap, 5′- and 3′-UTRs, the coding region optimization, and the poly(A) tail, has overcome the aforementioned obstacle,13–15 which facilitates its application in gene therapy. Comparing to other forms of therapeutic nucleic acids, mRNA agents demonstrated several advantages. For example, mRNA does not integrate into host genome, avoiding aberrant transcription and insertional mutagenesis.16 Meanwhile, its expression kinetics is predictable and consistent,15,17 while nuclear localization is not required before rapid protein production even in dividing and hard-to-transfect cells.18,19 Furthermore, mRNA is only transiently active and is completely biodegradable via metabolic pathways.20 More importantly, IVT mRNA contains much fewer elements than plasmid DNA in structure when expressing specific genes, thus greatly reducing the delivery difficulty. Thus, these properties make mRNA a safe and attractive genetic material for gene therapy. However, despite these superiorities, only a limited number of attempts have been reported to apply mRNA in cancer gene therapy study. This might largely attribute to its restricted therapeutic targets and limited delivery methods.
As a core component, delivery system is one of the main obstacles of mRNA-based gene therapy. Conventional none-viral gene vectors are usually developed for the binding and transportation of plasmid DNA, while additional optimizations are required to further protect and condense mRNA molecule. As widely acknowledged, mRNA is single stranded with a flexible linear structure, presenting difficulties to form a nanosized particle with the given gene vectors.21,22 Thus, a strong condensation effect from delivery system is essential. Meanwhile, vectors with high delivery capacity are necessary to achieve efficient transportation of compressed mRNA complex. Moreover, the delivery efficiency also has much to do with the length of mRNA itself and the type of target cells.23,24 Therefore, the design and selection of ideal delivery system is critical for mRNA-based gene delivery. Survivin-T34A was previously reported to be a potent agonist of endogenous survivin, resulting in caspase-dependent cell apoptosis. Its anticancer effects have been well evaluated on several cancer models and the desired therapeutic efficacy was achieved.25–27 In the present study, we attempted to deliver the survivin-T34A-encoded IVT mRNA with liposome-protamine lipoplex. Within this lipoplex, protamine was designed to condense IVT mRNA into nanosized particle and protect it from nuclease degradation in cytoplasm.28–30 Meanwhile, cationic liposomes (CLP) act as a vector for transport the above complex. We assume that efficient delivery of survivin-T34A mRNA into C26 colon cancer cell could result in ideal anticancer effect. To our best knowledge, this is the first report about survivinT34A mRNA-based cancer gene therapy study.
In this study, we characterized the prepared liposome-protamine-mRNA particle. Its overall anticancer abilities were evaluated through both local and systemic administration. The efficacy and safety of CLPP/mRNA and its plasmid counterparts were further compared.
Materials and methods
Materials
DOTAP were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Cholesterol and protamine sulfate were purchased by Sigma-Aldrich (St Louis, MO, USA). Other chemicals were purchased from Sigma-Aldrich unless otherwise noted. mMESSAGE mMACHINE™ T7 Transcription Kit and MEGAclear™ Transcription Clean-Up Kit, LipofectamineTM2000, DMEM and serums were purchased from Thermo Fisher Scientific (Waltham, MA, USA). C26 Mus musculus colon carcinoma cell line and 293T human embryonic kidney cell line were purchased from the American Type Culture Collection (ATCC) (Manassas, VA, USA). BALB/c mice were obtained from Beijing HFK Bio-technology Co. Ltd. (Beijing, China) and maintained under specific pathogen-free conditions. All animal procedures were approved and controlled by the Institutional Animal Care and Treatment Committee of Sichuan University and carried out according to the Animal Care and Use Guidelines of Sichuan University.
In vitro transcription of mRNA
A mMESSAGE mMACHINE™ T7 Transcription Kit was used to prepare survivin-T34A mRNA (mSur-T34A) by T7 polymerase-based in vitro transcription method. Briefly, the open-reading frame (ORF) of Survivin-T34A gene was amplified from pVAX1-survivin-T34A plasmid by PCR reaction with forward primer: TAA TAC GAC TCA CTA TAG GG (T7 promoter) A TGG GAG CTC CGG CGC TGC CCC A and reverse primer: GGG ATC TAG ATT AGG CAG CCA GCT GCT CAA TT. With PCR products as templates, the mRNA transcription process was conducted according to manufacturer’s manual. The MEGAclear™ Transcription Clean-Up Kit was used to further purify transcript mRNA according to the manufacturer’s instructions. The purified survivin-T34A mRNA was quantified by spectrophotometry and analyzed by agarose gel electrophoresis to confirm the synthesis of modified mRNA.
Preparation and characterization of CLPP/mRNA particles
CLP were prepared according to our previous reports.29 Briefly, DOTAP and cholesterol (1:1, mol/mol) were dissolved in chloroform and solvent was removed under rotary evaporation. The lipid film was rehydrated with distilled water under 50°C to form a CLP solution and stored in 4°C for further use. Liposome-protamine lipoplex was prepared to deliver IVT mRNA. Briefly, survivin-T34A mRNA was first incubated with protamine sulfate solution (1:1, w/w) for 10 mins. Then, CLPs were added to the mixture in a ratio of 1:1:1 (liposome:protamine:mRNA, w/w/w) followed by incubation at room temperature for 15 mins. The size distribution of liposome or CLPP/mRNA particle was characterized by dynamic light scattering (DLS) using a ZetaSizer Nano ZS (Malvern Instruments Ltd., Malvern, UK). The morphology analysis of prepared particles was also examined via transmission electron microscope (TEM) (H6009IV, Hitachi, Tokyo, Japan).
Gel retardation assay
The mRNA-binding ability of CLPP lipoplex was evaluated by agarose retarding assay. 1 μg of survivin-T34A mRNA was separately mixed with different mass ratios of CLPP as mentioned before. Electrophoresis was then performed on 1% (w/v) agarose gel stained with Golden ViewTM for 30 mins at 120 V. Gels were visualized and imaged using ChemiDoc Imagers.
Cytotoxicity assay
293T cells were plated at a density of 1×104 cells per well in 100 μL of DMEM medium. After incubation for 24 hrs, cells were treated with different concentrations of CLPP, polyethyleneimine (PEI25K), or LipofactamineTM2000 (lipo2K) for 24 hrs. Subsequently, 20 mL of MTT solution was added to each well and incubated at 37°C for 4 hrs. The formazan was solubilized by adding DMSO and shaken for 30 mins. The absorbance was read at 570 nm by the Spectramax M5 Microtiter Plate Luminometer (Molecular Devices, Sunnyvale, CA, USA). Absorbance of untreated cells was considered as 100%.
Cellular uptake of CLPP/mRNA particles in vitro
C26 cells were seeded into 24-well plate at a density of 1×105 cells per well 24 hrs before transfection. Enhanced GFP (EGFP) encoding mRNA (mEGFP, TriLink Biotechnologies, San Diego, CA) was purchased as a reporter gene to test protein expression. CLPP/mEGFP particles equivalent to 1 μg of mRNA or plasmid DNA encoding luciferase were added to each well in serum-free medium. PEI25K/mEGFP (1:1, mass ratio), Lipo2K/mEGFP (2:1, mass ratio) and equal amount of liposome or protamine were used as controls. Medium was then replaced with complete medium 4 hrs after transfection, pictures of each well were taken under a microscope and the transfection efficiency was determined by flow cytometry at different time point (NovoCyte Flow Cytometer, ACEA Biosciences, San Diego, CA, USA).
To detect the cellular internalization mechanism of CLPP/mRNA particles, C26 were pretreated with different inhibitors: chlorpromazine (10 μg/mL), amiloride (0.8 mM), wortmazine (8 μM), genistein (200 μM), M-β-CD (5 mM), cytochalasin D (10 mg/mL) or 2-deoxyglucose/NaN3 (DOG/NaN3, 50 mM/5mM). Transfection of CLPP/mEGFP particles was then performed for 4 hrs. 24 hrs post-transfection, the cellular internalization rates of CLPP/mEGFP particle in each group were determined by flow cytometry.
Antiproliferation assay
C26 cells were seeded into a 96-well plate at the density of 1.0×104 cells per well. Cells were then treated with particles equivalent to 0.5 μg of mRNA or plasmid DNA encoding survivin-T34A. 24 or 48 hrs post-transfection, cells were subjected to MTT cell proliferation assay to measure cell viability. This assay was repeated three times independently.
Clonogenic assay
Clonogenic assay was utilized to evaluate the antitumor efficiency of CLPP/mSur-T34A particles in vitro. Particles equivalent to 2 μg of survivin-T34A mRNA or plasmid DNA was administered to C26 cells in 6-well plates. Cells were then cultured for another 10 days to form clones. For staining, colonies were washed with PBS and stained with 10% crystal violet blue at room temperature for 15 mins. The number of clones in each well was then counted. All results were defined by t-test.
In vitro apoptosis assay
C26 cells were preseeded into a 6-well plate at a density of 2×105 cells per well. Cells were then transfected with CLPP/mSur-T34A particles (2 μg mRNA per well), CLP/pSur-T34A (2 μg DNA per well) and liposome-protamine (in equivalent amount with related complex) separately for 4 hrs. After 24 hrs or 48 hrs transfection, cells were analyzed by PI and Annexin V-FITC (Sigma-Aldrich, St Louis, MO, USA) staining. Apoptotic cancer cells were measured and calculated by flow cytometry (NovoCyte Flow Cytometer, ACEA Biosciences). The morphologic image of apoptotic cancer cells was taken under microscope.
Tumor growth inhibition assays in vivo
For abdominal cavity metastatic model, female BALB/c mice (6–8 weeks old) were intraperitoneally injected with 2×105 C26 cells. On day 3, mice were divided randomly into 4 groups (5 mice per group) and marked. CLPP/mSur-T34A particles (10 μg mRNA) or CLP/pSur-T34A complexes equivalent to 5 μg of DNA were injected intraperitoneally every day for 10 treatments. Mice receiving equivalent normal saline (NS) or liposome-protamine lipoplex (CLPP) were regarded as control groups. On day 20, all mice were sacrificed and the volumes of ascites in each group were collected and measured. The tumors were also immediately harvested for further analysis.
For subcutaneous tumor model, mice were inoculated with 1.5×106 C26 cells on day 0. When average tumor volume reached 50 mm3, mice were randomized into 4 groups (5 mice per group). CLPP/mSur-T34A particles equivalent to 10 μg of mRNA or CLP/pSur-T34A complexes equivalent to 5 μg of DNA were administrated intratumorally every day for 10 treatments. Tumor volume was calculated as (0.5× length × width2). On day 18, mice were killed by cervical vertebra dislocation, and their tumors were immediately harvested and analyzed.
For the pulmonary metastatic model, mice were intravenously injected with 200 μL of cell suspension containing 2×105 cells C26 cells on day 0. On day 3, mice were randomized into 4 groups (4 mice per group) and injected with NS, lipoplex (liposome/protamine, 10 μg/10 μg), CLP/pSur-T34A complexes (liposome/pDNA 5 μg/5 μg) or CLPP/mSur-T34A particles (liposome/protamine/mRNA, 10 μg/10 μg/10 μg) every day for a total of 10 treatments. On day 18, mice were killed and lungs were harvested. The metastatic nodules and lung weight of C26 colon carcinoma were calculated in each group.
RT-PCR analysis
Real-time polymerase chain reaction (RT-PCR) was employed to measure mRNA transcription level of tumor cells or tumor tissues. Total RNA was isolated using TRIzol™ Reagent (Thermo Fisher Scientific, USA) and individual cDNAs were synthesized with Takara kit (Dalian, China) following the manufacturer’s instructions. All primers are shown in Figure S1.
 | Figure S1 Primer sequences. |
Western blot analysis
Total proteins were extracted from tumor tissues, and protein concentrations were quantified with Bicinchoninic acid (BCA) protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA). 30 μg of protein was then separated by 12% SDS-PAGE gel electrophoresis and incubated with antibodies against caspase3, caspase7, caspase9 and GAPDH (Cell Signaling Technology, Inc., Boston, MA, USA) at 4°C overnight. Horseradish peroxidase (HRP)-conjugated corresponding secondary antibody and detected with an enhanced chemiluminescence detection kit (EMD Millipore, Billerica, MA, USA).
Histological analysis
Tumor tissue and main organs were harvested from in vivo experiment and embedded in paraffin. Tissue sections were dewaxed, rehydrated and subjected to Mayer’s HE staining. A commercially available TUNEL kit (Promega, Madison, WI, USA) was used to analyze the apoptotic cells in tumor sections according to the manufacturer’s protocol. For CD31 staining, tumor sections were blocked and subsequently incubated with anti-CD31 antibody (Abcam, Cambridge, MA, USA) at 4°C overnight. Alexa Fluor®488-conjugated secondary antibody was then applied. To analyze infiltrated immune cells within tumor tissues, tumor sections were stained with fluorescein isothiocyanate (FITC)-conjugated CD4, PE-conjugated CD8, PE-conjugated CD49b, AP-conjugated F4/80 (BD, Franklin Lakes, NJ, USA) for 1 hr at 4°C after antigen repair. To detect cytokines in tumor tissues, sections were pretreated with 0.5% triton for 20 mins, and then stained with PE-conjugated TNF-α, IFN-γ and IL-6 antibodies. Fluorescent image was visualized and captured by a fluorescence microscope (Olympus, Tokyo, Japan).
Lymph nodes from subcutaneous model were prepared and minced into small pieces to release immune cells. Collected cells were mixed with 4% paraformaldehyde, and then stained with FITC-conjugated CD4 antibody, PE-conjugated CD49b antibody or PE-conjugated CD3 antibody (BD, USA) for 30 mins at 4°C. For intracellular IFN-γ detection, cells were pretreated with 0.5% triton for 20 mins and stained with PE-conjugated IFN-γ antibody at 4°C. Related fluorescence was then analyzed by FACSCalibur flow cytometer (BD Biosciences) with FlowJo software (Treestar, Inc., Carlsbad, CA, USA).
Statistical analysis
Data were expressed as the means with 95% CIs. Statistical analysis was performed with two-tailed t-test or one-way ANOVA using Prism 5.0c Software (GraphPad Software, La Jolla, CA, USA). For all results, statistical significance was defined by a value of *P<0.05, **P<0.01, ***P<0.01.
Results
Preparation and characterization of CLPP/mRNA particle
In this study, a CLP-protamine lipoplex system was developed to deliver survivin-T34A mRNA. The CLP used in this work was prepared as previously described.31 According to our results, the prepared liposome had a size of 96.2±2.8 nm with a polydispersity index of 0.12±0.01 (Figure 1A). The shape of prepared CLPs was also observed by transmission electron photomicrographs (TEM), which was consistent with the results of DLS measurement above (Figure 1B). Survivin-T34A mRNA was prepared by T7 polymerase-based in vitro transcription method with purified PCR products. The successful synthesizing of capped Survivin-T34A mRNA with poly(A) tailing was confirmed by electrophoresis (Figure 1C).
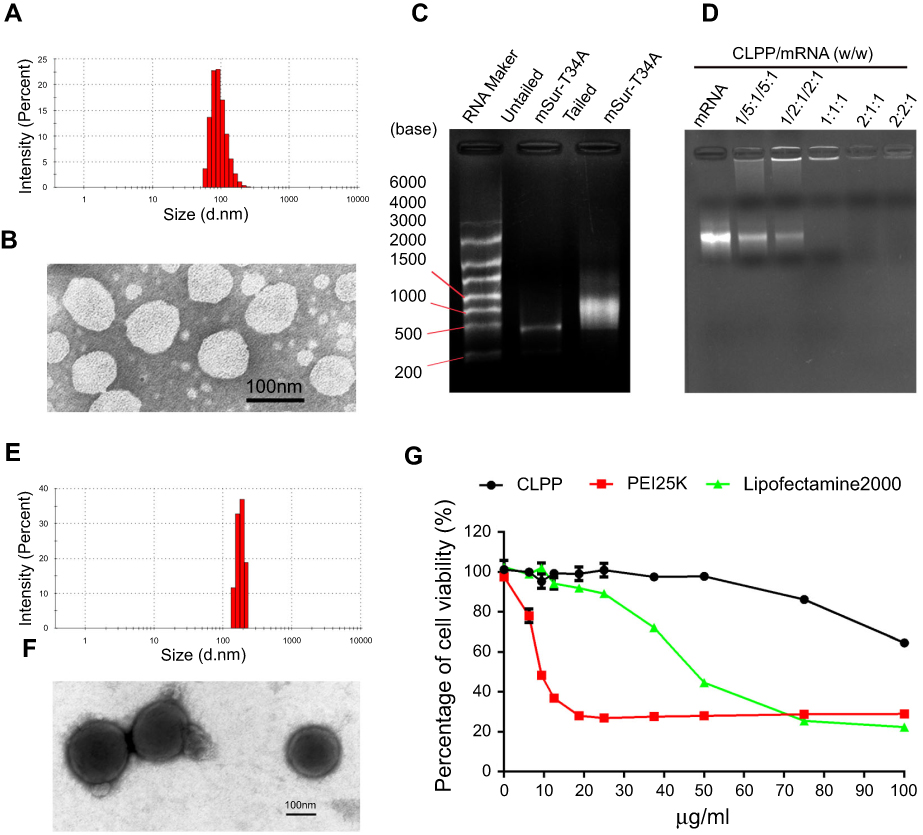 | Figure 1 Characterization of CLPP/mRNA particles. (A) Size distribution of prepared cationic liposome. (B) TEM image of cationic liposome. (C) Identify of survivin-T34A mRNA by electrophoresis. (D) Gel retarding assay of CLPP/mSur-T34A particle. (E) Size distribution of CLPP/mRNA particle. (F) TEM image of CLPP/mRNA particle, core-shell structures could be observed. (G) Cell viability assay of CLPP, PEI25K and Lipo2K on 293T cells. |
In our design, a CLP–protamine combination was utilized to condense and deliver IVT mRNA. To evaluate the binding ability of liposome-protamine lipoplex to survivin-T34A mRNA, a gel-retarding assay was performed. As shown in Figure 1D, when the molar ratio of liposome:protamine:mSur-T34A was 1:1:1, no bright mRNA band could be observed, suggesting that the negatively charged survivin-T34A mRNA was completely bound by CLPP lipoplex through electronic interaction. This ratio was therefore chosen for the following application in our study. According to our results, complexing of survivin-T34A mRNA with CLPP formed nanosized particles with an average size of 186.1±3.1 nm (Figure 1E). As shown by TEM image, highly condensed particles with lightly stained and ring-shaped coating structure were clearly observed (Figure 1F). Compared to CLP alone, this morphology suggested the formation of a core-shell structure in which the protamine-condensed mRNA was surrounded by lipid bilayer. Further to biophysical characterization, we then compared the cytotoxicity of CLPP lipoplex with PEI25K (“gold standard” transfection agent) and LipofactamineTM2000 in vitro. As shown in Figure 1G, PEI25K demonstrated a tremendous toxicity with IC50<10 μg/mL. Meanwhile, the IC50 value of LipofactamineTM2000 <50 μg/mL. However, CLPP lipoplex was much less toxic and their IC50 exceeded the concentration of 100 μg/mL, suggesting high biocompatibility.
CLPP lipoplex efficiently delivered mRNA in vitro
To evaluate the delivery ability of liposome-protamine lipoplex on IVT mRNA, EGFP-encoded mRNA was used as a reporter gene. According to our results, 6 hrs post-transfection, fluorescents could be detected in 12.1±0.9% of C26 cells. Transfection efficiencies of 24.6±1.5% and 40.3±1.6% were detected at the time point of 12 and 24 hrs post-transfection separately (Figure 2A), suggesting a quick gene expression mediated by mRNA. However, little fluorescent could be observed in liposome- or protamine-transfected well (Figure 2B and C). Comparing to PEI25K and LipofectamineTM2000, CLPP lipoplex demonstrated higher transfection efficiency than PEI25K, while less potent than LipofectamineTM2000. However, when referring to the mean fluorescence intensity calculated by flow cytometry, PEI25K resulted in the highest signal among these there (1.2 times over CLPP), while CLPP lipoplex was much more potent than LipofectamineTM2000 (1.8 times over Lipo2K). These results suggested that regarding mRNA delivery, CLPP is also an appropriated system. The transfection ability of CLPP on plasmid DNA was also evaluated. According to our results, PEI25K and LipofectamineTM2000 demonstrated a higher delivery capacity for plasmid DNA, suggesting that CLPP was more an optimized vector for mRNA (Figure S2A and B). These results indicated that CLP–protamine lipoplex could effectively deliver IVT mRNA into C26 cells in vitro.
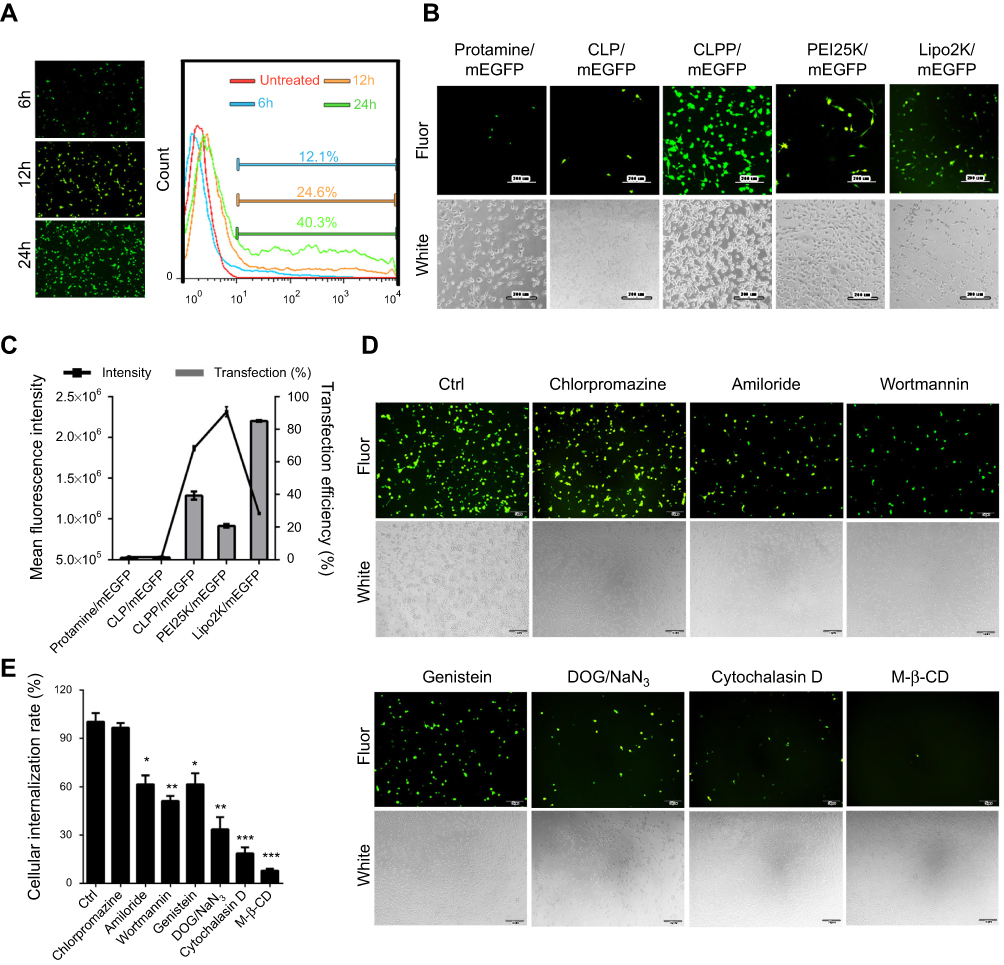 | Figure 2 CLPP lipoplex efficiently delivered mRNA in vitro. (A) Transfection efficiency of CLPP/mRNA particle at different time point on C26 cells. (B) Transfection efficiencies of different vectors delivered EGFP mRNA on C26 cells (scale bars, 200 μm). (C) Transfection efficiency and mean fluorescence intensity calculated by flow cytometry. (D) Study of cell uptake mechanism of CLPP/mRNA particle with various inhibitors (scale bars, 50 μm). (E) Cellular internationalization rates under inhibitors detected by flow cytometry. The results are shown as mean ± SEM, *P<0.05, **P<0.01, ***P<0.001 compared to the Ctrl group by two-tailed Student’s t-test. |
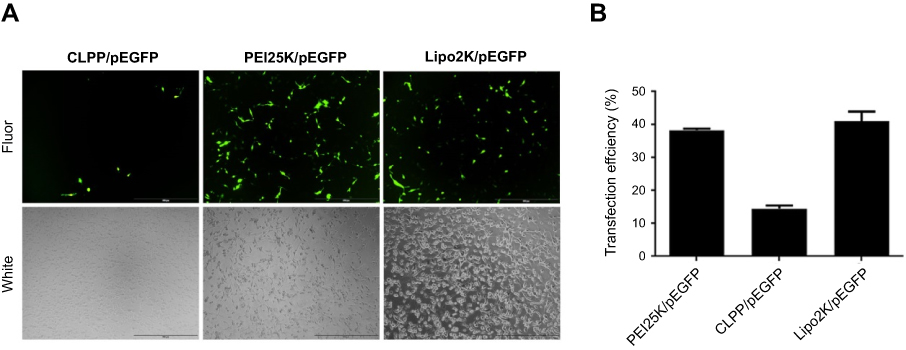 | Figure S2 Comparison of plasmid-based gene delivery by different vectors on C26 cells. (A) Images of transfected cells with different materials. (B) Transfection efficiencies calculated by flow cytometry. Abbreviation: Fluor, fluorescence. |
The endocytosis pathways involved in the internalization process of CLPP/mRNA particle was then studied. Before transfection, C26 cells were pretreated with known biochemical inhibitors of endocytosis pathways (caveolin-mediated endocytosis, clathrin-mediated endocytosis and macropinocytosis) in which caveolae-mediated endocytosis and macropinocytosis were also defined as lipid raft-mediated pathway. According to our results (Figure 2D and E), pretreated cells with 2-deoxyglucose/NaN3 (DOG/NaN3) and cytochalasin D resulted in a remarkable decrease in cellular internalization, suggesting the transportation of CLPP/mRNA particles was an energy-dependent process. Meanwhile, inhibitors of macropinocytosis (amiloride and wortmannin)32–34 and caveolin-mediated endocytosis (genistein) both resulted in >30% decrease in mRNA uptake. Particularly, a tremendous uptake inhibition of >96% was observed in M-β-CD treated groups, an inhibitor of lipid raft-mediated endocytosis.33,35 However, cells pretreated with chlorpromazine, an inhibitor of clathrin-mediated pathway, was almost unaffected.36 These results suggested that CLPP/mRNA particle was mainly internalized through lipid raft-mediated pathway.
CLPP/mSur-T34A particle efficiently inhibited tumor cell growth in vitro
The anticancer ability of CLPP delivered survivin-T34A-encoded mRNA was then evaluated on C26 cells by MTT assay. According to our results, cell proliferation was obviously inhibited after incubation with CLPP/mSur-T34A particle (Figure 3A). The inhibition rate reached >58% after 24 hrs. However, equivalent effect did not achieve by neither plasmid group nor other control groups. Meanwhile, 46% higher of inhibition rate could still be observed in CLPP/mSur-T34A group than plasmid group after 48 hrs (Figure S3A).
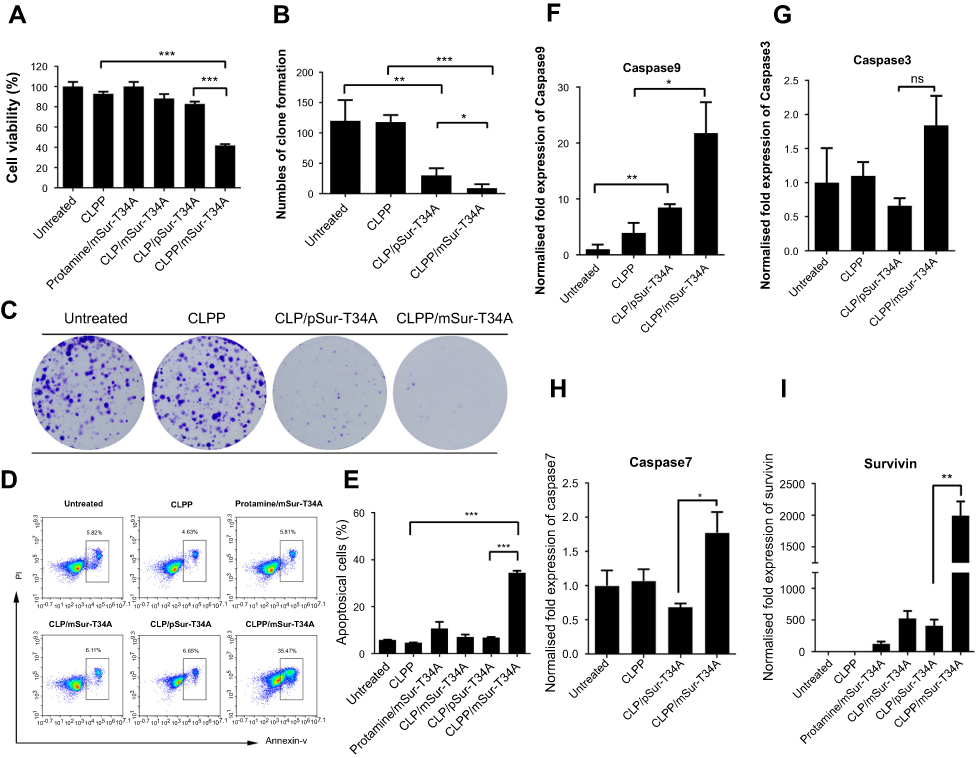 | Figure 3 CLPP/mSur-T34A particles inhibited the growth of C26 colon cancer cells in vitro. (A) Cell growth inhibition effect of CLPP/mSur-T34A particle evaluated by MTT assay. (B and C) Inhibition effect of CLPP/mSur-T34A particles was evaluated by clonogenic assay. (B) On 10 days post-transfection, the numbers of clones in each well were counted. (C) Brightfield image of each well. (D and E) CLPP-delivered survivin-T34A mRNA efficiently induced apoptosis in C26 cells. (F–I) 24 hrs post-transfection, the mRNA levels of survivin, caspase9, -3 and -7 were analyzed by RT-PCR (*P<0.05, **P<0.01, ***P<0.001, ns represents no significant difference). For all experiments, results are shown as mean ± SEM. |
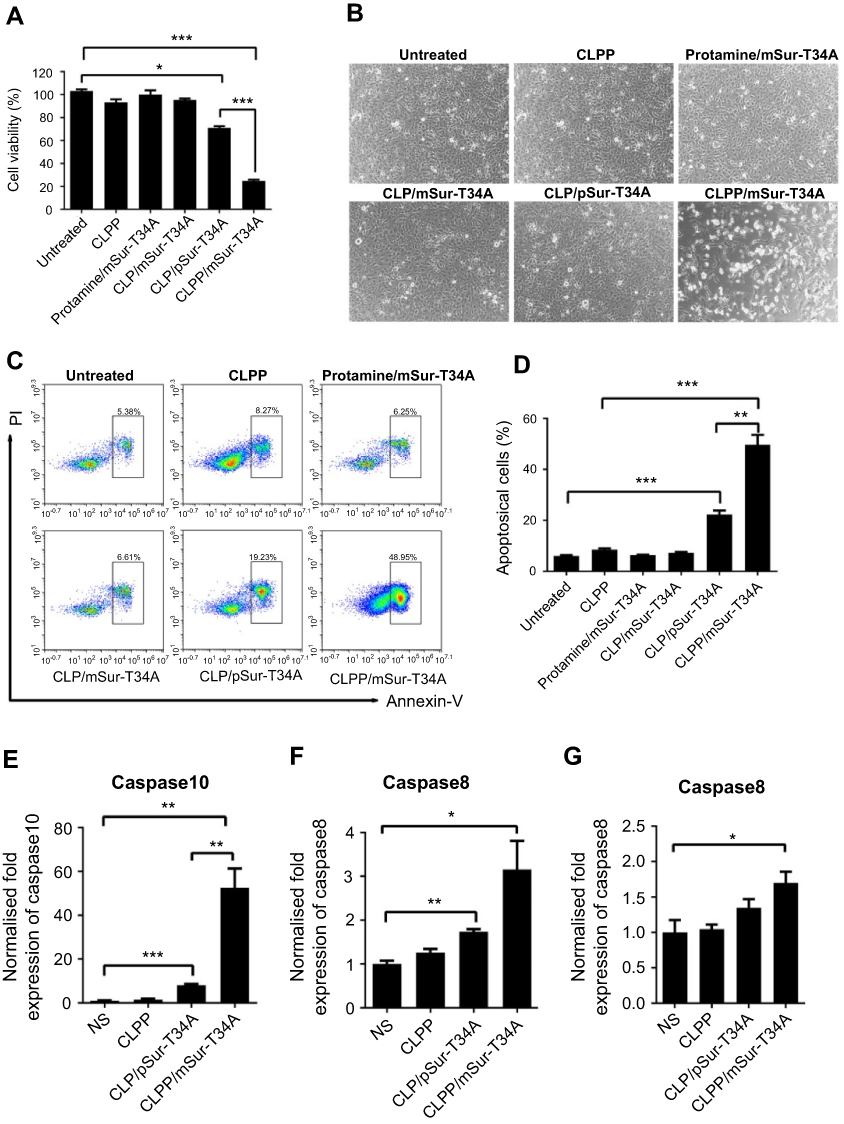 | Figure S3 CLPP/mSur-T34A particles efficiently inhibited the growth of C26 cancer cells in vitro. (A) Evaluation of cell growth inhibition by CLPP/mSur-T34A particles after 48 hrs. (B) The change of cell morphology in each group. (C and D) 48 hrs post transfection, CLPP-delivered survivin-T34A mRNA efficiently induced apoptosis in C26 cells. (E and F) In abdominal cavity metastases, treatment with CLPP/mSur-T34A particles increased the mRNA level of caspase8 and caspase10 in tumor tissue. (G) In subcutaneous xenograft model, treatment with CLPP/mSur-T34A particles increased the mRNA level of caspase8 in tumor tissue. All data are reported as mean ± SEM and ***P<0.001, **P<0.01, *P<0.05 (two-tailed Student’s t-test). Abbreviation: NS, normal saline. |
The antiproliferation ability of CLPP/mSur-T34A particle was also assessed by clonogenic assay. As shown in Figure 3B and C, compared to other groups, much fewer numbers of clones were formed in CLPP/mSur-T34A particle-treated cells. These results indicated that CLPP delivered survivin-T34A mRNA was more potent than equivalent amount of plasmid counterpart. Meanwhile, protamine- or liposome-delivered survivin-T34A mRNA was also inefficient, suggesting that the CLPP lipoplex combination was critical for mRNA delivery.
The apoptosis-inducing ability of survivin-T34A gene product by activating mitochondrial-dependent apoptosis has been previously reported.24,25 Thus, we then investigated whether antiproliferation effect of CLPP/mSur-T34A particle was a result of apoptosis, and PI/Annexin V staining was performed. As shown in Figure 3D and E, 24 hrs post-transfection, much higher apoptosis was detected by flow cytometry in CLPP/mSur-T34A particle-treated cells compared to other groups. Meanwhile, distinct apoptotic morphology, including round and translucent cell morphology as well as cell fragmentation, could be observed under microscope (Figure S3B). Forty-eight hours post-transfection, the apoptosis rate further increased to 49.7%, demonstrating superior to plasmid group (Figure S3C and S3D). Meanwhile, obvious elevation of mRNA level including caspase9, caspas3 and caspas7 was also detected by RT-PCR (Figure 3F–H), further indicating the activation of mitochondrial-dependent apoptosis pathway. In particular, comparing to other groups, CLPP/mSur-T34A particle resulted in much higher intracellular survivin mRNA level (4.8-folds over CLP/pSur-T34A group, Figure 3I), which might strongly contribute to its superior anticancer ability. These results illustrated that CLPP lipoplex could efficiently deliver Survivin-T34A mRNA into C26 cells in vitro, demonstrating potent antitumor effect by inducing apoptosis.
Local delivery of CLPP/mSur-T34A particle inhibited tumor growth in vivo
The anticancer capacity of CLPP/mSur-T34A particle was first assessed on C26 abdominal cavity metastases model. Tumor-bearing mice were treated with CLPP/mSur-T34A particle or CLP/pSur-T34A complex by intraperitoneal administration. According to our result, treatment with CLPP/mSur-T34A particle resulted in a significant inhibition of abdominal cavity metastases tumor growth (Figure 4A). As shown in Figure 4B, much lower weight of total metastatic nodules was observed in CLPP/mSur-T34A group (0.16±0.11 g) compared to 4.67±0.32 g in NS group, 5.12±0.26 g in CLPP group and 3.20±0.43 g in CLP/pSur-T34A group. Meanwhile, an obvious decrease was also detected in the ascites volume of CLPP/mSur-T34A particle-treated mice. In our results, the volume of ascites in mice treated with mSur-T34A particles was 0.17±0.03 mL, while that of NS group, CLPP group and CLP/pSur-T34A group was 1.23±0.1 mL, 0.90±0.15 mL and 0.80±0.10 mL, respectively (Figure 4C). Meanwhile, a subcutaneous model of C26 colon carcinoma was also set up to evaluate the therapeutic effect of CLPP/mSur-T34A particle. As shown by representative images of tumor tissues and tumor growth curve (Figure 4D and E), intratumoral injection of CLPP/mSur-T34A also resulted in significant therapeutic effect. According to Figure 4F, the average tumor weight of CLPP/mSur-T34A group was 0.12±0.07 g while that of NS group, CLPP group and CLP/pSur-T34A group was 0.74±0.14 g, 0.63±0.08 g and 0.32±0.05 g, respectively. Particularly, 8 doses later, overall tumor repression was achieved on one mouse in CLPP/mSur-T34A group (shown in Figure 4D).
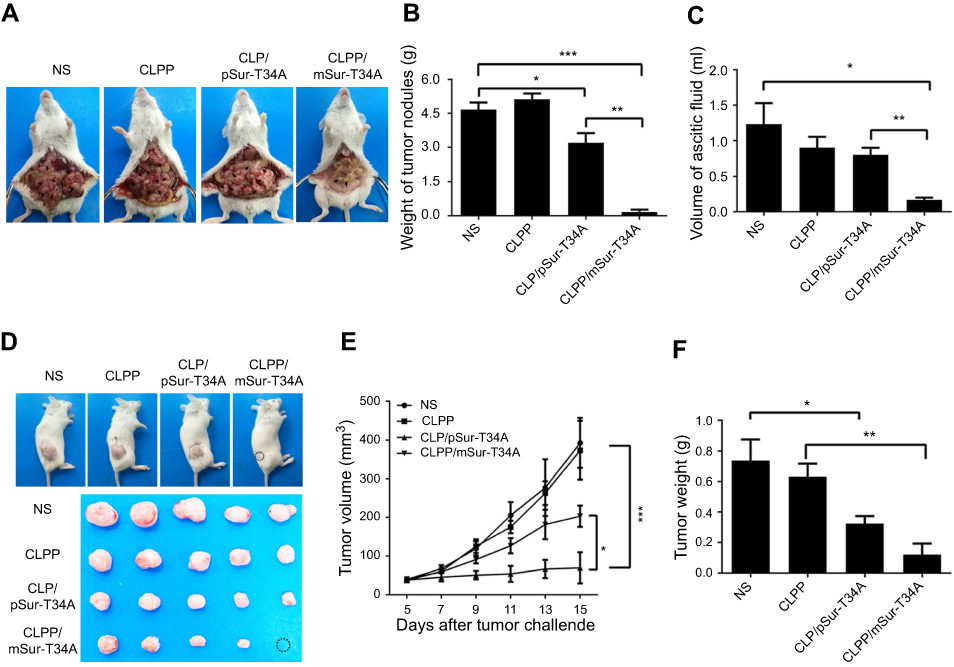 | Figure 4 Local delivery of CLPP/mSur-T34A particle inhibited tumor growth in two models. (A) Representative images of abdominal cavity metastases of C26 colon carcinoma (n=6 for each group). The arrows show the tumor nodes. (B) Average tumor weight in abdominal metastases model. (C) Average ascetic volume. (D) Representative images of subcutaneous xenograft of C26 colon cancer (n=4 for each group). A complete regression was observed in CLPP/mSur-T34A group (circle). (E) Tumor growth curves of each group(***P<0.001 vs NS, *P<0.05 vs CLP/pSur-T34A). (F) Average tumor weight. Error bars represent SEM for n=4 and *P<0.05 compared to NS group, **P<0.01 compared to CLPP group. Abbreviation: NS, normal saline. |
To verify whether apoptosis inducing was involved in above anticancer activities, TUNEL assay was performed on tumor tissues from both models. As shown in Figure 5A and D, obviously more positive apoptotic cells were observed in tumor tissue sections from CLPP/mSur-T34A group. Except for TUNEL assay, CD31 staining was also performed on tumor sections. Comparing to other groups, fewer microvessel density and smaller vessel volumes could be observed in tumor tissues form CLPP/mSur-T34A group (Figure 5A and D), suggesting that vessel formation was obviously attenuated. Meanwhile, according to western blot and RT-PCR results, both protein and mRNA levels of caspase9, caspase3 and caspase7 were markedly increased in tumor samples treated with CLPP/mSur-T34A and CLP/pSur-T34A group (Figure 5B and C). Increased expression of actived-caspase3 and -caspase7 in mRNA group than plasmid group were also detected by WB analysis. Besides, increased mRNA levels of caspase8 and caspase10 in CLPP/mSur-T34A group were also observed compared to other groups (Figure S3E and F), which was not observed in in vitro experiment. Similar results were observed in tumor tissues from subcutaneous model (Figures 5F and S3G). In the subcutaneous model, over 50-fold mRNA level of survivin was detected in CLPP/mSur-T34A-treated group than the NS group (Figure 5E), suggesting efficient mRNA delivery to tumor tissue. Moreover, significant elevated levels of TNF-α, IFN-γ and IL-6 mRNA expression were detected in CLPP/mSur-T34A-treated groups, with over 5-fold in metastases model (Figure 5C) and over 100-fold in xenograft model (Figure 5F) compared to NS group, respectively. Altogether, these results suggested that local administration of CLPP/mSur-T34A particles could efficiently inhibit the growth of C26 colon cancer model by inducing apoptosis and antiangiogenesis in vivo. Meanwhile, mRNA formulation demonstrated superior therapeutic effect over plasmid counterpart, which was consistent with our in vitro data. Besides, no obvious pathological changes of major organs in CLPP/mSur-T34A treatment were observed by microscopic examination (Figure S4), suggesting high safety.
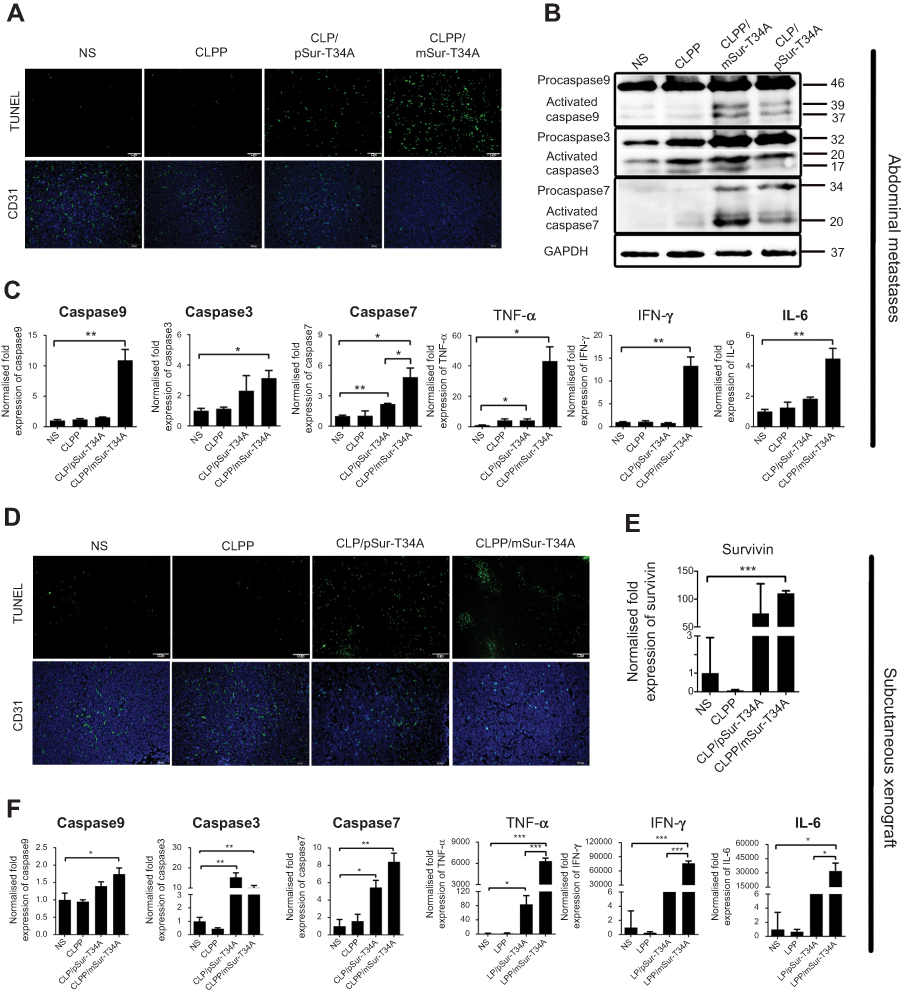 | Figure 5 CLPP/mSur-T34A particles induced a significant antitumor effect in vivo. (A) Apoptosis-inducing and antiangiogenesis effect detected by TUNEL assay and CD31 staining in tumor tissues in abdominal cavity metastatic model (scale bars, 50 μm). (B) CLPP/mSur-T34A effectively induced caspase-dependent tumor apoptosis. Protein expression of activated caspase9, -3 and -7 in tumor tissues was detected. (C) The mRNA expression of caspase9, -3 and -7 and increased releasing of cytokines including TNF-α, IFN-γ and IL-6 were detected by RT-PCR. (D) Apoptosis and vessels in tumor tissues also evaluated by TUNEL assay (up) and CD31 staining (down) in subcutaneous xenograft (scale bars, 100 μm). (E) Relative expression of survivin in tumor tissue of each group (***P<0.001 vs NS). (F) Elevated mRNA levels of caspase9, -7, -3, TNF-α, IFN-γ and IL-6 were measured by RT-PCR. All data are reported as mean ± SEM (Student’s t-test, ***P<0.001, **P<0.01, *P<0.05). Abbreviation: NS, normal saline. |
 | Figure S4 HE analysis of main organs from each treatment group, no obvious pathological changes were observed (magnification =200×). |
Systemic delivery of CLPP/mSur-T34A particle inhibited pulmonary metastatic model in vivo
A C26 pulmonary metastatic animal model was also utilized to test the antitumor efficacy of CLPP/mSur-T34A in vivo. Particularly, for this study, the CLPP/mSur-T34A particle was administrated by intravenous injection. According to our results, treatment with CLPP/mSur-T34A resulted in a significant inhibition of pulmonary metastatic tumor growth (Figure 6A). As shown by lung tissue section, the lungs of mice in CLPP/mSur-T34A group showed fewer pulmonary metastatic nodules than other groups (Figure 6B). CLPP/mSur-T34A particles exhibited a statistically significant reduction in the weight of lung comparing to other groups, as indicated with 0.19±0.01 g versus 0.42±0.03 g in NS group, 0.39±0.03 g in CLPP group and 0.36±0.07 g in CLP/pSur-T34A group (Figure 6C). The numbers of the pulmonary metastatic nodules in each group were also calculated, as presented in Figure 6D. The metastatic nodules in CLPP/mSur-T34A group were much fewer than other groups (mean number of 27 vs 122 in controls, ***P<0.001; 121 in CLPP group, ***P<0.001; 105 in CLP/pSur-T34A group, **P<0.01). The inhibition rate of CLPP/mSur-T34A treatment group reached 78%, which was 64% higher than plasmid-treated group (Figure 6E). This indicates that intravenous application of CLPP/mSur-T34A particles can efficiently inhibit the growth of pulmonary metastases model of C26 colon carcinoma in vivo.
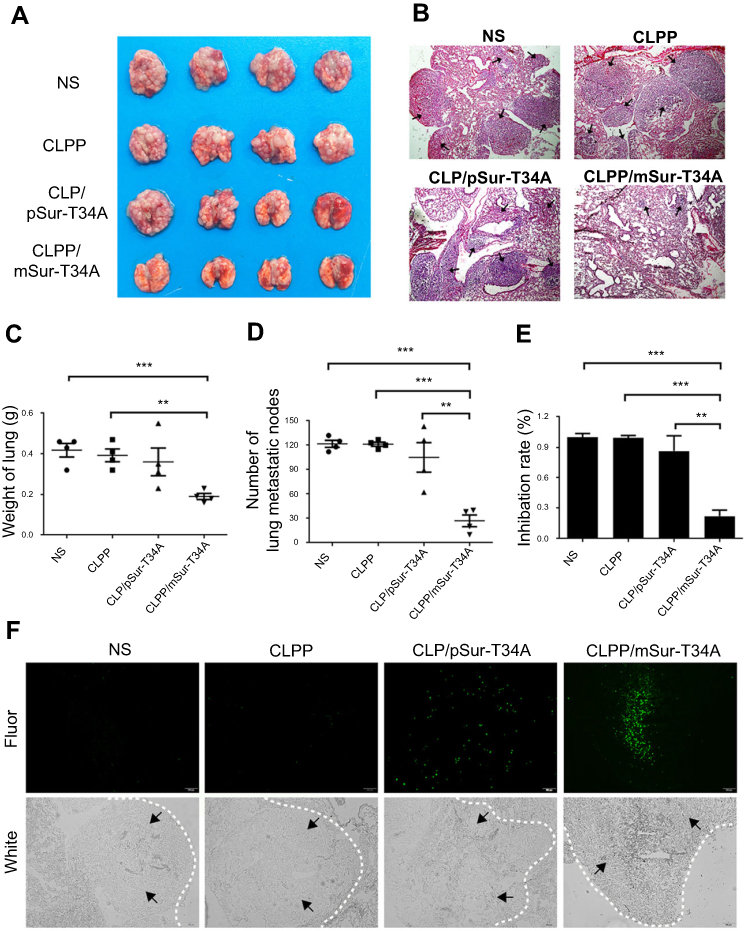 | Figure 6 Systemic delivery of CLPP/mSur-T34A particles inhibited pulmonary metastases of C26 colon cancer. (A) Representative images of lung tissues harvested from animals after systemic treatments (n=4 for each group). (B) HE staining of lungs bearing metastases of C26 colon carcinomas in each treatment group (scale bars, 200 μm). Arrows indicate metastases modules in lung tissue. (C) Weight of lungs bearing pulmonary metastases (***P<0.001 vs NS; **P<0.01 vs CLPP). (D) Number of pulmonary metastatic nodules in each treatment group. (E) Inhibition rate calculated by the number of pulmonary metastases according to the number of pulmonary metastases. In this experiment (n=4 for each group, ***P<0.001 vs NS, ***P<0.001 vs CLPP, **P<0.01 vs CLP/pSur-T34A). (F) Apoptosis inducing detected by TUNEL assay. Tumor-rich regions are indicated by arrow direction (scale bars, 100 μm). Abbreviation: NS, normal saline; Fluor, fluorescence. |
To evaluate the apoptosis induced by each treatment, TUNEL assay was performed on lung tissue samples. According to our results, CLPP/mSur-T34A group induced larger area of apoptosis than other groups (Figure 6F). Compared with CLPP/mSur-T34A particle, plasmid group showed lower antitumor effect and induced less apoptosis than mRNA group. These results demonstrated that the efficient anticancer ability of CLPP/mSur-T34A particles could be achieved both through local and systemic delivery. Meanwhile, CLPP/mSur-T34A particle also successfully demonstrated superior therapeutic capacity than its plasmid counterparts in these two administration methods.
CLPP/mSur-T34A activated antitumor immune response in microenvironment
As a primary immune-competent cell, tumor-infiltrating lymphocytes (TILs) are considered to be the manifestation of host antitumor reaction.37 According to our results, CLPP/mSur-T34A particles showed obvious antitumor efficiency in various C26 colon cancer models. Thus, we investigated whether the activation of TILs in tumor microenvironment was involved in the process. In this study, immunofluorescence was used to measure the levels of TNF-α, IFN-γ and IL-6 in tumor tissues. As shown in Figure 7A, immunofluorescence assay demonstrated enhanced secretion of TNF-α, IFN-γ and IL-6 cytokine in CLPP/mSur-T34A group. Furthermore, mRNA group resulted in higher cytokine production behavior than plasmid group, suggesting stronger immune cell activation in tumor microenvironment. Therefore, the infiltration of macrophages, CD4+ T cells, CD8+ T cells and NK cells in tumor samples was also evaluated in the study. According to our results, after the treatment of CLPP/mSur-T34A particle, the population of CD4+ T cells, CD8+ T cells, NK cells and macrophages was obviously increased in tumor area (Figure 7A and B). In addition, immune cells in lymph nodes from xenograft animal model were also analyzed by flow cytometry. As shown in Figure 7C, compared to NS group, the proportion of CD3+/CD4+/IFN-γ+ T cells increased by 2-fold in CLPP/mSur-T34A particle group. Besides, the increasing amounts of F4/80+ macrophages (4.8% vs 1.1% NS group) and CD3−/CD49b+ NK cells (1.03% vs 0.67% NS group) were also detected (Figure 7D–F). Our results further suggested that along with apoptosis-inducing, immune activation, especially tumor microenvironment regulation also contributed to the potential anti-cancer effect of CLPP delivered survivin-T34A mRNA.
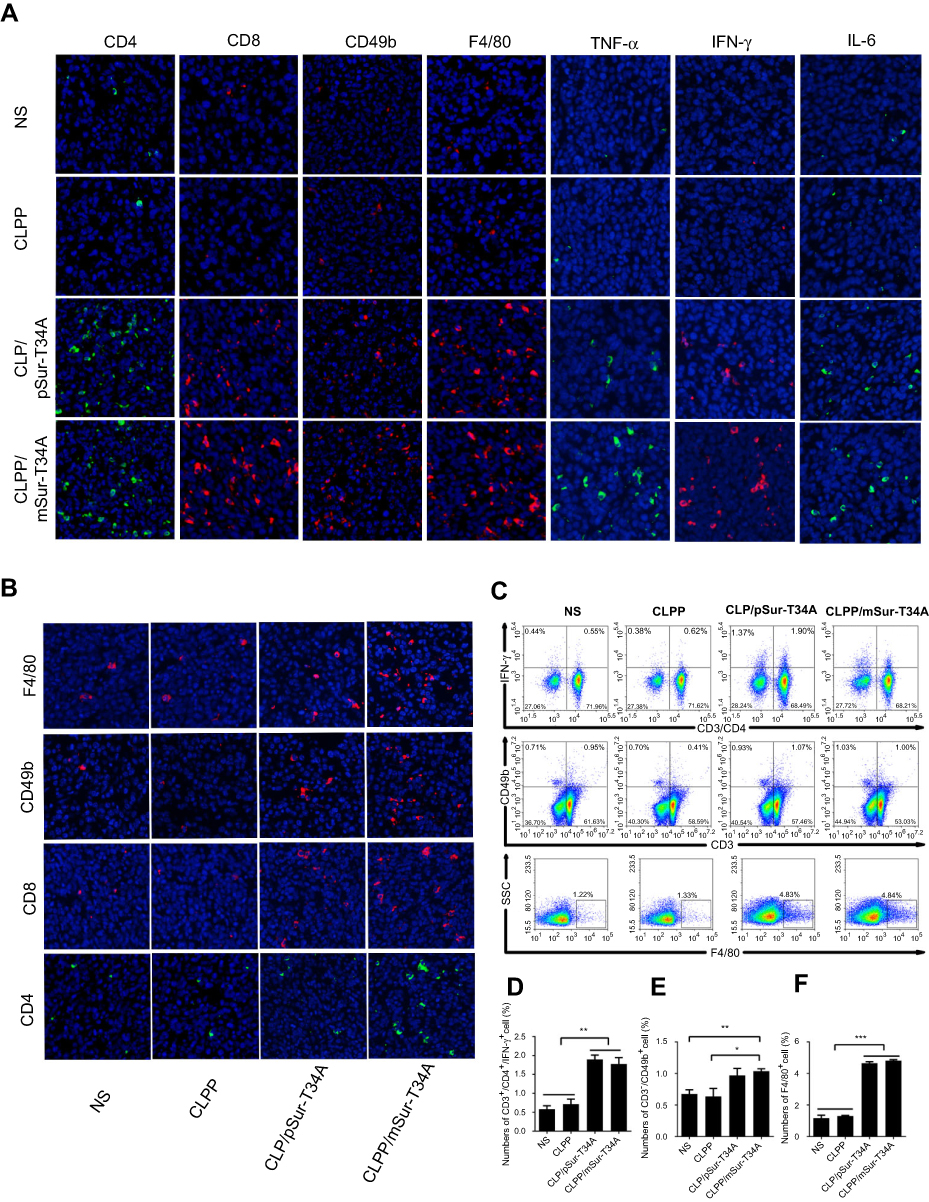 | Figure 7 Activation of antitumor immune response by CLPP/mSur-T34A particle. (A) The infiltration of immune cells in abdominal cavity metastatic tumor tissues were detected by immunofluorescence staining. (B) The fluorescent images of CD4+ T cells, CD8+ T cells, CD49b+ NK cells and F4/80+ macrophages in tumor tissues from subcutaneous xenograft tumor model. (C–F) Lymph node-derived immune cells from subcutaneous xenograft tumor model were analyzed. Flow cytometry analysis of CD3+/CD4+/IFN-γ+ T cell, NK cell and macrophages. All data are reported as mean ± SEM and ***P<0.001, **P<0.01, *P<0.05 (two-tailed Student’s t-test). Abbreviation: NS, normal saline. |
Discussion
In this work, a liposome-protamine lipoplex was developed to deliver survivin-T34A encoded mRNA (Figure 8), resulting in a new mRNA-based formulation for colon cancer gene therapy. The prepared nanosized CLPP/mRNA particles demonstrated obvious therapeutic effects on various models of C26 colon cancer both in vitro and in vivo. Particularly, local and systemic administration of CLPP/mRNA particle exhibited superior antitumor effect regarding its DNA plasmid counterpart with high safety. Our results suggested that liposome-protamine lipoplex-delivered survivin-T34A mRNA is a potential candidate for colon cancer therapy.
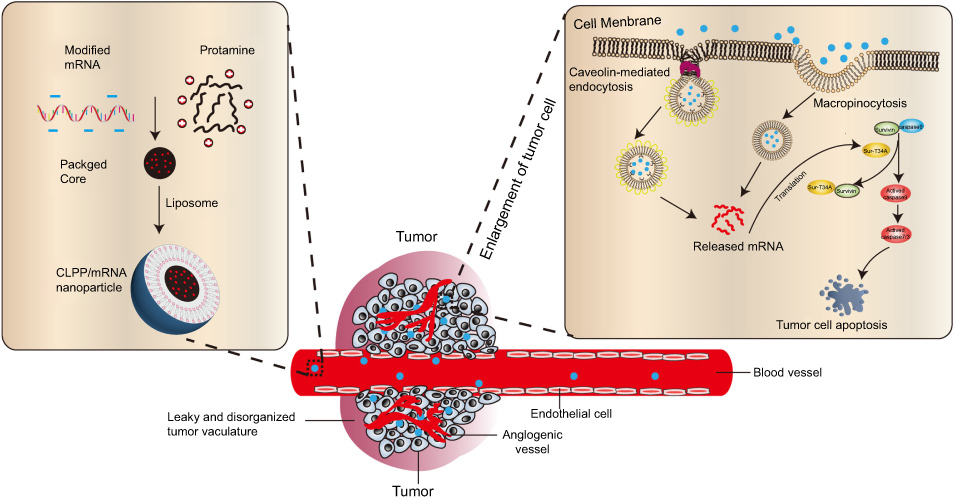 | Figure 8 Schematic view of CLPP-based mRNA delivery. |
It is widely acknowledged that delivery system plays a critical role in gene delivery, especially for mRNA. Efficient and safe delivery of therapeutic mRNAs remains one of the major challenges for their further application. The characteristics of mRNA with flexible linear structure and instability make it highly dependent on delivery system to further condense and protect mRNA.38–40 Most recently, mRNA-based therapeutics was benefited from the rapid progress of delivery methods. For example, Choi et al used the graphene oxide-polyethylenimine complexes (polymers) to deliver mRNAs encoding four reprogramming transcription factors and successfully generated induced pluripotent stem cells from adipose tissue-derived fibroblasts.41 Li et al applied the recombinant coat protein of bacteriophage MS2 to deliver granulocyte-macrophage colony stimulating factor mRNA and protected mice against prostate cancer.42 Other studies using lipid or lipid-derived particles for mRNA delivery were also reported.13,14,43 These efforts greatly contributed to the further development and application of therapeutic mRNA. Currently, mRNA-based therapeutics mainly focuses on ex vivo transfection and local administration in preclinical and clinical studies. However, it is crucial that therapeutic nucleic acid substances such as mRNAs could be administrated systemically, which would then facilitate the expansion of gene therapy application. Thus, improved vectors are always required. In the present study, the prepared liposome-protamine lipoplex demonstrated high mRNA delivery efficiency up to 40%, resulting in strong anticancer ability through i.p. and i.t. administration on C26 colon cancer models. More importantly, this formulation was also successfully applied in a systemic way by intravenous injection. According to our results, compared with the control group, i.v. administration of CLPP/survivin-T34A particles significantly reduced the incidence and development of lung metastasis with an inhibition rate of 78%. Meanwhile, no obvious pathological damage was observed in the main organs, suggesting its high safety. These results fully demonstrated its safety and delivery ability of mRNA during local and systemic cancer therapy, which might probably attribute to the two formulation components. Within the lipoplex, protamine condensed mRNA into a solid core, while liposome offered a lipid bilayer shell to form a core-shell structure. Upon delivery, this protective strategy prevented mRNA from degradation by ribozymes and promoted high gene expression efficiency. Therefore, the combination of liposome-protamine lipoplex is a good candidate for mRNA delivery with high potential.
As an alternative form of nucleic acid therapeutics, mRNA has demonstrated potential value in gene therapy application. Compared with plasmid DNA-based therapy, mRNA has a series of theoretical advantages such as compressible and flexible structure, rapid protein expression, high safety and reduced structural elements.14,17 These properties make mRNA an advanced choice for gene delivery. However, few studies have been reported regarding the comparison of plasmid DNA and mRNA in the issues of delivery and therapy, especially by multiple delivery methods. In this study, we compared the therapeutic effects of suicide gene survivin-T34A in the form of DNA and mRNA on various C26 colon cancer models. Our results showed that CLPP delivered survivin-T34A mRNA illustrated better antitumor effect over DNA group both in vivo and in vitro. Particularly, by intravenous injection, mRNA group showed an enhanced inhibiting on C26 lung metastasis tumor compared to plasmid group, showing high superiority. According to our results, after 10 treatments, the result was a 64% higher inhibition rate over in C26 metastasis nodule number. Meanwhile, 40.7% lower lung weight could be observed in the CLPP/mRNA group compared to its plasmid counterpart. Several reasons might account for the advances of mRNA-based gene therapy in our present work. For example, owing to their own structures, the size differences between DNA and mRNA might lead to divergences in gene delivery efficiencies. In this work, our results demonstrated that mRNA group was superior to DNA group in both apoptosis-inducing and antiproliferation in vitro. For this issue, inequivalent molar mass of coding sequence might be one explanation since plasmid has much more none-therapeutic sequences. Second, it takes more processes for plasmid DNA than mRNA before expected protein expression, since nuclear localization and transcription is necessary. This allows mRNA to induce its effect more rapidly than DNA. However, in another aspect, factors such as production difficulty, costs as well as the function of extra elements in plasmid should not be omitted when compared these two. Meanwhile, plasmid DNA might be more convenient for scaled production considering current technology for in vitro mRNA transcription. Nevertheless, it has been reported that CpGs products from plasmid backbone might stimulate immunogenicity along with gene expression. Thus, mRNA-based therapy not only offers a more flexible gene delivery strategy, but also provides a comparably safer choice regarding the “backbone effect” of plasmid DNA. Overall, our results successfully demonstrated that liposome-protamine lipoplex-delivered survivin-T34A mRNA was more potent than its plasmid counterpart, showing strong potential in further colon cancer therapy.
Abbreviations list
CLP, liposome; CLPP, liposome-protamine lipoplex; mSur-T34A, survivin-T34A mRNA; pSur-T34A, survivin-T34A plasmid DNA; mEGFP, EGFP mRNA; pEGFP, EGFP plasmid DNA; Lipo2K, LipofectamineTM2000; i.t., intratumoral injection; i.p., intraperitoneal injection; IVT, in vitro transcribed; IL-6, interleukin-6; TNF-α, tumor necrosis factor-α; IFN-γ, interferon-γ.
Acknowledgments
This work was supported by the National Major Scientific and Technological Special Project for “Significant New Drugs Development” (No. 2018ZX09733001), the National Key Specialty Construction Project of Clinical Pharmacy (No. 30305030698) and the Science Foundation of Chengdu (2016-HM01-00438-SF).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi:10.3322/caac.21387
2. Oldhamrobert K. Cancer biotherapy: more than immunotherapy. Cancer Biother Radiopharm. 2017;32(4):111–114. doi:10.1089/cbr.2017.28999.old
3. Robison JG, Smith CL. Therapeutic massage during chemotherapy and/or biotherapy infusions: patient perceptions of pain, fatigue, nausea, anxiety, and satisfaction. Clin J Oncol Nurs. 2016;20(2):E34–E40. doi:10.1188/16.CJON.E34-E40
4. Gupta HB, Clark CA, Yuan B, et al. Tumor cell-intrinsic PD-L1 promotes tumor-initiating cell generation and functions in melanoma and ovarian cancer. Signal Transduct Target Ther. 2016;1(1):16030. doi:10.1038/sigtrans.2016.30
5. Senapati S, Mahanta AK, Kumar S, Maiti P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct Target Ther. 2018;3(1):7. doi:10.1038/s41392-017-0004-3
6. Kotterman MA, Schaffer DV. Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet. 2014;15(7):445–451. doi:10.1038/nrg3742
7. Naldini L. Gene therapy returns to centre stage. Nature. 2015;526(7573):351–360. doi:10.1038/nature15818
8. Kotterman MA, Chalberg TW, Schaffer DV. Viral vectors for gene therapy: translational and clinical outlook. Annu Rev Biomed Eng. 2015;17:63–89. doi:10.1146/annurev-bioeng-071813-104938
9. Ulmer JB, Mason PW, Geall A, Mandl CW. RNA-based vaccines. Vaccine. 2012;30(30):4414–4418. doi:10.1016/j.vaccine.2012.04.060
10. Rodriguez-Gascon A, del Pozo-Rodriguez A, Solinis MA. Development of nucleic acid vaccines: use of self-amplifying RNA in lipid nanoparticles. Int J Nanomedicine. 2014;9:1833–1843. doi:10.2147/IJN.S39810
11. Ulmer JB, Geall AJ. Recent innovations in mRNA vaccines. Curr Opin Immunol. 2016;41:18–22. doi:10.1016/j.coi.2016.05.008
12. Whitworth KM, Cecil R, Benne JA, et al. Zygote injection of RNA encoding Cre recombinase results in efficient removal of LoxP flanked neomycin cassettes in pigs. Transgenic Res. 2018;27(2):167–178. doi:10.1007/s11248-018-0064-3
13. Midoux P, Pichon C. Lipid-based mRNA vaccine delivery systems. Expert Rev Vaccines. 2015;14(2):221–234. doi:10.1586/14760584.2015.986104
14. Grabbe S, Haas H, Diken M, Kranz LM, Langguth P, Sahin U. Translating nanoparticulate-personalized cancer vaccines into clinical applications: case study with RNA-lipoplexes for the treatment of melanoma. Nanomedicine (Lond). 2016;11(20):2723–2734. doi:10.2217/nnm-2016-0275
15. Yamamoto A, Kormann M, Rosenecker J, Rudolph C. Current prospects for mRNA gene delivery. Eur J Pharm Biopharm. 2009;71(3):484–489. doi:10.1016/j.ejpb.2008.09.016
16. Youn H, Chung J-K. Modified mRNA as an alternative to plasmid DNA (pDNA) for transcript replacement and vaccination therapy. Expert Opin Biol Ther. 2015;15(9):1337–1348. doi:10.1517/14712598.2015.1057563
17. Geall AJ, Mandl CW, Ulmer JB. RNA: the new revolution in nucleic acid vaccines. Semin Immunol. 2013;25(2):152–159. doi:10.1016/j.smim.2013.05.001
18. Guo C, Manjili MH, Subjeck JR, Sarkar D, Fisher PB, Wang X-Y. Therapeutic cancer vaccines: past, present, and future. Adv Cancer Res. 2013;119:421–475. doi:10.1016/B978-0-12-407190-2.00007-1
19. Schalper KA, Velcheti V, Carvajal D, et al. In situ tumor PD-L1 mRNA expression is associated with increased TILs and better outcome in breast carcinomas. Clin Cancer Res. 2014;20(10):2773–2782. doi:10.1158/1078-0432.CCR-13-2702
20. Sahin U, Kariko K, Tureci O. mRNA-based therapeutics—developing a new class of drugs. Nat Rev Drug Discov. 2014;13(10):759–780. doi:10.1038/nrd4278
21. Persano S. A self-assembled non-viral vector as potential platform for mRNA-based vaccines. Transl Biomed. 2017;8(3). doi:10.21767/2172-0479.100119
22. Pollard C, De Koker S, Saelens X, Vanham G, Grooten J. Challenges and advances towards the rational design of mRNA vaccines. Trends Mol Med. 2013;19(12):705–713. doi:10.1016/j.molmed.2013.09.002
23. Li B, Zhang X, Dong Y. Nanoscale platforms for messenger RNA delivery. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2018;11(2):e1530. doi:10.1002/wnan.1530.
24. Tavernier G, Andries O, Demeester J, Sanders NN, De Smedt SC, Rejman J. mRNA as gene therapeutic: how to control protein expression. J Control Release. 2011;150(3):238–247. doi:10.1016/j.jconrel.2010.10.020
25. Bi Y, Zhang Y, Cui C, Ren L, Jiang X. Gene-silencing effects of anti-survivin siRNA delivered by RGDV-functionalized nanodiamond carrier in the breast carcinoma cell line MCF-7. Int J Nanomedicine. 2016;11:5771–5787. doi:10.2147/IJN.S117611
26. Duan X, Wang P, Men K, et al. Treating colon cancer with a suicide gene delivered by self-assembled cationic MPEG-PCL micelles. Nanoscale. 2012;4(7):2400–2407. doi:10.1039/c2nr30079f
27. Zhang L, Gao X, Men K, et al. Gene therapy for C-26 colon cancer using heparin-polyethyleneimine nanoparticle-mediated survivin T34A. Int J Nanomedicine. 2011;6:2419–2427. doi:10.2147/IJN.S23582
28. Persano S, Guevara ML, Li Z, et al. Lipopolyplex potentiates anti-tumor immunity of mRNA-based vaccination. Biomaterials. 2017;125:81–89. doi:10.1016/j.biomaterials.2017.02.019
29. Kauffman KJ, Webber MJ, Anderson DG. Materials for non-viral intracellular delivery of messenger RNA therapeutics. J Control Release. 2016;240:227–234. doi:10.1016/j.jconrel.2015.12.032
30. McKinlay CJ, Vargas JR, Blake TR, et al. Charge-altering releasable transporters (CARTs) for the delivery and release of mRNA in living animals. Proc Natl Acad Sci U S A. 2017;114(4):E448–E456. doi:10.1073/pnas.1614193114
31. Chen X, Wang X, Wang Y, et al. Improved tumor-targeting drug delivery and therapeutic efficacy by cationic liposome modified with truncated bFGF peptide. J Control Release. 2010;145(1):17–25. doi:10.1016/j.jconrel.2010.03.007
32. Koivusalo M, Welch C, Hayashi H, et al. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J Cell Biol. 2010;188(4):547–563. doi:10.1083/jcb.200908086
33. Jia H-R, Zhu Y-X, Xu K-F, Liu X, Wu F-G. Plasma membrane-anchorable photosensitizing nanomicelles for lipid raft-responsive and light-controllable intracellular drug delivery. J Control Release. 2018;286:103–113. doi:10.1016/j.jconrel.2018.07.027
34. Lacerda L, Russier J, Pastorin G, et al. Translocation mechanisms of chemically functionalised carbon nanotubes across plasma membranes. Biomaterials. 2012;33(11):3334–3343. doi:10.1016/j.biomaterials.2012.01.024
35. Hu Q, Gu G, Liu Z, et al. F3 peptide-functionalized PEG-PLA nanoparticles co-administrated with tLyp-1 peptide for anti-glioma drug delivery. Biomaterials. 2013;34(4):1135–1145. doi:10.1016/j.biomaterials.2012.10.048
36. Bewersdorff T, Vonnemann J, Kanik A, Haag R, Haase A. The influence of surface charge on serum protein interaction and cellular uptake: studies with dendritic polyglycerols and dendritic polyglycerol-coated gold nanoparticles. Int J Nanomedicine. 2017;12:2001–2019. doi:10.2147/IJN.S124295
37. Shiao SL, Ganesan AP, Rugo HS, Coussens LM. Immune microenvironments in solid tumors: new targets for therapy. Genes Dev. 2011;25(24):2559–2572. doi:10.1101/gad.169029.111
38. Lee K, Yu P, Lingampalli N, Kim HJ, Tang R, Murthy N. Peptide-enhanced mRNA transfection in cultured mouse cardiac fibroblasts and direct reprogramming towards cardiomyocyte-like cells. Int J Nanomedicine. 2015;10:1841–1854. doi:10.2147/IJN.S75124
39. Li J, Wang W, He Y, et al. Structurally programmed assembly of translation initiation nanoplex for superior mRNA delivery. ACS Nano. 2017;11(3):2531–2544. doi:10.1021/acsnano.6b08447
40. Li J, He Y, Wang W, Wu C, Hong C, Hammond PT. Polyamine-mediated stoichiometric assembly of ribonucleoproteins for enhanced mRNA delivery. Angew Chem (Int Ed Engl). 2017;56(44):13709–13712. doi:10.1002/anie.201707466
41. Choi HY, Lee T-J, Yang G-M, et al. Efficient mRNA delivery with graphene oxide-polyethylenimine for generation of footprint-free human induced pluripotent stem cells. J Control Release. 2016;235:222–235. doi:10.1016/j.jconrel.2016.06.007
42. Li J, Sun Y, Jia T, Zhang R, Zhang K, Wang L. Messenger RNA vaccine based on recombinant MS2 virus-like particles against prostate cancer. Int J Cancer. 2014;134(7):1683–1694. doi:10.1002/ijc.28482
43. Zou S, Scarfo K, Nantz MH, Hecker JG. Lipid-mediated delivery of RNA is more efficient than delivery of DNA in non-dividing cells. Int J Pharm. 2010;389(1–2):232–243. doi:10.1016/j.ijpharm.2010.01.019
Supplementary materials
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.