Back to Journals » International Journal of Nanomedicine » Volume 11
Liquid crystal nanoparticle formulation as an oral drug delivery system for liver-specific distribution
Authors Lee DR, Park JS, Bae I, Lee Y, Kim BM
Received 24 September 2015
Accepted for publication 17 December 2015
Published 3 March 2016 Volume 2016:11 Pages 853—871
DOI https://doi.org/10.2147/IJN.S97000
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Dong Ryeol Lee,1,2 Ji Su Park,1 Il Hak Bae,1 Yan Lee,1 B Moon Kim1
1Department of Chemistry, College of Natural Sciences, Seoul National University, Seoul, Republic of Korea; 2Technology Development Center, BASF Company Ltd., Hwaseong, Gyeonggi-do, Republic of Korea
Abstract: Liquid crystal nanoparticles have been utilized as an efficient tool for drug delivery with enhanced bioavailability, drug stability, and targeted drug delivery. However, the high energy requirements and the high cost of the liquid crystal preparation have been obstacles to their widespread use in the pharmaceutical industry. In this study, we prepared liquid crystal nanoparticles using a phase-inversion temperature method, which is a uniquely low energy process. Particles prepared with the above method were estimated to be ~100 nm in size and exhibited a lamellar liquid crystal structure with orthorhombic lateral packing. Pharmacokinetic and tissue distribution studies of a hydrophobic peptide-based drug candidate formulated with the liquid crystal nanoparticles showed a five-fold enhancement of bioavailability, sustained release, and liver-specific drug delivery compared to a host–guest complex formulation.
Keywords: LCNP, PIT, sustained release, bioavailability
Introduction
In recent years, hydrophobic peptide-based therapeutics have been investigated intensively owing to their many benefits compared with low molecular weight drugs.1,2 These benefits include extremely high potency achieved from structure-based drug design, selectivity, broad spectrum of application, lower toxicity, vast chemical and biological diversity, and ease of discovery at the peptide and nucleic acid levels.1–5 Although many hydrophobic peptide-based therapeutics have been developed, most of them have shortcomings, such as poor metabolic stability, poor oral bioavailability, rapid clearance, and poor solubility.6 To overcome these obstacles, new drug delivery systems have been intensively studied during the last decade and many successful results have been reported. These systems include liposomes,7–10 PEGylated (polyethylene glycosylated) peptides,11,12 lipid nanocapsules,13,14 solid lipid nanoparticles,15 liquid crystal nanoparticles (LCNPs),16 and polymeric nanocapsules.17 Many hydrophobic peptide-based therapeutics have been approved on account of the new drug delivery systems. However, there are still unresolved challenges related to some persistent drawbacks, including the instability of vesicles for liposome and lipid nanocapsules,18,19 as well as the limited application of PEGylation.11
Among the reported drug delivery systems, LCNPs have received increasing attention because of the advantages they offer compared to other systems. For example, LCNPs are often incorporated with biocompatible and non-toxic excipients compared to polymeric nanoparticles; hence, oral delivery of LCNPs is possible.20–23 In a similar fashion to the protection afforded to the biodegradable drugs by polymeric nanoparticles, the liquid crystalline structure of LCNPs can protect active ingredients from the harsh gastrointestinal conditions.24,25 Additionally, sustained drug release may be achieved with drugs incorporated in LCNPs,26 allowing for a reduction in drug toxicity, which can be elevated at high drug concentrations.27 However, LCNPs have not been widely applied, particularly owing to the high costs associated with the massive energy input required in the manufacturing process, which includes high-shear homogenization and high-pressure emulsification or ultrasonication.16,28 Since the introduction of lipid nanocapsules with the phase inversion temperature (PIT) method,29–31 numerous topical, oral, and pulmonary applications of the nanocapsules have been implemented.32–34 The benefits of the PIT method include low energy and cost inputs, short realization time, obviation of organic solvents, and versatile application for lipophilic drugs. The lipid nanocapsules can also circumvent a number of drawbacks associated with conventional drug delivery systems.31 In addition, these nanocapsules improve drug bioavailability and drug stabilization, and the PEGylation of the particle surface prevents interaction with serum components, which allows for improved pharmacokinetic (PK) profile and prolonged residence time.35 However, it has been challenging to ensure that the capsules are stable against flocculation, which leads to an increase in vesicle sizes and emulsion instability.36 From our studies on the lipid nanocapsules prepared with the PIT method, we hypothesized that preparing LCNPs using the PIT method could be a solution to these limitations because particle aggregation can be prevented by the liquid crystal structure of LCNPs, which is stabilized by the bulky chains of the surfactants.37 Moreover, we envisioned that the LCNPs could be used as a liver-specific delivery system, avoiding clearance by the mononuclear phagocyte system, because the surface of the LCNPs is covered with the PEG chains and the size of the LCNPs can be set to ~100 nm.38,39
In this study, we report a novel and economic drug delivery system using LCNPs prepared by the PIT method incorporating a new hepatitis C virus NS5A inhibitor, BMK-20113. BMK-20113 is a very hydrophobic peptide-based drug candidate possessing an excessively high inhibitory activity against the NS5A protein (half maximal effective concentration, EC50=0.26 nM).40 However, it exhibited a rather undesirable PK profile in rats.40 The composition of LCNPs incorporating BMK-20113 and the manufacturing process were optimized by varying the quantities/proportions of surfactants to melting lipids and then observing the phase behavior at the PIT of the complex. The physicochemical properties of the LCNPs were evaluated using conventional methods, including small- and wide-angle X-ray diffraction (SAXD and WAXD), particle size analysis, differential scanning calorimetry (DSC), and transmission electron microscopy (TEM). The PK profiles and tissue distribution of BMK-20113 incorporated into the LCNPs were compared with those obtained from the examination of the compound incorporated in a conventional host–guest complex (HGC), which was previously utilized in PK studies of BMK-20113.40
Material and methods
Materials
BMK-20113 was synthesized in our lab and its structure and activity were described in our previous report.40 Emulgade® SE-PF, tetradecyl tetradecanoate (Cetiol® MM, myristyl myristate), polyethylene glycol (PEG)-15 hydroxystearate (Solutol® HS 15), and PEG-12 cetostearyl ether (Eumulgin® B1, ceteareth-12) were kindly provided by BASF SE (Dusseldorf, Germany). 2-Hydroxypropyl-β-cyclodextrin (HP-β-CD) was purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Carbopol 980 NF was purchased from Shanghai Chineway Pharmaceutical Technology (Shanghai, People’s Republic of China). The water used in the sample preparation, high-performance liquid chromatography (HPLC), and liquid chromatography-mass spectrometry (LC-MS/MS) procedures and the isopropyl alcohol used in the HPLC procedure were of HPLC grade and were purchased from Thermo Fisher Scientific (Waltham, MA, USA). All other reagents were of analytical grade and were used as supplied.
Preparation of LCNPs and HGC
The compositions of the LCNP formulations are described in Table 1 and the PIT method used for preparing the formulations is as follows: first, the lipid (composed of lipids, drug, and surfactants) and aqueous portions were heated separately at ~85°C. The aqueous portion was then added to the lipid portion and stirred until the hazy mixture turned semi-transparent. The mixture was then cooled to 25°C in a water bath containing ice for 1 minute to obtain the LCNPs. The PIT was measured with a conductivity meter (Cond 6+; Eutech Instruments Pte Ltd, Singapore), which measures the conductivity change at the emulsion inversion zone.
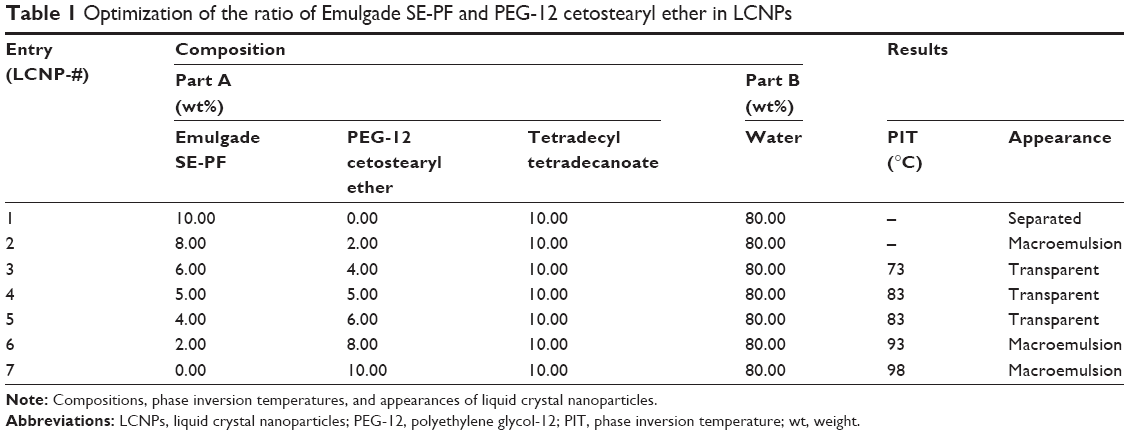 | Table 1 Optimization of the ratio of Emulgade SE-PF and PEG-12 cetostearyl ether in LCNPs |
LCNP formulations with the compositions described in Table 2 were prepared for the PK and structure analysis. The procedure for the preparation of these formulations was the same as that described above for the LCNPs. For the HGCs, 20.0 g of HP-β-CD was slowly added to 100 mL of purified water to form a solution, which was vortexed until clear. Then 0.5 mL of PEG-15 hydroxystearate was added to 0.25 mL of dimethyl sulfoxide incorporated with 5 mg of BMK-20113, and this solution was mixed until clear. The BMK-20113 solution was added to 4.25 mL of the HP-β-CD solution and then vortexed until clear. The prepared LCNPs and HGC were stored at 4°C for further evaluation.
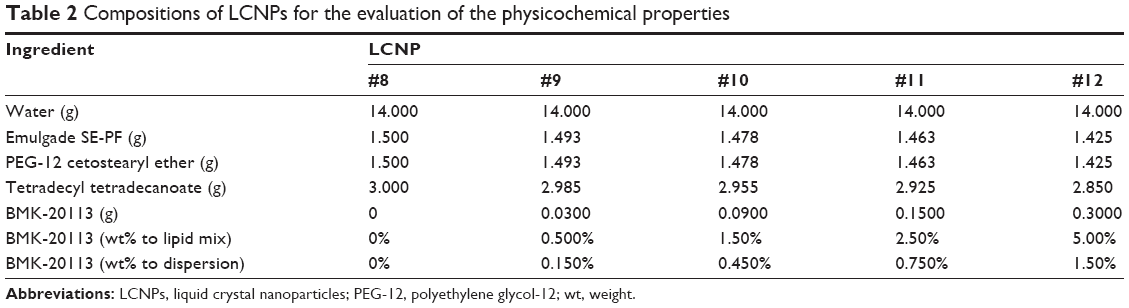 | Table 2 Compositions of LCNPs for the evaluation of the physicochemical properties |
SAXD and WAXD analysis
The structures of the LCNPs were characterized using SAXD and WAXD analyses carried out on a SAXSpace (Anton-Paar, Graz, Austria) at the National Instrumentation Center for Environmental Management (NICEM, Seoul, Korea). The scattering intensity was estimated as a function of the scattering vector q which is defined as:
|
|
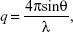
where θ and λ stand for the scattering angle and wavelength, respectively. The distances between the planes of the LCNP surface (d) were calculated from the obtained indexing peaks in the SAXD scattering patterns using Bragg’s equation,
|
|
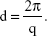
The calculated inter-planar distances were multiplied by the peak ratio of the particular peak. The packing structure of the LCNPs was determined using WAXD. The scattering patterns were compared with the patterns previously reported by Bouwstra et al.41
DSC analysis
The thermal properties of the LCNPs were investigated at the NICEM using a DSC-Q1000 (TA Instruments, Crawley, UK) equipped with a thermal analysis data system for the baseline correction, transition temperature, and calculation of the transition heat. The samples were first cooled to 20°C and then heated to 80°C at a heating rate of 2°C/minute under a constant flow of nitrogen gas.
TEM analysis
A 0.10 mL aliquot of the LCNP dispersion was diluted in 1.0 mL of water or hydrochloric acid-potassium chloride buffer solution (0.2 M, pH 1.5) at 25°C. The diluted dispersion was dropped onto CF300-Cu carbon film (Electron Microscopy Sciences, Hatfield, PA, USA) and then dried for 1 day under reduced pressure. The TEM images were obtained on JEOL EM-2010 and JEM-3010 microscopes (JEOL, Tokyo, Japan) at an accelerating voltage of 200 kV.
Particle size analysis
The particle size was evaluated using dynamic light scattering, which yields the mean particle size and particle size distribution. Approximately 0.2 mL of the LCNP dispersion was diluted in 10 mL of purified water at 25°C. Then 3 mL of the diluted dispersion was added to a ϕ21 cylinder cell in a particle size analyzer (DLS-8000HL; Otsuka Electronics, Osaka, Japan), which was equipped with a 10 mW helium–neon laser. The evaluation was performed 30 times for each dispersion at a detection angle of 90° and the measurement was repeated three times.
In vitro dialysis
To avoid the potential precipitation of BMK-20113, a modified assay was used, as previously reported by Hua.42 To prepare the donor solution, 1.5 mL of the HGC dispersion was mixed with 4 mL of 0.5 wt% carbopol gel and then dispersed into 38.5 mL of the dissolution medium, which was 50 mM phosphate-buffered saline (pH 6.5). To prepare the LCNP dispersion donor solution, 0.2 mL of LCNP-#11 was mixed with 4 mL of 0.5 wt% carbopol gel and then dispersed into 39.8 mL of the dissolution medium. A total of 11 mL of the donor solution was added to a dialysis bag (10 kDa molecular weight cut-off, Thermo Fisher Scientific) and this was suspended in 90 mL of the acceptor solution, which was the dissolution medium. At scheduled intervals, 400 μL of the acceptor solution was collected for HPLC assay and an equal volume of fresh dissolution medium was added to maintain a constant volume. The concentration of BMK-20113 in the collected sample was determined by HPLC.
PK studies
The Sprague Dawley rats (184–238 g body weight) used in the study were supplied by Sippr-BK Lab Animal Ltd (Shanghai, People’s Republic of China) as shown in Table S1. The animal studies were performed at Sundia MediTech Co. Ltd. The institutional animal ethics care and use committee of Sundia approved the animal protocols and the studies were performed in accordance with the guidelines of the committee. For the intravenous (IV) and oral (PO) dose group, the nominal concentration of the dosing solutions was 1.0 mg/mL. The study was designed as described in Table 3 and conducted in parallel with the two formulations (HGC and LCNP-#11) as described in Table 2. Individual doses were calculated based on pre-treatment body weights recorded on the day of dose administration. The IV dose was administered via the tail vein as a bolus injection, and each animal received 5 mL of the solution per kg of body weight. For the PO dose, each animal was administered 10 mL of the solution per kg of body weight via a gavage tube. Blood (0.2–0.3 mL) was collected in polypropylene tubes containing ethylenediaminetetraacetic acid (EDTA)-K2 as an anticoagulant, at different time points and then stored on wet ice until processed for plasma by centrifugation. The samples were centrifuged at 6,000 rpm for 8 minutes within 1 hour of blood collection. The plasma samples were then stored at ~−20°C until analyzed by LC-MS/MS with an internal standard. The lower limit of quantification of BMK-20113 was 1.0 nmol/L. PK parameters were calculated with WinNonlin® 6.3 through the use of non-compartmental PK analysis method.
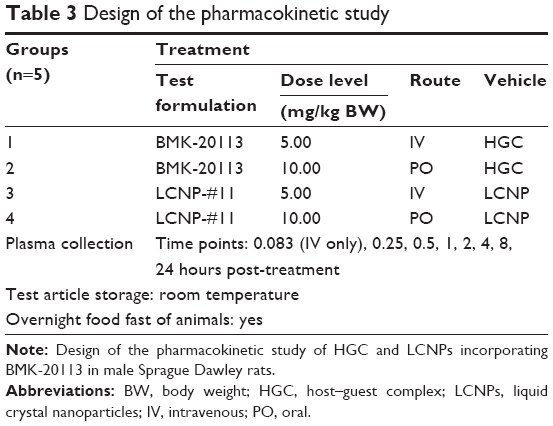 | Table 3 Design of the pharmacokinetic study |
Tissue distribution studies
Six Sprague Dawley rats (190–216 g in body weight) supplied by Sippr-BK Lab Animal Ltd. were used in this study. The nominal concentration of the dose solution was 1.0 mg/mL. For the study, six male Sprague Dawley rats were divided into two groups: the animals in group 1 and group 2 were given a single IV dose with HGC and LCNP-#11, respectively, at a nominal dose of 5 mg/kg as a 1 mg/mL solution. Individual doses were calculated based on pre-dose body weights recorded on the day of dose administration. For IV dose, administered amounts via tail vein as a bolus injection, are presented in Table S2. Samples of the cerebrospinal fluid (CSF), blood (0.150–0.200 mL), lung, liver, kidney, spleen, skin, and muscle were collected into polypropylene tubes containing EDTA-K2 as the anticoagulant and stored on wet ice until processed for plasma by centrifugation. The samples were centrifuged within 30 minutes of blood collection at 6,000 rpm for 8 minutes and the plasma samples were then stored in a freezer until analyzed by LC-MS/MS. The tissues were homogenized with five volumes of phosphate-buffered saline to obtain a tissue suspension.
LC-MS/MS analysis in the body distribution study
LC-MS/MS analysis of plasma, kidney, liver, skin, and spleen samples was carried out using a Shimadzu LC-20AD coupled with an AB Sciex API 4000 (Shimadzu, Kyoto, Japan). Standard curve samples (20 μL), quality control samples and rat plasma samples were mixed with 60 μL of acetonitrile containing internal standard (200 ng/mL of tolbutamide or 50 ng/mL of propranolol) in Eppendorf tubes. After vortexing the mixture for 1 minute, the mixture was centrifuged for 10 minutes at 13,000 rpm at 4°C and then the supernatant (50 μL) was transferred to a 96-well plate which was pre-added in 150 μL of water. After shaking the samples for 10 minutes, 10 μL of sample was injected into LC-MS/MS. The separation was carried out on a Thermo Betasil C18 column (2.1×50 mm, 5 μm particle size) with 0.1% trifluoroacetic acid in water (A) and acetonitrile (B) as the mobile phase at a flow rate of 0.5 mL/minute. Method: 10% B (0–0.5 minutes), 10%–95% B (0.5–1.1 minutes), 95% B (1.1–1.4 minutes), 95%–10% B (1.4–1.5 minutes), 10% B (1.5–2.2 minutes). Mass transitions: 260.3/116.2 for propranolol (internal standard); 761.4/289.1 for BMK-20113.
The lung, muscle, and CSF samples were analyzed by LC-MS/MS using a Shimadzu LC-20AD coupled with an AB Sciex API 4000. The other analytical conditions are the same as those used for the plasma, kidney, liver, skin, and spleen samples.
Results
Preparation and physicochemical properties of LCNPs
The estimated PIT values and appearances of the LCNPs prepared with varying compositions are shown in Table 1. The ratio of Emulgade® SE-PF to PEG-12 cetostearyl ether was optimized by varying the ratio of the surfactant to the fixed concentrations of the surfactant mixture (10 wt%) and tetradecyl tetradecanoate (10 wt%). Semi-transparent dispersions were evident when the ratios of Emulgade SE-PF and PEG-12 cetostearyl ether were in the range of 2:1–2:3 and the phase inversion of the compositions appeared above 80°C. Based on previous reports that the stability of the formulation was improved as the PIT increased, a 1:1 ratio of Emulgade SE-PF and PEG-12 cetostearyl ether, which showed the highest PIT, was selected.30 As the composition of surfactant mix, tetradecyl tetradecanoate and water was varied, semi-transparent nanodispersions were obtained at ratios of the dotted area illustrated in Figure 1, which was plotted based upon the results shown in Table S3. The dispersion with composition #8 in Figure 1 and Table 2 (LCNP-#8) was selected for further evaluation because it was incorporated with the highest amount of the lipid mix (30 wt% to the dispersion) comprising tetradecyl tetradecanoate (15 wt%) and the surfactant mix (15 wt%) compared with the other dispersions. The phase behavior of dispersion LCNP-#8 on heating is described in Figure 2 with the estimation of electric current value. The electric current value decreased from ~140 μS at 80°C to below 20 μS at 95°C; hence, the PIT was determined as 88°C, which is the median value of 80°C and 95°C.30 BMK-20113 was incorporated in LCNP-#8 at 0.500, 1.50, 2.50, and 5.00 wt% to the lipid complex, as illustrated in Table 2. The 2.50 wt% sample of LCNP-#11 was selected for further PK studies because precipitation was observed with the 5.00 wt% sample (LCNP-#12). Drug stability during the heating process was evaluated using HPLC, and no drug decomposition was observed, as shown in Figure S1. BMK-20113 is practically insoluble in water40 and is therefore assumed to be partitioned in the lipid phase of the LCNP dispersion.
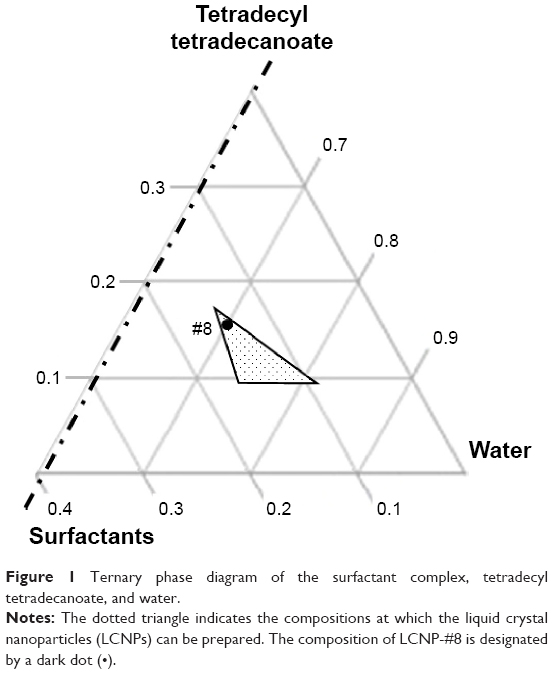 | Figure 1 Ternary phase diagram of the surfactant complex, tetradecyl tetradecanoate, and water. |
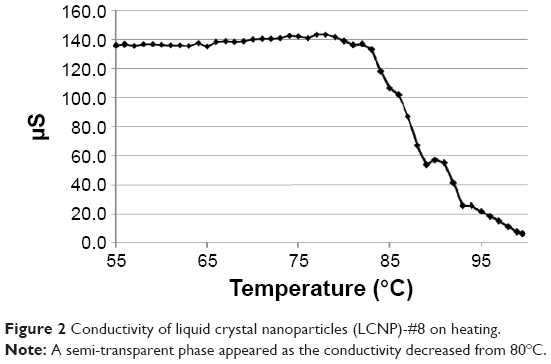 | Figure 2 Conductivity of liquid crystal nanoparticles (LCNP)-#8 on heating. |
The SAXD intensity (y-axis) is plotted against the scattering vector (x-axis) in Figure 3A. Since the diffraction patterns are composed of sequential peaks, the LCNPs can be considered to have a lamellar liquid crystal structure. There is a correlation between the peaks positioned at 0.65 and 1.3 (nm−1) with:
2Q1 = Q2, | (3) |
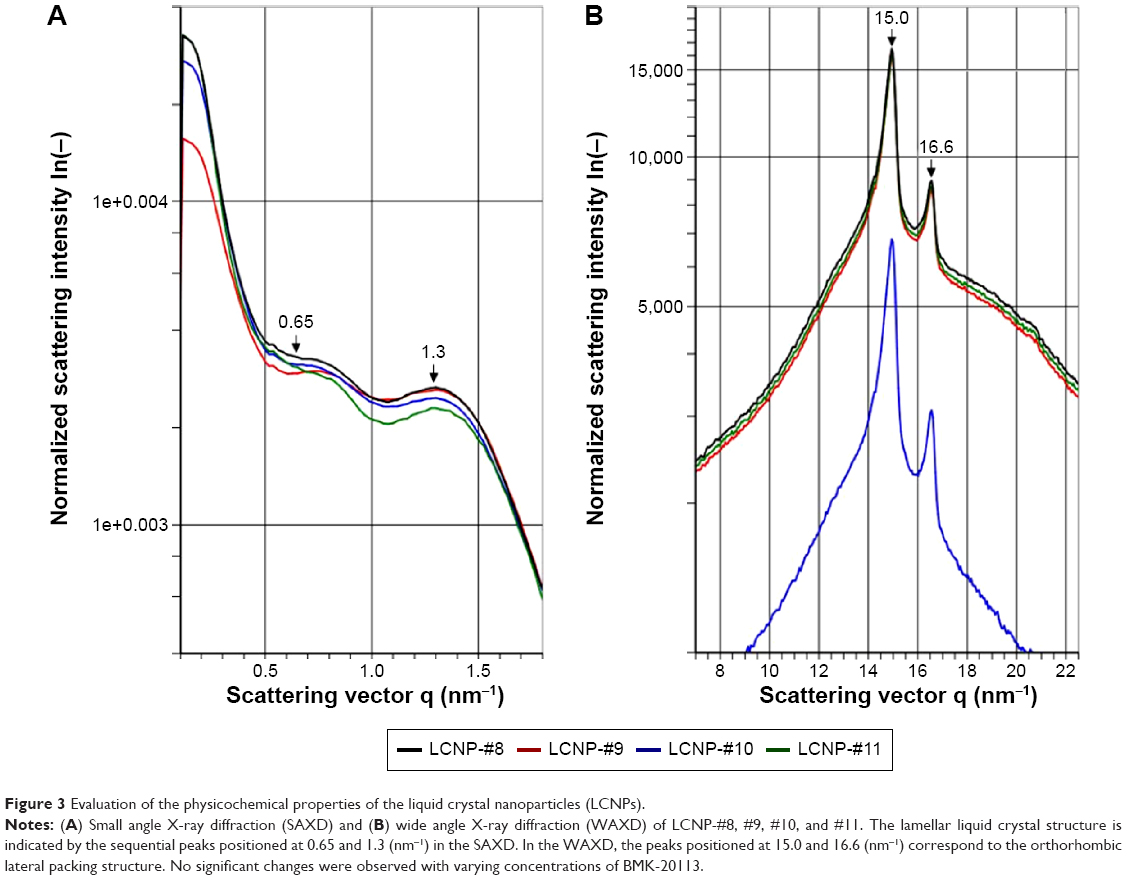 | Figure 3 Evaluation of the physicochemical properties of the liquid crystal nanoparticles (LCNPs). |
where Q1 stands for 0.65 and Q2 stands for 1.3. Therefore, the distance between the two phases is calculated to be ~9.7 nm by using Bragg’s equation,
|
|
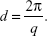
From the results of the SAXD, it can be assumed that the surface of the LCNPs comprises a lipid lamellar phase with a periodicity of 9.7 nm. As shown in Figure 3B, the scattering vector values in the WAXD are 15.0 and 16.6 (nm−1); therefore, the distances between the hydrocarbon chains are 0.419 and 0.379 nm, respectively, according to Bragg’s equation. These values are comparable with the orthorhombic lateral packing values, which are 0.41 and 0.37 nm, respectively, as reported by Bouwstra et al.41 There were no differences in the distance values as the concentration of BMK-20113 increased from 0 to 5.00 wt% in the lipid phase. From the results of SAXD and WAXD, it can be presumed that the LCNPs are composed of a lipid lamellar liquid crystal with orthorhombic lateral packing, as shown in Figure 4.
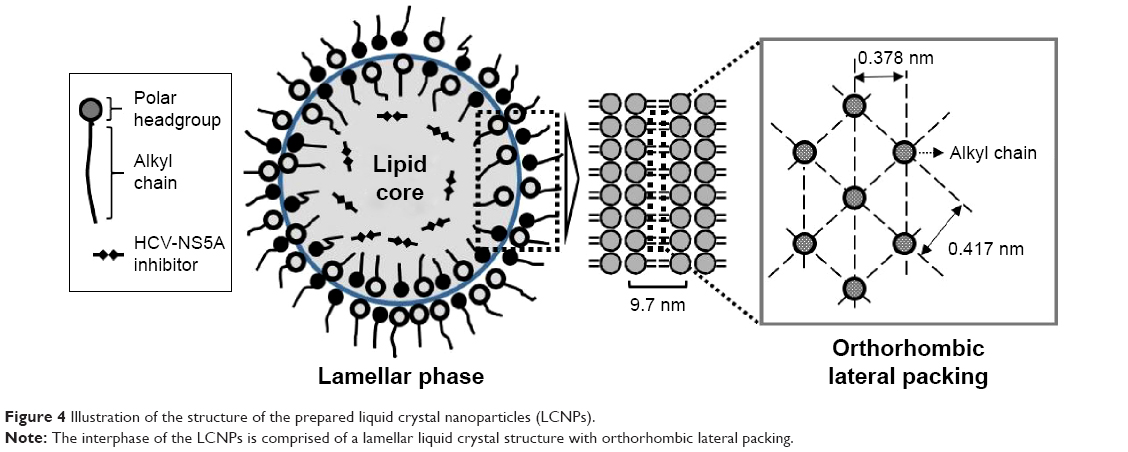 | Figure 4 Illustration of the structure of the prepared liquid crystal nanoparticles (LCNPs). |
The DSC results are shown in Figure 5. The melting peak of the LCNPs without BMK-20113 was observed at ~35°C, which is lower than those observed for tetradecyl tetradecanoate (43.79°C), Emulgade SE PF (39.21°C), and PEG-12 cetostearyl ether (38.27°C), as shown in Figure S2. The enthalpy values of the LCNPs incorporated with 0, 0.50, 1.50, and 2.50 wt% BMK-20113 to the lipid mix were estimated to be 34.3, 34.3, 34.0, and 34.1 J/g, respectively.
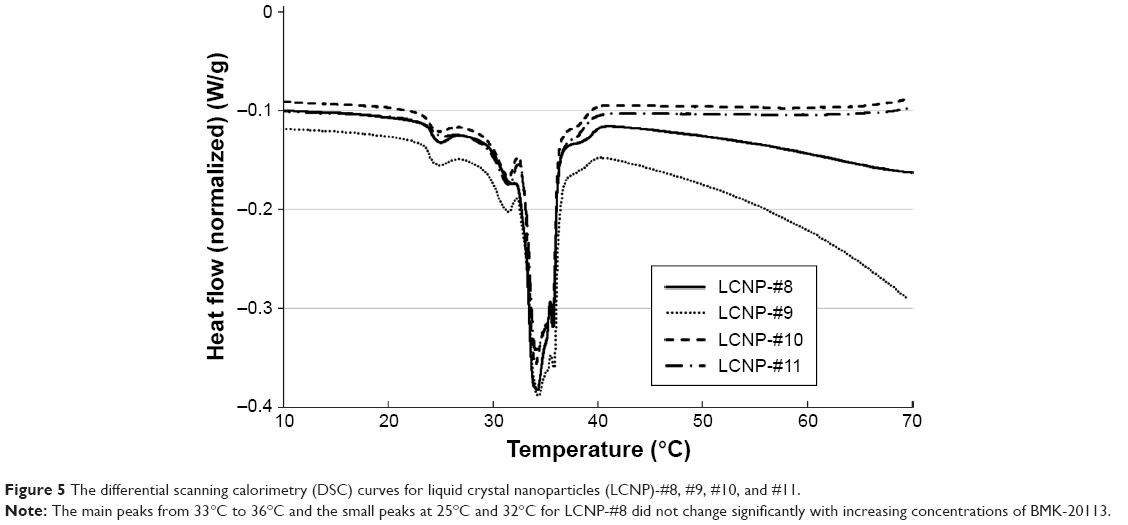 | Figure 5 The differential scanning calorimetry (DSC) curves for liquid crystal nanoparticles (LCNP)-#8, #9, #10, and #11. |
TEM images of the LCNPs incorporating BMK-20113 in 0, 0.50, 1.50, and 2.50 wt% to the lipid mix are shown in Figure 6. The sizes of LCNPs are from 70 to 130 nm and there were no significant differences in the particle shape at all tested concentrations of BMK-20113. However, the particle size increased with increasing concentrations of BMK-20113 from 0 to 2.50 wt%. There was no drug expulsion based on the TEM evaluation, which detects the drug recrystallization as shown in Figure S3. Additionally, the shapes and sizes of LCNP-#8 and #11 in an acidic condition (pH 1.5) were evaluated with TEM to ensure the stability in PO. As shown in Figure S4, the morphological properties of LCNP-#8 and #11 in the acidic condition were comparable with those of LCNP-#8 and #11 in the neutral condition.
 | Figure 6 Transmission electron microscopy (TEM) images of liquid crystal nanoparticles (LCNP)-#8, #9, #10, and #11. |
To evaluate the stability of the LCNP dispersions, particle size analysis was performed using dynamic light scattering at scheduled time intervals, and the results are illustrated in Figure 7. There were no significant changes in the particle size when stored at 4°C from day 1 after preparation for up to 2 months. However, the particle size of the LCNPs with 2.50 wt% BMK-20113 was larger than those with other concentrations. At 37°C, there was no increase in the particle size at 0 and 0.50 wt% BMK-20113. However, a significant increase in size was observed in the LCNPs with 2.50 wt% BMK-20113 after 2 weeks. The LCNPs with 1.50 wt% BMK-20113 showed a slight increase in size after 2 weeks. However, this was comparable with the values at 1 and 2 months. This result suggests that the size increase is not accelerated until after 2 months.
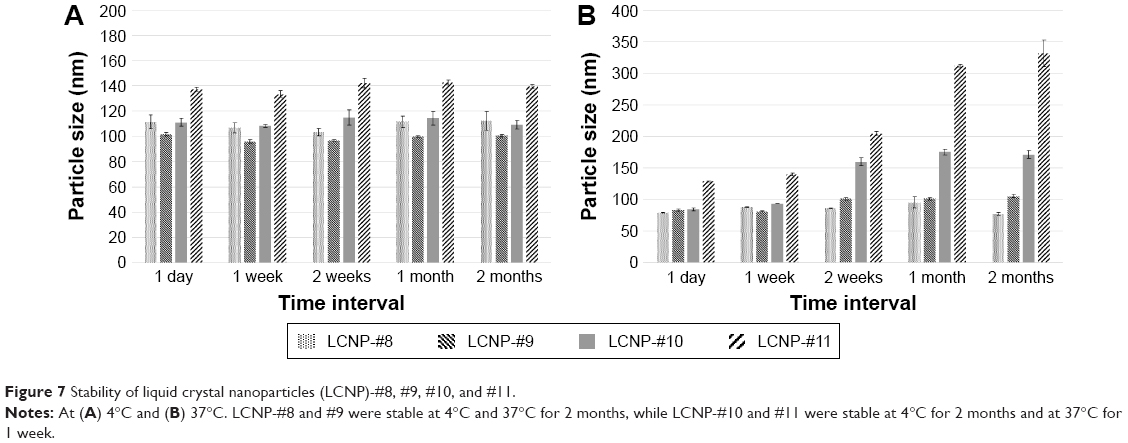 | Figure 7 Stability of liquid crystal nanoparticles (LCNP)-#8, #9, #10, and #11. |
PK studies
The in vitro release patterns of BMK-20113 from the HGC and LCNPs are shown in Figure 8. For the HGC, 3.87% of the BMK-20113 was released within 3 hours, followed by 3.51% in the next 69 hours. The evaluation of LCNP-#11 showed that 19.5% of BMK-20113 was released within 12 hours, with a 1.65%/hour release rate (R2=0.995). This was followed by 32.0% release in the next 60 hours, with a 0.518%/hour release rate in a sustained manner (R2=0.955).
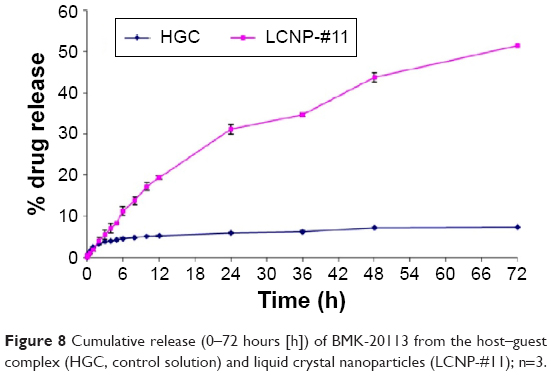 | Figure 8 Cumulative release (0–72 hours [h]) of BMK-20113 from the host–guest complex (HGC, control solution) and liquid crystal nanoparticles (LCNP-#11); n=3. |
The results of the PK studies following IV and PO administration of HGC and LCNP-#11 in rats are shown in Table 4 and Figure 9. Significant differences were observed between HGC and LCNPs in the maximum concentration (Cmax), clearance rate (CLz/F), release pattern, and bioavailability (F) of BMK-20113. With PO administration, the Cmax value with the LCNPs was two-fold higher than that with HGC, and the peptide-based drug in the LCNPs was released in a sustained manner. In addition, a sudden increase in the drug concentration was observed with the HGC with PO administration. A significant increase (five-fold) in F value was observed with PO administration of LCNPs compared with the HGC.
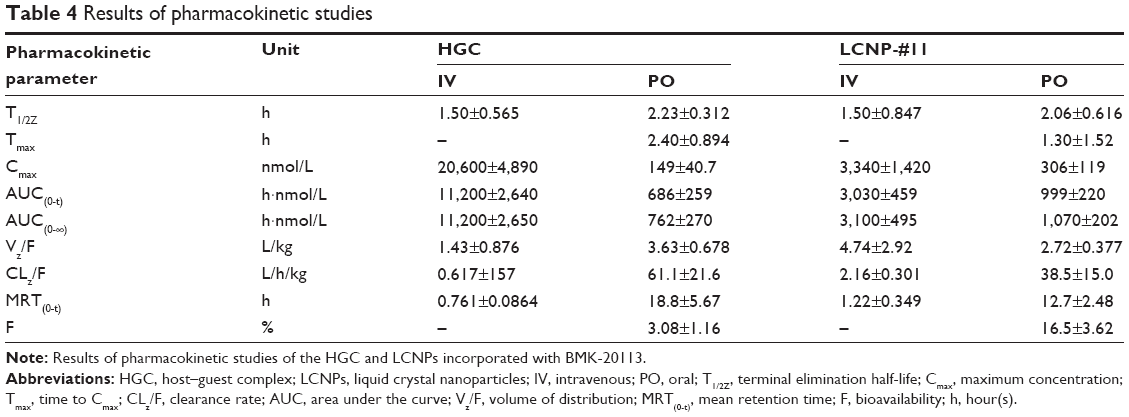 | Table 4 Results of pharmacokinetic studies |
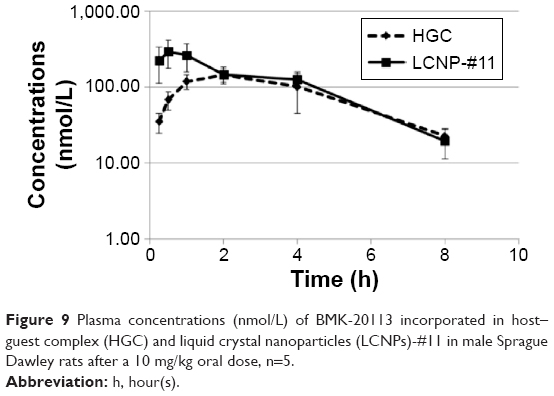 | Figure 9 Plasma concentrations (nmol/L) of BMK-20113 incorporated in host–guest complex (HGC) and liquid crystal nanoparticles (LCNPs)-#11 in male Sprague Dawley rats after a 10 mg/kg oral dose, n=5. |
Tissue distribution studies
The tissue distributions of BMK-20113 after IV administrations of the test compound in the HGC and LCNPs are described in Figure 10. The plasma concentration of BMK-20113 after dosing with the LCNP formulation was lower than that resulting from the HGC formulation, in line with the results shown in Table 4, which presumably results from the prolonged retention of LCNPs in the tissues. As shown in Figure 10A, the concentration of BMK-20113 after dosing with the LCNP formulation is the highest in the liver, while after dosing with the HGC formulation the concentration of BMK-20113 is higher in the kidney, plasma, and lung than in the liver. Furthermore, the liver/plasma distribution ratio in the LCNP formulation is more than ten-fold higher than that in the HGC formulation, as shown in Figure 10B. Regarding the distribution to the kidney and lung, the ratios in the LCNP formulation are higher than those in the HGC formulation; however, the drug concentrations in the LCNP formulation are lower than those in the HGC formulation. This shows that BMK-20113 in the LCNP formulation is specifically distributed into the liver, while that in the HGC formulation is distributed into the plasma, kidney, liver, and lung nonspecifically. Additionally, the concentration in CSF is very low with the LCNP formulation, less than one seventh of that observed with the HGC formulation, which indicates that the LCNP formulation does not allow noticeable blood–brain barrier penetration.
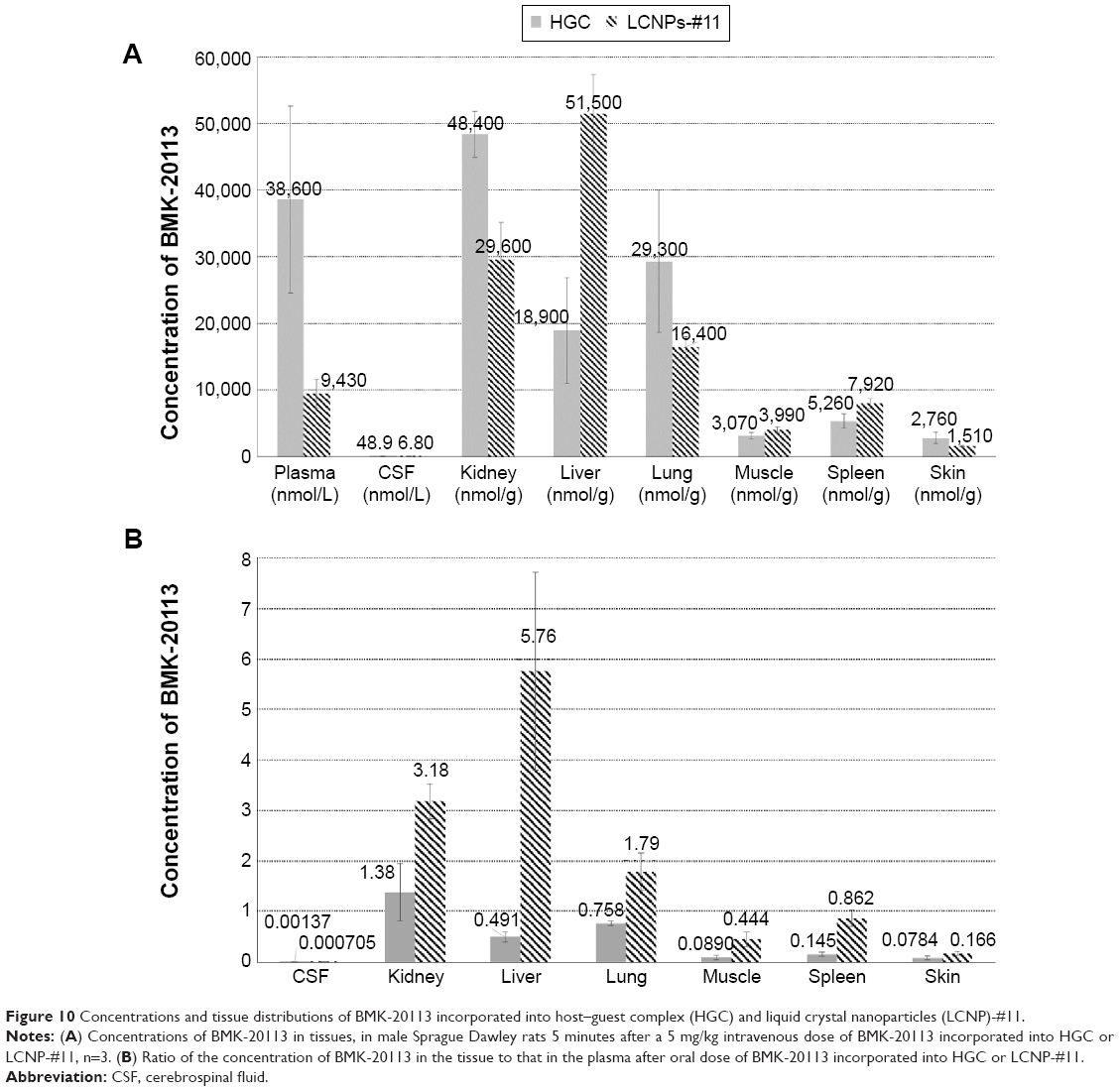 | Figure 10 Concentrations and tissue distributions of BMK-20113 incorporated into host–guest complex (HGC) and liquid crystal nanoparticles (LCNP)-#11. |
Discussion
The LCNPs in this study were prepared by simple heating of the complex to 85°C, followed by rapid cooling to 25°C in a minute. Therefore, the energy input required for the preparation was much lower than that required for high-pressure homogenization and ultrasonication, which are used in the conventional preparation. Therefore, the industrial preparation of LCNPs using the method in this study could be more convenient than that with conventional methods. As illustrated in Figure 2, the conductivity was ~140 μS at 80°C and with a temperature increase to 95°C, it fell below 20 μS. During the conductivity change period, the appearance of the complex changed from turbid to semi-transparent. This change in appearance signified that the complex phase had changed from the melt-lipid in water emulsion to a bicontinuous phase.43 The solubility of BMK-20113 in the lipid phase was improved as the temperature increased. Therefore, the dissolution process for BMK-20113 was endothermic, and recrystallization or aggregation of BMK-20113 could occur at low temperatures. At 4°C, there was no aggregation and recrystallization of the poorly water-soluble BMK-20113;40 therefore, it can be concluded that BMK-20113 was encapsulated in the liquid crystal structure of the LCNPs. One major concern with the heating process during the preparation of lipid nanocapsules is the degradation of the drug during the process. There was no change in the HPLC analysis of BMK-20113 during the preparation of the LCNPs compared to the original BMK-20113 in tetrahydrofuran, which confirmed the stability of BMK-20113 under the heating conditions, as shown in Figure S1.
The SAXD analysis revealed that the surface of the LCNPs is composed of a lipid lamellar phase with a periodicity of 9.7 nm. In the WAXD analysis, the alkyl chains of octadecanol, hexadecanol, and the surfactants are positioned at distances of 0.419 and 0.379 nm, respectively, which means that the LCNPs are composed of orthorhombic lateral packing, as illustrated in Figure 4. The lamellar liquid structure is considered suitable for drug delivery because the sustained drug release was reported to be possible with the multilayer structured carrier.44,45 There was no difference in the SAXD and WAXD patterns with increasing concentrations of BMK-20113. Therefore, it can be concluded that the lateral packing of lipids at the surface of the LCNPs was not concentration-dependent. It can also be predicted that the controlled release of the drug was possible because of the layer-by-layer erosion of the drug from the lamellar liquid crystal structure.45
The DSC evaluation of the LCNPs showed a main peak at 35°C, which is lower than the melting peaks of excipients (Figure 5 and S2). As reported by Montenegro et al,46 the melting peak of cetyl palmitate could be lower than the peak of the cetyl palmitate bulk. This lower melting point could be attributed to the nano-size of the particles and the non-crystallized, super-cooled state of the core of the LCNPs.46,47 The state of tetradecyl tetradecanoate in the LCNP layer in this study was nano-sized and could also be the super-cooled form resulting from the rapid cooling process. This phenomenon may explain the decrease in the melting point.
Because the melting peak of the LCNPs is observed at 34°C–36°C, the particle could melt after oral dosing and the coalescence of the particles could follow. Because the coalescence results in destabilization of the formulation, it is not suitable for oral administration. In order to ensure the stability under the PK evaluation conditions, the stability of the LCNPs was evaluated using dynamic light scattering at 4 and 37°C, as shown in Figure 7. It should be noted that the stability at 37°C was important to ensure the stability of the vehicle under the conditions of the PK study. At 4°C, there was no significant change in particle size for 2 months, which means that the LCNPs are stable under that condition. However, an increase was observed in the particle size when the LCNPs were stored at 37°C. The particle size of the LCNPs was preserved for up to 1 week; however, it significantly increased 2 weeks after the incorporation of 1.50 and 2.50 wt% BMK-20113 to the lipid mix (LCNP-#10 and #11, respectively). It can be assumed that the addition of BMK-20113 to the LCNPs at a concentration higher than 1.50 wt% (LCNP-#10) affects the amorphous property of the core, which presumably leads to the acceleration of coalescence and Ostwald ripening. Furthermore, the lipids in the LCNPs are converted to liquid form above 35°C, as shown by the DSC results. Therefore, the coalescence and Ostwald ripening could be stimulated at 37°C, thereby increasing the particle size and destabilizing the LCNPs. However, the stability of the LCNPs at 37°C is acceptable for the PK study since the test is performed within 2 to 3 days. The acceptable stability of the LCNPs at 37°C can be rationalized based upon the retardation of coalescence because of the repulsive forces produced by the PEG group of PEG-12 cetostearyl ether.48 Because of the repulsive forces, the surface layer comprising the surfactant can act as a physical barrier, therefore, the formulation can be stable for 1 week. As for the stability in the acidic condition, the particle shapes and sizes of LCNP-#8 and #11 were comparable with those in the neutral condition. Therefore, it can be assumed that the LCNPs are durable under the “stomach environment”.
As shown in Figure 8, sustained release of BMK-20113 incorporated in the LCNPs was observed until 72 hours, compared with the HGC formulation, which showed a very low drug release rate. Because the evaluation was carried out below the saturation point of BMK-20113, it clearly indicates that the sustained release is possible with the LCNPs. The results can be explained by the structure of the LCNPs, as determined by SAXD and WAXD. These analyses showed that the drug release can be controlled by the layer-by-layer erosion of the orthorhombic lateral packing structure of the LCNPs at 37°C.45 Conversely, the HGC is composed of HP-β-CD as the host incorporated with the peptide-based therapeutic BMK-20113, which is the guest with a cyclic structure. The structure is efficient for incorporating the hydrophobic drug with the hydrophobic site of HP-β-CD; however, the drug release can be delayed or inhibited if the affinity of the bond between the HP-β-CD and the peptide-based drug candidate is too strong.49 Additionally, the loading efficiency of the BMK-20113 was 25.0 mg in 1.00 g of the LCNPs, which is much higher compared with that for the HGC at 5.88 mg in 1.00 g of the complex. The enhanced efficiency of the drug loading can be explained by the amorphous state of the LCNP core. This complex core is composed of tetradecyl tetradecanoate, cetyl palmitate, glyceryl stearate, hexadecanol, and octadecanol, which provides more room for the encapsulation of BMK-20113.50
In the PK study shown in Table 4, the bioavailability of the BMK-20113 incorporated in the LCNPs is more than five times higher than that with the HGC. In addition, the terminal half-life (T1/2Z) of the LCNP–BMK-20113 complex was comparable with that of the HGC–BMK-20113 complex. The mean retention time of the PO dosing was higher for the HGC compared to the LCNP, as shown by the sudden increase in drug concentration observed at 1, 2, and 4 hours in the HGC (Figure 9). Therefore, the half-life and mean retention time could have been retarded during the evaluation of the HGC. The amounts of BMK-20113 released from the HGC at 1, 2, and 4 hours were about twice as high as that at 0.25 hours. Conversely, sustained release of BMK-20113 was observed from the LCNPs and the concentration was maintained at a constant level from 0.25 to 4 hours, which suggests that the drug release was uniform. With these results, it can be concluded that drug delivery with the LCNPs is more efficient than with the HGC. These results are in agreement with the predicted properties obtained from the dialysis, SAXD, and WAXD analysis. The improvement in the bioavailability of the BMK-20113 incorporated in the LCNPs is presumably attributable to the multilayer structure of LCNPs, which offers sustained drug release.44 In addition, the liquid crystal structure protects the drug within the gastrointestinal tract, thereby enhancing its stability because the drug is encapsulated in the lipid core. Moreover, the outer layer is PEGylated with ceteareth-12 and ceteareth-20, which function as a stealth layer under gastrointestinal conditions; therefore, the BMK-20113 incorporated into the LCNPs is more stable than with the HGC.35,51
From the tissue distribution study, it can be reasoned that the hydrophobic peptide-based drug is delivered specifically to the liver when it is incorporated into the LCNPs. As described in the results, the distribution into the liver with LCNP-#11 is more than ten-fold higher than with HGC. The concentrations in the kidney and lung from the LCNP formulation are higher than those from the HGC formulation. However, the higher values are not because of selective delivery to the kidney and lung by the LCNPs, but because of the lower plasma concentration with the LCNP formulation compared to the HGC formulation, as shown in Figure 10A. Therefore, it can be concluded that the LCNPs are a more liver-specific drug delivery system than the HGC. The liver-specific drug delivery can be rationalized with the mechanism of lipid metabolism.38,39 In the reported mechanism, the emulsions can be metabolized via the comparable metabolism pathway of chylomicron or removed from the plasma by the cells of the mononuclear phagocyte system when the emulsions are recognized as foreign materials.38 If the LCNPs are recognized as a foreign material, then the concentration of BMK-20113 should be significantly reduced in the blood within 30 minutes or less and should be increased in both the liver and spleen because of the mononuclear phagocyte system, the Kupffer cells of the liver, and the macrophages of the spleen.38 However, the concentrations are preserved at a certain level until 1 hour after IV and 4 hours after PO administration, as shown in Table 4 and Figure 9. Another plausible mechanism for liver-specific delivery is that the LCNPs can be metabolized via the lipid emulsion metabolism pathway. As it was reported in the drug delivery system with lipid emulsions, the emulsion can be recognized as artificial chylomicrons.52 Because the LCNPs melt at body temperature, it can also be recognized as artificial chylomicrons which enable the specific accumulation into the liver. Furthermore, in the body distribution study, the concentration in the spleen with the LCNP formulation is not significantly different from that with the HGC formulation, while the difference in the liver is significant, as shown in Figure 10A. From the results, it can be seen that the LCNPs are specifically delivered to the liver by avoiding clearance by the mononuclear phagocyte system, presumably because of the PEG groups of the surfactants and the reduced particle size.
Conclusion
In this study, the LCNPs incorporated with the peptide-based therapeutics were prepared using the PIT method, which required low energy input with a short processing time. The structure of the LCNPs was determined to be orthorhombic lateral packing with a lamellar liquid crystal structure, and the particle sizes were ~100 nm. The stability of the LCNPs was confirmed with the particle size analysis at 4°C. The BMK-20113 incorporated in the LCNPs was stable during the PIT procedure and the loading efficiency of the LCNPs was 2.5 wt% for the lipid mix and 0.75 wt% for the dispersion, which was higher than that for the HGC. The dialysis test confirmed the sustained release of the drug incorporated in the LCNPs. The PK study showed that the F value of the LCNP–BMK-20113 complex increased five-fold compared with that for the HGC–BMK-20113 complex. The improvement in the F value and sustained release are in agreement with the predicted properties of the liquid crystal structure of the LCNPs. The liquid crystal structure facilitated the controlled release of the drug and the F value was improved because the LCNPs were stable under the gastrointestinal conditions owing to the PEGylated surface, which acts as a stealth layer. Furthermore, it was revealed in the tissue distribution study that the LCNPs are a liver-specific drug delivery system.
Acknowledgments
This research was supported by the Korean Ministry of Education, Science and Technology (grant number 2012M3A7B4049644, Nano Material Development Program through the National Research Foundation of Korea, NRF).
Disclosure
The authors report no conflicts of interest in this work.
References
Craik DJ, Fairlie DP, Liras S, Price D. The future of peptide-based drugs. Chem Biol Drug Des. 2013;81(1):136–147. | ||
Kaspar AA, Reichert JM. Future directions for peptide therapeutics development. Drug Discov Today. 2013;18(17–18):807–817. | ||
Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. 2008;7(1):21–39. | ||
Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Synthetic therapeutic peptides: science and market. Drug Discov Today. 2010;15(1–2):40–56. | ||
Collins I, Workman P. New approaches to molecular cancer therapeutics. Nat Chem Biol. 2006;2(12):689–700. | ||
Tice CM, Singh SB. Evolution of Diverse Classes of Renin Inhibitors through the Years. In: Ghosh AK, editors. Aspartic Acid Proteases as Therapeutic Targets. Weinheim, Germany: Wiley-VCH; 2010:297–324. | ||
Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–160. | ||
Martins S, Sarmento B, Ferreira DC, Souto EB. Lipid-based colloidal carriers for peptide and protein delivery – liposomes versus lipid nanoparticles. Int J Nanomedicine. 2007;2(4):595–607. | ||
Mohanraj VJ, Barnes TJ, Prestidge CA. Silica nanoparticle coated liposomes: a new type of hybrid nanocapsule for proteins. Int J Pharm. 2010;392(1–2):285–293. | ||
Niu M, Lu Y, Hovgaard L, Wu W. Liposomes containing glycocholate as potential oral insulin delivery systems: preparation, in vitro characterization, and improved protection against enzymatic degradation. Int J Nanomedicine. 2011;6:1155–1166. | ||
Veronese FM, Pasut G. PEGylation, successful approach to drug delivery. Drug Discov Today. 2005;10(21):1451–1458. | ||
Roberts MJ, Bentley MD, Harris JM. Chemistry for peptide and protein PEGylation. Adv Drug Deliver Rev. 2002;54:459–476. | ||
Hirsjarvi S, Belloche C, Hindre F, Garcion E, Benoit JP. Tumour targeting of lipid nanocapsules grafted with cRGD peptides. Eur J Pharm Biopharm. 2014;87(1):152–159. | ||
Watnasirichaikul S, Davies NM, Rades T, Tucker IG. Preparation of biodegradable insulin nanocapsules from biocompatible microemulsions. Pharm Res. 2000;17(6):684–689. | ||
Almeida AJ, Runge S, Müller RH. Peptide-loaded solid lipid nanoparticles (SLN): influence of production parameters. Int J Pharm. 1997;149(2):255–265. | ||
Zeng N, Gao X, Hu Q, et al. Lipid-based liquid crystalline nanoparticles as oral drug delivery vehicles for poorly water-soluble drugs: cellular interaction and in vivo absorption. Int J Nanomedicine. 2012;7:3703–3718. | ||
Kumar M, Gupta D, Singh G, et al. Novel polymeric nanoparticles for intracellular delivery of peptide Cargos: antitumor efficacy of the BCL-2 conversion peptide NuBCP-9. Cancer Res. 2014;74(12):3271–3281. | ||
Huynh NT, Passirani C, Saulnier P, Benoit JP. Lipid nanocapsules: a new platform for nanomedicine. Int J Pharm. 2009;379(2):201–209. | ||
Park K, Kwon IC, Park K. Oral protein delivery: Current status and future prospect. React Funct Polym. 2011;71(3):280–287. | ||
Tangso KJ, Lindberg S, Hartley PG, Knott R, Spicer P, Boyd BJ. Formation of liquid-crystalline structures in the bile salt-chitosan system and triggered release from lamellar phase bile salt-chitosan capsules. ACS Appl Mater Interfaces. 2014;6(15):12363–12371. | ||
Chen Y, Ma P, Gui S. Cubic and hexagonal liquid crystals as drug delivery systems. Biomed Res Int. 2014;2014:815981. | ||
Murgia S, Falchi AM, Mano M, et al. Nanoparticles from lipid-based liquid crystals: emulsifier influence on morphology and cytotoxicity. J Phys Chem B. 2010;114(10):3518–3125. | ||
Spillmann CM, Naciri J, Algar WR, Medintz IL, Delehanty JB. Multifunctional liquid crystal nanoparticles for intracellular fluorescent imaging and drug delivery. ACS Nano. 2014;8(7):6986–6997. | ||
Swarnakar NK, Thanki K, Jain S. Lyotropic liquid crystalline nanoparticles of CoQ10: implication of lipase digestibility on oral bioavailability, in vivo antioxidant activity, and in vitro-in vivo relationships. Mol Pharm. 2014;11(5):1435–1449. | ||
Swarnakar NK, Thanki K, Jain S. Bicontinuous cubic liquid crystalline nanoparticles for oral delivery of Doxorubicin: implications on bioavailability, therapeutic efficacy, and cardiotoxicity. Pharm Res. 2014;31(5):1219–1238. | ||
Misra R, Mohanty S. Sustained release of methotrexate through liquid-crystalline folate nanoparticles. J Mater Sci Mater Med. 2014;25(9):2095–2109. | ||
Mei Z, Li X, Wu Q, Hu S, Yang X. The research on the anti-inflammatory activity and hepatotoxicity of triptolide-loaded solid lipid nanoparticle. Pharmacol Res. 2005;51(4):345–351. | ||
Madheswaran T, Baskaran R, Thapa RK, et al. Design and in vitro evaluation of finasteride-loaded liquid crystalline nanoparticles for topical delivery. AAPS PharmSciTech. 2013;14(1):45–52. | ||
Heurtault B, Saulnier P, Pech B, Proust JE, Benoit JP. A novel phase inversion-based process for the preparation of lipid nanocarriers. Pharm Res. 2002;19(6):875–880. | ||
Izquierdo P, Esquena J, Tadros TF, et al. Formation and stability of nano-emulsions prepared using the phase inversion temperature method. Langmuir. 2002;18(1):26–30. | ||
Anton N, Benoit JP, Saulnier P. Design and production of nanoparticles formulated from nano-emulsion templates-a review. J Control Release. 2008;128(3):185–199. | ||
Zhou H, Yue Y, Liu G, et al. Characterisation and Skin Distribution of Lecithin-Based Coenzyme Q10-Loaded Lipid Nanocapsules. Nanoscale Res Lett. 2010;5(10):1561–1569. | ||
Peltier S, Oger JM, Lagarce F, Couet W, Benoit JP. Enhanced oral paclitaxel bioavailability after administration of paclitaxel-loaded lipid nanocapsules. Pharm Res. 2006;23(6):1243–1250. | ||
Hureaux J, Lagarce F, Gagnadoux F, et al. Lipid nanocapsules: ready-to-use nanovectors for the aerosol delivery of paclitaxel. Eur J Pharm Biopharm. 2009;73(2):239–246. | ||
Song H, Zhang J, Han Z, et al. Pharmacokinetic and cytotoxic studies of pegylated liposomal daunorubicin. Cancer Chemother Pharmacol. 2006;57(5):591–598. | ||
Yoon G, Park JW, Yoon I-S. Solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs): recent advances in drug delivery. J Pharm Invest. 2013;43(5):353–362. | ||
Tongcher O, Sigel R, Landfester K. Liquid crystal nanoparticles prepared as miniemulsions. Langmuir. 2006;22(10):4504–4511. | ||
Rossi J, Leroux J-C. Principles in the Development of Intravenous Lipid Emulsions. In: Wasan KM, editor. Role of Lipid Excipients in Modifying Oral and Parenteral Drug Delivery: Basic Principles and Biological Examples. Hoboken, New Jersey: Wiley-Interscience; 2006:88–123. | ||
Rensen PC, Herijgers N, Netscher MH, Meskers SC, Van Eck M, Van Berkel TJ. Particle size determines the specificity of apolipoprotein E-containing triglyceride-rich emulsions for the LDL receptor versus hepatic remnant receptor in vivo. J Lipid Res. 1997;38(6):1070–1084. | ||
Bae IH, Choi JK, Chough C, et al. Potent Hepatitis C Virus NS5A Inhibitors Containing a Benzidine Core. ACS Med Chem Lett. 2014;5(3):255–258. | ||
Bouwstra JA, Gooris GS, Dubbelaar FE, Ponec M. Phase behavior of stratum corneum lipid mixtures based on human ceramides: the role of natural and synthetic ceramide 1. J Invest Dermatol. 2002;118(4):606–617. | ||
Hua S. Comparison of in vitro dialysis release methods of loperamide-encapsulated liposomal gel for topical drug delivery. Int J Nanomedicine. 2014;9:735–744. | ||
Fernandez P, André V, Rieger J, Kühnle A. Nano-emulsion formation by emulsion phase inversion. Colloids Surf A. 2004;251(1–3):53–58. | ||
Guillaume O, Garric X, Lavigne JP, Van Den Berghe H, Coudane J. Multilayer, degradable coating as a carrier for the sustained release of antibiotics: Preparation and antimicrobial efficacy in vitro. J Control Release. 2012;162(3):492–501. | ||
Hwang JJ, Iyer SN, Li LS, Claussen R, Harrington DA, Stupp SI. Self-assembling biomaterials: liquid crystal phases of cholesteryl oligo(L-lactic acid) and their interactions with cells. Proc Natl Acad Sci U S A. 2002;99(15):9662–9667. | ||
Montenegro L, Campisi A, Sarpietro MG, et al. In vitro evaluation of idebenone-loaded solid lipid nanoparticles for drug delivery to the brain. Drug Dev Ind Pharm. 2011;37(6):737–746. | ||
Westesen K, Bunjes H. Do nanoparticles prepared from lipids solid at room temperature always possess a solid lipid matrix? Int J Pharm. 1995;115(1):129–131. | ||
Tadros TF. Emulsion Formation, Stability, and Rheology. In: Tadros TF, editor. Emulsion Formation and Stability. Weinheim, Germany: Wiley-VCH; 2013:1–75. | ||
Hirayama F, Uekama K. Cyclodextrin-based controlled drug release system. Adv Drug Deliv Rev. 1999;36(1):125–141. | ||
Müller RH, Radtke M, Wissing SA. Nanostructured lipid matrices for improved microencapsulation of drugs. Int J Pharm. 2002;242(1–2):121–128. | ||
Amoozgar Z, Yeo Y. Recent advances in stealth coating of nanoparticle drug delivery systems. Wiley Interdisciplinary Reviews Nanomedicine and Nanobiotechnology. 2012;4(2):219–233. | ||
Tamilvanan S. Oil-in-water lipid emulsions: implications for parenteral and ocular delivering systems. Prog Lipid Res. 2004;43(6):489–533. |
Supplementary materials
General information
High-performance liquid chromatography analyses were performed with an HP1100 system (Agilent Technologies, Santa Clara, CA, USA), comprised of HP Chemstation software, an auto sampler, a quaternary pump, and a photodiode array detector. C18 Vydac 218TP54 column 250×4.6 mm in diameter (5 μm particle size) was employed for the analysis. The mobile phase for the analysis was 0.1% trifluoroacetic acid in water (A) and acetonitrile (B) at a flow rate of 1 mL/min at 20°C. Method: 100% A and 0% B (0 minutes), 100% B (10 minutes), 100% B (20 minutes), 0% B (22 minutes), 0% B (25 minutes).
 | Table S1 Body weights and dose volumes of Sprague Dawley rats for the pharmacokinetic study |
 | Table S2 Body weights and dose volumes of Sprague Dawley rats for the tissue distribution study |
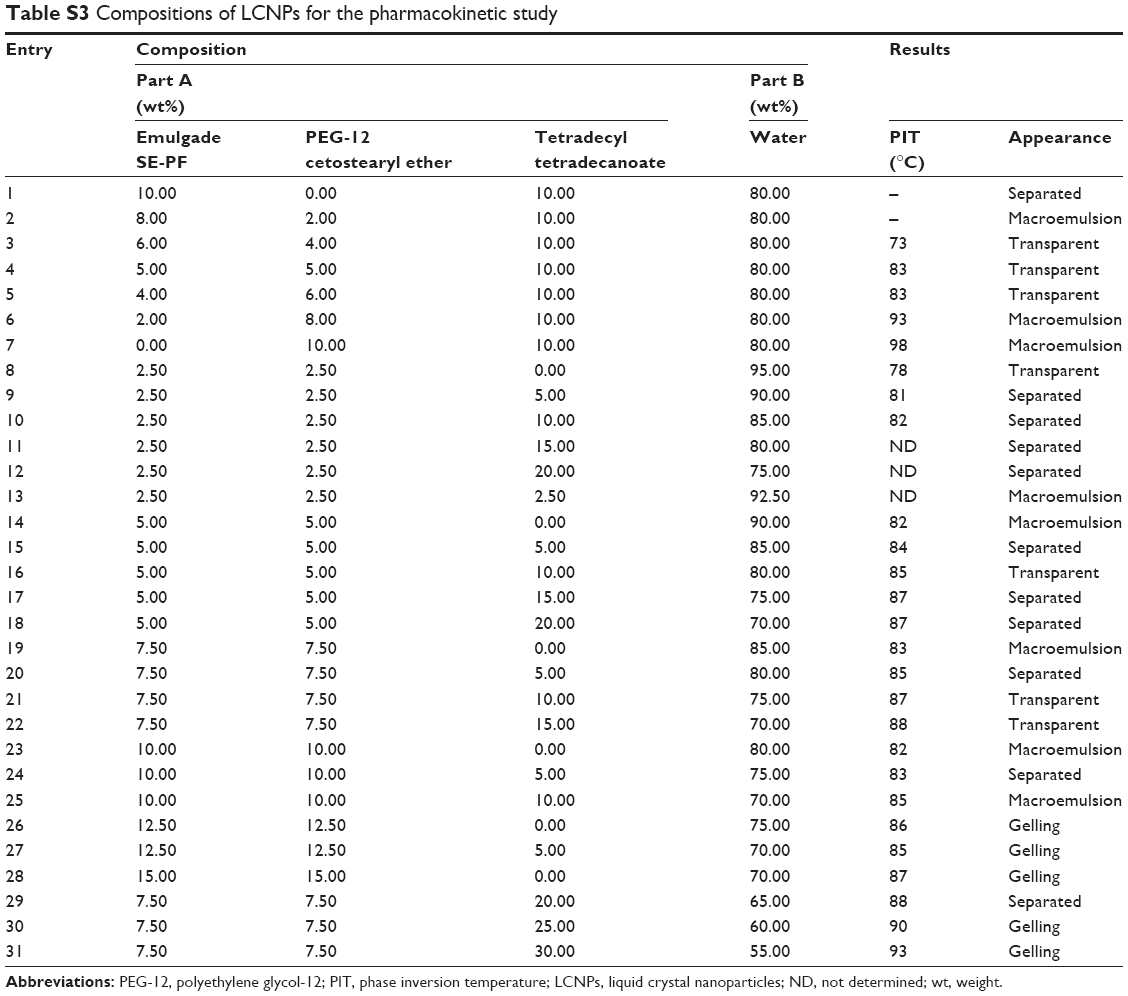 | Table S3 Compositions of LCNPs for the pharmacokinetic study |
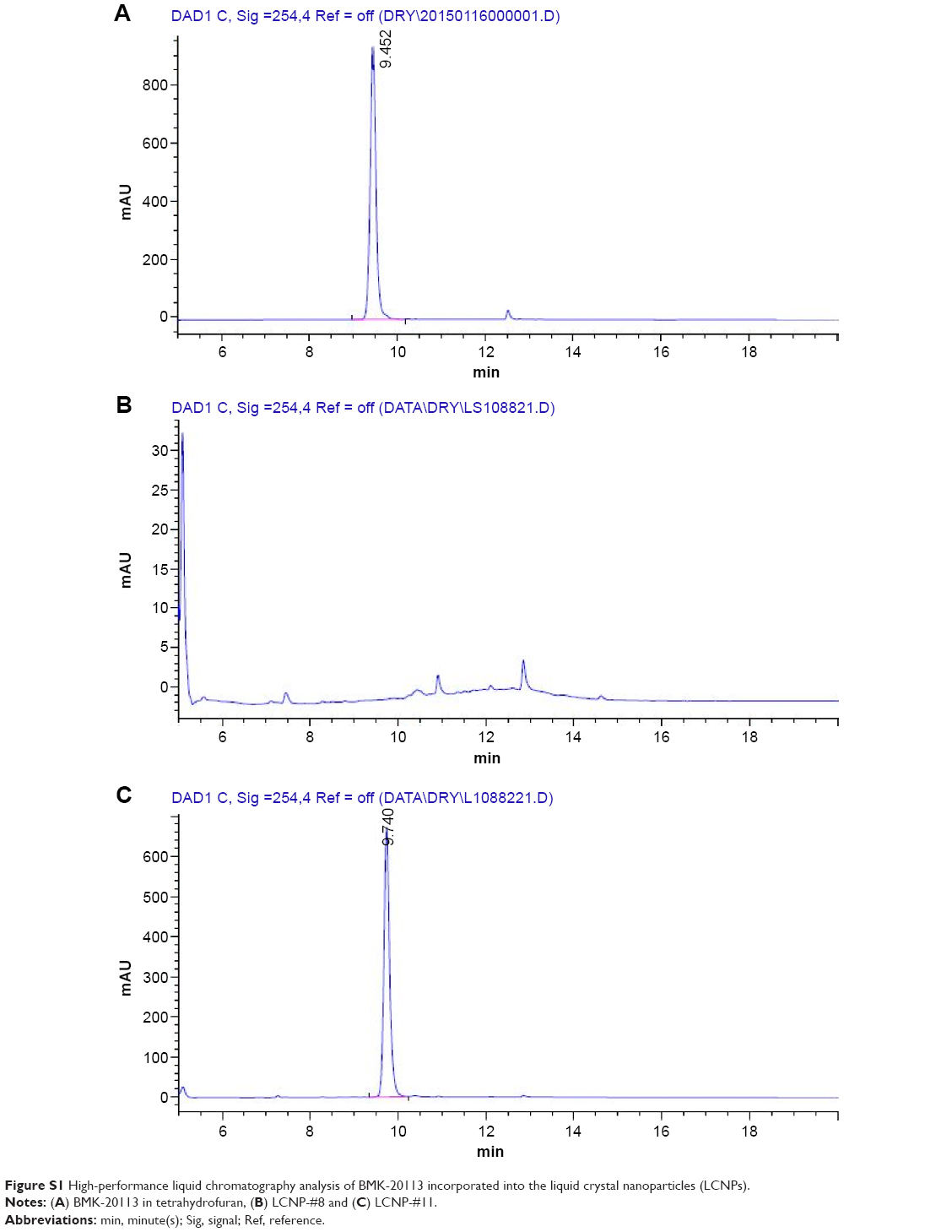 | Figure S1 High-performance liquid chromatography analysis of BMK-20113 incorporated into the liquid crystal nanoparticles (LCNPs). |
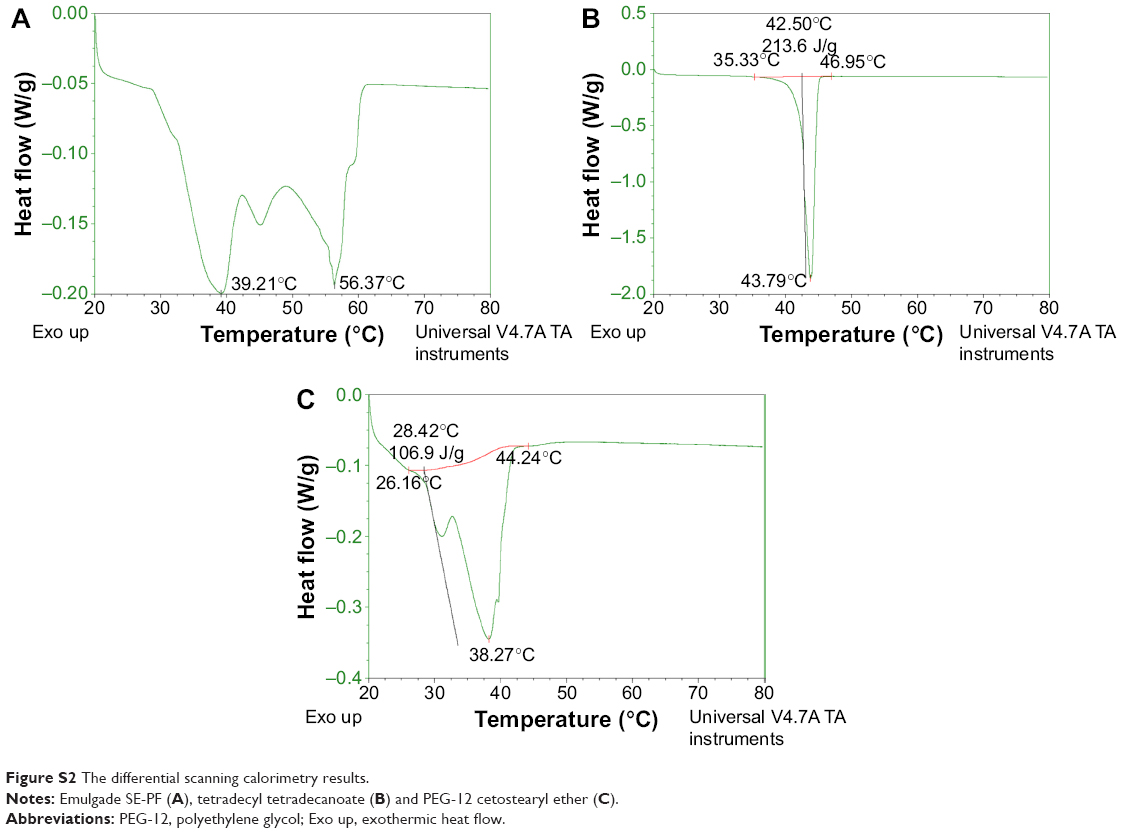 | Figure S2 The differential scanning calorimetry results. |
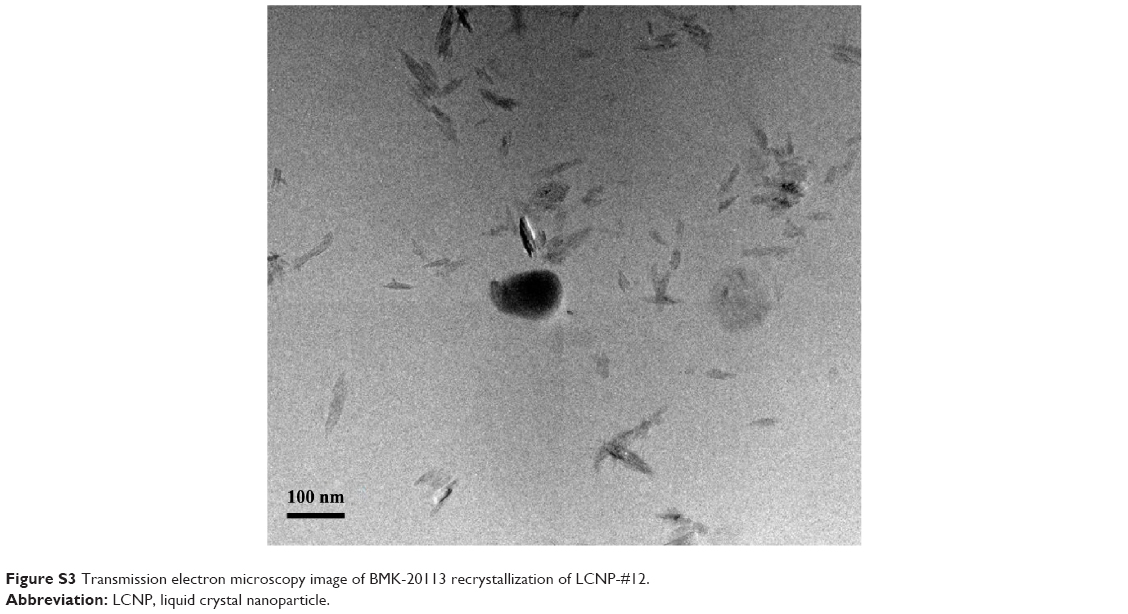 | Figure S3 Transmission electron microscopy image of BMK-20113 recrystallization of LCNP-#12. |
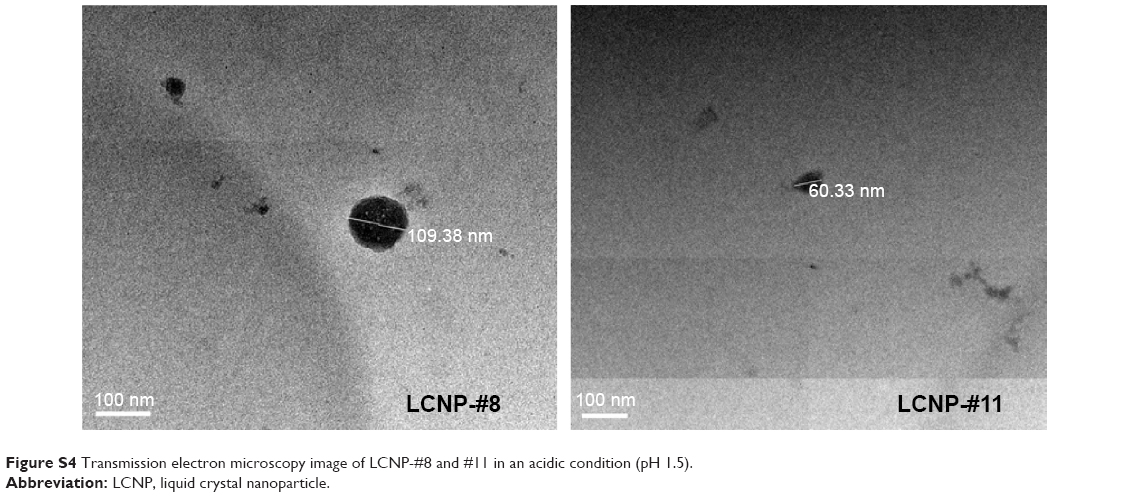 | Figure S4 Transmission electron microscopy image of LCNP-#8 and #11 in an acidic condition (pH 1.5). |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.