Back to Journals » International Journal of Nanomedicine » Volume 12
Liposomes derivatized with multimeric copies of KCCYSL peptide as targeting agents for HER-2-overexpressing tumor cells
Authors Ringhieri P, Mannucci S, Conti G, Nicolato E, Fracasso G ![]() , Marzola P
, Marzola P ![]() , Morelli G, Accardo A
, Morelli G, Accardo A ![]()
Received 26 May 2016
Accepted for publication 29 September 2016
Published 13 January 2017 Volume 2017:12 Pages 501—514
DOI https://doi.org/10.2147/IJN.S113607
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Paola Ringhieri,1 Silvia Mannucci,2 Giamaica Conti,2 Elena Nicolato,2 Giulio Fracasso,3 Pasquina Marzola,4 Giancarlo Morelli,1 Antonella Accardo1
1Department of Pharmacy and Interuniversity Research Centre on Bioactive Peptides (CIRPeB), University of Naples “Federico II”, Napoli, 2Department of Neurological Biomedical and Movement Sciences, 3Section of Immunology, Department of Medicine, 4Department of Informatics, University of Verona, Verona, Italy
Abstract: Mixed liposomes, obtained by coaggregation of 1,2-dioleoyl-sn-glycero-3-phosphocholine and of the synthetic monomer containing a gadolinium complex ([C18]2DTPA[Gd]) have been prepared. Liposomes externally decorated with KCCYSL (P6.1 peptide) sequence in its monomeric, dimeric, and tetrameric forms are studied as target-selective delivery systems toward cancer cells overexpressing human epidermal growth factor receptor-2 (HER-2) receptors. Derivatization of liposomal surface with targeting peptides is achieved using the postmodification method: the alkyne-peptide derivative Pra-KCCYSL reacts, through click chemistry procedures, with a synthetic surfactant modified with 1, 2, or 4 azido moieties previously inserted in liposome formulation. Preliminary in vitro data on MDA-MB-231 and BT-474 cells indicated that liposomes functionalized with P6.1 peptide in its tetrameric form had better binding to and uptake into BT-474 cells compared to liposomes decorated with monomeric or dimeric versions of the P6.1 peptide. BT-474 cells treated with liposomes functionalized with the tetrameric form of P6.1 showed high degree of liposome uptake, which was comparable with the uptake of anti-HER-2 antibodies such as Herceptin. Moreover, magnetic MRI experiments have demonstrated the potential of liposomes to act as MRI contrast agents.
Keywords: anti-HER2 liposomes, target peptide, KCCYSL peptide, breast cancer, click chemistry, branched peptides
Introduction
Human epidermal growth factor receptor-2 (HER-2 or ErbB-2) is a 185 kDa transmembrane glycoprotein with an N-terminal extracellular ligand binding domain and an intracellular tyrosine kinase domain. It belongs to a family of receptors that includes four structurally related members: HER-1 (ErbB-1 or EGFR), HER-2, HER-3 (ErbB-3), and HER-4 (ErbB-4).1 HER-2 plays a central role in many biological processes mediated by EGFR family and is expressed at baseline levels in normal cells. Its overexpression is associated with a variety of cancers, such as breast and ovarian cancers (approximately 25%–30% of all reported cases), gastric cancer (6%–35%), and prostate cancer.2 This high incidence in many tumors, together with the extreme accessibility of its extracellular domain, renders HER-2 an attractive candidate for the development of selective anticancer drugs. The humanized monoclonal antibody anti-HER-2 (Herceptin or trastuzumab), which binds the extracellular ligand binding domain of the receptor, is actually the most successful therapy available in clinics for the treatment of HER-2-positive breast cancers. Unfortunately, a significant number of patients are resistant to the treatment initially or become resistant eventually.3
Herceptin molecule or its fragments (Fab or ScFv) have been also used to decorate nanoparticles or liposomes for the selective delivery of anticancer drugs such as doxorubicin4 or paclitaxel5 to HER-2-positive tumors. Compared with large molecules such as antibodies and their derivatives, smaller peptide molecules appear to be more attractive targeting moieties because of their less expensive and simple production, lack of immunogenicity, chemical stability, and flexibility in the choice of conjugation procedures to the carrier’s surface.2 Actually, only three peptide sequences, AHNP, LTVSPWY, and KCCYSL (called P6.1), have been found to selectively recognize HER-2. The first one is an anti-HER-2 peptide mimetic obtained by rational design by Park et al6 and is based on the structure of Herceptin,7 whereas the other two peptides were derived from phage display libraries and chosen via affinity selection.8,9 The P6.1 peptide, derivatized with different metal chelators (DOTA, NOTA, CB-TE2A, DAP) and labeled with radionuclide metal ions (111In, 64Cu, 99mTc), has been successfully used for in vivo imaging of HER-2 overexpressing tumor models.10–13 Single-positron emission computed tomography/computed tomography studies showed that radioconjugated peptides were able to accumulate specifically at the tumor site, with Kd ranging from 30 to 45 nM, with minimal nonspecific retention in other organs. This peptide sequence has also been proposed for the selective delivery of supramolecular carriers such as copolymeric micelles14 or pH-tunable liposomes15 for diagnostic or therapeutic applications.
Here, we report the synthesis, the structural characterization, and the in vitro binding properties of novel, target-selective liposome-based delivery systems. Mixed liposomes, composed of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and the amphiphilic gadolinium complex (C18)2DTPA(Gd), were externally decorated with the P6.1 peptide sequence. Liposomes with the dimeric or tetrameric form of P6.1 externally were also prepared and characterized to evaluate the effect of multimeric copies of the peptide on the in vitro receptor binding ability. The binding properties of functionalized liposomes were tested in vitro in two breast cancer cell lines, namely, MD-MB-231, characterized by low expression of HER-2, and BT-474 that overexpresses HER-2. Several papers have reported the capacity of peptides synthesized in their branched form to enhance resistance toward proteases and to increase linear peptide biological activity through multivalent binding.16–18 Recently, some evidence that these properties are also maintained when the peptide is bounded on the external liposomal surface have been demonstrated.19,20 Liposomes were chosen as the vector since they can accommodate large quantities of relevant therapeutic drugs in their lumen. Moreover, the addition of a large amount of the amphiphilic gadolinium complex (C18)2DTPA(Gd) to the liposomal formulation allows its use a potential contrast agent for magnetic resonance imaging (MRI). Our preliminary results demonstrate the binding and the uptake of liposomes exposing the tetrameric form of P6.1 on BT-474 cells and also demonstrate their in vitro and in vivo contrast agent properties in MRI; these two features make the described delivery system an appealing candidate for future investigation so as to develop a target-selective theranostic agent.
Materials and instrumentation
Fmoc-protected amino acid derivatives, coupling reagents, and Rink amide 4-methylbenzhydrylamine (MBHA) resin were purchased from Calbiochem-Novabiochem (Laufelfingen, Switzerland). Fmoc-L-propargylglycine (Fmoc-Pra-OH) was bought from Neosystem (Strasbourg, France). The 14-azido-5-oxo-3,9,12-trioxa-6-azatetradecan-1-oic acid (N3-Peg(9)-COOH) was bought from Iris Biotech GmbH (Marktredwitz, Germany), and diethylenetriamine-N,N,N″,N″-tetra-tert-butyl acetate-N′-acetic acid [DTPA(OtBu)4] was bought from CheMatech (Dijon, France). DOPC and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Rho-PE) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). N,N-dioctadecylsuccinamic acid was synthesized according to the manufacturer’s instruction.21 Sephadex G-50 columns were purchased from GE Healthcare Europe GmbH (Milan, Italy). All other chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA), Fluka (Buchs, Switzerland), or LabScan (Stillorgan, Dublin, Ireland) and were used as received, unless otherwise stated. Preparative high-performance liquid chromatography (HPLC) was carried out on an LC8 Shimadzu HPLC system (Shimadzu Corporation, Kyoto, Japan) equipped with a ultraviolet lambda-Max Model 481 detector. For the P6.1 peptide, a Phenomenex (Torrance, CA, USA) C18 (300 Å, 250×21.20 mm, 5 μ) column eluted with H2O/0.1% trifluoroacetic acid (TFA) (A) and CH3CN/0.1% TFA (B) from 5% to 70% over 20 min at a flow rate of 20 mL min−1, while for all the surfactant scaffolds, a Phenomenex C4 (300 Å, 250×21.20 mm, 5 μ) column eluted with H2O/0.1% TFA (A) and CH3CN/0.1% TFA (B) from 20% to 95% over 20 min at 20 mL min−1 flow rate was used. Purity and identity of the products were assessed by analytical liquid chromatography/mass spectrometry (LC/MS) analyses by using Finnigan Surveyor MSQ single quadrupole electrospray ionization (Finnigan; Thermo Electron Corporation, San Jose, CA, USA), column: C18-Phenomenex eluted with H2O/0.1% TFA (A) and CH3CN/0.1% TFA (B) from 5% to 70% over 30 min for P6.1 peptide, C4-Phenomenex eluted with H2O/0.1% TFA (A) and CH3CN/0.1% TFA (B) from 20% to 95% over 20 min, both at a flow rate of 0.8 mL min−1. Ultraviolet–visible measurements were performed on a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific Inc; Wilmington, DE, USA) equipped with a 1.0 cm quartz cuvette (Hellma, Müllheim, Germany).
Solid-phase synthesis
Synthesis of the P6.1-alkyne peptide derivative
Pra-Lys-Cys-Cys-Tyr-Ser-Leu-NH2 (Pra-P6.1), schematically presented in Figure 1, was synthesized in solid phase under standard conditions by using the Fmoc/tBu strategy. Rink amide MBHA resin (0.73 mmol g−1, 0.1 mmol, 0.140 g) was used as a polymeric support. Fmoc-protected amino acids (4 equivalent relative to resin loading) were coupled according to the benzotriazol-1-yl-oxy-tris-pyrrolidinophosphonium (PyBop)/1-hydroxybenzotriazole (HOBt)/N,N-diisopropylethylamine (DIPEA) method. In brief, Fmoc amino acid (4 equivalent), PyBOP (4 equivalent), HOBt (4 equivalent), and DIPEA (8 equivalent) were dissolved in DMF and added to the resin. The Fmoc protecting group was removed with 30% piperidine in DMF (v/v), and all couplings were performed twice for 1 h. Fmoc-Pra-OH was coupled once for 45 min with 2 equivalents of PyBop/HOBt and 4 equivalents of DIPEA. The Fmoc protecting group on the N-terminus of propargylglycine was removed by washing the resin five times with 40% piperidine and 2% 1,8-diazabicyclo[5.4.0]undec-7-ene in DMF (v/v) for 10 min. At the end of the synthesis, the peptide was fully deprotected and cleaved from the resin with a cleavage cocktail containing TFA (94%), triisopropylsilane (TIS, 1%), ethanedithiol (EDT, 2.5%), and water (2.5%) as scavengers at room temperature for 2 h; then, it was precipitated with ice-cold ethyl ether, filtered, dissolved in water, and lyophilized. The crude alkyne derivative was purified by reverse-phase HPLC (RP-HPLC). The final product yield was 65%, and it was analyzed by LC/MS.
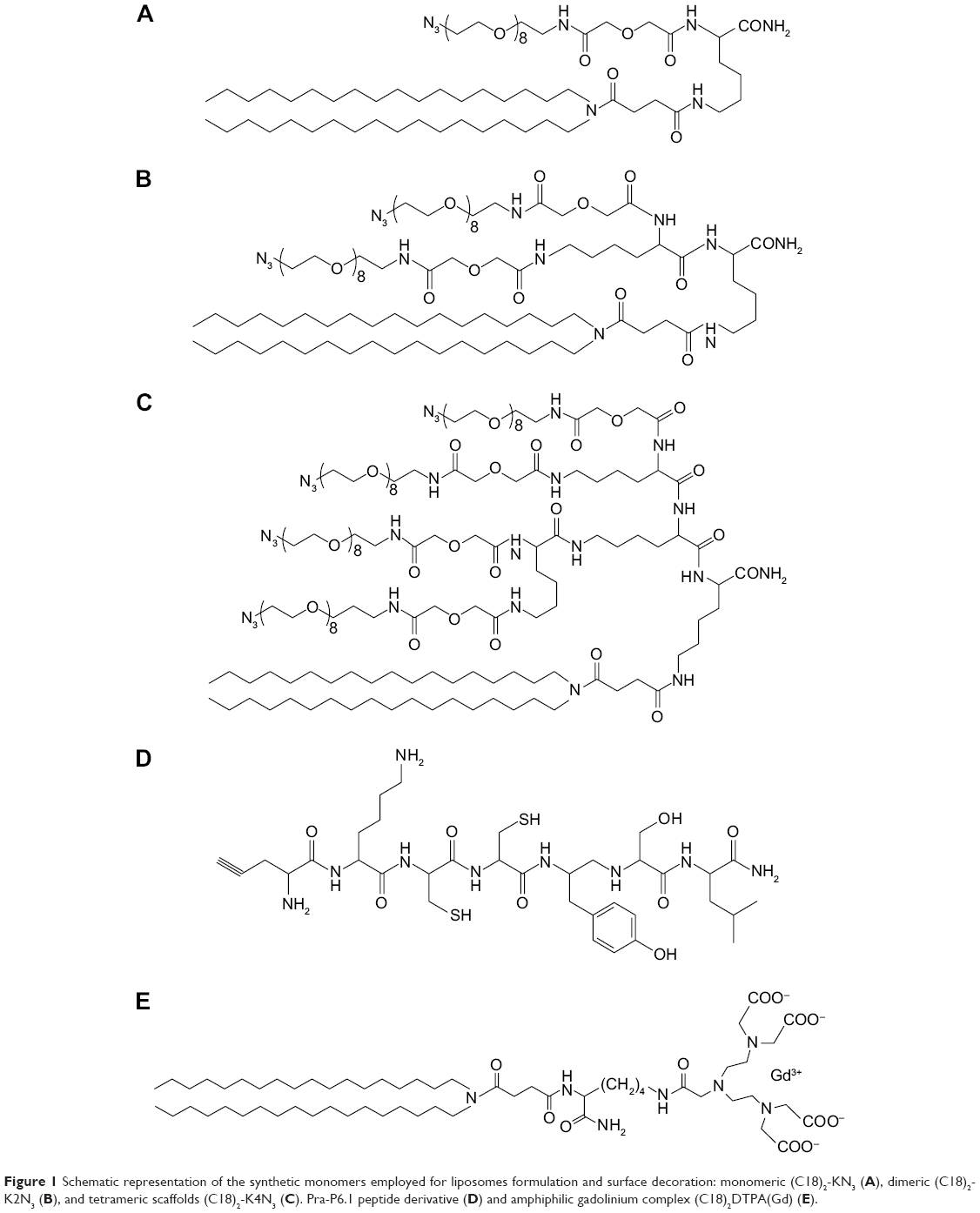 | Figure 1 Schematic representation of the synthetic monomers employed for liposomes formulation and surface decoration: monomeric (C18)2-KN3 (A), dimeric (C18)2-K2N3 (B), and tetrameric scaffolds (C18)2-K4N3 (C). Pra-P6.1 peptide derivative (D) and amphiphilic gadolinium complex (C18)2DTPA(Gd) (E). |
Pra-P6.1: MS (ESI+): Rt =11.21 min, m/z (%) calcd for C35H52N9O9S2 [M + H+]:806.9 amu; found: 808.0 uma.
Synthesis of the surfactant scaffolds
Synthesis of (C18)2-KN3 {[Azide-Peg9-Lys[C(O)CH2CH2C(O)N-(C18H37)2]-amide}
(C18)2-KN3 mono-azido amphiphilic derivative, schematically depicted in Figure 1B, was synthesized according to experimental procedures reported previously.22
Synthesis of (C18)2-K2N3 {(Azide-Peg9)2-Lys-Lys[C(O)CH2CH2C(O)N-(C18H37)2]-amide} and (C18)2-K4N3, {[(Azide-Peg9)2-Lys]2-Lys-Lys[C(O)CH2CH2C(O)N-(C18H37)2]-amide}
(C18)2-K2N3 and (C18)2-K4N3 di- and tetra-azido amphiphilic derivatives, presented in Figure 1C and 1D, were synthesized (Scheme 1) according to the standard procedures described in the “Solid-phase synthesis” section for Pra-P6.1 peptide. The Rink amide MBHA resin (substitution 0.73 mmol g−1, scale 0.2 mmol, 0.280 g) was used as the solid-phase support. After Fmoc removal from the resin and the coupling of Fmoc-Lys(Mtt)-OH, the resin was washed four times with dichloromethane (DCM), and then the 4-methyltrityl (Mtt) protecting group on the Nε-amino functional group of lysine was removed by treatment with DCM-TIS-TFA (94/5/1, v/v/v, 5.0 mL) for 2 min. This procedure was repeated several times until the solution became colorless. Then, the resin was washed three times with DCM and three times with DMF, and N,N-dioctadecylsuccinamic acid (0.249 g, 0.4 mmol) was coupled according to the previously published procedure.23 The Fmoc-Lys(Fmoc)-OH residue was coupled successively on the principal chain. The two Fmoc protecting groups, on the Nα and Nε, were then simultaneously removed by washing the resin five times with 30% piperidine in DMF (v/v) for 10 min. Then, the peptide-resin was split in two reactors (each one containing 0.1 mmol of peptide-resin), and the synthesis was manually completed. For the synthesis of the dimeric scaffold (C18)2-K2N3, N3-Peg(9)-COOH was coupled with DMF overnight using two equivalents of the azido-PEGylated derivative for each NH2 group. For the synthesis of the tetrameric scaffold (C18)2-K4N3, the peptide chain was further elongated by coupling another lysine residue both on the α and the ε amino groups; then, the scaffold was completed by coupling two azido-PEGylated moieties for each N-terminal lysine. At the end of the synthesis, the scaffolds were cleaved from the resin using a TFA/TIS/DTT/H2O mixture (92.5/2.5/2.5/2.5, v/v/v/v) at room temperature for 2 h, followed by precipitation with ice-cold water. The sample was then filtered, suspended in water, and lyophilized.
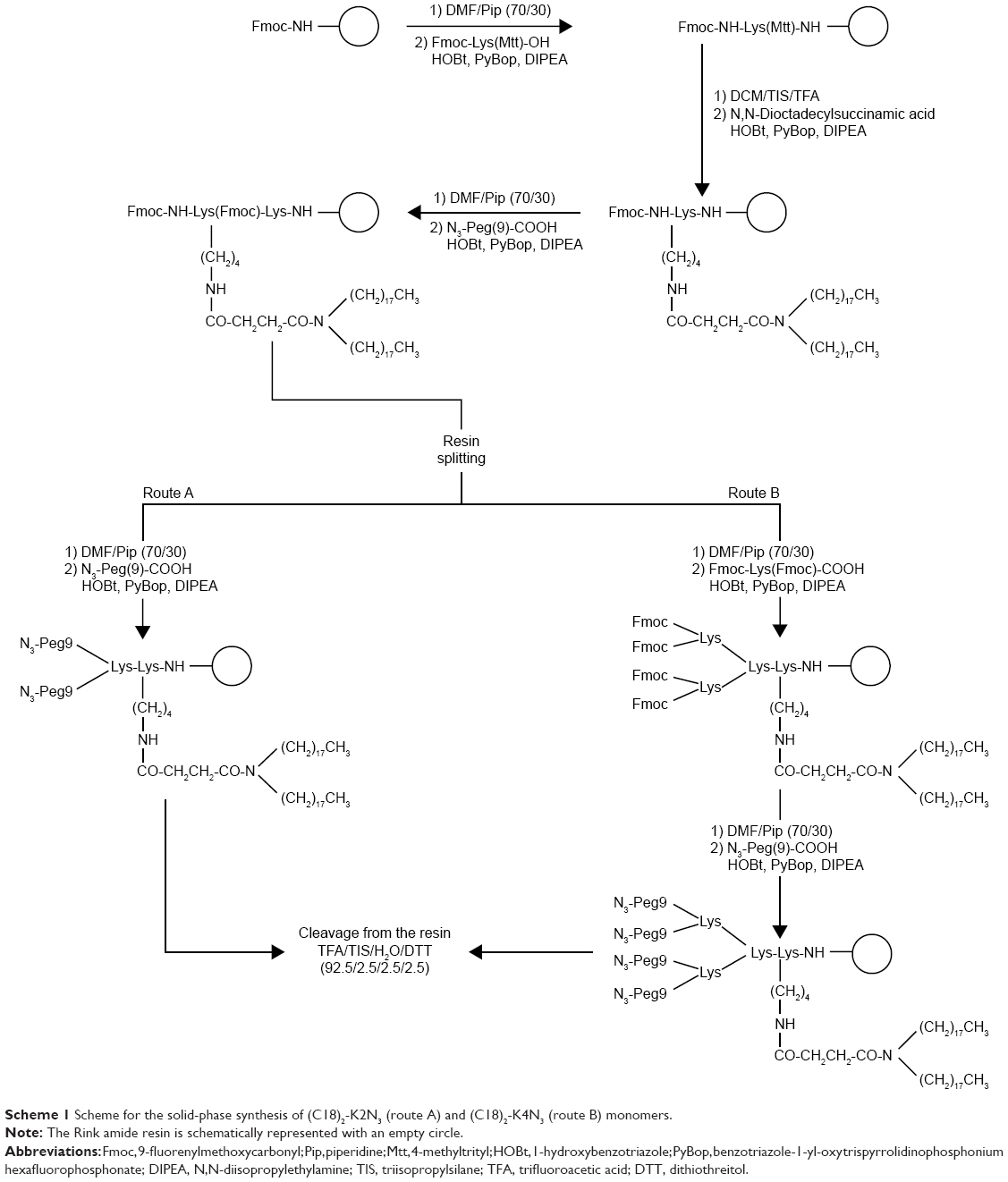 | Scheme 1 Scheme for the solid-phase synthesis of (C18)2-K2N3 (route A) and (C18)2-K4N3 (route B) monomers. |
(C18)2-K2N3: 1H NMR (400 MHz, CDCl3/CD3OD 50:50): d=4.43 (m, 1H; CH Lysα), 4.38 (m, 1H; CH Lysα), 4.1 (m, 8H; OCH2CONH, OCH2CONH), 3.70 (s, 56H; OCH2CH2O), 3.4 (m, 8H; N3CH2CH2O, NHCH2CH2O), 3.25–3.20 (m, 4H; NCH2), 3.24 (t, 4H; CH2Lysε), 3.22 (t, 4H; N3CH2CH2O), 2.64–2.49 (m, 4H; NHCOCH2CH2CO), 1.71 (m, 4H; CH2Lysβ), 1.57 (m, 4H; CH2Lysδ),1.49 (m, 4H; CH2Lysγ), 1.40 (m, 4H; RCH2CH3), 1.26 (m, 60H; CH2 aliphatic), 0.88 ppm (t, 6H; CH3); MS (ESI+): m/z (%) calcd for C96H184N14O26 [M + H+]:1950.5; found: [M + H+]: 1951.7; [M + 2H+]/2: 976.4.
(C18)2-K4N3: 1H NMR (400 MHz, CDCl3/CD3OD 50:50): d=4.40 (m, 4H; CH Lysα), 4.1 (m, 16H; OCH2CONH, OCH2CONH), 3.70 (s, 112H; OCH2CH2O), 3.4 (m, 16H; N3CH2CH2O, NHCH2CH2O), 3.25–3.20 (m, 4H; NCH2), 3.24 (t, 8H; CH2Lysε), 3.22 (t, 8H; N3CH2CH2O), 2.64-2.49 (m, 4H; NHCOCH2CH2CO), 1.71 (m, 8H; CH2Lysβ), 1.57 (m, 8H; CH2Lysδ),1.49 (m, 8H; CH2Lysγ), 1.40 (m, 4H; RCH2CH3), 1.26 (m, 60H; CH2 aliphatic), 0.88 ppm (t, 6H; CH3); MS (ESI+): m/z (%) calcd for C152H288N26O50 [M + H+]:3279.7; found: [M + 2H+]/2: 1641.3; [M + 3H+]/3: 1094.5.
Synthesis of (C18)2DTPA(Gd) {(C18H37)2NCO(CH2)2COLys[DTPA(Gd)]CO-NH2} amphiphilic gadolinium complex
The synthesis and the complexation with Gd(III) ions were performed according to the previously described procedure.24
Liposome preparation
All liposomal solutions (nonfunctionalized liposomes [Lipo-NT], monomeric [Lipo-M], dimeric [Lipo-D], and tetrameric [Lipo-T]) were prepared in 100 mM phosphate buffer at pH 7.4 with 0.9% wt NaCl by the thin film method.25 Briefly, the amphiphiles were dissolved in 2 mL of a MeOH/CHCl3 (50/50, v/v) mixture, and then the organic solvents were discarded under a gentle stream of N2 to obtain a thin film. The relative amount of each surfactant (expressed in mol%) used in the film preparation is reported in Table 1. The film was rehydrated in the phosphate buffer at 4 mM total lipid concentration, and the resulting suspension was sonicated for 30 min to allow liposome formation. Subsequently, all the liposomes were extruded 10 times at room temperature using a thermobarrel extruder system (Northern Lipids Inc., Vancouver, BC, Canada) under nitrogen through a polycarbonate membrane (Nucleopore Track Membrane 25 mm, Whatman, Brentford, UK) with 0.1 μm pore size. Liposomes labeled with rhodamine were prepared by adding 1% mol of Rho-PE to the phospholipid mixture during the lipid film preparation phase.
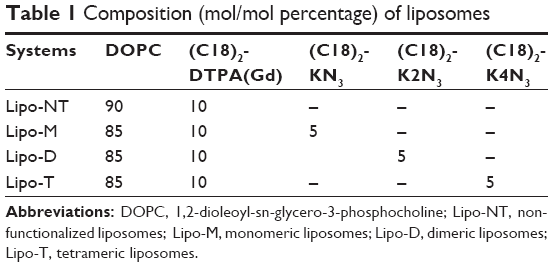 | Table 1 Composition (mol/mol percentage) of liposomes |
‘‘Click’’ chemistry reaction on liposomes
Lipo-M, Lipo-D, and Lipo-T were externally functionalized with several copies of P6.1 peptide using the click chemistry reaction. For the reaction, 1 equivalent of the peptide derivative Pra-P6.1, 2 equivalents of sodium L-ascorbate, and 0.5 equivalent of CuCl2 with respect to the azido moiety were added to each liposomal suspension in the exact order reported. The reaction mixture was stirred at 40°C for 30 min and then left overnight at room temperature. Unreacted peptide and copper ions were removed by gel-filtration chromatography using a Sephadex G-50 column pre-equilibrated with 100 mM phosphate buffer at pH 7.4 with 0.9% wt NaCl. The amount of peptide effectively linked to the liposomal surface was estimated indirectly from the concentration of free Pra-P6.1 eluted from the column by using an RP-HPLC calibration curve. The percentage of functionalization was calculated as the ratio of bound P6.1 to the total amount used for the reaction.
Dynamic light scattering characterization
Liposomes were characterized by dynamic light scattering (DLS using a Zetasizer Nano ZS (Malvern Instruments, Westborough, MA, USA) equipped with a noninvasive backscatter system that allows measurements with a 173° backscatter angle. Settings for the determination of the mean diameter and polydispersity index (PI) of all the liposomal suspensions were as follows: measurement position (mm): 4.65; attenuator: 8; temperature: 25°C; cell: disposable sizing cuvette. DLS samples were prepared at 2.0×10−4 M concentration and centrifuged at room temperature at 13,000 rpm for 5 min. For each batch, hydrodynamic radius and size distribution were taken as the mean of three measurements, and the final values were calculated as the mean of three different batches.
Cells
Human breast cancer cell lines MDA-MB-231 and BT-474 were purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2 and 90% of humidity; cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Invitrogen, NY, USA) and antibiotics (0.1 mg mL−1 streptomycin and 100 units mL−1 penicillin G, Sigma-Aldrich). HER-2 expression on MDA-MB-231 and BT-474 cells was assessed by flow cytometry.
In vitro viability assay
Viability of the MDA-MB-231 and BT-474 cancer cells was assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) test. Cells were plated at a density of 5×103 cells per well in 96-well plates and incubated at 37°C in a mixture of air and 5% CO2. After 24 h, the medium was replaced with fresh medium containing 0.02, 0.04, and 0.1 mM of sterilized liposomes, which correspond to a Gd-complex concentration of 0.002, 0.004, and 0.01 mM, respectively. After 1, 3, and 6 h of incubation, 100 μL of MTT solution (at 5 mg mL−1 concentration, Sigma, Italy) was added to each well; the plates were then incubated for an additional 4 h (37°C, 5% CO2). Afterward, the formazan crystals were dissolved in 100 μL of dimethyl sulfoxide (DMSO) (Sigma, Italy). The multiwell plate was placed into a monochromator (ChroMate Awareness Technology) for the measurement of absorbance at 570 and 630 nm. Three measurements of optical density (OD) were recorded for each sample, and cell viability (CV)% was calculated using the following equation: CV% = (ODsample/ODcontrol) ×100.
In vitro binding assay
BT-474 and MDA-MB-231 cell lines were detached from culture flasks using 1% trypsin for 5 min and harvested in 15 mL sterile conic tubes. Then, they were pelleted, counted, and suspended in sterile phosphate-buffered saline (PBS). Then, cell lines were treated with liposomes ([Gd-complex] =0, 0.002, 0.004, 0.01, 0.02, 0.2, 0.5 mM) functionalized with monomeric, dimeric, and tetrameric forms of P6.1 and labeled with rhodamine. Incubation conditions were 4°C for 60 min so as to evaluate liposome binding to cell surface while avoiding their internalization. After incubation, the binding capability of liposomes was determined using a flow cytometer BD FACSCanto II (BD Biosciences, San Jose, CA, USA), equipped with three different lasers. For this experiment, the excitation wavelength was set at 488 nm and the emission wavelength at 550–570 nm, based on the fluorescence properties of rhodamine. Lipo-NT were used as the control.
In vitro uptake assay
BT-474 and MDA-MB-231 cell lines, cultured in adhesion, cells were seeded in 24-well plates and treated at 37°C with 0.04 mM liposomes ([Gd-DOTA] =0.004 mM) functionalized with the tetrameric forms of P6.1 and labeled with rhodamine. Cells were monitored at different time points: 0, 15, 30, 45, and 90 min. The specificity of uptake was determined by flow cytometry performed using a BD FACSCanto II apparatus with excitation and emission wavelengths at 488 and 550–570 nm, respectively. The same cancer cells incubated with anti-Herceptin fluorescein isothiocyanate (FITC)-labeled antibody were used as positive control.
MRI
The efficiency of liposomes functionalized with the tetrameric forms of P6.1 to act as MRI contrast agents was evaluated by in vitro relaxivity measurements and in vivo imaging studies. For in vitro relaxivity measurements, longitudinal and transversal nuclear relaxation times, T1 and T2, respectively, were measured as a function of the Gd(III) concentration in aqueous solutions at 4.7 T (200 MHz) using a Bruker MRI tomograph (Bruker Biospin, Rheinstetten, Germany). Briefly, 5 samples containing different Gd(III) concentrations (1, 0.5, 0.25, 0.125, and 0.0625 mM) were prepared in water. T1 and T2 relaxation times were measured by using a rapid acquisition with relaxation enhancement (RARE) image sequence with variable relaxation time (RAREVTR). Images were acquired with 8 echoes (echo times ranging between 9.5 and 76 ms) and 15 relaxation times (ranging between 241 and 5,000 ms). Three slices with 2 mm slice thickness, field of view (FOV) =5×5 cm2, and 64×64 matrix size were acquired. To obtain quantitative relaxivity values, the longitudinal and transversal relaxation rates (1/Ti) were plotted as a function of Gd-concentration according to the relationship:26
1/Timeas =1/Tiwater + ri × CGd, |
where i=1, 2; Timeas indicates the relaxation time measured; Tiwater indicates the relaxation time of plain water; ri denotes the relaxivity; and CGd indicates the millimolar concentration of Gd. The longitudinal and transversal relaxivities were calculated by linear best-fitting of these reported relationships to experimental data.
In vivo MR images were acquired using 5 mice (C57BL/6 male mice; Harlan Laboratories, Indianapolis, IN, USA) that were intravenously injected with liposomes at a dose corresponding to 100 μmol kg−1 of Gd. Animal handling was performed according to the protocols approved by the Animal Care and Use Committee of the University of Verona (CIRSAL) and by the Italian Ministry of Health, in strict adherence to the European Communities Council (86/609/EEC) directives. Images were acquired using a T1-weighted RARE sequence with TR =300 ms, TE =10.7 ms, RARE factor =4, slice thickness =1 mm, interslice distance =4 mm, FOV =6×3 cm2, and matrix size 256×128. Images were acquired at different time points (up to 30 min) after injection of liposomes.
Results and discussion
Design and synthesis
Many synthetic strategies for the derivatization of liposomal surface with targeting synthetic peptides have been proposed until now.27 Peptide sequences can be introduced directly during liposome formulation using a peptide amphiphile (PA) or by grafting the liposomal surface after the preparation with a method well-known in literature as the postliposomal modification method.27 This last method is particularly appealing for functionalization of liposomes with peptides in their dimeric or multimeric form.22 The anchorage of multimeric copies of the peptide on the liposomes can be achieved using activated functional groups available on the peripheral side of the liposome for covalent or noncovalent binding of the peptide. Many couples of functional groups (such as maleimide/thiol, biotin/avidin, triphosphines/azides, and azides/alkyn) have been proposed to achieve a specific reaction. Cu(I)-catalyzed Huisgen cycloaddition (CuAAC), also known as “click-chemistry” reaction, is particularly appealing because of its regiospecificity, chemoselectivity, and orthogonality to a wide variety of other functional groups. In our synthesis, DOPC-based liposomes were doped with a low amount (5 mol%) of an amphiphilic monomer such as (C18)2-KN3, (C18)2-K2N3, or (C18)2-K4N3, as reported in Figure 1A–C. These monomers contain one, two, or four PEGylated azido groups, respectively. Liposomes prepared with M, D, and T azido derivatives are here schematically indicated as Lipo-M, Lipo-D, and Lipo-T, respectively. Derivatization of the liposomal surface with targeted P6.1 peptide was achieved by using Pra-P6.1 derivative (Figure 1D) in which the peptide moiety contains a propargylglycine at the N-terminus. All the reactants [Pra-P6.1, (C18)2-KN3, (C18)2-K2N3, (C18)2-K4N3, and (C18)2-DTPA(Gd)], schematized in Figure 1, were synthesized on Rink amide resin by SPPS with Fmoc/tBu chemistry according to the standard protocols. Chemical synthesis of dimeric and tetrameric azido scaffolds, (C18)2-K2N3 and (C18)2-K4N3, was performed according to the reaction depicted in Scheme 1.
Liposome formulation and characterization
Mixed liposomes DOPC/(C18)2-DTPA(Gd) (90/10 mol/mol) and the analogs containing 5 mol% of PEGylated azido monomers (Table 1 shows the molar composition of the aggregates) were obtained according to the standard thin lipid film procedure in which the amphiphiles are dissolved in a mixture of organic solvents and are then removed under a N2 stream. Then, the film was hydrated by addition of 0.1 M phosphate buffer 0.9% wt NaCl at pH 7.4, sonicated, and extruded through a polycarbonate membrane with 100 nm pore size. The real number of functional azido groups, exposed on the liposomal surface, was checked using a fluorescent derivative (7-nitrobenzofurazan-propargylglycine) as previously described by Tarallo et al.28 As expected, ~50% of the theoretical azido groups were available on the outer liposomal surface for the click-chemistry reaction. One, two, or four copies of P6.1 peptide were bound on the monomeric, dimeric, or tetrameric azido scaffold to obtain Lipo-M6, Lipo-D6, and Lipo-T6 liposomes (Figure 2). After the click-chemistry reaction, liposomes were purified from unreacted alkyne peptide molecules and copper ions by gel-filtration chromatography. The effective yield of the reaction (95% after 12 h at room temperature) was calculated using the RP-HPLC calibration curve as the ratio of unreacted P6.1-alkyne to its starting amount. The mean diameters, the diffusion coefficients (D), and the PIs of all the liposomal formulations, before and after the click-chemistry reaction, were assessed by DLS. The nontargeted formulation (NT in Figure 2) has also been prepared and characterized. Distribution functions of liposomes are reported in Figure 3, and their structural data are summarized in Table 2. All aggregate solutions present a monomodal distribution due to a translational diffusion process, which could be attributed to liposome aggregates. According to the literature, the size of mixed liposomes DOPC/(C18)2-DTPA(Gd), at 90/10 molar ratio, was not substantially different from the size of pure DOPC liposomes, indicating that the insertion of a small amount of the amphiphilic gadolinium complex in the lipid mixture does not significantly influence the size of the DOPC liposomes29 (data not shown). Further introduction of (C18)2-KN3 or (C18)2-K2N3 PEGylated azido-function in Lipo-NT did not produce substantial deviation in the diameter values (Figure 3 and Table 1). On the other hand, the introduction of the more hindered (C18)2-K4N3 amphiphilic monomer caused a slight increase in the liposomal size (~10%). After the click-chemistry reaction, distribution functions of all liposomes showed an increase of the mean diameter (Figure 3) of 15%, 25%, and 35% for Lipo-M6, Lipo-D6, and Lipo-T6, respectively.
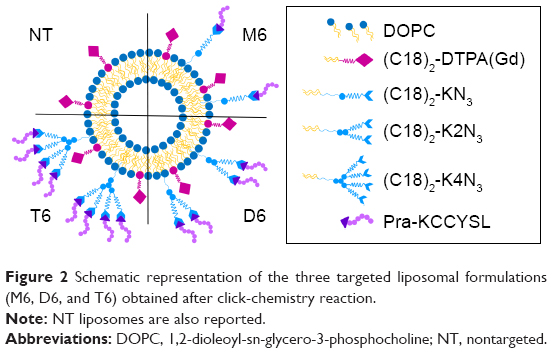 | Figure 2 Schematic representation of the three targeted liposomal formulations (M6, D6, and T6) obtained after click-chemistry reaction. |
 | Figure 3 DLS plots of liposomes decorated with monomeric (A), dimeric (B), and tetrameric (C) forms of the P6.1 peptide at 25°C. |
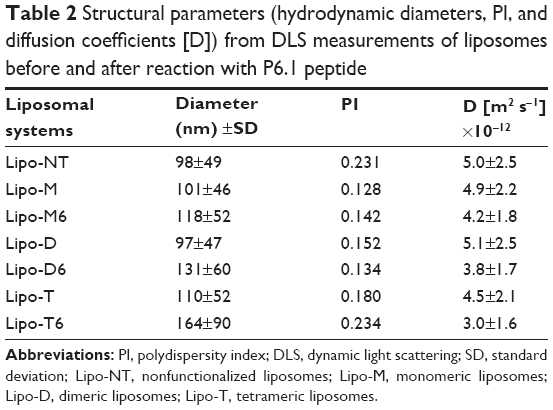 | Table 2 Structural parameters (hydrodynamic diameters, PI, and diffusion coefficients [D]) from DLS measurements of liposomes before and after reaction with P6.1 peptide |
Viability test
In vitro cell viability was assessed after incubation of BT-474 and MDA-MB-231 cancer cells with sterilized liposomes at three concentrations (0.002, 0.004, and 0.01 mM) and for different time periods (1, 3, and 6 h). Data are reported in Figure 4 and show that all the investigated liposome batches did not cause statistically significant effect on cell viability on both cell lines, thus indicating the absence of toxicity.
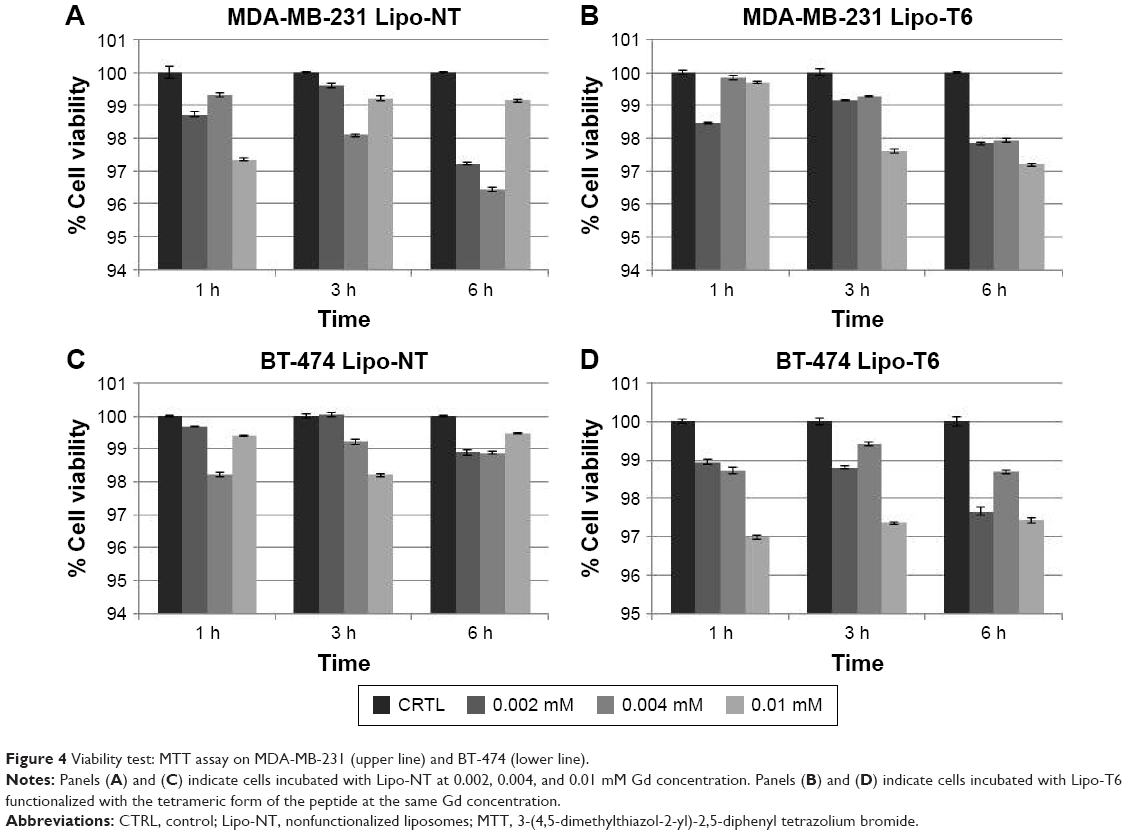 | Figure 4 Viability test: MTT assay on MDA-MB-231 (upper line) and BT-474 (lower line). |
Binding assay
The expression of HER-2 in MDA-MB-231 and BT-474 cells was preliminarily tested by flow cytometry using an anti-Herceptin FITC-conjugated antibody and its isotype control (IgG1-FITC conjugated). BT-474 cells were characterized by substantially higher expression (from 2.5 to 10 times higher) of HER-2 than MDA-MB-231 cells. The binding of targeted and untargeted liposomes to MDA-MB-231 and BT-474 cell surface was studied at low temperature (4°C) to avoid the internalization process. Results are shown in Figure 5. P6.1-peptide-targeted liposomes, labeled with rhodamine fluorophore, showed different behaviors when incubated with BT-474 or MDA-MB-231 cells. Liposomes functionalized with the tetrameric form (Lipo-T6) of the peptide displayed the most efficient binding to the BT-474 cancer cell line that overexpresses HER-2. Other liposomes, functionalized with the monomeric and dimeric form of the peptide, recognized BT-474 cancer cells, but their fluorescence was lower. The Lipo-T6 fluorescence signal showed a tendency to reach a plateau at concentrations of gadolinium complex ranging between 0.004 and 0.01 mM, attributable to the specific binding to the target antigen; at concentrations greater than 0.01 mM, the signal started to increase linearly, indicating a nonspecific binding (Figure S1). At the highest investigated concentrations, 0.2 and 0.5 mM, the mean fluorescence signal was very high and was ascribable to a nonspecific binding (data not shown). When the same experiment was performed on MDA-MB-231 cell line, the Lipo-T6 fluorescence signal showed a tendency toward a plateau at liposome concentrations higher than that used with BT-474 cancer cells (ie, between 0.01 and 0.02 mM).
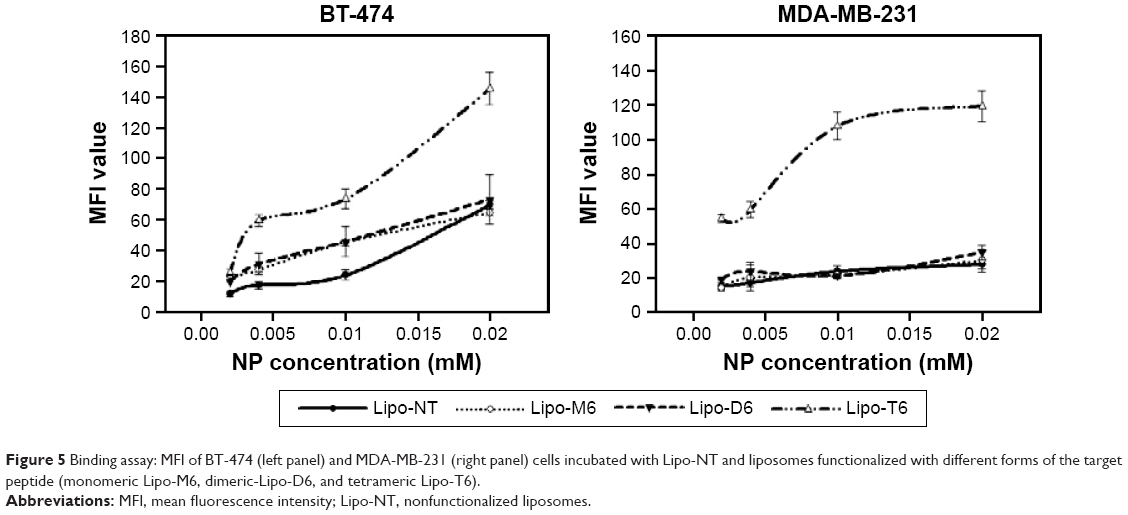 | Figure 5 Binding assay: MFI of BT-474 (left panel) and MDA-MB-231 (right panel) cells incubated with Lipo-NT and liposomes functionalized with different forms of the target peptide (monomeric Lipo-M6, dimeric-Lipo-D6, and tetrameric Lipo-T6). |
Uptake assay
Binding and uptake are relevant for therapeutic applications of liposomes. Therefore, the uptake assay was performed at 37°C to investigate the internalization of liposomes functionalized with the tetrameric form of the peptide. In this case, the mean fluorescence was determined at 37°C and at different time intervals of incubation. As a positive control, the same cell lines were incubated with an anti-HER-2 Herceptin monoclonal antibody labeled with FITC (Figure 6). For BT-474 cancer cells, the mean fluorescence was not substantially different from the mean fluorescence obtained with Herceptin, the monoclonal antibody currently used to detect HER-2 overexpressing cancer cells. Using MDA-MB-231 cancer cells, the mean fluorescence was more than 10 times lower than in BT-474 cancer cells and was comparable to the mean fluorescence obtained with Herceptin. So, this uptake assay demonstrates that liposomes functionalized with the tetrameric form of P6.1 have uptake properties for BT-474 comparable with those of Herceptin. These results were also confirmed by fluorescence microscopy (Figure S2).
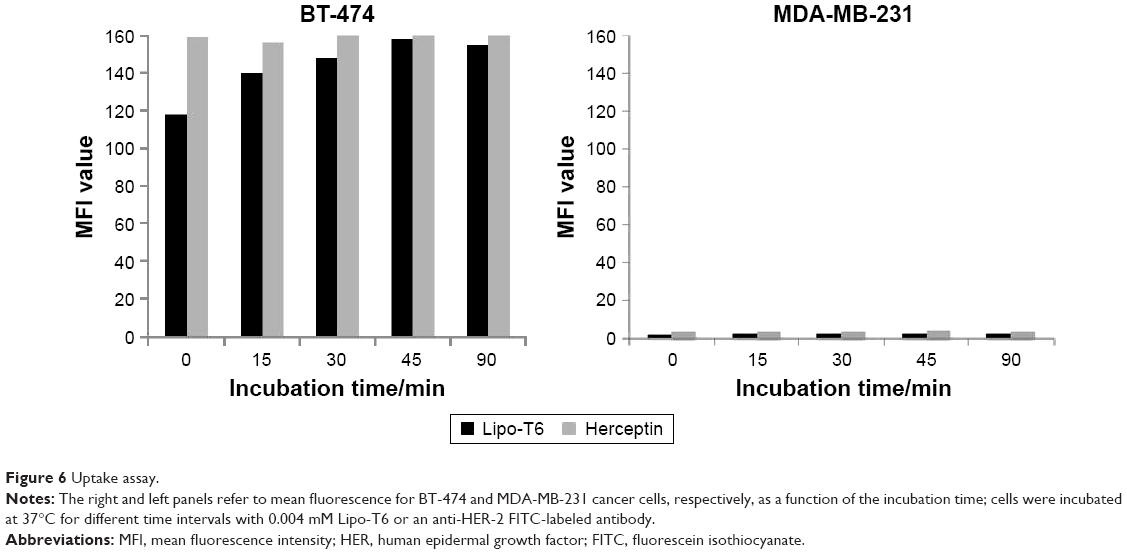 | Figure 6 Uptake assay. |
MRI
Longitudinal and transversal relaxation rates of liposome solutions are reported in Figure 7 (upper panel) as a function of Gd concentration; longitudinal and transversal relaxivities were obtained by the slope of the best fitting straight lines of experimental data: r1 =3.9 mM−1s−1 and r2 =27.0 mM−1s−1. The longitudinal relaxivity is relatively low for a Gd-based macromolecular contrast agent but is in agreement with values reported for longitudinal relaxivity in similar liposomal complexes containing Gd(III) at high frequency.30,31 Representative images of the abdominal region of mice acquired before and at 15 and 30 min after the injection of liposomes are shown in Figure 7 (lower panel). The increase in signal intensity due to the presence of Gd is clearly detectable in blood, liver, and kidneys. These MRI results, although preliminary, demonstrate the ability of these liposomal Gd complexes to be detected in vivo in MRI and pave the way for their use as contrast agents for MRI.
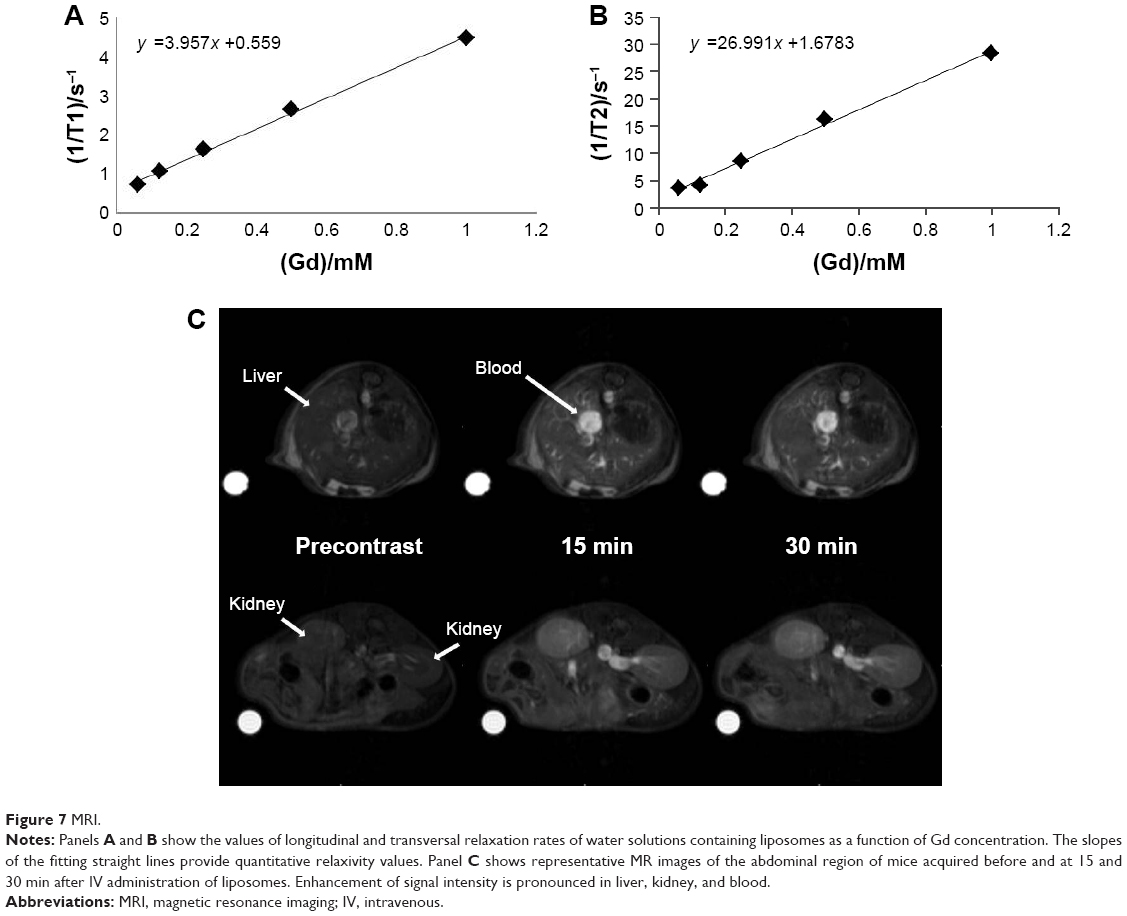 | Figure 7 MRI. |
Conclusion
Mixed liposomes externally decorated with P6.1 peptide sequence in its monomeric, dimeric, and tetrameric forms have been proposed as target-selective delivery systems for cancer cells overexpressing HER-2 receptors.
Derivatization of liposomal surface with targeting peptides was achieved using the postmodification method. The viability assays performed to assess the biological properties of these derivatized liposomes showed no toxicity effects. Moreover, preliminary in vitro data led us to conclude that liposomes functionalized with P6.1 peptide in its tetrameric form give better binding and uptake results in cells overexpressing HER-2 in comparison to vectors decorated with monomeric or dimeric versions of the P6.1 peptide. Finally, in vitro and in vivo MRI experiments demonstrated the potential of these liposomes to be used as contrast agents for MRI.
Acknowledgments
We are indebted to the Italian Minister for Research (MIUR) for financial support under FIRB ‘RINAME’ RBAP114AMK project. GF gratefully acknowledges the Italian Minister of Health RF-2010-2305526 grant for supporting this work. The authors thank Leopoldo Zona for excellent technical support in recording NMR spectra.
Disclosure
The authors report no conflicts of interest in this work.
References
Park JW, Kirpotin DB, Hong K, et al. Tumor targeting using anti-her2 immunoliposomes. J Control Release. 2001;74(1–3):95–113. | ||
Tai W, Mahato R, Cheng K. The role of HER2 in cancer therapy and targeted drug delivery. J Control Release. 2010;146(3):264–275. | ||
Kawamoto M, Horibe T, Kohno M, Kawakami K. HER2-targeted hybrid peptide that blocks HER2 tyrosine kinase disintegrates cancer cell membrane and inhibits tumor growth in vivo. Mol Cancer Ther. 2013;12(4):384–393. | ||
Bandekar A, Karve S, Chang MY, Mu Q, Rotolo J, Sofou S. Antitumor efficacy following the intracellular and interstitial release of liposomal doxorubicin. Biomaterials. 2012;33(17):4345–4352. | ||
Shigehiro T, Kasai T, Murakami M, et al. Efficient drug delivery of paclitaxel glycoside: a novel solubility gradient encapsulation into liposomes coupled with immunoliposomes preparation. PLoS One. 2014;9(9):e107976–e107976. | ||
Park BW, Zhang HT, Wu C, et al. Rationally designed anti-HER2/neu peptide mimetic disables P185HER2/neu tyrosine kinases in vitro and in vivo. Nat Biotechnol. 2000;18(2):194–198. | ||
Yang Z, Tang W, Luo X, et al. Dual-ligand modified polymer-lipid hybrid nanoparticles for docetaxel targeting delivery to Her2/neu overexpressed human breast cancer cells. J Biomed Nanotechnol. 2015;11(8):1401–1417. | ||
Shadidi M, Sioud M. Identification of novel carrier peptides for the specific delivery of therapeutics into cancer cell. FASEB J. 2003;17(2):256–258. | ||
Karasseva NG, Glinsky VV, Chen NX, Komatireddy R, Quinn TP. Identification and characterization of peptides that bind human ErbB-2 selected from a bacteriophage display library. J Protein Chem. 2002;21(4):287–296. | ||
Kumar SR, Gallazzi FA, Ferdani R, Anderson CJ, Quinn TP, Deutscher SL. In vitro and in vivo evaluation of 64Cu-radiolabeled KCCYSL peptides for targeting epidermal growth factor receptor-2 in breast carcinomas. Cancer Biother Radiopharm. 2010;25(6):693–703. | ||
Kumar SR, Quinn TP, Deutscher SL. Evaluation of an 111In-radiolabeled peptide as a targeting and imaging agent for ErbB-2 receptor-expressing breast carcinomas. Clin Cancer Res. 2007;13(20):6070–6079. | ||
Deutscher SL, Figueroa SD, Kumar SR. 111In-labeled KCCYSL peptide as an imaging probe for ErbB-2-expressing ovarian carcinomas. J Labelled Comp Radiopharm. 2009;52(14):583–590. | ||
Zhang X, Cabral P, Bates M, et al. In vitro and in vivo evaluation of [99mTc(CO)3]-radiolabeled ErbB-2-targeting peptides for breast carcinoma imaging. Current Radiopharm. 2010;3(4):308–321. | ||
Buckway B, Wang Y, Ray A, Ghandehari H. Overcoming the stromal barrier for targeted delivery of HPMA copolymers to pancreatic tumors. Int J Pharm. 2013;456(1):202–211. | ||
Bandekar A, Zhu C, Gomez A, Menzenski MZ, Sempkowski M, Sofou S. Masking and triggered unmasking of targeting ligands on liposomal chemotherapy selectively suppress tumor growth in vivo. Mol Pharm. 2013;10(1):152–160. | ||
Bracci L, Falciani C, Lelli B, et al. Synthetic peptides in the form of dendrimers become resistant to protease activity. J Biol Chem. 2003;278(47):46590–46595. | ||
Falciani C, Fabbrini M, Pini A, et al. Synthesis and biological activity of stable branched neurotensin peptides for tumor targeting. Mol Cancer Ther. 2007;6(9):2441–2448. | ||
Liu S. Radiolabeled cyclic RGD peptide bioconjugates as radiotracers targeting multiple integrins. Bioconjug Chem. 2015;26(8):1413–1438. | ||
Falciani C, Accardo A, Brunetti J, et al. Target-selective drug delivery through liposomes labeled with oligobranched neurotensin peptides. ChemMedChem. 2011;6(4):678–685. | ||
Falciani C, Brunetti J, Lelli B, et al. Nanoparticles exposing neurotensin tumor-specific drivers. J Pep Sci. 2013;19(4):198–204. | ||
Schmitt L, Dietrich C, Tampe R. Synthesis and characterization of chelator-lipids for reversible immobilization of engineered proteins at self-assembled lipid interfaces. J Am Chem Soc. 1994;116(19):8485–8491. | ||
Accardo A, Ringhieri P, Tesauro D, Morelli G. Liposomes derivatized with tetrabranched neurotensin peptides via click chemistry reactions. New J Chem. 2013;37(11):3528–3534. | ||
Tesauro D, Accardo A, Gianolio E, et al. Peptide derivatized lamellar aggregates as target-specific MRI contrast agents. ChemBioChem. 2007;8(8):950–955. | ||
Accardo A, Morisco A, Gianolio E, et al. Nanoparticles containing octreotide peptides and gadolinium complexes for MRI applications. J Pept Sci. 2011;17(2):154–162. | ||
Ringhieri P, Diaferia C, Galdiero S, Palumbo R, Morelli G, Accardo A. Liposomal doxorubicin doubly functionalized with CCK8 and R8 peptide sequences for selective intracellular drug delivery. J Pept Sci. 2015;21(5):415–425. | ||
Masotti A, Pitta A, Ortaggi G, et al. Synthesis and characterization of polyethylenimine-based iron oxide composites as novel contrast agents for MRI. MAGMA. 2009;22(2):77–87. | ||
Feldborg LN, Jølck RI, Andresen TL. Quantitative evaluation of bioorthogonal chemistries for surface functionalization of nanoparticles. Bioconjug Chem. 2012;23(12):2444–2450. | ||
Tarallo R, Accardo A, Falanga A, et al. Clickable functionalization of liposomes with the gH625 peptide from Herpes simplex virus type I for intracellular drug delivery. Chem Eur J. 2011;17(45):12659–12668. | ||
Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev. 1999;51(4):691–743. | ||
Filippi M, Remotti D, Botta M, Terreno E, Tei L. GdDOTAGA(C18)2: an efficient amphiphilic Gd(iii) chelate for the preparation of self-assembled high relaxivity MRI nanoprobes. Chem Commun. 2015;51(98):17455–17458. | ||
Gløgård C, Stensrud G, Hovland R, Fossheim SL, Klaveness J. Liposomes as carriers of amphiphilic gadolinium chelates: the effect of membrane composition on incorporation efficacy and in vitro relaxivity. Int J Pharm. 2002;233(1–2):131–140. |
Supplementary materials
Specific binding of Lipo-T6 to BT-474 cells
The specific binding curve of Lipo-T6 to BT-474 cells was obtained by subtracting the mean fluorescence intensity (MFI) value of Lipo-NT from the corresponding MFI value of Lipo-T6; the data reported in Figure S1 clearly demonstrated that Lipo-T6-specific binding reaches a plateau between 0.004 and 0.01 mM of liposome concentration (data obtained after incubation for 60 min at 4°C). The increasing trend in MFI value between 0.01 and 0.02 mM of Lipo-T6 concentration is due to the nonspecific binding.
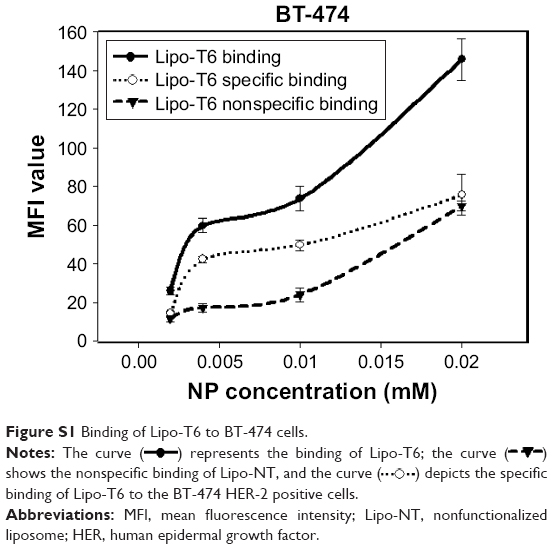 | Figure S1 Binding of Lipo-T6 to BT-474 cells. |
 ) represents the binding of Lipo-T6; the curve (
) represents the binding of Lipo-T6; the curve ( )
) ) depicts the specific binding of Lipo-T6 to the BT-474 HER-2 positive cells.
) depicts the specific binding of Lipo-T6 to the BT-474 HER-2 positive cells.Confocal microscopy analysis of Lipo-T6 uptake by BT-474 and MDA-MB-231 cancer cells
Confocal microscopy was performed using both BT-474 and MDA-MB-231 cell lines. Cells were incubated with anti-CD340 (Herceptin) fluorescein isothiocyanate-conjugated commercial antibody or with Rhodamine-Lipo-T6 for 3 h at 37°C and 5% CO2. After incubation, cells were fixed with 10% of buffered formalin, washed three times with sterile phosphate-buffered saline, and mounted with 4′,6-diamidino-2-phenylindole (DAPI) mounting medium. Cells were analyzed using Leica Microscope SP5 and observed at 40× magnification.
As clearly shown in Figure S2, both anti-Herceptin antibody and Lipo-T6 were taken up by BT-474 tumor cells. The efficiency of MDA-MB-231 cell uptake was substantially lower when treated with both anti-Herceptin and Lipo-T6. Therefore, confocal microscopy confirmed the results obtained with flow cytometry reported in Figure 6.
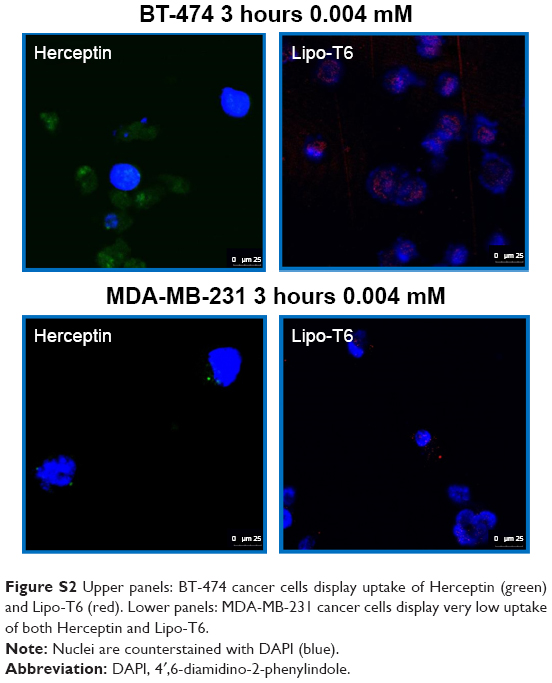 | Figure S2 Upper panels: BT-474 cancer cells display uptake of Herceptin (green) and Lipo-T6 (red). Lower panels: MDA-MB-231 cancer cells display very low uptake of both Herceptin and Lipo-T6. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.