Back to Journals » International Journal of Nanomedicine » Volume 11
Lipid nanoparticles for targeted siRNA delivery – going from bench to bedside
Authors Zatsepin T ![]() , Kotelevtsev Y, Koteliansky V
, Kotelevtsev Y, Koteliansky V
Received 18 February 2016
Accepted for publication 20 April 2016
Published 5 July 2016 Volume 2016:11 Pages 3077—3086
DOI https://doi.org/10.2147/IJN.S106625
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Timofei S Zatsepin,1–3 Yuri V Kotelevtsev,1 Victor Koteliansky1,2
1Center of Functional Genomics, Skolkovo Institute of Science and Technology, 2Department of Chemistry, Lomonosov Moscow State University, 3Production Department, Central Research Institute of Epidemiology, Moscow, Russia
Abstract: This review covers the basic aspects of small interfering RNA delivery by lipid nanoparticles (LNPs) and elaborates on the current status of clinical trials for these systems. We briefly describe the roles of all LNP components and possible strategies for their improvement. We also focus on the current clinical trials using LNP-formulated RNA and the possible outcomes for therapy in the near future. Also, we present a critical analysis of selected clinical trials that reveals the common logic behind target selection. We address this review to a wide audience, especially to medical doctors who are interested in the application of RNA interference–based treatment platforms. We anticipate that this review may spark interest in this particular audience and generate new ideas in target selection for the disorders they are dealing with.
Keywords: RNA therapeutics, siRNA, mRNA, lipid nanoparticle, targeted delivery, clinical trial
Introduction
Development of novel therapeutics is a ceaseless process as public heath becomes more and more valuable in the society. Today, small molecules and monoclonal antibodies are main players in drug development, but RNA therapeutics is foreseen to soon become a breakthrough technology. RNA therapeutics is advantageous when current techniques fail due to inefficiency of the treatment, complicated schemes of therapy, or newly developed resistance to the drug due to mutational escape. The Food and Drug Administration has recognized the importance of this new approach: two oligonucleotide-based drugs are already approved (pegaptanib1,2 and kynamro3,4). Fast track status has been awarded to studies testing small interfering RNAs (siRNAs) for viral infections, and efficacy studies are being conducted in nonhuman primates, while the safety studies are performed in humans. This siRNA-based revolutionary approach can soon come to the market, depending on the results of several active Phase III clinical trials.5,6 This review will focus on siRNA delivery by lipid nanoparticles (LNPs) and the current status of clinical trials for these systems. In the first section, we will describe the different components forming the LNPs and discuss the possible routes for their improvement. We will then report current clinical trials using LNP-formulated RNA and the possible outputs for therapy in the near future. Finally, we will present a critical analysis of selected clinical trials, which would reveal the common logic behind target selection, and demonstrate some examples of lessons learned from failed trials.
LNPs for RNA delivery: structure and requirements
During the last 5 years, siRNA, microRNA (miRNA) mimetics, antagomirs, and messenger RNA (mRNA) have made tremendous progress as potential therapeutics.7–10 However, targeted delivery and endosome escape still remain the major challenges for RNA drug development. siRNAs are short (19–21 nucleotide), double-stranded RNAs that enter the natural RNA interference (RNAi) mechanism and degrade complementary mRNAs by the use of complicated protein machinery. As a result, one can reversibly knock down in vivo a protein of interest (80%–90% decrease) at doses of siRNAs approximately 1–2 mg/kg. Antagomirs sterically block miRNAs by the formation of duplexes, which influences posttranscriptional regulation of gene expression.11 Artificial mRNAs are used for controlled transient expression of proteins in cancer immunotherapeutics and vaccination.10 Today, there are several efficient systemic delivery strategies for RNAs that are presently in clinical trial: LNPs, N-acetylgalactosamine conjugates, and dynamic polyconjugates (DPCs).6,12,13 Also, there are techniques for local delivery or topical application of RNAs.14,15
Delivery systems based on cationic lipids (liposomes and LNPs) are the most commonly used nonviral vectors for RNA delivery, both in academic studies and clinical trials.16–18 The basic principle of LNP formation is self-assembly that is an integral property of smart nanosystems. Hundreds of lipids have been developed to improve transfer efficiency and stability and decrease immune response and renal escape. Since the first iterations of LNPs, lipids have evolved a lot; from the common 1,2-dioleoyl-3-trimethylammonium-propane/1,2-dioleoyl-sn-glycero-3-phosphoethanolamine combination to more sophisticated structures (Figure 1), they have become much less toxic and are biodegradable and pH sensitive (smart). From a chemical viewpoint, there are two main structural improvements that make the new lipids more efficient vehicles for siRNA: multiply unsaturated alkyl chains promote significant destabilization of intracellular membrane bilayer,19 while protonation of the ionizable dimethylaminopropyl group only during lisosome maturation improves LNP initial fusion with the membrane.20 Moreover, lipidoids, or lipid-like materials, were shown to be very efficient carriers for siRNA. For example, C12-200 (Figure 1) is suitable for multiple animal species, such as rodents and nonhuman primates.21,22 One of the main benefits of lipidoids is the decreased amount of helper lipids needed for effective delivery.
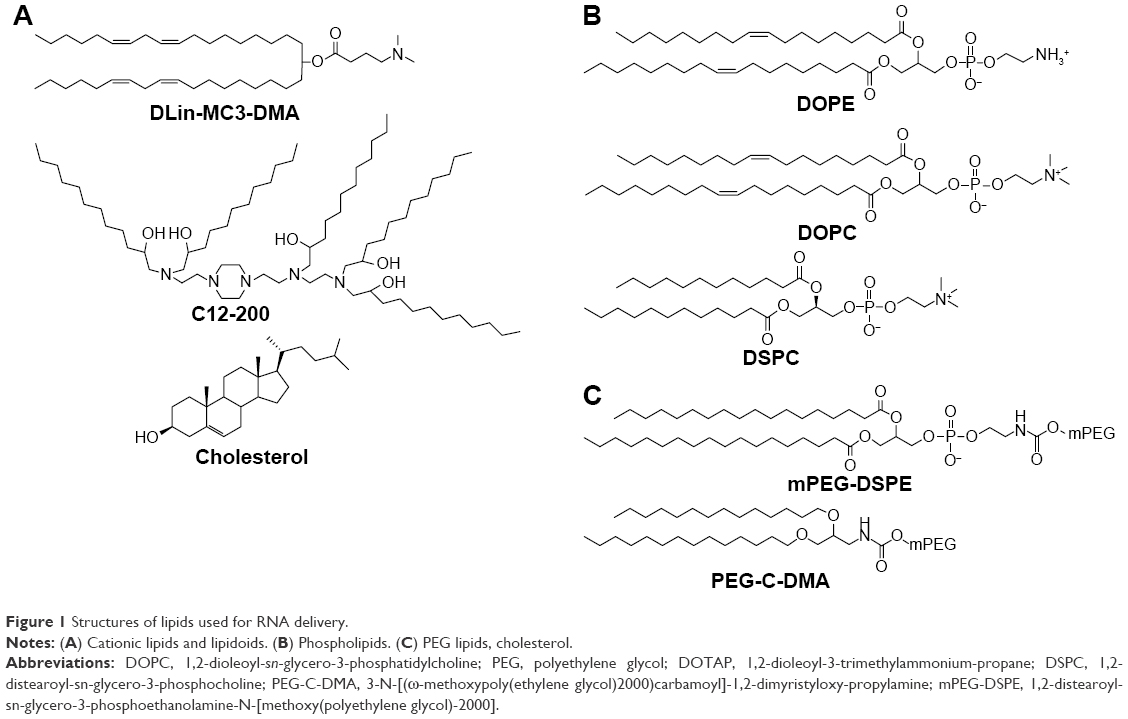 | Figure 1 Structures of lipids used for RNA delivery. |
By themselves, cationic lipids are inadequate delivery vehicles for siRNA, and therefore, the so-called helper lipids should be added to form the LNP.23 Among them are fusogenic phospholipids (Figure 1B), which enhance the transfection activity of LNPs by destabilizing the lipid bilayer structure in cell membranes, and polyethylene glycol (PEG) lipids (Figure 1C), which decrease the immune response by increasing colloidal stability and shielding LNPs from macrophages.24 However, high surface content of PEG lipids decreases cellular internalization of LNPs by hindering membrane destabilization; so, an optimal proportion should be found. To overcome this problem, one can decrease molar percentage or use modified variants of PEG lipids that weakly anchor to LNPs and are gradually lost during circulation.25 Another key component of LNP is cholesterol, which stabilizes nanoparticles by filling the gaps between lipids26 and enhances the activity of cationic lipids.27 A common scheme of siRNA–LNP formation includes several steps. The first step is the formation of complexes of siRNA with ionizable cationic lipids in 40% ethanol solution with low ionic strength and pH approximately 4. As siRNAs are rather short in comparison to plasmids, they should be truly covered by lipids to achieve stable incorporation into LNP. Then, these nucleating structures are covered by helper lipids, cholesterol, and finally by PEG-lipids that lead to the formation of LNPs with embedded siRNA (Figure 2). Most of the parameters such as siRNA:lipids ratio, concentrations, and temperature should be optimized empirically.
 | Figure 2 Schematic structure of lipid nanoparticles. |
To decrease glomerular filtration, renal clearance, and generation of immune response, one should control the size (optimal 50–100 nm) and surface charge of the LNPs. The size of LNPs is controlled mainly by concentration of the components during mixing and the fraction of PEG lipids contained in the mixture. Work performed on LNPs also revealed that neutral carriers are more efficient during circulation in the blood and penetration in tumor tissues, while cationic carriers are preferable during cellular uptake and endosomal escape.28 Indeed, first-generation LNPs were formed during formulation by self-assembly and were disrupted in endosomes, which led to the degradation of RNA molecules in the endosomes rather than RNA release in the cytosol. These results generated the development of more complicated pH-sensitive LNPs that change their charge in endosomes, thus improving RNA escape after penetration into the cell. However, even in this case, only 1%–2% of the total siRNA is released into the cytosol.29 Moreover, approximately 70% of all internalized nanoparticles are rejected outside the cell by recycling.30 Hence, future small molecules developed to enhance the uptake and endosome release of LNPs can really propel the use of RNA therapeutics.31,32 Another prospective effector for LNPs is melittin, a known pore-forming peptide33 that was successfully used for RNA delivery directly34 or as a masked part of the polymer carrier for RNA.35 Finally, incorporation of metal nanoparticles in LNPs could improve disruption of the endosome membrane under alternating magnetic fields.36
As we mentioned earlier, the main problem in targeted delivery of RNA, besides the escape from endosomes after cell penetration, is the specific and efficient delivery to the appropriate cell types of the tissue of interest. Attachment of targeting molecules (N-acetylgalactosamine – a ligand of ASGPR receptor,24,37 hyaluronan that specifically binds the CD44 receptor38) and antibodies39–42 to the nanoparticle is the main concept presently used for directing the LNP to the cell or the tissue of interest.
Native siRNAs are easily degraded by nucleases in biological fluids. So, considerable chemical modification for in vivo applications using siRNAs is a necessity. The most common modifications for siRNA used in LNP delivery are 2′-O-methyl, 2′-fluoro and phosphorothioate (Figure 3). More intricate modifications are used to increase RNA stability in conjugates, as RNA itself is less sterically protected than when enveloped by LNP.43 In most cases, data obtained on the activity of modified siRNA in vitro are easily translated in vivo; however, in vivo toxicity and siRNA stability (degradation pattern) should be thoroughly studied.
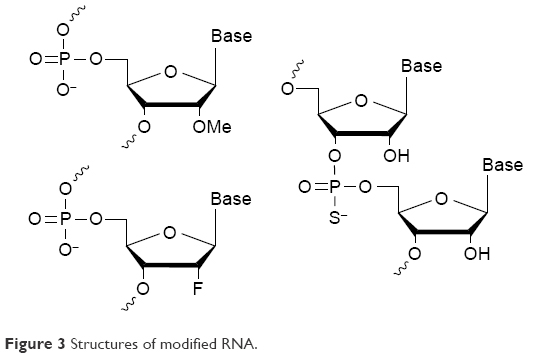 | Figure 3 Structures of modified RNA. |
Most of the LNP-formulated siRNAs and mRNAs are administered by intravenous (IV) injection. However, high stability of LNPs allows for subcutaneous administration in the case of delivery to the liver,44 which significantly simplifies their future application as therapeutics.
Toxicity and immunogenicity of siRNA–LNP in Phase I clinical trials
LNPs are the carriers most often used in clinical trials for siRNA-based therapeutics, not only because of their obvious advantages over other existing delivery vehicles, such as ease of preparation and versatility, but also their biocompatibility, biodegradability, and accumulated record of clinical use. Unfortunately, being synthetic and quite “unnatural”, lipids can be toxic and promote immune response at high doses.45 These negative effects are significantly lower in the newer versions of lipids and polymers; however, immunogenicity and toxicity of LNPs can still be an obstacle in clinical application, particularly if the required dose is high or if frequent repetitive application is required.
Novel lipids selected by advanced screening13,21,46 facilitate clinical trials, but application of similar LNPs does not guarantee the success of novel siRNA therapeutics. For example, the clinical trial for the treatment of hypercholesterolemia using siRNA/LNPs to target ApoB production was terminated due to immunogenicity (NCT00927459). On the contrary, no serious adverse effects were reported in the ALN-PCS02 trial with similar LNP by Alnylam Pharmaceuticals, targeting the proprotein convertase subtilisin/kexin type 9 (PCSK9) transcript (NCT01437059). This recent study is particularly encouraging, as it demonstrates that selection of proper lipid components can overcome earlier reported toxicity, immunogenicity such as the release of tumor necrosis factor-α, interferon-γ, interleukin-6, interleukin-12, and damage to the liver tissue resulting in elevated levels of transaminases (alanine transaminase, aspartate transaminase), leukopenia, and thrombocytopenia.47–49
We want to emphasize that application of siRNA/LNP in clinical studies should be based on predictive animal models and understanding the physiology. Also, sharing data is a key point for successful translation of LNP from a bench scale into clinical studies.
Principles and examples of RNA target selection for LNP–siRNA therapeutics
Intervention with RNAi is based either on direct loss of function or gain of function. In the first case, RNAi causes downregulation of overproduced proteins that cause pathological changes, for example, reducing the production of angiotensinogen by the liver during preeclampsia. Gain of function of specific protein can be achieved by removal of negative regulatory feedback. Both approaches are currently being tested in clinical trials in three major areas: antiviral approaches, anticancer therapies, and genetic disorders, but the success of the therapy relies on careful target selection. In this section, we will discuss the common logic behind efficient target selection. All data on current clinical trials of siRNA/LNP are summarized in Table 1.
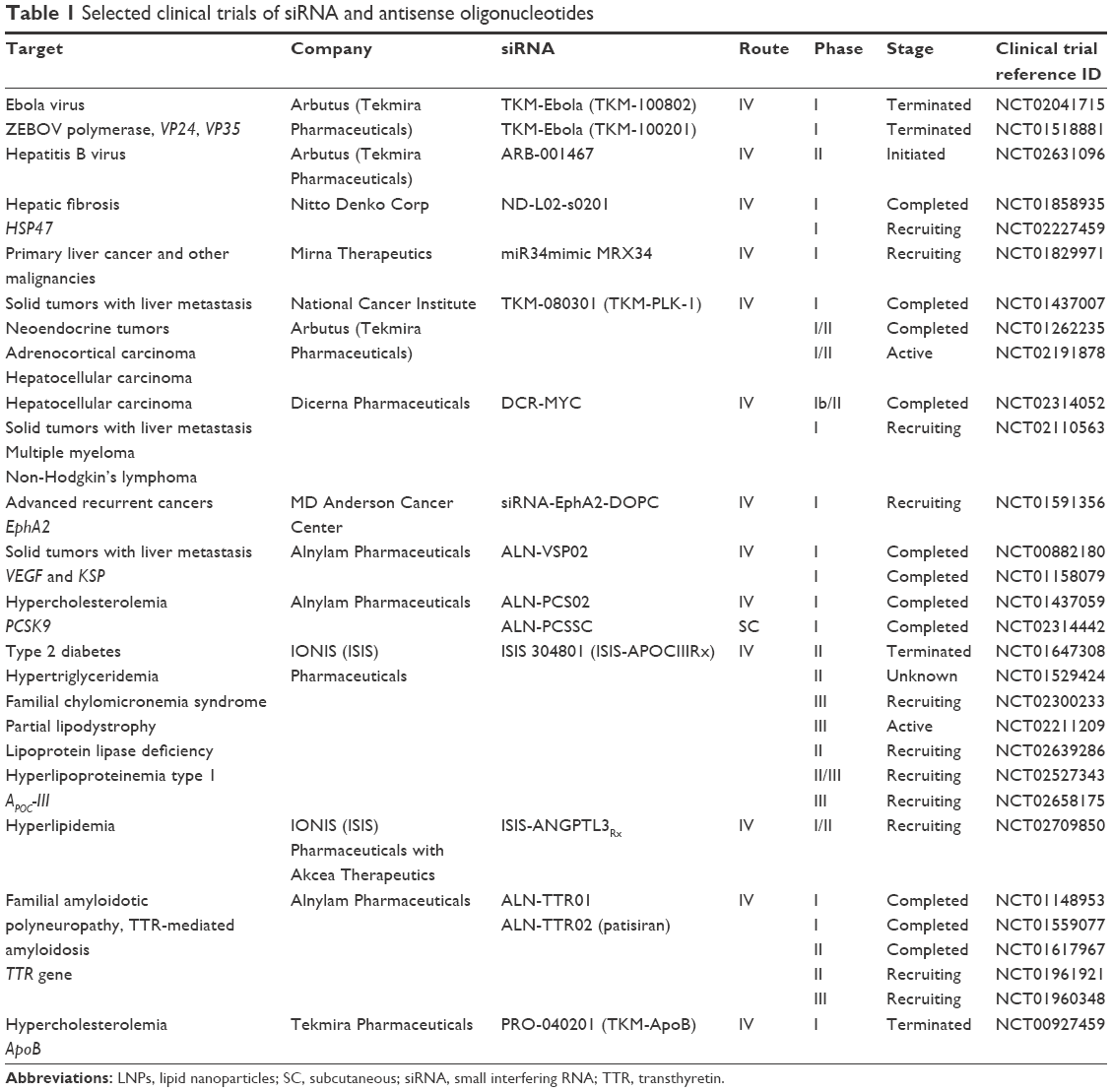 | Table 1 Selected clinical trials of siRNA and antisense oligonucleotides |
Application of the RNAi approach to combat viral infections has several specific advantages. First, targeting several gene products simultaneously will reduce the possibility of accumulated mutations and reinfection with the stable form of the virus. Second, reducing the expression of cell surface antigens, such as hepatitis B surface antigen in hepatitis B virus (HBV), diminishes immunosuppression and activates the T-cell response, leading to seroconversion and eventually functional cure.30 Finally, fast response to new strains of viruses by minimal changes of siRNA sequences can be provided with this technology.
Promising results have been obtained using siRNA-loaded LNPs in treating liver cancer. Existing LNP-based delivery protocols point toward clinically important siRNA targets expressed in the liver. Also, clinical studies in humans demonstrated little adverse effect to LNPs in the liver or any general immune response to modified siRNA. Therefore, the success of siRNA drug development depends mostly on the careful selection of the drug target expressed in the liver.
Most of the studies targeting cancer-associated pathways with an RNAi approach are at the discovery stage and some researchers express doubts about the accessibility of nonhepatic tumors to LNP-formulated RNAs. Increased permeability of the blood vessels for proteins and particles <20 nm unfortunately does not translate into permeability to LNPs. Hence, the efficacy of local delivery in the tumor remains a challenge. Target selection in cancer is also not as straightforward as it may seem to be. Numerous pathways are activated in cancer; however, blocking an individual pathway usually does not lead to death of a tumor. Nonetheless, enormous unmet needs and a reduced threshold for acceptable severity of adverse effects guarantee the interest of pharmaceutical companies in the development of anticancer drugs. Several pharmaceutical companies demonstrated willingness to bring RNAi therapeutics to Phase I/Phase II clinical trials. Currently, these trials provide the much needed hope for aggressive cancers resistant to existing treatment protocols. Continued success depends on the improvement of efficient delivery technologies while maintaining a good safety profile. If successful, this will attract investments needed for Phase III trials.
RNA therapeutics can be used not only for direct inhibition of key genes of interest but also as a helper for small-molecule drugs or monoclonal antibodies. For example, inhibition of Testican-1 by siRNA restores sensitivity to lapatinib in the treatment of human epidermal growth factor receptor 2-positive gastric cancer.50 Another interesting example is the simultaneous application of siRNA and 5-fluorouracil in hepatic metastasis of colon cancer, which reduced the incidence of liver metastases.51
Antiviral therapy
Arbutus Biopharma Corporation recently published the efficacious treatment of macaques in the early stages of infection by Ebola (<3 days after infection) using IV LNP injections with siRNA (TKM-Ebola formulation).52 However, in the later clinical trials, adverse toll-like receptors-mediated immune response presumably provoked by the LNPs was observed in some patients during dose escalation, causing the termination of the studies at Phase I. The adverse effect was seen during initial injections of high doses, but subsequent injections were well tolerated (NCT02041715). Large-scale animal trials are expected to provide solutions to this problem.
Continuous growth of the population infected with HBV and hepatitis C virus urges the development of RNAi-based treatments. Today, a number of efficient small-molecule drugs exist: nucleoside analogs entecavir or tenofovir for HBV and sofosbuvir or simeprevir for hepatitis C virus. However, strict adherence to the prescribed daily/weekly schedule is necessary for the success of the treatment, while RNAi acts for months, which represents a significant improvement in quality of life. Arbutus Biopharma Corporation developed LNP-based siRNAs targeting three different loci of the gene coding for the HBV surface antigen present in chronically infected patients. This preparation is entering a Phase I trial. Targeting this surface antigen is particularly important, as reduction of its expression can lead to ablation of immune suppression and stimulation of T-cell response of the patient. For the treatment of HBV, two Phase II clinical trials led by Arrowhead Research Corporation are presently ongoing, testing the RNAi dynamic conjugate (DPC) ARC-520 in combination with approved nucleoside analogs (NCT02065336 and NCT02349126). In the case of DPC, the duration of the effect is shorter than that achieved with the LNP-based Arbutus formulation, which might be a disadvantage of the otherwise very promising ARC-520.53–55
Cancer
The miRNA miR-34 is a well-known tumor suppressor, which inhibits cell cycle progression and induces cancer cell death. In their Phase I trials and safety and dose-escalation studies, NCT01829971 Mirna Therapeutics used LNPs (Smarticle LNP) for the delivery of MRX34. This treatment has been well tolerated in 52 patients who received two injections per week during 3 weeks, followed by 1 week off, with a maximal tolerated dose of 110 mg/m2. Another cohort of patients received daily administration for 5 days with 2 weeks off until disease progression or intolerance. Two patients (one patient with primary hepatocellular carcinoma [HCC], metastatic to the lung, and another patient with melanoma, metastatic to the lymph nodes) with advanced, metastatic stage IV cancer have achieved 30% tumor shrinkage after treatment with MRX34.56
Another important target for the treatment of cancer is polo-like kinase 1 (PLK1), a protein that is often overexpressed in cancer and whose inhibition results in reduced cell division.57 Inhibition of PLK1 expression prevents the tumor cell from completing the cell division, resulting in apoptosis. Arbutus Biopharma Corporation has completed Phase I studies and is currently conducting Phase II trials in patients with gastrointestinal neuroendocrine tumors, adrenocortical carcinoma, and HCC (NCT01437007, NCT01262235, NCT02191878). Phase I trials resulted in a dose-dependent effect and achieved stable disease and partial response in five out of six patients, in addition to a 19.3% reduction in tumor size.58 This disease model is ideal for LNPs because of the natural biodistribution of the particles in the neuroendocrine tumors. This makes the adrenal cortex and similar structures possible candidates for LNPs. Arbutus Biopharma Corporation is currently evaluating TKM-PLK1 in an expansion cohort with 20 patients who have HCC. This study is designed to evaluate TKM-PLK1’s safety and efficacy in treating HCC patients.
MYC is one of the first discovered classical oncogenes essential for the growth of many tumors, including HCC.59 Dicerna is presently conducting Phase I studies with DCR-MYC, a Dicer substrate-based RNAi targeting the MYC oncogene, in combination with proprietary EnCore Dicerna’s liposomal delivery technology,60 in 72 patients with various cancers, including both solid and hematological cancers, and particularly HCC (NCT02314052, NCT02110563). Patients undergo IV administration once a week for 2 weeks, followed by a week of rest with the potential for follow-up administration. Tumor penetration is the major issue with the EnCore technology, and this is addressed in the clinical trial.
EphA2 is a cell surface receptor tyrosine kinase involved in neuronal cell migration during the development of embryo and is essentially absent in adult tissues.61 Its overexpression in cancer is associated with poor clinical outcome.62 EphA2 knockdown in mice reduced tumorigenicity in breast and pancreatic cancer63 and resulted in reduced tumor growth in a mouse model of ovarian cancer when injected twice a week during 3 weeks and combined with chemotherapeutics. MD Anderson Cancer Center is conducting a Phase I study involving 40 patients with advanced solid tumors, undergoing IV-administration of 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine neutral liposomes loaded with EphA2 siRNA twice weekly for 3 weeks (NCT01591356).61 Antineoplastic activity was reported, associated with reduced transcription and translation of EphA2.
Noncancer diseases
Experiments with knockdown of PCSK9 give an excellent example of the gain-of-function approach.64 Selection of PCSK9 was prompted by clinical cases of genetic disorders with gain of function of this protease causing hypercholesterolemia or, on the contrary, loss-of-function mutations resulting in very low levels of plasma cholesterol. It was further revealed that PCSK9 activity has a strong influence on cholesterol levels through degradation of major hepatocyte receptors or low density lipoprotein (LDL) clearing receptors in hepatocytes. PCSK9 binds hepatocyte LDL clearing receptors both intracellularly and extracellularly, leading to their lysosomal degradation.65,66 Patients treated with siRNA against PCSK9 formulated in LNPs had lower LDL-C levels by 60% in the absence of statins, an effect that was sustained for more than 3 months with a single dose of 10 mg/kg (NCT02314442). This success story can be considered the first published clinical data proving the efficacy of a new class of well-tolerated drugs. LDL levels can potentially be effectively controlled by other classes of drugs such as statins, fibrins, bile acid binding resins, niacin, and cholesterol adsorption inhibitors. Moreover, monoclonal antibodies against PCSK9 have also been found to be almost as effective as siRNA.67 However, there is a strong opinion that RNAi-based drugs will find a niche even in this competitive market, since existing treatments leave a significant number of patients with poorly controlled LDL levels, even at the highest tolerated doses of existing drugs.
Other tricky targets are apolipoprotein C3 (apoC3) in hypertriglyceridemia and angiopoietin-like 3 protein (ANGPTL3) in the genetic forms of mixed hyperlipidemia and severe hypertriglyceridemia.68 The strategy for the selection of apoC-III for RNAi targeting resembles that implemented in the selection of PCSK9. ApoC-III is located on the surface of triglyceride-rich lipoproteins, including very low density lipoprotein (VLDL), chylomicrons, and their remnants. In two genetic studies, subjects with loss-of-function mutations in the apoC-III gene presented 39%–44% lower levels of triglycerides and ~40% lower risk of coronary heart disease.69 Patients with a dysfunctional apoC-III have lower cardiovascular risk. Knocking down apoC-III using antisense oligonucleotides reduced apoC-III and triglycerides but increased the “good” facilitating effect of high density lipoprotein-cholesterol, without inducing hepatic steatosis.70 Genetic studies also pointed out the ANGPTL3 gene as a potential target for RNAi intervention. Mutations in this gene cause familial hypobetalipoproteinemia type 2,65 and ANGPTL3-deficient patients feature an insulin-sensitive phenotype with lower plasma insulin and glucose levels. This gene encodes a family of secreted proteins expressed predominantly in the liver. Once translated into protein, it is further processed into an N-terminal peptide and a C-terminal fibrinogen chain. The N-terminal chain is a regulator of lipid metabolism and lipoprotein secretion, while the C-terminal chain may be involved in angiogenesis. Silencing ANGPTL3 in cells improves glucose uptake and shifts the secretion of VLDL from VLDL1 to poor VLDL2 during insulin stimulation.71
Primary hyperoxaluria type I (PH1) is a rare recessive disorder (5 per 1 million births) caused by a mutation in the alanine glyoxylate aminotransferase (AGT) protein, which is encoded by the AGXT gene and is involved in breakdown of hydroxyproline. AGT deficiency causes buildup of oxalate, the metabolite that characterizes PH1 and causes irreversible kidney damage. Ultimate treatment of PH1 requires both liver and kidney transplant. The strategy of Dicerna was to inhibit the liver enzyme glycolate oxidase (GO), which is responsible for converting glycolate into glyoxylate, the precursor of oxalate. LNP RNAi inhibition of GO in a mouse knockout model of PH1 has nearly normalized oxalate levels. In primates, 85% reduction of GO activity was achieved with 0.3 mg/kg of siRNA, an encouraging result for future Phase I clinical trials. At present, there are no small-molecule inhibitors of GO, and due to the orphan status of the disease, such drugs are not expected to appear in the near future. Hence, RNAi therapy gives a real opportunity for the treatment of this rare disease.
Finally, there are targets that present direct pathology-causing proteins. For example, transthyretin (TTR) is implicated in a rare genetic disease caused by amyloidosis of TTR (ATTR).72,73 A mutated variant of this protein causes amyloid deposition in nerve endings and in the heart. However, it is synthesized by the liver and, hence, is an accessible target for siRNA. Therefore, choosing TTR as a drug target is very straightforward. In patients, LNP formulations ALN-TTR01 and ALN-TTR02 suppressed the production of both mutant and nonmutant forms of TTR, establishing proof of concept for RNAi therapy targeting mRNA transcribed from a disease-causing gene (NCT01148953 and NCT01559077). This project is now in Phase III clinical trials, the most advanced stage for an LNP-based therapy (NCT01960348). The trials, led by Alnylam Pharmaceuticals, will serve to evaluate efficacy and safety, with a goal of achieving improved neuropathy impairment scores over placebo with 0.3 mg/kg administration of patisiran every 3 weeks for 18 months in up to 200 patients (NCT01960348).46 This project is in the discovery stage; however, if successful, it can provide a cure for preeclampsia, a disease with unmet efficacious treatment options.74
Conclusion
It can be concluded that the present techniques of RNA delivery using LNPs guarantee effective knockdown of any RNA target in the liver. Moreover, there are minimal adverse effects related to the delivery vehicle or the siRNAs themselves. Therefore, the success of siRNA-based drug development in the liver depends mostly on proper target selection. The last 3 years were extremely productive in translation of fundamental studies and preclinical research of RNAi-based therapies into clinical trials. Research in the fields of cancer and antiviral therapies demonstrated the safety and efficacy of an approach based on LNP delivery of siRNA in Phase I/II clinical trials, paving the road for RNAi therapies for the treatment of genetic and metabolic diseases. Alnylam Pharmaceuticals’ PCSK9 studies have demonstrated a competitive edge over existing small molecules and biological intervention in the treatment of hypercholesterolemia. The TTR program is anticipated to receive the first Food and Drug Administration approval for clinical application in the nearest future. Urgent unmet medical needs, particularly in the treatment of patients with viral infections and patients with refractive aggressive cancers, stimulate the development of novel delivery strategies. There is no doubt that we are at the verge of transformation of medical practice by a widespread application of RNAi-based treatment for many human disorders.
Acknowledgments
This work was supported by the Russian Scientific Foundation (grant no 14-34-00017). The authors are grateful to Dominique Leboeuf for critical reading of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Vinores SA. Pegaptanib in the treatment of wet, age-related macular degeneration. Int J Nanomedicine. 2006;1(3):263–268. | ||
U.S. Food and Drug Administration. FDA Approves New Drug Treatment for Age-Related Macular Degeneration [press release]. 2004 [December 20]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2004/ucm108385.htm. Accessed December 1, 2015. | ||
Tse MT. Regulatory watch: Antisense approval provides boost to the field. Nature Rev Drug Disc. 2013;12:179. | ||
U.S. Food and Drug Administration. FDA approves new orphan drug Kynamro to treat inherited cholesterol disorder [press release]. 2013 [January 29]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm337195.htm. Accessed December 1, 2015. | ||
Zuckerman JE, Davis ME. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat Rev Drug Discov. 2015;14(12):843–856. | ||
Bobbin ML, Rossi JJ. RNA interference (RNAi)-based therapeutics: delivering on the promise? Annu Rev Pharmacol Toxicol. 2016;56:103–122. | ||
Lam JK, Chow MY, Zhang Y, Leung SW. siRNA versus miRNA as therapeutics for gene silencing. Mol Ther Nucleic Acids. 2015;4:e252. | ||
Chakraborty C, Wen ZH, Agoramoorthy G, Lin CS. Therapeutic microRNA delivery strategies with special emphasis on cancer therapy and tumorigenesis: current trends and future challenges. Curr Drug Metab. 2016;17(5):469–477. | ||
Wittrup A, Lieberman J. Knocking down disease: a progress report on siRNA therapeutics. Nat Rev Genet. 2015;16(9):543–552. | ||
Sahin U, Karikó K, Türeci Ö. mRNA-based therapeutics – developing a new class of drugs. Nat Rev Drug Discov. 2014;13(10):759–780. | ||
Kumar V, Mahato RI. Delivery and targeting of miRNAs for treating liver fibrosis. Pharm Res. 2015;32(2):341–361. | ||
Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. 2013;12(11):967–977. | ||
Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15(8):541–555. | ||
Sarett SM, Nelson CE, Duvall CL. Technologies for controlled, local delivery of siRNA. J Control Release. 2015;218:94–113. | ||
Fujita Y, Kuwano K, Ochiya T. Development of small RNA delivery systems for lung cancer therapy. Int J Mol Sci. 2015;16(3): 5254–5270. | ||
Singh Y, Tomar S, Khan S, et al. Bridging small interfering RNA with giant therapeutic outcomes using nanometric liposomes. J Control Release. 2015;220(Pt A):368–387. | ||
Leung AK, Tam YY, Cullis PR. Lipid nanoparticles for short interfering RNA delivery. Adv Genet. 2014;88:71–110. | ||
Hope MJ. Enhancing siRNA delivery by employing lipid nanoparticles. Ther Deliv. 2014;5(6):663–673. | ||
Wan C, Allen TM, Cullis PR. Lipid nanoparticle delivery systems for siRNA-based therapeutics. Drug Deliv Transl Res. 2014;4(1): 74–83. | ||
Maier MA, Jayaraman M, Matsuda S, et al. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol Ther. 2013;21(8):1570–1578. | ||
Love KT, Mahon KP, Levins CG, et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc Natl Acad Sci U S A. 2010;107(5):1864–1869. | ||
Novobrantseva TI, Borodovsky A, Wong J, et al. Systemic RNAi-mediated gene silencing in nonhuman primate and rodent myeloid cells. Mol Ther Nucleic Acids. 2012;1:e4. | ||
Cheng X, Lee RJ. The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv Drug Deliv Rev. 2016;99(Pt A):129–137. | ||
Kumar V, Qin J, Jiang Y, et al. Shielding of lipid nanoparticles for siRNA delivery: impact on physicochemical properties, cytokine induction, and efficacy. Mol Ther Nucleic Acids. 2014;3:e210. | ||
Xia Y, Tian J, Chen X. Effect of surface properties on liposomal siRNA delivery. Biomaterials. 2016;79:56–68. | ||
Dabkowska AP, Barlow DJ, Hughes AV, Campbell RA, Quinn PJ, Lawrence MJ. The effect of neutral helper lipids on the structure of cationic lipid monolayers. J R Soc Interface. 2012;9(68):548–561. | ||
Tenchov BG, MacDonald RC, Siegel DP. Cubic phases in phosphatidylcholine-cholesterol mixtures: cholesterol as membrane “fusogen”. Biophys J. 2006;91(7):2508–2516. | ||
Ernsting MJ, Murakami M, Roy A, Li SD. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J Control Release. 2013;172(3):782–794. | ||
Gilleron J, Querbes W, Zeigerer A, et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol. 2013;31(7):638–646. | ||
Sahay G, Querbes W, Alabi C, et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat Biotechnol. 2013;31(7):653–658. | ||
Gilleron J, Paramasivam P, Zeigerer A, et al. Identification of siRNA delivery enhancers by a chemical library screen. Nucleic Acids Res. 2015;43(16):7984–8001. | ||
Osborn MF, Alterman JF, Nikan M, et al. Guanabenz (Wytensin™) selectively enhances uptake and efficacy of hydrophobically modified siRNAs. Nucleic Acids Res. 2015;43(18):8664–8672. | ||
Hou KK, Pan H, Schlesinger PH, Wickline SA. A role for peptides in overcoming endosomal entrapment in siRNA delivery – a focus on melittin. Biotechnol Adv. 2015;33(6 Pt 1):931–940. | ||
Hou KK, Pan H, Ratner L, Schlesinger PH, Wickline SA. Mechanisms of nanoparticle-mediated siRNA transfection by melittin-derived peptides. ACS Nano. 2013;7(10):8605–8615. | ||
Wooddell CI, Rozema DB, Hossbach M, et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol Ther. 2013;21(5):973–985. | ||
Golovin YI, Gribanovsky SL, Golovin DY, et al. Towards nanomedicines of the future: Remote magneto-mechanical actuation of nanomedicines by alternating magnetic fields. J Control Release. 2015;219:43–60. | ||
Akinc A, Querbes W, De S, et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol Ther. 2010;18(7):1357–1364. | ||
Cohen ZR, Ramishetti S, Peshes-Yaloz N, et al. Localized RNAi therapeutics of chemoresistant grade IV glioma using hyaluronan-grafted lipid-based nanoparticles. ACS Nano. 2015;9(2):1581–1591. | ||
McCaskill J, Singhania R, Burgess M, et al. Efficient biodistribution and gene silencing in the lung epithelium via intravenous liposomal delivery of siRNA. Mol Ther Nucleic Acids. 2013;2:e96. | ||
Hajdu P, Chimote AA, Thompson TH, Koo Y, Yun Y, Conforti L. Functionalized liposomes loaded with siRNAs targeting ion channels in effector memory T cells as a potential therapy for autoimmunity. Biomaterials. 2013;34(38):10249–10257. | ||
Ramishetti S, Kedmi R, Goldsmith M, et al. Systemic gene silencing in primary T lymphocytes using targeted lipid nanoparticles. ACS Nano. 2015;9(7):6706–6716. | ||
Weinstein S, Toker IA, Emmanuel R, et al. Harnessing RNAi-based nanomedicines for therapeutic gene silencing in B-cell malignancies. Proc Natl Acad Sci U S A. 2016;113(1):E16–E22. | ||
Ku SH, Jo SD, Lee YK, Kim K, Kim SH. Chemical and structural modifications of RNAi therapeutics. Adv Drug Deliv Rev. Epub 2015 Nov 5. | ||
Chen S, Tam YY, Lin PJ, Leung AK, Tam YK, Cullis PR. Development of lipid nanoparticle formulations of siRNA for hepatocyte gene silencing following subcutaneous administration. J Control Release. 2014;196:106–112. | ||
Landesman-Milo D, Peer D. Transforming nanomedicines from lab scale production to novel clinical modality. Bioconjug Chem. Epub 2016 Jan 20. | ||
Semple SC, Akinc A, Chen J, et al. Rational design of cationic lipids for siRNA delivery. Nat Biotechnol. 2010;28(2):172–176. | ||
Tousignant JD, Gates AL, Ingram LA, et al. Comprehensive analysis of the acute toxicities induced by systemic administration of cationic lipid:plasmid DNA complexes in mice. Hum Gene Ther. 2000;11(18):2493–2513. | ||
Zhang JS, Liu F, Huang L. Implications of pharmacokinetic behavior of lipoplex for its inflammatory toxicity. Adv Drug Deliv Rev. 2005;57(5):689–698. | ||
Xue HY, Liu S, Wong HL. Nanotoxicity: a key obstacle to clinical translation of siRNA-based nanomedicine. Nanomedicine (Lond). 2014;9(2):295–312. | ||
Kim HP, Han SW, Song SH, et al. Testican-1-mediated epithelial-mesenchymal transition signaling confers acquired resistance to lapatinib in HER2-positive gastric cancer. Oncogene. 2014;33(25):3334–3341. | ||
Zhou D, Zhang YI, Liang D, et al. Effect of combination therapy of siRNA targeting growth hormone receptor and 5-fluorouracil in hepatic metastasis of colon cancer. Oncol Lett. 2015;10(6):3505–3509. | ||
Geisbert TW, Lee AC, Robbins M, et al. Postexposure protection of non-human primates against a lethal Ebola virus challenge with RNA interference: a proof-of-concept study. Lancet. 2010;375(9729):1896–1905. | ||
Yuen MF, Chan HL, Given BD, Hamilton J, Schluep T, David L. Phase II, dose ranging study of ARC-520, a siRNA-based therapeutic, in patients with chronic hepatitis B virus infection. Hepatology. 2015;62:1385A. | ||
Yuen MF, Chan HL, Given BD, et al. ARC-520 produces deep and durable knockdown of viral antigens and DNA in a phase II study in patients with chronic hepatitis B. Hepatology. 2015;62:1385A. | ||
Gish RG, Yuen MF, Chan HL, et al. Synthetic RNAi triggers and their use in chronic hepatitis B therapies with curative intent. Antiviral Res. 2015;121:97–108. | ||
Beg MS, Brenner A, Sachdev J, et al. Safety, tolerability, and clinical activity of MRX34, the first-in-class liposomal miR-34 mimic, in patients with advanced solid tumors. Mol Cancer Ther. 2015;14:C43. | ||
Liu X. Targeting polo-like kinases: a promising therapeutic approach for cancer treatment. Transl Oncol. 2015;8(3):185–195. | ||
Ramanathan RK, Hamburg SI, Borad MJ, et al. A phase I dose escalation study of TKM-080301, a RNAi therapeutic directed against PLK1, in patients with advanced solid tumors. Cancer Res. 2013;73(8 Suppl):LB-289. | ||
Abrams M, Ganesh S, Ying B, et al. EnCore-LNP mediated tumor delivery of MYC and CTNNB1 dicer substrate RNAs (DsiRNAs). Mol Cancer Res. 2015;13(10 Suppl):B20. | ||
Ganesh S, Ying B, Koser M, et al. Systemic delivery of CTNNB1 dicer-substrate siRNAs (DsiRNAs) leads to efficient oncogene silencing in diverse tumor types of extra hepatic origin. Cancer Res. 2015;75(15 Suppl):3533. | ||
Landen CN Jr, Chavez-Reyes A, Bucana C, et al. Therapeutic EphA2gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res. 2005;65(15):6910–6918. | ||
Shen H, Rodriguez-Aguayo C, Xu R, et al. Enhancing chemotherapy response with sustained EphA2 silencing using multistage vector delivery. Clin Cancer Res. 2013;19(7):1806–1815. | ||
Ozcan G, Ozpolat B, Coleman RL, Sood AK, Lopez-Berestein G. Preclinical and clinical development of siRNA-based therapeutics. Adv Drug Deliv Rev. 2015;87:108–119. | ||
Fitzgerald K, Frank-Kamenetsky M, Shulga-Morskaya S, et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, Phase 1 trial. Lancet. 2014;383(9911):60–68. | ||
Horton JD, Cohen JC, Hobbs HH. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem Sci. 2007;32(2):71–77. | ||
Poirier S, Mayer G, Poupon V, et al. Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: evidence for an intracellular route. J Biol Chem. 2009;284(42):28856–28864. | ||
Zhang XL, Zhu QQ, Zhu L, et al. Safety and efficacy of anti-PCSK9 antibodies: a meta-analysis of 25 randomized, controlled trials. BMC Med. 2015;13:123. | ||
Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjærg-Hansen a. loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371(1):32–41. | ||
Graham MJ, Lee RG, Bell TA 3rd, et al. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ Res. 2013;112(11):1479–1490. | ||
Wang X, Wang D, Shan Z. Clinical and genetic analysis of a family diagnosed with familial hypobetalipoproteinemia in which the proband was diagnosed with diabetes mellitus. Atherosclerosis. 2015;239(2):552–556. | ||
Tikka A, Soronen J, Laurila PP, Metso J, Ehnholm C, Jauhiainen M. Silencing of ANGPTL 3 (angiopoietin-like protein 3) in human hepatocytes results in decreased expression of gluconeogenic genes and reduced triacylglycerol-rich VLDL secretion upon insulin stimulation. Biosci Rep. 2014;34(6):e00160. | ||
Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819–829. | ||
Butler JS, Chan A, Costelha S, et al. Preclinical evaluation of RNAi as a treatment for transthyretin-mediated amyloidosis. Amyloid. 2016;23(2):1–10. | ||
Haase N, Foster D, Bercher J, et al. RNAi therapeutics targeting human angiotensinogen (hAGT) ameliorate preeclamptic sequelae in an established transgenic rodent model for preeclampsia. Hypertension. 2014;64:A666. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.