Back to Journals » OncoTargets and Therapy » Volume 8
Links between cancer stem cells and epithelial– mesenchymal transition
Authors Wang S, Jiang J, Liang X, Tang Y
Received 6 July 2015
Accepted for publication 17 September 2015
Published 16 October 2015 Volume 2015:8 Pages 2973—2980
DOI https://doi.org/10.2147/OTT.S91863
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Daniele Santini
Sha-sha Wang,1,2 Jian Jiang,1,2 Xin-hua Liang,1,2 Ya-ling Tang1,3
1State Key Laboratory of Oral Diseases, 2Department of Oral and Maxillofacial Surgery, 3Department of Oral Pathology, West China Hospital of Stomatology, Sichuan University, Chengdu, People’s Republic of China
Abstract: The epithelial–mesenchymal transition (EMT) has been reported to be an important program that is often activated during the process of cancer invasion and metastasis. Cancer stem cells (CSCs) that can initiate and maintain cancer are also involved in invasion and metastasis of cancer. Recently, insights into the molecular mechanisms and functional features of mesenchymal cells have been greatly colored by findings that some of them have been endowed with the self-renewal trait associated with normal tissue stem cells and CSCs. Among cancer cells experiencing EMT, only some of the most competent CSCs will succeed in planting in another organ. In this paper, we review the molecular mechanism behind the link of EMT and CSCs in cancer progression.
Keywords: epithelial–mesenchymal transition (EMT), EMT regulators, cancer stem cells (CSCs), miRNAs, metastasis
Introduction
Tumor metastasis is an intricate sequential process that requires that a discrete population of tumor cells own the capacity to intravasate from the primary tumor into systemic circulation, survive in circulation, extravasate at a distant site, and proliferate in a second microenvironment. Despite many years of basic and clinical research aimed at curbing cancer, metastasis is still a challenging issue and remains the leading cause of cancer-related deaths worldwide.1 Recently, epithelial–mesenchymal transition (EMT) and cancer stem cells (CSCs) have been shown to play an important role in cancer metastasis. This review discusses the changes in cancer cells from EMT to acquisition of CSC properties to clarify the molecular mechanisms behind the invasion and metastasis of cancer, which will provide insights for the prevention of tumor metastasis.
EMT in cancer cell invasion and metastasis
During cancer progression, some cancer cells from the primary tumor may reactivate a latent embryonic program known as EMT, which has been thought to be a necessary step in tumor invasion and metastasis.2 Through EMT, the transformed epithelial cells can obtain mesenchymal traits that seem to contribute to metastasis. Individual cancer cell with a mesenchymal phenotype possesses the ability to cross endothelial barriers and enter blood and lymphatic circulations. Once cancer cells reach their foreign tissue, they no longer encounter the signals that they experienced in the primary tumor, and will invert to an epithelial phenotype via mesenchymal–epithelial transition (MET).3 Zinc-finger transcriptional factors, including SNAIL, SLUG, TWIST, ZEB1, SIP1, and E47, play a critical role in inducing EMT through the inhibition of E-cadherin.4–6 A large number of pathways, such as TGFβ, Wnt, NF-κB, Notch, integrins, and tyrosine-kinase receptors (EGF, FGF, HGF, PDGF, IGF), among others, have been shown to be crucial in leading to EMT. Functional interaction between these pathways might result in signal amplification and induce EMT and metastasis.
CSCs in cancer cell invasion and metastasis
The concept of CSCs was first put forward in liquid tumors (myeloma and leukemia) when only a small percentage (1%–4%) of cancer cells were observed to proliferate extensively and form colonies.7,8 Currently, a growing body of evidence supports the view that cancers are diseases driven by a subpopulation of self-renewing CSCs, which have been found in hematopoietic9,10 and solid tumors, including brain cancer,11 breast cancer,12 head and neck cancer,13,14 colon cancer,15 lung cancer,16 prostate cancer,17–19 and ovarian cancer.20 CSCs have the capacity to differentiate, self-renew, acquire drug resistance, anchor independently, and migrate.21 They can generate diverse tumor cells to maintain long-term growth and self-renewal to sustain their own population. A variety of developmental signaling pathways, such as the Wnt, Notch, Hedgehog, BMP, FGF, IGF, and TGFβ pathways, are known to affect stem cell self-renewal and differentiation.22 The most widely used method for identifying CSCs involves sorting viable cells based on the expression of surface markers, such as CD10, CD24, CD44, CD133, Bmi-1, SCF, ABCG2, c-Kit, ALDH1, Oct3/4, Sox2, Notch-1, Nanog, nestin, p63, and α2β1-integrin.23–25 CSCs have been shown to initiate and sustain primary tumor growth, drive seeding, and establish metastases at distal sites,26 and targeting their eradication may hold promise for ultimately effective cancer treatment.
Relationship between EMT and CSCs
The association of CSCs with EMT in cancer was established only recently, as similarities in these two fields were noted for contributing to tumor recurrence, metastasis, and drug resistance. EMT has been confirmed to play a critical role in tumor metastasis and recurrence, which have been shown to be tightly linked with the function of CSCs.27–30 However, the molecular mechanism through which cells with EMT transform stem-like cells remains to be addressed.
Reports have demonstrated that cells undergoing EMT can acquire stem cell-like characteristics, which indicated an interesting conjunction between EMT and stem cells.31,32 Breast epithelial cells induced into EMT have been shown to have similarity with mesenchymal stem cells in gene-expression profile, multidirectional differentiation, and ability to migrate toward wound sites.33 Mani et al found that EMT induction in human mammary epithelial cells could lead to the acquisition of mesenchymal morphology and the expression of mesenchymal markers, which increased the CD44+/high/CD24−/low subpopulation with stem cell properties. They also found that transformed human mammary epithelial cells showed effective tumor-initiating ability through EMT.27 Similar results were found by Morel et al34 and Dyck et al.35 Gupta et al showed that the induction of EMT in transformed HMLER breast cancer cells increased the population of CD44+/high/CD24−/low cells, which enhanced a ~100-fold greater mammosphere-forming ability than their epithelial phenotypic cells and increased drug resistance related to the biology of CSCs.36 It has also been reported that cancer-associated fibroblast-induced EMT in prostate carcinoma cells overexpressed stem cell markers, as well as formed spheres and self-renewed.37 This suggested that stem cells can adopt a mesenchymal phenotype without losing their pluripotency, and mesenchymal status seems to be a condition to regain pluripotency. However, widespread consensus on their description and definition is still lacking. In the present review, we bring together the current evidence leading to an increased understanding of the connection between EMT and CSCs.
Signaling pathways linking EMT and CSCs
The signaling pathway links between EMT and the gain of CSC properties are still not explicit; however, the formation of EMT and CSCs has been shown to be a dynamic process, and it is triggered by multiple cellular signaling pathways, such as TGFβ, Wnt/β-catenin, Hedgehog, Notch, and others (Figure 1).38–41
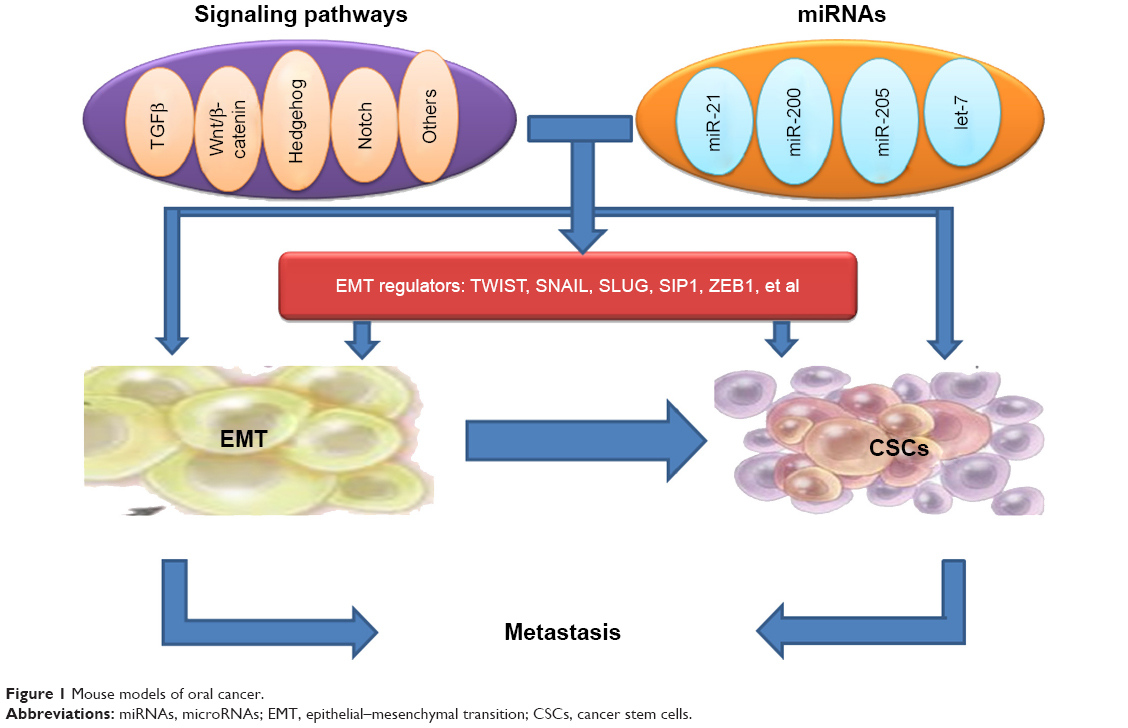 | Figure 1 Mouse models of oral cancer. |
TGFβ signaling
TGFβ is a multifunctional cytokine, as well as one of the major EMT inducers.42 Some groups summarized the complexity of TGFβ signaling during hepatocarcinogenesis, specifically as related to β2-spectrin loss and malignant stem cell transformation.43–45 Recently, the induction of EMT by TGFβ has been linked to the acquisition of tumor-initiating stem cells (TISCs) in breast cancer. Consistent with this finding, van der Horst et al found that mesenchymal liver cancer with EMT demonstrated TISC characteristics, such as tumor-sphere formation. They also found that TGFβ induced EMT and TISC characteristics through the upregulation of SNAIL and Nanog.46 Another study has also shown that the gene-expression profile of the human mammary epithelial cell line introduced by EMT inducers, including TGFβ, closely aligns with a stem cell-like expression profile.47 As additional evidence linking EMT to TISCs, TGFβ regulates Nanog expression in human embryonic stem cells.48,49 Usually, TGFβ works together with Wnt, Hedgehog, Notch, and Ras signaling pathways to induce complete EMT.50 However, Tang et al reported that in transformed human breast epithelial cells, TGFβ stimulation reduced the stem cell-like properties, and TGFβ inhibition increased the size of the CSC population and promoted tumorigenesis by another mechanism that was independent of direct effects on proliferation.51 Therefore, more work is needed to address these contradictory results on the role of TGFβ signaling in the regulation of tumor-initiating properties and EMT.
Wnt/β-catenin signaling
The Wnt/β-catenin signaling pathway can adjust stem cell renewal and be involved in EMT induction in cancer. Loss of the Wnt antagonist SFRP1 results in the activation of Wnt signaling, EMT, and stem cell-like properties, including the CD44+/high/CD24−/low signature.52,53 It has been found that overexpression of the homeobox protein Six1 in the mouse mammary gland produces highly aggressive tumors with an EMT phenotype, stem cell features, and activated Wnt signaling,54 providing in vivo evidence for the emergence of cells with combined EMT–CSC phenotypes. CD24 is a direct target of Wnt signaling and Six1 in mammary epithelial cells, which regulates the population of progenitor cells during EMT induction and obtaining of stem-like traits.55,56 DiMeo et al discovered that the inhibition of Wnt signaling could reduce the capacity of cancer cells to self-renew, and downregulated the expression of SLUG and TWIST.57 Moreover, constitutively activated β-catenin signaling predisposes to tumorigenesis and leads to excessive stem cell proliferation.58–60 Nuclear β-catenin is confined to the invasive front of colorectal cancer, and can be regarded as a marker of EMT in vivo.61 CD44, a downstream target of the β-catenin signaling pathway, correlates with the activation of β-catenin in TWIST-overexpressing cells. The treatment of Wnt3a can induce the activation of β-catenin and the induction of CD44, suggesting that EMT initiates and primes β-catenin activation, and this activation can be further synergized by the Wnt ligand from the microenvironment of the tumor.62 These results together suggest that the treatment of targeting the Wnt/β-catenin pathway can inhibit the stem cell-like properties associated with EMT.
Hedgehog signaling
Hedgehog signaling has been found to relate to the formation of CSCs and EMT.40–63 Reports showed that Hedgehog signaling played a critical role in the maintenance of TISCs and Bmi-1, which may directly mediate Hedgehog signaling in order to confer a self-renewal capacity in TISCs.64,65 The downregulation of Hedgehog signaling by the inhibitors of Hedgehog inhibits CSCs and EMT, accompanied with downregulation of SNAIL and upregulation of E-cadherin, cutting down the invasion and metastasis of pancreatic cancer.66,67
Notch signaling
The Notch signaling pathway has been shown to contribute to EMT induction and regulate asymmetric cell-fate decision in human mammary stem cells.68,69 Many reports have described the close connections between transcription factors regulated by Notch and pathways known to control stem cell function, indicating that Notch is a shared signaling pathway, and may link cancer EMT and CSCs.70
Regulation of CSCs by EMT regulators
Recent evidence suggests that the expression of certain genes involved in CSCs are influenced by transcription factors of EMT, implicating EMT as potential factors involved in stem cell maintenance. The link between EMT regulators and CSCs points to CSCs as the molecular and cellular explanation for the relationship between EMT and cancer metastasis (Figure 1).
TWIST
Some groups showed that cells induced to undergo EMT (by ectopic expression of SNAIL, TWIST, or TGFβ treatment) acquired a CD44+/high/CD24−/low signature, similar to a small population of breast CSCs in xenograft models that had been identified to possess a unique ability to form tumors.27,71 Moreover, Vesuna et al demonstrated the direct involvement of TWIST in producing a breast CSC phenotype through downregulation of CD24.72 Another experiment showed that upregulation of TWIST induced EMT in HeLa and MCF7 cells accompanying the gain of stem cell markers, such as overexpression of CD44 and ALDH1.62 Mani et al further reported that the induction of nontumorigenic, immortalized human mammary epithelial cells by TWIST or SNAIL led to the loss of epithelium and the acquisition of mesenchyme concomitant with the acquisition of a CD44+/high/CD24−/low expression pattern.27 Similar results were also found by Patel et al.73 It has also been reported that overexpression of TWIST2 in mammary epithelial cells and breast cancer cells enhanced the size and number of CD44+/high/CD24−/low stem-like cell populations and the self-renewal capabilities of stem-like cells.74
At the molecular level, TWIST1 directly stimulates the expression of Bmi-1,75,76 which encodes a polycomb-group protein that maintains self-renewal through repression of the p16INK4a–ARF locus. The direct activation of Bmi-1 expression by TWIST1 has been revealed by different assays, such as transient transfection, electrophoretic mobility shift, and chromatin immunoprecipitation.21,76 TWIST1 not only directly activates Bmi-1 expression but also cooperates with Bmi-1 to mediate cancer stemness and EMT.77
SNAIL
Loss of SNAIL in mesenchymal cells can cause downregulated Nanog promoter luciferase activity and the loss of self-renewal characteristics in vitro, which verifies the direct role of SNAIL in some TISC traits. In mesenchymal cells post-EMT, SNAIL can directly control Nanog expression, and loss of SNAIL can control tumor growth without influencing tumor initiation. Inhibition of SNAIL can result in the downregulation of Nanog, Bmi-1, and CD44 and the loss of self-renewal, as evidenced by decreased tumor-sphere formation.78 Recent studies have demonstrated that some members of the SNAIL family can confer an EMT phenotype to breast epithelial cells, which can change the cell phenotype from CD44−/low/CD24+/high to CD44+/high/CD24−/low.27,34 In human colorectal carcinoma tissues, SNAIL regulates expression of IL8 and other genes to induce CSC activities.79
SLUG
SLUG overexpression in MCF-7 cells generates CD44+/high/CD24+/high cells with enhanced mammosphere-forming ability.80 SLUG highly expressed in basal type breast cancers also tends to express higher levels of stem cell-associated genes, such as CD133, BMI1, and KIT.81 Moreover, SLUG−/− embryonic fibroblasts show reduced expression of several genes linked to self-renewal and chromatin remodeling.82 It has also been reported that SNAIL and SLUG induce the expression of stem-like promoting genes, such as NANOG, KLF4, and TCF4, to mediate radio- and chemoresistance to ovarian cancer cells.83 SLUG has been regarded as a critical regulator of epithelial cell identity in breast development and cancer.84
Other regulators
Liu and Dean suggested that tumor-sphere cells express ABCG2 on their surface, excluded Hoechst dye, had a CD44+/high/CD24−/low cell-surface pattern, and overexpressed the EMT transcription factor ZEB1. More importantly, knockdown of ZEB1 blocked formation of these reprogrammed cells.85 Recently, YB1 has been shown to promote SNAIL, TWIST, and SIP1, together with the upregulation of the stem cell markers p63, CD44, and CD10, thus appearing to link the acquisition of the EMT and CSC phenotypes.86 Moreover, the molecular targets of YB1 in the MDAMB-231 and SUM149 breast cancer cell lines have been shown to include the stem cell-associated markers CD44 and CD49, as well as c-Kit, Bmi-1, and members of the Wnt and Notch signaling pathways.87 Further study demonstrated that the LBX1 protein transcriptionally targets ZEB1, SIP1, SNAIL, and TGFβ2. Accordingly, ectopic LBX1 expression in mammary epithelial cells induces EMT, with a concordant increase in mammosphere formation and the proportion of CD44+/high/CD24−/low cells.88 The homeobox transcription factor Six1 (when activated in transgenic mice) induces an EMT-like conversion and causes mammary gland cancer by increasing the population of cells in mouse mammary tumors displaying CSC markers.89 Together, these findings seem consistent with the notion of regulators of EMT as potential factors involved in stem cell maintenance.
On the other hand, CSC markers can inversely regulate EMT transcription factors. Yu et al recently showed that Bmi-1 played a major role in the maintenance of stemness and the metastatic ability of head and neck squamous cell carcinoma (HNSCC) CSCs by regulation of SNAIL expression.90 Hu et al demonstrated that downregulation of Oct4 induces an EMT via enhancement of Ca2+ influx in breast cancer cells.91 Moreover, Oct4 and Nanog controlled the EMT of lung adenocarcinoma cells through activating SLUG.92
miRNAs linking EMT with stem cell signatures
MicroRNAs (miRNAs) have been shown to regulate the formation of CSCs and the acquisition of the EMT phenotype.93,94 The discovery of miRNAs has complicated the molecular networks regulating EMT and stemness in cancer metastasis (Figure 1).95,96 miRNAs, small noncoding RNA molecules, can lower gene expression through interacting with seed sequences located in the 3′UTR of multiple target messenger RNAs, which leads to translational repression and degradation of messenger RNAs.97
It has been reported that pancreatic cancer cells with the EMT phenotype display stem-like cell features and promote clonogenic and sphere-forming ability and tumorigenicity in mice, which is associated with the downregulation of miR-200 and/or the let-7 family. Reversal of EMT by reexpression of miR-200 suppresses the prostasphere-forming ability of EMT-type cells and decreases the expression of Notch1 and Lin28B. Loss of Lin28B adds let-7 expression and represses self-renewal capability. These data suggest that miR-200 and let-7 could link cancer stem-like cells with EMT.29 The miR-200 family can regulate the processes of EMT by targeting the proteins ZEB1 and ZEB2,98 but also relates to stem-like cell signatures by regulating Bmi-1.95,96 Moreover, EMT and stem cell-like properties associated with miR-205 and miR-200 epigenetic silencing manifest early during carcinogen-induced transformation of human lung epithelial cells.99,100 In claudinlow SUM159 cells, the expression of miR-93 induces MET associated with reduction in TGFβ signaling and downregulates multiple stem cell regulatory genes including SOX4, JAK1, AKT3, EZH1, and HMGA2, resulting in CSC depletion.101 In another experiment, Han et al demonstrated that the inhibition of miR-21 reverses EMT and CSC phenotypes of cancer cells by targeting PTEN via inactivation of the AKT and ERK1/2 pathways.102 Targeting of CSCs and EMT-phenotypic cells through selectively changing the expression of specific miRNAs toward removing cancer metastasis and recurrence will help to determine novel therapeutic strategies. The potential synergistic combination of natural compounds that affect critical miRNAs, such as curcumin or epigallocatechin-3-gallate with chemotherapeutic agents will be particularly promising.103
Potential application of CSCs and EMT in cancer biology
The potential application of the identification of this link between CSCs and EMT has just begun, though it is useful. Some groups have tried to apply the molecular mechanism of the link between CSC and EMT to carry on the initial application of cancer treatment. Loss of the tumor suppressor p53 in mammary epithelial cells has been shown to induce EMT and enrich CSCs through repression of miR200c, suggesting that the p53–miR200c pathway can be activated to suppress EMT-associated CSCs to treat cancer.104 Overexpression of FoxM1 in pancreatic cancer is responsible for the acquisition of EMT and CSC phenotypes, which is in part mediated through the regulation of miR-200b. More importantly, these processes could be easily attenuated by genistein, a natural chemopreventive agent.105 Knockdown of TFAP2C in luminal breast carcinoma cells induces EMT with morphological and phenotypic changes characterized by a loss of luminal-associated gene expression and a concomitant gain of basal-associated gene expression, suggesting that TFAP2C has an important role in regulated luminal-specific genes and may be a viable therapeutic target in breast cancer.106 Sumoylation inhibitors facilitate MET activity of the transcriptional factor TFAP, clears the CD44+/high/CD24−/low cell population characterizing basal cancers, and inhibits tumor outgrowth of basal cancer xenografts. These findings establish a critical role for sumoylation in the potential application of the link between CSCs and EMT.107 The G9a protein could induce EMT and CSC-like properties in HNSCC, and targeting the G9a–Snail axis may represent a novel strategy for the treatment of metastatic HNSCC.108
Conclusion
EMT has been emerging as one of the hottest medical science topics, and the role of EMT in cancer explains part of the mechanism of the initial step of metastasis. The concept of CSCs has been established as a subpopulation of cells within a tumor entirely responsible for tumorigenesis. With the discovery of more molecular knowledge of CSCs and EMT, novel therapeutic strategies could be designed to target CSCs and EMT cells to add drug sensitivity, which will thereby delay tumor progression and metastasis. Taken together, CSCs and EMT seem to be an axis of evil in cancer, for which better understanding may contribute to the appearance of new therapeutic platforms.
Acknowledgments
We apologize to those colleagues whose work we could not reference directly due to space constraints. This work was supported by National Natural Science Foundation of China grants (81361120399, 81321002, 81272961, 81572650, and 81372891) and by State Key Laboratory of Oral Diseases Special Funded Projects (SKLOD201512).
Disclosure
The authors report no conflicts of interest in this work.
References
Fan S, Tang QL, Lin YJ, et al. A review of clinical and histological parameters associated with contralateral neck metastases in oral squamous cell carcinoma. Int J Oral Sci. 2011;3(4):180–191. | ||
Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7(2):131–142. | ||
Wu Y, Zhou BP. Inflammation: a driving force speeds cancer metastasis. Cell Cycle. 2009;8(20):3267–3273. | ||
Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7(6):415–428. | ||
Ye X, Tam WL, Shibue T, et al. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature. 2015;525(7568):256–260. | ||
Yang MH, Wu KJ. TWIST activation by hypoxia inducible factor-1 (HIF-1): implications in metastasis and development. Cell Cycle. 2008;7(14):2090–2096. | ||
Park CH, Bergsagel DE, McCulloch EA. Mouse myeloma tumor stem cells: a primary cell culture assay. J Natl Cancer Inst. 1971;46(2):411–422. | ||
Bruce WR, Van Der Gaag H. A quantitative assay for the number of murine lymphoma cells capable of proliferation in vivo. Nature. 1963;199(4888):79–80. | ||
Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–737. | ||
Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645–648. | ||
Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. | ||
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. | ||
Ailles L, Prince M. Cancer stem cells in head and neck squamous cell carcinoma. Methods Mol Biol. 2009;568:175–193. | ||
Prince ME, Sivanandan R, Kaczorowski A, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. 2007;104(3):973–978. | ||
Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445(7123):111–115. | ||
Eramo A, Lotti F, Sette G, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15(3):504–514. | ||
Gu G, Yuan J, Wills M, Kasper S. Prostate cancer cells with stem cell characteristics reconstitute the original human tumor in vivo. Cancer Res. 2007;67(10):4807–4815. | ||
Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67(3):1030–1037. | ||
Hermann PC, Huber SL, Herrler T, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1(3):313–323. | ||
Zhang S, Balch C, Chan MW, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68(11):4311–4320. | ||
Maugeri-Saccà M, Vigneri P, De Maria R. Cancer stem cells and chemosensitivity. Clin Cancer Res. 2011;17(15):4942–4947. | ||
Wu KJ. Direct activation of Bmi1 by Twist1: implications in cancer stemness, epithelial-mesenchymal transition, and clinical significance. Chang Gung Med J. 2011;34(3):229–238. | ||
Zhang S, Balch C, Chan MW. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68(11):4311–4320. | ||
Yadav AK, Sahasrabuddhe AA, Dimri M, Bommi PV, Sainger R, Dimri GP. Deletion analysis of BMI1 oncoprotein identifies its negative regulatory domain. Mol Cancer. 2010;9:158. | ||
Biddle A, Liang X, Gammon L, et al. Cancer stem cells in squamous cell carcinoma switch between two distinct phenotypes that are preferentially migratory or proliferative. Cancer Res. 2011;71(15):5317–5326. | ||
May CD, Sphyris N, Evans KW, Werden SJ, Guo W, Mani SA. Epithelial-mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res. 2011;13(1):202. | ||
Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. | ||
Zhi Y, Mou Z, Chen J, et al. B7H1 expression and epithelial-to-mesenchymal transition phenotypes on colorectal cancer stem-like cells. PLoS One. 2015;10(8):e0135528. | ||
Garg M. Urothelial cancer stem cells and epithelial plasticity: current concepts and therapeutic implications in bladder cancer. Cancer Metastasis Rev. Epub 2015 Sep 2. | ||
Chang YW, Su YJ, Hsiao M, et al. Diverse targets of β-catenin during the epithelial-mesenchymal transition define cancer stem cells and predict disease relapse. Cancer Res. 2015;75(16):3398–3410. | ||
Radisky DC, LaBarge MA. Epithelial-mesenchymal transition and the stem cell phenotype. Cell Stem Cell. 2008;2(6):511–512. | ||
Scheel C, Weinberg RA. Phenotypic plasticity and epithelial-mesenchymal transitions in cancer and normal stem cells? Int J Cancer. 2011;129(10):2310–2314. | ||
Battula VL, Evans KW, Hollier BG, et al. Epithelial-mesenchymal transition-derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem Cells. 2010;28(8):1435–1445. | ||
Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 2008;3(8):e2888. | ||
Dyck HG, Hamilton TC, Godwin AK, Lynch HT, Maines-Bandiera S, Auersperg N. Autonomy of the epithelial phenotype in human ovarian surface epithelium: changes with neoplastic progression and with a family history of ovarian cancer. Int J Cancer. 1996;69(6):429–436. | ||
Gupta PB, Onder TT, Jiang G, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138(4):645–659. | ||
Giannoni E, Bianchini F, Masieri L, et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010;70(17):6945–6956. | ||
Singh A, Greninger P, Rhodes D, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15(6):489–500. | ||
Wang Z, Li Y, Kong D, et al. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009;69(6):2400–2407. | ||
Sarkar FH, Li Y, Wang Z, Kong D. Pancreatic cancer stem cells and EMT in drug resistance and metastasis. Minerva Chir. 2009;64(5):489–500. | ||
Wang H, Wu J, Zhang Y, et al. Transforming growth factor β-induced epithelial-mesenchymal transition increases cancer stem-like cells in the PANC-1 cell line. Oncol Lett. 2012;3(1):229–233. | ||
Amin R, Mishra L. Liver stem cells and TGF-β in hepatic carcinogenesis. Gastrointest Cancer Res. 2008;2(4 Suppl):S27–S30. | ||
Thenappan A, Li Y, Kitisin K, et al. Role of transforming growth factor β signaling and expansion of progenitor cells in regenerating liver. Hepatology. 2010;51(4):1373–1382. | ||
Mishra L, Derynck R, Mishra B. Transforming growth factor-β signaling in stem cells and cancer. Science. 2005;310(5745):68–71. | ||
Tang Y, Katuri V, Dillner A, Mishra B, Deng CX, Mishra L. Disruption of transforming growth factor-β signaling in ELF β-spectrin-deficient mice. Science. 2003;299(5606):574–577. | ||
van der Horst G, van den Hoogen C, Buijs JT, et al. Targeting of α(v)-integrins in stem/progenitor cells and supportive microenvironment impairs bone metastasis in human prostate cancer. Neoplasia. 2011;13(6):516–525. | ||
Muraoka-Cook RS, Shin I, Yi JY, et al. Activated type I TGFβ receptor kinase enhances the survival of mammary epithelial cells and accelerates tumor progression. Oncogene. 2006;25(24):3408–3423. | ||
Xu RH, Sampsell-Barron TL, Gu F, et al. NANOG is a direct target of TGFβ/activin-mediated SMAD signaling in human ESCs. Cell Stem Cell. 2008;3(2):196–206. | ||
Greber B, Lehrach H, Adjaye J. Control of early fate decisions in human ES cells by distinct states of TGFβ pathway activity. Stem Cells Dev. 2008;17(6):1065–1077. | ||
Fuxe J, Vincent T, Garcia de Herreros A. Transcriptional crosstalk between TGF-β and stem cell pathways in tumor cell invasion: role of EMT promoting Smad complexes. Cell Cycle. 2010;9(12):2363–2374. | ||
Tang B, Yoo N, Vu M, et al. Transforming growth factor-β can suppress tumorigenesis through effects on the putative cancer stem or early progenitor cell and committed progeny in a breast cancer xenograft model. Cancer Res. 2007;67(18):8643–8652. | ||
van den Brink GR, Bleuming SA, Hardwick JC, et al. Indian Hedgehog is an antagonist of Wnt signaling in colonic epithelial cell differentiation. Nat Genet. 2004;36(3):277–282. | ||
Douard R, Moutereau S, Pernet P, et al. Sonic Hedgehog-dependent proliferation in a series of patients with colorectal cancer. Surgery. 2006;139(5):665–670. | ||
McCoy EL, Iwanaga R, Jedlicka P, et al. Six1 expands the mouse mammary epithelial stem/progenitor cell pool and induces mammary tumors that undergo epithelial-mesenchymal transition. J Clin Invest. 2009;119(9):2663–2677. | ||
Taipale J, Chen JK, Cooper MK, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406(6799):1005–1009. | ||
Maeda O, Kondo M, Fujita T, et al. Enhancement of GLI1-transcriptional activity by β-catenin in human cancer cells. Oncol Rep. 2006;16(1):91–96. | ||
DiMeo TA, Anderson K, Phadke P, et al. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res. 2009;69(13):5364–5373. | ||
Zhang B, Yang Y, Shi X, et al. Proton pump inhibitor pantoprazole abrogates adriamycin-resistant gastric cancer cell invasiveness via suppression of Akt/GSK-β/β-catenin signaling and epithelial-mesenchymal transition. Cancer Lett. 2015;356(2 Pt B):704–712. | ||
Warrier S, Bhuvanalakshmi G, Arfuso F, Rajan G, Millward M, Dharmarajan A. Cancer stem-like cells from head and neck cancers are chemosensitized by the Wnt antagonist, sFRP4, by inducing apoptosis, decreasing stemness, drug resistance and epithelial to mesenchymal transition. Cancer Gene Ther. 2014;21(9):381–388. | ||
Yang N, Hui L, Wang Y, Yang H, Jiang X. Overexpression of SOX2 promotes migration, invasion, and epithelial-mesenchymal transition through the Wnt/β-catenin pathway in laryngeal cancer Hep-2 cells. Tumour Biol. 2014;35(8):7965–7973. | ||
Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, β-catenin and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009;28(1–2):151–166. | ||
Li J, Zhou BP. Activation of β-catenin and Akt pathways by Twist are critical for the maintenance of EMT associated cancer stem cell-like characters. BMC Cancer. 2011;11:49. | ||
Batsaikhan BE, Yoshikawa K, Kurita N, et al. Cyclopamine decreased the expression of Sonic Hedgehog and its downstream genes in colon cancer stem cells. Anticancer Res. 2014;34(11):6339–6344. | ||
Liu S, Dontu G, Mantle ID, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66(12):6063–6071. | ||
Rangwala F, Omenetti A, Diehl AM. Cancer stem cells: repair gone awry? J Oncol. 2011;2011:465343. | ||
Feldmann G, Fendrich V, McGovern K, et al. An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol Cancer Ther. 2008;7(9):2725–2735. | ||
Feldmann G, Dhara S, Fendrich V, et al. Blockade of Hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. 2007;67(5):2187–2196. | ||
Zhang X, Zhao X, Shao S, et al. Notch1 induces epithelial-mesenchymal transition and the cancer stem cell phenotype in breast cancer cells and STAT3 plays a key role. Int J Oncol. 2015;46(3):1141–1148. | ||
Kotiyal S, Bhattacharya S. Breast cancer stem cells, EMT and therapeutic targets. Biochem Biophys Res Commun. 2014;453(1):112–116. | ||
Creighton CJ, Chang JC, Rosen JM. Epithelial-mesenchymal transition (EMT) in tumor-initiating cells and its clinical implications in breast cancer. J Mammary Gland Biol Neoplasia. 2010;15(2):253–260. | ||
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. | ||
Vesuna F, Lisok A, Kimble B, Raman V. Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia. 2009;11(12):1318–1328. | ||
Patel SA, Ndabahaliye A, Lim PK, Milton R, Rameshwar P. Challenges in the development of future treatments for breast cancer stem cells. Breast Cancer (Dove Med Press). 2010;2:1–11. | ||
Fang X, Cai Y, Liu J, et al. Twist2 contributes to breast cancer progression by promoting an epithelial-mesenchymal transition and cancer stem-like cell self-renewal. Oncogene. 2011;30(47):4707–4720. | ||
Martin A, Cano A. Tumorigenesis: Twist1 links EMT to self-renewal. Nat Cell Biol. 2010;12(10):924–925. | ||
Yang MH, Hsu DS, Wang HW, et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat Cell Biol. 2010;12(10):982–992. | ||
Wu CY, Hung JJ, Wu KJ. Linkage between Twist1 and Bmi1: molecular mechanism of cancer metastasis/stemness and clinical implications. Clin Exp Pharmacol Physiol. 2012;39(8):668–673. | ||
You H, Ding W, Dang H, Jiang Y, Rountree CB. c-Met represents a potential therapeutic target for personalized treatment in hepatocellular carcinoma. Hepatology. 2011;54(3):879–889. | ||
Hwang WL, Yang MH, Tsai ML, et al. SNAIL regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology. 2011;141(1):279–291, 291.e1–e5. | ||
Bhat-Nakshatri P, Appaiah H, Ballas C, et al. SLUG/SNAI2 and tumor necrosis factor generate breast cells with CD44+/CD24− phenotype. BMC Cancer. 2010;10:411. | ||
Lim E, Vaillant F, Wu D, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15(8):907–913. | ||
Bermejo-Rodríguez C, Pérez-Caro M, Pérez-Mancera PA, Sánchez-Beato M, Piris MA, Sánchez-García I. Mouse cDNA microarray analysis uncovers SLUG targets in mouse embryonic fibroblasts. Genomics. 2006;87(1):113–118. | ||
Kurrey NK, Jalgaonkar SP, Joglekar AV, et al. Snail and Slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells. 2009;27(9):2059–2068. | ||
Phillips S, Kuperwasser C. SLUG: critical regulator of epithelial cell identity in breast development and cancer. Cell Adh Migr. 2014;8(6):578–587. | ||
Liu Y, Dean DC. Tumor initiation via loss of cell contact inhibition versus Ras mutation: do all roads lead to EMT? Cell Cycle. 2010;9(5):897–900. | ||
Evdokimova V, Tognon C, Ng T, et al. Translational activation of Snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial-mesenchymal transition. Cancer Cell. 2009;15(5):402–415. | ||
To K, Fotovati A, Reipas KM, et al. Y-box binding protein-1 induces the expression of CD44 and CD49f leading to enhanced self-renewal, mammosphere growth, and drug resistance. Cancer Res. 2010;70(7):2840–2851. | ||
Yu M, Smolen GA, Zhang J, et al. A developmentally regulated inducer of EMT, LBX1, contributes to breast cancer progression. Genes Dev. 2009;23(15):1737–1742. | ||
Gavert N, Ben-Ze’ev A. Coordinating changes in cell adhesion and phenotype during EMT-like processes in cancer. F1000 Biol Rep. 2010;2:86. | ||
Yu CC, Lo WL, Chen YW, et al. Bmi-1 regulates Snail expression and promotes metastasis ability in head and neck squamous cancer-derived ALDH1 positive cells. J Oncol. 2011;2011:609259. | ||
Hu J, Qin K, Zhang Y, et al. Downregulation of transcription factor Oct4 induces an epithelial-to-mesenchymal transition via enhancement of Ca(2+) influx in breast cancer cells. Biochem Biophys Res Commun. 2011;411(4):786–791. | ||
Chiou SH, Wang ML, Chou YT, et al. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res. 2010;70(24):10433–10444. | ||
Garofalo M, Croce CM. Role of microRNAs in maintaining cancer stem cells. Adv Drug Deliv Rev. 2015;81:53–61. | ||
Yu D, Shin HS, Lee YS, Lee YC. miR-106b modulates cancer stem cell characteristics through TGF-β/Smad signaling in CD44-positive gastric cancer cells. Lab Invest. 2014;94(12):1370–1381. | ||
Wellner U, Schubert J, Burk UC, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11(12):1487–1495. | ||
Shimono Y, Zabala M, Cho RW, et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009;138(3):592–603. | ||
Garzon R, Fabbri M, Cimmino A, Calin GA, Croce CM. MicroRNA expression and function in cancer. Trends Mol Med. 2006;12(12):580–587. | ||
Gregory PA, Bert AG, Paterson EL, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10(5):593–601. | ||
Tellez CS, Juri DE, Do K, et al. EMT and stem cell-like properties associated with miR-205 and miR-200 epigenetic silencing are early manifestations during carcinogen-induced transformation of human lung epithelial cells. Cancer Res. 2011;71(8):3087–3097. | ||
Wiklund ED, Bramsen JB, Hulf T, et al. Coordinated epigenetic repression of the miR-200 family and miR-205 in invasive bladder cancer. Int J Cancer. 2011;128(6):1327–1334. | ||
Liu S, Patel SH, Ginestier C, et al. MicroRNA93 regulates proliferation and differentiation of normal and malignant breast stem cells. PLoS Genet. 2012;8(6):e1002751. | ||
Han M, Liu M, Wang Y, et al. Antagonism of miR-21 reverses epithelial-mesenchymal transition and cancer stem cell phenotype through AKT/ERK1/2 inactivation by targeting PTEN. PLoS One. 2012;7(6):e39520. | ||
Wang Z, Li Y, Ahmad A, et al. Targeting miRNAs involved in cancer stem cell and EMT regulation: an emerging concept in overcoming drug resistance. Drug Resist Updat. 2010;13(4–5):109–118. | ||
Chang CJ, Chao CH, Xia W, et al. P53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13(3):317–323. | ||
Bao B, Wang Z, Ali S, et al. Over-expression of FoxM1 leads to epithelial-mesenchymal transition and cancer stem cell phenotype in pancreatic cancer cells. J Cell Biochem. 2011;112(9):2296–2306. | ||
Cyr AR, Kulak MV, Park JM, et al. TFAP2C governs the luminal epithelial phenotype in mammary development and carcinogenesis. Oncogene. 2015;34(4):436–444. | ||
Bogachek MV, Chen Y, Kulak MV, et al. Sumoylation pathway is required to maintain the basal breast cancer subtype. Cancer Cell. 2014;25(6):748–761. | ||
Liu S, Ye D, Guo W, et al. MT-mediated metastasis and maintenance of cancer stem cell-like characters in head and neck squamous cell carcinoma. Oncotarget. 2015;6(9):6887–6901. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.