Back to Journals » International Journal of Nanomedicine » Volume 11
Ligand-based targeted therapy: a novel strategy for hepatocellular carcinoma
Authors Li M ![]() , Zhang W, Wang B, Gao Y, Song Z, Zheng QC
, Zhang W, Wang B, Gao Y, Song Z, Zheng QC
Received 24 June 2016
Accepted for publication 19 September 2016
Published 31 October 2016 Volume 2016:11 Pages 5645—5669
DOI https://doi.org/10.2147/IJN.S115727
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Min Li,1 Weiyue Zhang,2 Birong Wang,3 Yang Gao,1 Zifang Song,1 Qi Chang Zheng1
1Department of Hepatobiliary Surgery, Union Hospital, 2The First Clinic Institute, Tongji Medical College, Huazhong University of Science and Technology, 3Department of Breast and Thyroid Surgery, Puai Hospital, Wuhan, The People’s Republic of China
Abstract: Hepatocellular carcinoma (HCC) is the most common primary liver cancer with high morbidity and mortality worldwide. Chemotherapy is recommended to patients with intermediate or advanced stage cancer. However, the conventional chemotherapy yields low desired response rates due to multidrug resistance, fast clearance rate, nonspecific delivery, severe side effects, low drug concentration in cancer cells, and so on. Nanoparticle-mediated targeted drug delivery system can surmount the aforementioned obstacles through enhanced permeability and retention effect and active targeting as a novel approach of therapeutics for HCC in recent years. The active targeting is triggered by ligands on the delivery system, which recognize with and internalize into hepatoma cells with high specificity and efficiency. This review focuses on the latest targeted delivery systems for HCC and summarizes the ligands that can enhance the capacity of active targeting, to provide some insight into future research in nanomedicine for HCC.
Keywords: targeted therapy, hepatocellular carcinoma, ligand, drug delivery system, nanoparticle
Introduction
Hepatocellular carcinoma (HCC), the most common primary liver cancer, represents the sixth most common cancer worldwide, which results in the third cause of death from cancer per year.1–3 The epidemiology, risk factors, and pathogenesis were summarized in a previous highlight article.4 According to the Barcelona Clinic Liver Cancer staging system, patients with intermediate or advanced stage cancer are recommended local or systemic chemotherapy.1,2,5,6 However, conventional chemotherapy yields low objective response rates. Some randomized trials showed that the response rates of combined chemotherapy have been slightly increased, but those patients did not gain survival benefit.7 Sorafenib (Nexavar®; Bayer Pharmaceuticals, Berlin, Germany) is a small multikinase inhibitor which blocks several tyrosine protein kinases such as vascular endothelial growth factor receptors 1, 2, and 3 and platelet-derived growth factor receptor β, as well as Raf kinases and intracellular serine/threonine kinases.8,9 Unfortunately, the median survival of patients treated with sorafenib was just prolonged ~3 months.10 It is known that the undesired outcome is associated with multidrug resistance (MDR),11 fast drug elimination from bloodstream, narrow therapeutic window, nonspecific delivery, a range of side effects, low drug concentration in targeted cells, and aggregation formation due to their poor solubility, and so forth.12 Thus, development of some new approach of anticancer agents is an imperative task to improve the therapeutic efficacy of HCC.
In recent years, nanoparticle (NP)-mediated targeted drug delivery system (NTDDS) has attracted researchers’ great interest as a novel approach of therapeutics for HCC. The NTDDS involves three essential components: a potent therapeutic such as chemotherapeutic agent, gene, or photosensitizer; a rational delivery vehicle to transport therapeutics in high concentration into tumor cells with long circulation time and excellent stability; and a surface ligand that mediates delivery vehicle to interact with and internalize into hepatoma cells with high specificity and efficiency.13–15 Targeted delivery could overcome the deficiencies of conventional administration routes as mentioned earlier. As NPs for targeted delivery of therapeutics and small interfering RNAs (siRNA) in HCC have been reviewed in a recent review,16 this review focuses on the latest NTDDS for HCC and summarizes the ligands that can specifically bind with the matching receptors on hepatoma cell membrane and trigger the receptor-mediated endocytosis, to provide some insight into future research in nanomedicine for HCC.
Enhanced permeability and retention (EPR) effect in HCC
Tumor angiogenesis is a process in which new blood vessels in tumor tissue form from the existing ones. The angiogenesis is induced by various growth factors such as hypoxia inducible factor,17 vascular endothelial growth factor (VEGF),18 platelet-derived growth factor,19 and basic fibroblast growth factor (bFGF)20 because more oxygen and nutrients need to be provided to the tumor mass by dedicated blood supply to sustain fast growth beyond a size of 2 mm3.21 However, the tumor vasculature is dramatically different from normal tissue blood vessels. The abnormal vascular architecture is the pathophysiological basis of passive targeting in that drug delivery systems accumulate in tumor mass much more than they do in normal tissues.22 For example, Kaminskas et al23 investigated the pharmacokinetics and targeting capability of PEGylated dendrimer with 12 nm in diameter and PEGylated liposome with 89 nm in hepatoma cell-bearing mice model, demonstrating that both the systems could promote increased tumor targeting of doxorubicin (DOX) and enhance tumor regression through EPR effect. In 1986, Gerlowski and Jain24 and Matsumura and Maeda25 first defined the phenomenon as EPR effect. The abnormal tumor vasculatures include extensive angiogenesis, inefficient lymphatic drainage, slow venous return, as well as defective vascular architectures such as discontinuous endothelial lining, lack of smooth muscle cells and pericytes, fenestrations, and so on.26,27 The abnormal vasculature in HCC tissue was observed using scanning electron microscopy in Dufour’s laboratory.28
The EPR effect mainly depends on the size of drug delivery system. The drug delivery system is eliminated by liver, kidney, and reticuloendothelial system (RES), which is based on their physicochemical properties.29,30 Kidneys filter various metabolites and wastes from the blood into urine through the glomerulus, the pores of which are ~10 nm.31 Thus, NPs with hydrodynamic diameters <10 nm will be cleared by the kidneys.32 On the contrary, NPs >100 nm are removed from blood through RES such as macrophage cells in the liver and spleen.33 It is reported that the NPs with 30-200 nm in hydrodynamic diameter accumulate with high efficiency in many solid tumors by EPR effect.34 Distinguishingly, the fenestrations in the liver sinusoidal endothelium facilitate the substrate transfer into space of Disse between the liver sinusoid and hepatocytes in normal liver. The diameter of the fenestrations is ~78 ± 12 nm in wild-type mice.35 Therefore, the size of NTDDS for HCC should be considered in order to avoid them entering into the space of Disse. Furthermore, long circulation time of the NPs without being eliminated in bloodstream is also extremely significant for their accumulation into tumor tissues through the EPR effect.36,37 In normal liver, the Kupffer cells, also known as stellate macrophages, are located inside the sinusoids, thus avoiding the capture by Kupffer cells is very important for designing NTDDS for HCC. Many studies have demonstrated that NPs with more hydrophobic surface are more liable to be cleared by liver, spleen, and lungs,38 whereas hydrophilic surface could make the NPs escape macrophage capture.36 To achieve prolonged blood circulation, a strategy of surface functionalization has been developed by coating hydrophilic polymer such as poly(ethylene glycol) (PEG),39,40 poly(vinyl pyrrolidone),41 and so on on the surface of the NPs and by designing block copolymers with hydrophilic and hydrophobic domains.42
Ligand-based active targeting
As described in Figure 1, the NTDDS could disperse in tumor mass through feeding arteries and then could accumulate into tumor interstitial fluid through fenestration by EPR effect. More importantly, the specific NP–cell surface interactions play a critical role in facilitating internalization of NPs into targeting tumor cells.43 The receptor-mediated endocytosis, an approach of active targeting, is one of the most common strategies for HCC to further improve the targeting property.44,45 Fortunately, some proteins and molecules are overexpressed on the surface of hepatoma cells or intratumoural angiogenesis compared to normal cells in Figure 2, thus their ligands, including (poly)saccharides (Table 1), vitamins (Table 2), antibodies (Table 3), peptides (Table 4), aptamers (Tables 4 and 5), transferrin (Tf) (Table 6), other small molecules, growth factors, and so on (Table 7), were utilized to decorate drug delivery system in order to enhance the recognition between NPs and tumor cells. Subsequently, the NTDDS was internalized into cells by receptor-mediated endocytosis triggered by the ligands on the surface of NPs, and then the therapeutics in NTDDS was released into cytoplasm in order to kill the tumor cells.
 | Figure 1 The schematic diagram of ligand-based targeted therapy of NTDDS for HCC through EPR effect and active targeting. |
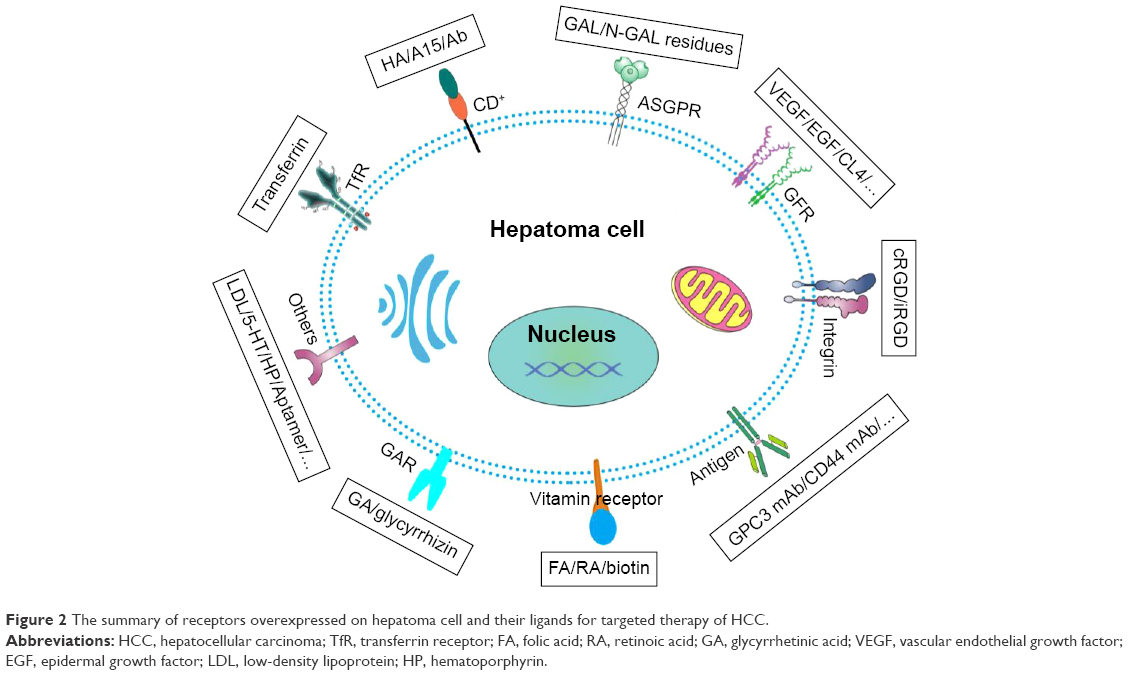 | Figure 2 The summary of receptors overexpressed on hepatoma cell and their ligands for targeted therapy of HCC. |
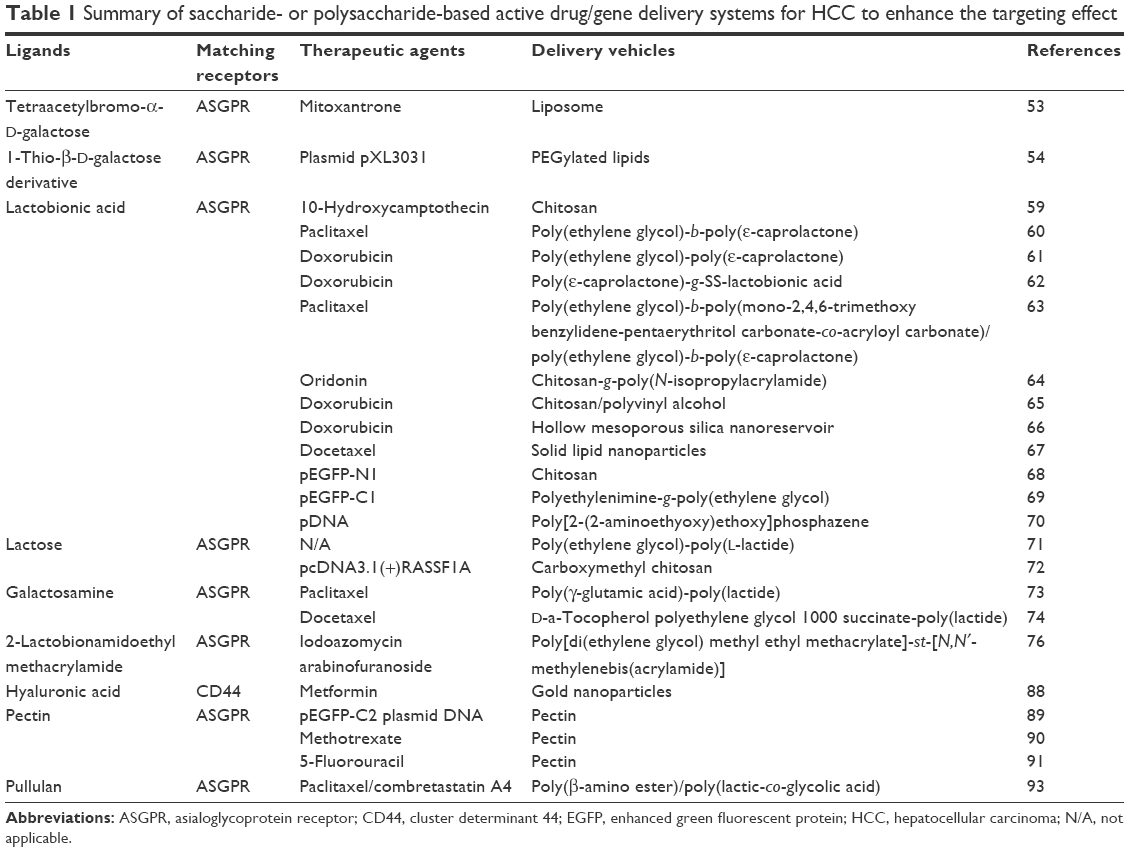 | Table 1 Summary of saccharide- or polysaccharide-based active drug/gene delivery systems for HCC to enhance the targeting effect |
 | Table 2 Summary of vitamin-based active drug/gene delivery systems for HCC to enhance the targeting effect |
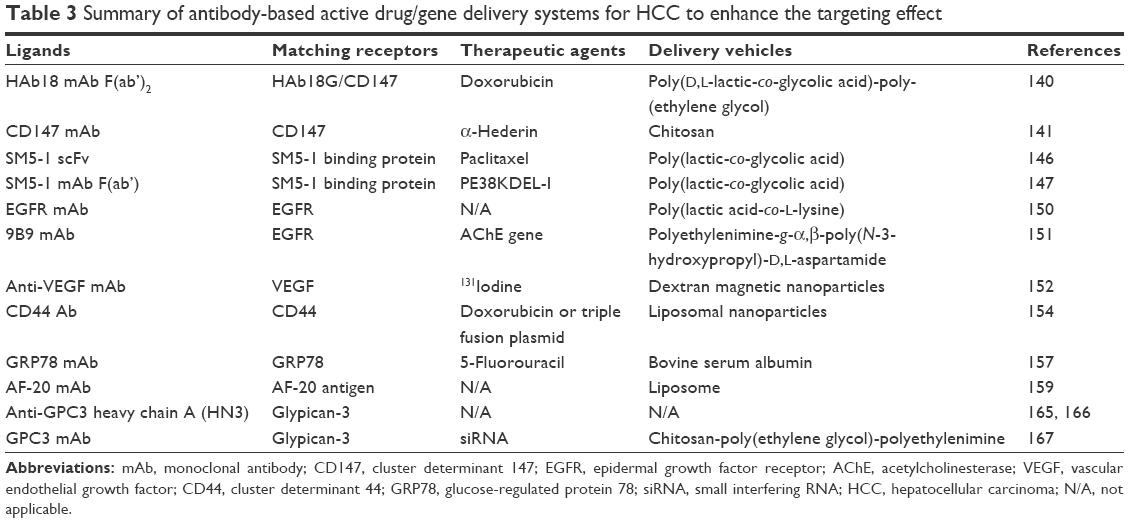 | Table 3 Summary of antibody-based active drug/gene delivery systems for HCC to enhance the targeting effect |
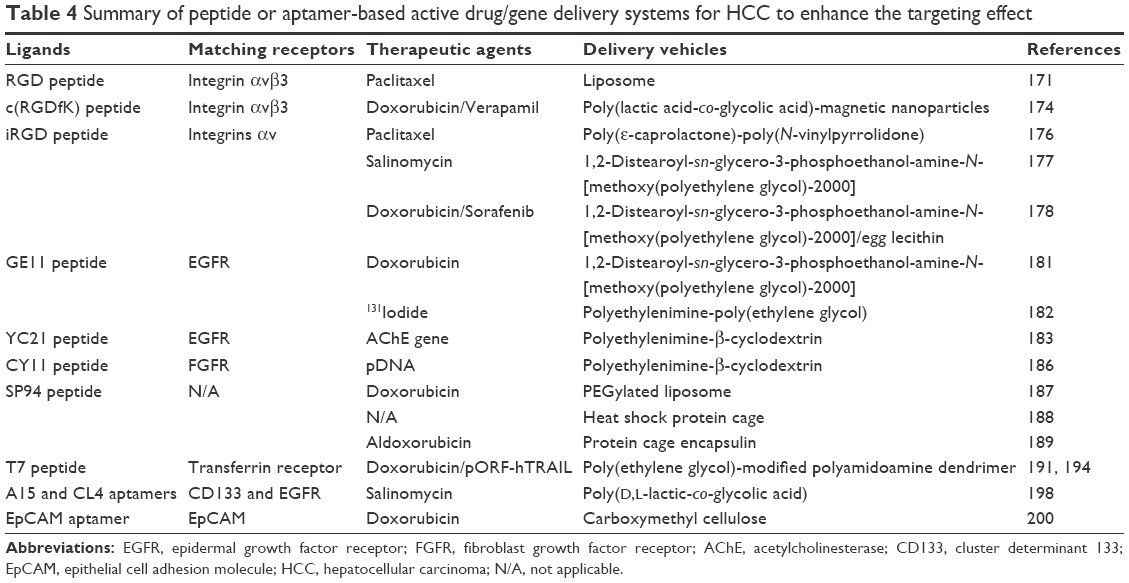 | Table 4 Summary of peptide or aptamer-based active drug/gene delivery systems for HCC to enhance the targeting effect |
 | Table 5 The sequence of aptamers reported in previous publications for HCC |
 | Table 6 Summary of transferrin-based active drug/gene delivery systems for HCC to enhance the targeting effect |
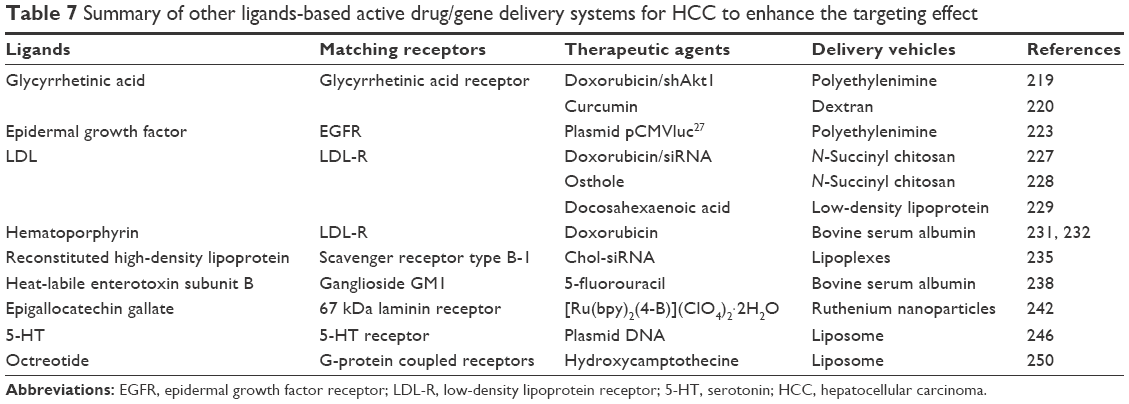 | Table 7 Summary of other ligands-based active drug/gene delivery systems for HCC to enhance the targeting effect |
Saccharide- or polysaccharide-based active targeting
Saccharides
Various glycans attached to protein molecules as glycoproteins and proteoglycans on the exterior surface of cancer cells are often upregulated as compared with the healthy cells, which renders saccharides or polysaccharides as potential ligands for liver-targeting drug delivery.46,47 Particularly, the asialoglycoprotein receptor (ASGPR), also known as “The Ashwell-Morell Receptor,” was first isolated and characterized by Baenziger and Maynard.48 It is known that the ASGPR is the most common target site of NTDDS for HCC because of the high expression on hepatocytes and hepatoma cells but minimal expression on extrahepatic cells.49–51 For example, the in vitro ASGPR analysis showed that the hepatoma cell HepG2 expresses 76,000 ASGPRs/cell with a high density on membrane.49 These receptors can recognize and bind D-galactose (Gal) and N-acetylgalactosamine residues with high specificity and efficiency. The common ligand moieties to ASGPR in previous literatures were enumerated in Figure 3. The centroid consisted of six amino acids in H1 subunit of ASGPR which forms an active ligand-binding site.49 The ASGPR is a calcium-dependent receptor, that is to say, calcium is essential for recognition and interaction between ASGPR and ligand moieties.52
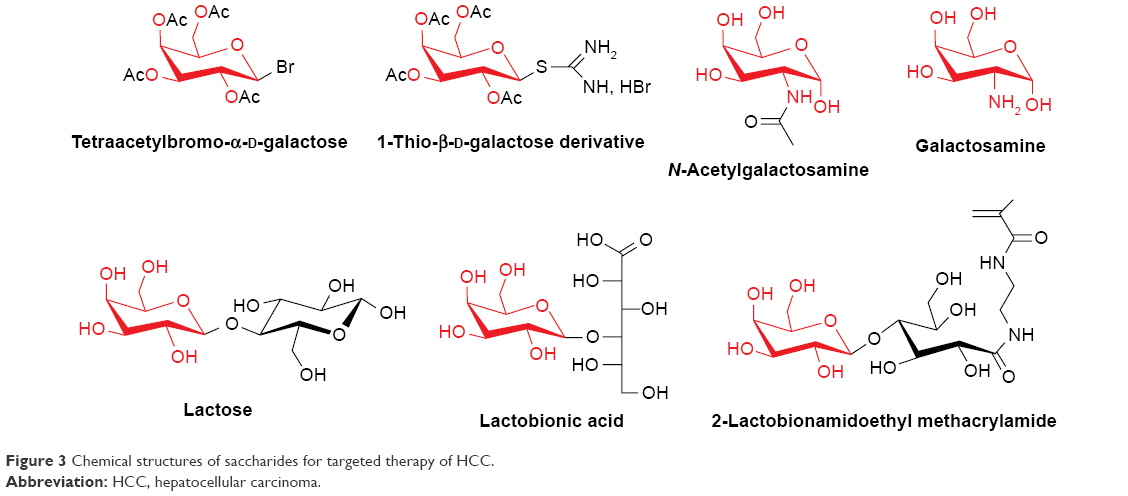 | Figure 3 Chemical structures of saccharides for targeted therapy of HCC. |
Galactosylated polymers were developed as a drug/gene carrier system for targeted delivery in HCC therapy.53,54 Zhang et al53 coupled Pluronic P123 with tetraacetylbromo-α-D-galactose (Gal-P123) through Koenigs–Knorr reaction and then fabricated mitoxantrone (MX)-loaded Gal-P123-modified liposomes (MX-LPG) for targeting cancer cells and reverse of MDR in HCC. Compared with free MX, MX-LPG had 2.3-fold higher cytotoxicity in hepatoma Huh-7 cells and a 14.9-fold increased intracellular MX accumulation in breast cancer resistance protein-overexpressing MDCKII cells in vitro. Moreover, MX-LPG strengthened the capability of antitumor and tumor targeting in mice bearing orthotopic xenograft HCC tumors in vivo. All the results indicated that Gal moiety is a potential active ligand for targeted therapy in HCC. Gal-decorated gene carrier was also developed to investigate the transfection efficiency, demonstrating that galactosyl ligand could enhance the validity of targeted gene transfer for HCC.54
Lactobionic acid (LA), comprising gluconic acid and Gal moiety, is the common ligand for hepatoma-targeted delivery.55–57 The Gal residues on drug delivery system can recognize and bind specifically to the ASGPR on the hepatoma cells,58,59 thus they facilitate drug delivery into the cells, which are inhibited and killed by therapeutic agents in delivery vehicles. Zhong’s group developed a series of Gal-directed hepatoma-targeting delivery system to enhance the accumulation of anticancer drugs into and antitumor activity toward hepatoma cells.60–63 The anticancer drugs were transported into and released in hepatoma cells through receptor-mediated endocytosis between Gal residues and ASGPR on the cells. Consequently, these Gal-directed hepatoma-targeting delivery systems inhibited the proliferation of the human hepatoma cells in vitro and tumor growth in vivo with enhanced efficiency compared with nontargeting delivery systems or free drugs. Galactosylated chitosan (GC) was conjugated with other polymers to enhance the drug-loading capacity and uptake of drug into hepatoma cells.64,65 By coincidence, the targeting agent LA was immobilized on the surface of hollow mesoporous silica nanoreservoirs through linking with β-cyclodextrin.66 Besides, a large number of research studies reported that LA-modified delivery system could enhance the uptake of drugs or genes into hepatoma cells as a novel therapeutic strategy for HCC.67–70
Lactose is a disaccharide sugar composed of Gal and glucose groups. In aqueous solution, the glucose group becomes the chain structure, which can react with amino group.51,71,72 The Gal group yet maintains a stable ring structure. In addition, some research studies indicated that only a small amount of NPs without galactosamine was internalized into hepatoma cells, whereas the galactosamine-modified NPs lead to high targeting to hepatic tumor and facilitate their cellular uptake.73,74 Hence, lactose and galactosamine are also potential ligands to ASGPR. To develop galactosylated polymer for drug and gene delivery to hepatoma cells and hepatocytes, Narain’s group synthesized 2-lactobionamidoethyl methacrylamide including Gal residues,75,76 which could be used in composing copolymer through reversible addition-fragmentation chain transfer polymerization technique.
The endocytotic pathway involved in the ASGPR-mediated endocytosis of NPs was investigated through pretreating hepatoma cells with inhibitors of clathrin-mediated endocytosis (ClME), caveolae-mediated endocytosis (CaME), and micropinocytosis. The experiments demonstrated that galactosylated NPs stimulate intracellular uptake by ClME and CaME.59,67 On the other hand, the “cluster effect” has impact on the receptor–ligand interaction.77 It is confirmed that the Gal density on drug delivery system is important for effective recognition by ASGPR and internalization into hepatoma cells.78,79 Definitely, the uptake capacity is enhanced with increase in the ratio of Gal moieties. The space between Gal moieties and vehicle surface is also another parameter of targeting specificity. The adequate PEG spacer could further increase the targeting capacity of ASGPR-overexpressing hepatoma cells and thus improve the antitumor efficacy.80 The route of administration is another concern. A recent study demonstrated that galactosylated polymer carrier still retained the capacity to enhance the accumulation of chemotherapeutic agent sorafinib into liver through oral administration compared with the non-targeting polymer carrier.81
Polysaccharides
In addition to saccharides, hyaluronic acid (HA), a natural biodegradable and biocompatible linear polysaccharide, is a multifunctional glycosaminoglycan composed of D-glucuronic acid and N-acetyl-D-glucosamine, linked through alternating β-1,4 and β-1,3 glycosidic bonds in Figure 4,82,83 and has been utilized in arthritis treatment, tissue augmentation, ocular surgery, and so forth.82 Various HA receptors including cluster determinant 44 (CD44), receptor for hyaluronate-mediated motility, HA receptor for endocytosis (HARE), and lymphatic vessel endothelial hyaluronan receptor-1 were found in different tissues for different biological functions.82 In recent years, HA has also been investigated as a targeting moiety of NTDDS for cancer therapy because of the overexpression of CD44 in various cancer cells.84–86 A recent study demonstrated that HA could actively mediate NPs into hepatoma cells. For instance, the surface modification of superparamagnetic iron oxide (SPIO) NPs was performed by conjugating HA, and the in vitro magnetic resonance (MR) imaging of CD44+ HepG2 cells and in vivo MR imaging of mice HepG2 cell-bearing tumor xenografts confirmed the high efficiency and targeting of liver carcinoma.87 Furthermore, a HA-capping gold NPs (AuNPs) loaded with metformin (MET) were developed through the amide bond formation between carboxyl groups of HA on AuNPs and amine group of MET,88 in which the HA serves as a targeting agents for liver cancer cells. The cell apoptosis assay indicated that the drug system pronounced higher apoptotic behavior in HepG2 cells than CD44− NIH 3T3 cells due to the higher cellular uptake in HepG2 cells through HA-CD44-mediated endocytosis. All the studies suggest that the HA could be a promising targeting agents for drug delivery in HCC therapy. On the other hand, the interaction of HA and HARE receptors on liver sinusoidal endothelial cells occurs, thus HA could hypothetically be beneficial for liver targeting.82 Consequently, it is a new problem that how to escape the endocytosis of liver sinusoidal endothelial cells when the HA-functioned NTDDS targeted to HCC are injected into blood stream. As a polyanionic polymer, HA can be used as the constituent of pH-sensitive NTDDS owing to its degradability by hyaluronidase (HAase) widely distributed in the acidic tumor extracellular matrix, which is discussed further in the following subsection of dual-ligand-mediated active targeting.
 | Figure 4 Chemical structures of polysaccharides for targeted therapy of HCC. |
Pectin, another polysaccharide with anion, water solubility, and non-toxicity, has attracted attention for targeted therapy in HCC.89–91 It is a natural linear polymer mainly consisting of α-(1-4)-linked D-polygalacturonic acid residues in Figure 4 and extracted from citrus peels or apple pomaces. The D-polygalacturonic acid is an oxidized form of Gal, thus the pectin displays active targeting ability to hepatoma cells contributing to the specific interaction between galacturonic acid residues and ASGPR on the cells. The pEGFP-C2 plasmid DNA (pDNA), encoding enhanced green fluorescent protein (EGFP), was loaded into pectinate micro/nanoparticles through ionotropic gelation by Opanasopit et al.89 The transfection efficiency of pEGFP-C2 pectinate NPs in hepatoma Huh7 cells was examined, displaying that the NPs were capable of mediating the transfection into Huh7 cells compared with free pDNA, and the extent of transfection depended on the ratio of pectin to DNA and the amount of pectinate NPs. The conjugation and release of chemotherapeutic agent methotrexate (MTX) in pectinate NPs were investigated in a subsequent study.90 The results verified that pectin can deliver MTX to HepG2 cells with high efficiency and specificity and exhibited sustained drug release. Recently, in vitro and in vivo evaluation of pectinate NPs loaded with anticancer drug 5-fluorouracil (5-FU) for HCC was carried out, and the pectin served as drug delivery vector of chemotherapy agent and natural targeting ligand to ASGPR in this NTDDS.91 The content of 5-FU in HepG2 cells incubated with the nano-system was significantly higher than that in cells treated with free 5-FU due to the high efficiency of NTDDS. The ASGPR-mediated recognition and subsequent endocytosis of pectinate NPs was proved by blocking the ASGPR on HepG2 cells by free Gal, indicating that it is a promising platform for targeted therapy of HCC through specific binding of galacturonic acid residues of pectin and ASGPR on hepatoma cells.
Another polysaccharide pullulan, consisting of maltotriose units connected by α-1,4 and α-1,6 glycosidic bonds, is also a natural ligand for ASGPR, thus it conduces to HCC-targeted drug delivery.92 Mediating through interaction between polysaccharide backbone of pullulan and ASGPR, the pullulan-coated drug carrier significantly inhibited hepatoma cell proliferation and migration, as well as tumor growth and angiogenesis.93
Vitamin-based active targeting
Vitamins are a series of organic compounds and vital nutrients that all living cells require for their survival. Rapid proliferation of tumor cells, in particular, requires certain vitamins in excess such as folate, biotin, retinoic acid (RA), and dehydroascorbic acid (DHAA) to sustain their rapid growth. The chemical structures of these vitamins are presented in Figure 5. Compared to the normal cells, the receptors involved in the uptake of the vitamins are thus upregulated on tumor cell surface. Consequently, these vitamin receptors serve as beneficial target substrates for tumor-targeted drug delivery.
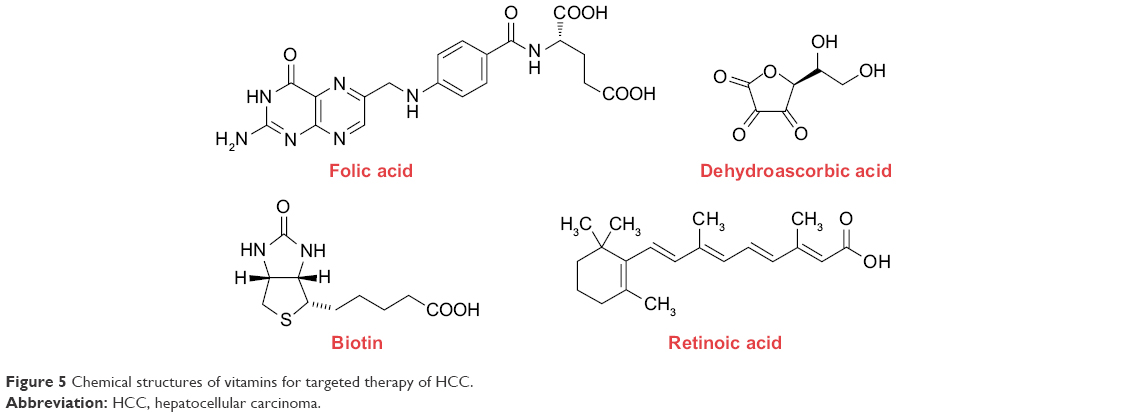 | Figure 5 Chemical structures of vitamins for targeted therapy of HCC. |
Folate
Folate or folic acid (FA), referred to as water-soluble vitamin B9, vitamin M, and vitamin Bc, is required by eukaryotic cells for facilitating the transfer of one-carbon units from donor molecules into vital biosynthetic pathways such as methionine, purine, and pyrimidine biosynthesis as various coenzymes.94,95 Besides, it plays an important role in the interconversion of serine and glycine, as well as histidine catabolism.95 Unfortunately, animal cells need to capture exogenous folates to sustain life because of lacking key enzymes of the biosynthetic pathway themselves.94 The receptor-mediated endocytosis is the main mechanism of the cellular internalization through folate receptors (FRs) with a high affinity (KD ~10−10 M).96
Natural product triptolide has been proved to be highly effective against many tumor cells including cholangiocarcinoma,97 pancreatic cancer,98 HCC,99 and so on. However, the clinical applications have been limited by poor solubility and extreme side effects. Hence, Ling et al13 synthesized smart pH-sensitive nanoformulated triptolide (Nf-Trip) coated with folate as a targeted therapeutic strategy for HCC. The hepatoma cells BEL-7402 with positive FRs and normal hepatocyte MIHA with negative FRs were selected to investigate the in vitro cellular uptake. When incubated with Nf-Trip, the MIHA cells showed very little cellular uptake, whereas the BEL-7404 cells were highly efficient. However, the FRs on BEL-7404 cells were competitively blocked by an excess amount of free FA, the cellular uptake of Nf-Trip dramatically decreased, suggesting that folate is an excellent hepatoma cell-specific ligand that effectively facilitate the endocytosis of Nf-Trip. In HCC orthotopic xenograft model, the biodistribution in vivo showed that a large amount of specific accumulation of the Nf-Trip was detected in the liver tumor tissue. Moreover, the Nf-Trip reduced tumor burden and improved survival without systemic toxicity.
Although FA-functionalized drug delivery loaded with small molecular drugs such as DOX and docetaxel could induce tumor cell apoptosis,100,101 hepatoma cells may advance several mechanisms to resist apoptosis. Downregulation of antiapoptotic genes enhanced the sensitivity of hepatoma cells to chemotherapeutic agents by RNA interference technology.101–104 Therefore, Cao et al103 developed a multifunctional drug carrier that co-delivered DOX and siRNA against the antiapoptotic BCL-2 gene. The DOX and BCL-2 siRNA was much more efficiently transferred into BEL-7402 cells by the FA-modified nanocomplex than the nontargeting one. Moreover, the DOX-induced cell death was enhanced by the co-delivery of BCL-2 siRNA, indicating that co-loading siRNA and chemotherapeutic agent in a targeted drug delivery enables simultaneous delivery of specific siRNA and drug into hepatoma cell with synergistic effect in antitumor activity, which was also proved by another study.101 Co-delivering two chemotherapeutic agents in a drug delivery is alternative strategy to overcome MDR of HCC. For example, multifunctional DOX loaded-folate-chitosan self-assembly micellar NPs co-delivered pyrrolidinedithiocarbamate, a NF-κB inhibitor, to achieve specific targeting and to surmount the DOX MDR.105
Tumor metastasis remains the major obstacle for the improvement in the long-term survival after HCC resection.106,107 Intrahepatic metastasis is the most frequent, followed by extrahepatic pulmonary metastasis.108 NP-mediated targeted therapy is a promising antimetastatic strategy for pulmonary metastasis. FA and paclitaxel (PTX) containing composite micelles (FA-M[PTX]) were fabricated by co-assembling the ligand FA polymer conjugate and chemotherapeutic agent PTX polymer conjugate.109 Compared with M[PTX] and free MTX with equivalent dose of PTX, the pulmonary metastasis in intravenously injected murine hepatoma 22 bearing BALB/c mice models was efficiently inhibited and the survival time was significantly prolonged by FA-M[PTX].
Multifunctional delivery systems that carry therapeutic and diagnostic imaging agents with FA are emerging concept for effective targeted therapy. The diagnostic imaging agents contain fluorophores, quantum dots (QDs),110 small-molecule paramagnetic agents,111,112 SPIO,113–117 and so forth. Wang et al110 designed a smart multifunctional polymeric micelle for targeted therapy of HCC. The pH-sensitive polymeric vehicle turns PTX release “off” at neutral environment but “on” inside acidic lysosomes, and the QD encapsulation tracks pH-tunable drug release behavior and monitors the therapeutic effect. Moreover, the targeting capacity of the micelle was enhanced through the specific interaction and internalization with high affinity between ligand FA on micelle and FRs on human hepatoma cells, which was proved by cellular uptake in vitro and tumor growth inhibition in vivo. Multifunctional NPs encapsulated with SPIO is another approach to monitor the tumor progression using magnetic resonance imaging (MRI). A large quantity of FA-functioned polymeric NPs loaded with SPIO was developed,113–116 which mediates by FA–FRs interaction; hence, these nanosystems could specifically accumulate in the tumor tissue and inhibit the growth of tumor.
Biotin
Biotin, also known as vitamin H, vitamin B7, or coenzyme R, is one of the water-soluble B complex vitamin families and is a growth promotor at the cellular level.118 It is reported that the biotin receptors are overexpressed more than the FRs in various cancer cells such as leukemia, colon, mastocytoma, lung, renal, and breast cancer cells.118 A study demonstrated that modification of drug delivery with biotin is an effective pattern to enhance cell specificity against the cancer cells overexpressed with biotin receptors on the cell surfaces and to accelerate the internalization of the drug delivery into the targeted cancer cells through receptor-mediated endocytosis.119 To verify the potential value of biotin for targeted liver neoplasms, Mishra and Jain120 designed biotin-modified erythrocytes loaded with MTX by combining with N-hydroxysuccinimide ester of biotin. In vivo study showed that the MTX level of liver administrated with biotinylated erythrocytes was increased ~3-fold compared with free MTX and 1.8-fold compared with nonbiotinylated erythrocytes at 1 h after injection into rats, indicating that this drug system could be used to place hepatic arterial catheters for locoregional treatment of liver neoplasms.
In order to improve the cancer-targeting activity and internalization of pullulan acetate (PA) NPs, Na et al121 synthesized the biotinylated PA through coupling biotin with PA between carboxyl groups of biotin and hydroxyl groups of PA. The conjugated polymers self-assemble to form the biotinylated PA NPs through a diafiltration method. The cellular uptake investigation exhibited that the biotinylated PA NPs were strongly internalized into HepG2 cells, while only slight PA NPs were absorbed in to cells. Moreover, the efficiency of internalization was enhanced with increasing biotin content.
In our previous work, the biotin-decorated fluorescent silica NPs loaded with aggregation-induced emission fluorophores (FSNP-biotin), a series of molecules emitting strong fluorescence in aggregated structure but non-fluorescence or weak fluorescence in disperse state,122,123 were fabricated to verify the targeting efficiency and mechanism of endocytosis into hepatoma cells.124 After 3 h of staining, strong fluorescence was emitted from the hepatoma cells BEL-7402 with overexpression of biotin receptors, whereas dim fluorescence was observed from normal liver cells LO2 with low expression of biotin receptors. To prove the mechanism of ligand–receptor interactions or the occurrence of receptor-mediated endocytosis, the biotin receptors on BEL-7402 cell surface were blocked through the incubation of free biotin prior to staining. The fluorescence in these cells was markedly decreased. All results also indicate that biotin can enhance the cellular uptake by hepatoma cells as a ligand.
RA
RA is a metabolite of vitamin A that underlies the functions of vitamin A with a vital role in the regulation of cell proliferation and development.125 Additionally, RA serves as an anti-HCC agent by regulating apoptosis and differentiation, as well as modulating the sensitivity of tumor cells to the innate immune response.126 Among the RA receptor subtypes α, β, and γ, the level of mRNA and protein of RA receptor α in HCC tissue was dramatically higher than those in normal liver tissue, and the expression of RA receptor α mRNA and protein was higher than that of RA receptor β and γ in HCC tissue, suggesting that the RA receptor α is the dominant receptor in HCC.127 The RA-decorated chitosan-albumin NPs were developed using an ionic coacervation method by Varshosaz et al.128 The HepG2 cells incubated with RA-decorated chitosan-albumin NPs exhibited rapid internalization and accumulation by cells, while just inefficient uptake of nontargeted chitosan-albumin NPs was observed. The targeting efficiency was investigated by cell proliferation assay. The higher growth inhibitory effect was observed in cells treated with RA-decorated chitosan-albumin NPs loaded with DOX compared with chitosan-albumin NPs loaded with DOX. All the results indicate that the RA could mediate the nanocarrier into HCC cells as a potential ligand.
DHAA
Transporter-mediated pathway is more rapid and efficient in the transportation of small molecules compared to receptor-mediated transport. Recently, several transporters have been proved to be upregulated on the tumor cell surface to sustain the excess nutritional requirement of tumors.129,130 Tumor cells avidly consume glucose for proliferation in hypoxic environment, termed the Warburg effect.131 Glucose transporter isoform 1 (GLUT1), a representative member of the GLUT family, transports D-glucose into cells across the membrane and maintains an appropriate D-glucose concentration in cells.132 It has been reported that GLUT1 is overexpressed on hepatoma cells. DHAA can be specifically recognized by GLUT1 contributing to its structural similarity to D-glucose133 and rapidly reduced to ascorbic acid (AA) in cells.134 Therefore, Guo et al135 developed DHAA-modified PEG-pLys-pPhe nanomicelle for efficient therapy of HCC. The cellular uptake in vitro showed that the internalization of DHAA-modified PEG-pLys-pPhe nanomicelle was significantly enhanced compared with that of PEG-pLys-pPhe nanomicelle and also demonstrated that the DHAA was mainly responsible for actively recognizing and binding to hepatoma cells BEL-7402 through GLUT1. Moreover, the in vivo imaging study proved that the micellar system possessed high tumor-targeting capacity, because of facilitating gradual accumulation in tumor cells. In addition, they also found that the transport capacity of GLUT1 would get to saturation state when the ratio of DHAA on micelles exceeded the number of GLUT1 on cell surface.
Antibody-based active targeting
The conjugation of NPs with antibodies, which combines the properties of the NPs with the specific and selective recognition ability of the antibodies to the antigens on the surface of tumor cells, is a successful targeting strategy for hepatic malignancy.136,137 For instance, iodine (131I) metuximab injection (Licartin®), a 131I-labeled HAb18G/CD147-specific monoclonal antibody (mAb) F(ab’)2 fragment, has been approved for the treatment of primary HCC by the China State Food and Drug Administration.138 The HAb18G/CD147, an antigen for being homologous to CD147, is highly expressed on HCC cells and tissues and can bind to the bivalent fragment HAb18 F(ab’)2 of HAb18 mAb with high affinity.139 Jin et al140 fabricated the DOX-poly(D,L-lactic-co-glycolic acid)-poly(ethylene glycol) (DOX-PLGA-PEG) micelle and further decorated with bivalent fragment HAb18 F(ab’)2 to improve the therapeutic effect for HCC, demonstrating that the cellular uptake was enhanced through antigen–antibody recognition between the micelle and tumor cells, and the therapeutic action was improved due to higher accumulation of DOX in tumor cells. Recent research also demonstrated that CD147 could mediate cellular internalization of NPs through the caveolae-dependent pathway and lysosomal escape with high specificity.141
SM5-1 is a mouse mAb, which binds to a SM5-1 binding membrane protein of ~230 kDa.142 The membrane protein is specifically expressed on HCC, melanoma, and breast cancer cells, indicating that it is a promising binding site of NPs for diagnosis and therapy of HCC.143–145 Kou et al146 fabricated negatively charged PTX-loaded poly(lactide-co-glycolide) (PLGA) NPs and then successfully coated them with cationic polypeptide polylysine fused to SM5-1 single-chain antibody (SM5-1 scFv), which was derived from SM5-1 mAb. The results demonstrated that the nanosystem retained high specific affinity to SM5-1 binding protein and could induce specific and efficient death of SM5-1 binding protein-positive Ch-hep-3 cells. In addition, the F(ab’) fragments of SM5-1 mAb, also known as SM5-1 mAb F(ab’), could bind to the SM5-1 binding protein. Gao et al also developed PE38KDEL type I mutant-loaded PLGA NPs conjugated with SM5-1 mAb F(ab’).147 The results showed that the NPs were specifically internalized into Ch-hep-3 cells with high efficiency and exhibited potent cytotoxicity of hepatoma cells.
The epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase overexpressed in some solid tumors such as HCC,148 and some studies proved that the upregulation of EGFR was positively correlated with the tumor progression.149 The human hepatoma cells can be effectively inhibited through blocking the signal transduction of EGFR. On the other hand, the EGFR can also be used as a mediator for the targeted delivery system. The EGFR mAb-modified poly(lactic acid-co-L-lysine) (PLA-PLL) NPs were fabricated by Liu et al,150 which were internalized into hepatoma cells with more efficiency and specificity mediated by ligand–receptor recognition. 9B9 mAb is another anti-EGFR/EGFRv III mAb, which can specifically react with EGFR/EGFRv III antigen on hepatoma cells. Wang et al151 designed biodegradable cationic therapeutic gene AChE-loaded-polyethylenimine-grafted-α,β-poly(N-3-hydroxypropyl)-DL-as-partamide (PHPA-PEI) NPs and then conjugated with 9B9 mAb to enhance the HCC-targeting abilities and therapeutic efficiency. The in vivo SMMC-7721 tumor-bearing mice administrated with intraperitoneal injection demonstrated that the tumor inhibition rate of PHPA-PEI/AChE NPs with 9B9 mAb was threefold that of PHPA-PEI/AChE NPs without 9B9 mAb contributing to high interaction and internalization between 9B9 mAb on the surface of NPs and EGFR on hepatoma cells, which indicates that 9B9 mAb is a potential delivery ligand for liver cancer gene therapy.
Dextran magnetic NPs labeled with 131I and anti-VEGF mAb (Sc7269) were prepared for radioimmunotherapy of liver cancer.152 The nanocomplex could accumulate in tumor tissues through both intratumoral and intravenous injections, whereas very low radioactivity in normal tissues was detected, indicating that targeting to VEGF such as VEFG165, VEGF189, and VEGF121 secreting from liver cancer cells is feasible for HCC therapy.
CD44 is a cell surface glycoprotein involved in immune recognition, in cell–cell and cell–matrix interactions, in cell migration, and so on, is associated with the tendency to vascular invasion, and may have implications for metastasis and poor prognosis in patients with HCC.153 Wang et al154 developed anti-CD44 antibody-mediated liposomal NPs loaded with chemotherapy drug DOX or triple fusion suicide genes, including the herpes simplex virus–truncated thymidine kinase, renilla luciferase, and red fluorescent protein, for the treatment of HCC by targeting cancer stem cells (CSCs) overexpressing CD44. The results demonstrated that the nanocarrier could target liver tumor cells and monitor the response to therapy in vivo.
Glucose-regulated protein 78 (GRP78), also known as binding immunoglobulin protein (BiP) or heat shock 70 kDa protein 5 (HSPA5), is overexpressed on the cell surface in HCC.155,156 GRP78 mAb-conjugated bovine serum albumin (BSA) loaded with 5-FU was developed to promote 5-FU into HCC cells and inhibit the adhesion, invasion, and metastasis of HCC,157 which was verified by cell administrations.
The AF-20 mAb has a high affinity for AF-20 antigen, which was found to distribute uniformly on 15/15 HCC tissues tested but was not evident on adjacent normal liver or in most normal human tissues.158 Moradpour et al159 synthesized immunoliposomes by coupling AF-20 with liposomes containing carboxyfluorescein. AF-20-conjugated targeted liposomes (AF20TL) were found to bind to all human cancer cell lines examined, including FOCUS, HuH-7, HepG2, Hep3B, SK-HEP-1 human HCC cell lines, and other human cancer cell lines expressing the AF-20 antigen, whereas control liposomes conjugated with C7-57 showed virtually no binding toward these cell lines. Competitive inhibition experiments conducted on FOCUS, HuH-7, and Hep3B cells using AF-20 mAb proved the specificity of the interaction of AF20TL with target cells.
It is known that glypican-3 (GPC3) is a member of the glypican family of heparin sulfate proteoglycans on some tumor cells.160 A study showed that GPC3 was not detectable in hepatocytes from normal liver tissue and benign liver lesions but expressive in 72% of HCCs.161 Therefore, several anti-GPC3 mAbs have been produced for immunotherapy of HCC, but none of them can inhibit cell proliferation or induce apoptosis.161–164 Feng et al165 identified a human heavy chain variable domain antibody (HN3) targeting GPC3 by phage display technology and proved that HN3 could recognize a unique conformational epitope in the GPC3 core protein with high affinity. The HN3 inhibits cell growth in several hepatoma cell models in vitro and significantly inhibits the growth of HCC tumor-bearing node mice in vivo. Additionally, Hanaoka et al166 compared the HN3 and anti-GPC3 YP7, a whole IgG antibody, with regard to their relative therapeutic effects, demonstrating that the HN3 possesses parallel high tumor accumulation but superior homogeneity within the tumor tissue compared with YP7. Due to the high affinity and specificity of GPC3 mAb and the high expression of GPC3 on HCC cells, a GPC3 mAb-functionalized theranostic nanovector loaded with siRNA against luciferase was developed for targeted delivery of siRNA to HCC,167 indicating that the GPC3 is a target substrate for cancer nanomedicines in HCC.
Peptide-based active targeting
Arg-Gly-Asp peptide (RGD), a tripeptide with selectivity and affinity for integrins, regulates cell–cell and cell–extracellular matrix interactions and can be utilized as an effective targeting motif in cancer therapy since it can achieve dual targeting for both angiogenic endothelial cells and several tumor cells through the integrin αvβ3.168,169 Moreover, RGD peptides enjoy the advantage of minimal risk of immune reactivity, simple and inexpensive synthesis, and tight control over ligand presentation.170 To confirm the expression level of the integrin αvβ3 on hepatoma cells, this study detected them by immunofluorescence staining. As shown in Figure 6, the fluorescent intensity of HepG2 cells is much higher than that of normal LO2 cells and HK-2 cells, indicating that the integrin αvβ3 is overexpressed on the hepatoma cells. Chen et al171 developed PTX-containing liposomes (LP) modified by RGD to enhance the targeting specificity and therapeutic effect, finding higher cellular uptake efficacy and anti-proliferative activity in HepG2 cells and greater anti-tumor effect with enhanced tumor penetration in nude mouse HCC xenograft models compared with LP-PTX without RGD. To improve the biological properties and pharmacokinetics of RGD, it undergoes cyclization, N-methylation, variation of stereochemical configuration or other kinds of modification.172 Compared with linear RGD, cyclic RGD manifests enhanced binding selectivity and affinity for the integrin αvβ3 and is less susceptible to biodegradation.173 Cyclo(Arg-Gly-Asp-D-Phe-Lys) (c(RGDfK)) peptide was employed by Shen et al to synthesize cRGD-DOX/VER-MNP-PLGA NPs for targeted cancer therapy.174 The in vitro cytotoxicity investigation showed that cRGD-targeted DOX/VER-MNP-PLGA NPs led to lower cell viability in HepG2 cells than cRGD-unconjugated NPs, indicating that cRGD enhanced the targeting efficiency of the NPs. Moreover, the result of biodistribution studies confirmed the integrin αvβ3 specificity of the cRGD-conjugated DOX/VER-MNP-PLGA NPs in vivo while suggesting that MNP improved the targeting by cRGD.
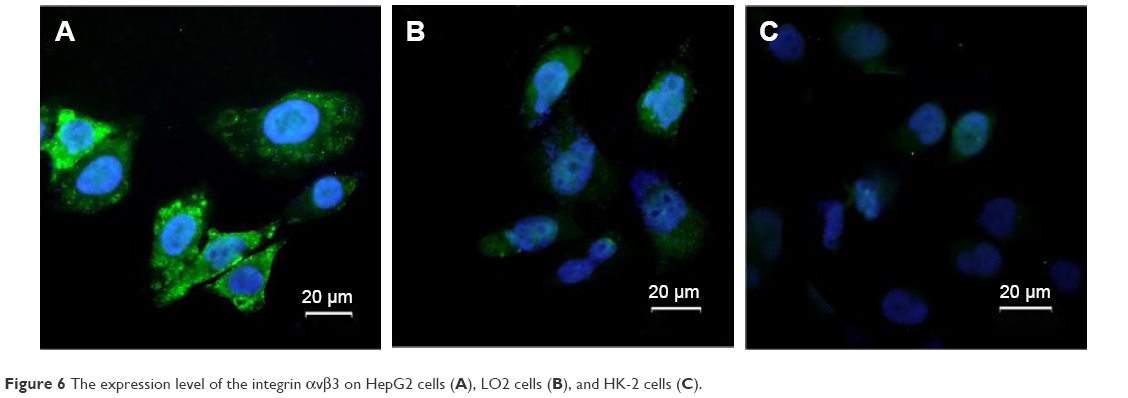 | Figure 6 The expression level of the integrin αvβ3 on HepG2 cells (A), LO2 cells (B), and HK-2 cells (C). |
Internalizing Arg-Gly-Asp peptide (iRGD) combines the tumor-homing capability of RGD and the tissue penetrating property of C-end Rule, enabling the targeting of extravascular tumor parenchyma.175 The mechanism of the iRGD includes three steps: the RGD motif targets αv integrins on tumor endothelium, then iRGD undergoes a proteolytic cleavage acquiring the ability to bind to neurophilin-1 to achieve tissue penetration.175 Linear iRGD (CRGDRCPDC) was conjugated to poly(ε-caprolactone)-poly(N-vinylpyrrolidone) (PCL-PVP) NPs by Zhu et al.176 The result of cytotoxicity and uptake investigation indicated that the iRGD-conjugated PCL-PVP NPs possessed higher cytotoxicity than the unconjugated NPs against murine hepatic H22 cell at 12 and 24 h after incubation. Through in vivo studies on H22-transplanted solid tumor model, they further demonstrated the superior antitumor efficacy of iRGD-conjugated NPs over the unconjugated counterparts. Moreover, immunofluorescence staining and near-infrared fluorescence imaging confirmed the capability of iRGD to facilitate the accumulation and penetration of NPs in tumor. The in vitro and in vivo studies on iRGD (CRGDKGPDC)-conjugated DSPE-PEG2000 nanomicelles (M-SAL-iRGD) were developed by Mao et al,177 which corroborated the functions of iRGD. Selective toxicity toward liver CSCs within the HepG2 cell population was found using M-SAL-iRGD compared with using M-SAL or free salinomycin (SAL). The result of in vivo anti-CSC assay further proved this. The targeting capacity of iRGD was also corroborated by a recent report.178
GE11, one of the 11 antigens in the Gebrich blood group system with relatively high prevalence,179 binds specifically and efficiently to EGFR leading to elevated internalization level while being safe for its much lower mitogenic activity compared with EGF.180 GE11 was incorporated on DOX-loaded liposomes as a targeting moiety by Tang et al.181 Compared with the untargeted liposomes, GE11-modified liposomes exhibited improved cellular uptake by human hepatoma cell SMMC-7721 and enhanced antitumor efficacy in SMMC-7721 HCC xenograft models though their blood circulation time decreased. LPEI-PEG-GE11/NIS polyplexes were developed by Klutz et al182 using linear polyethylenimine (LPEI), PEG, the synthetic EGFR-specific peptide GE11, and a sodium iodide symporter (NIS) expressing plasmid. LPEI-PEG-GE11/NIS polyplexes led to a 22-fold increase in iodide uptake in HCC HuH7 cells as well as high tumor-specific iodide accumulation, which inhibited tumor growth and increased survival in HCC xenograft bearing nude mice. YC21 (GEL) peptide, another EGFR targeting oligopeptide composed of 21 amino acid units, is GE11 peptide with a linker sequence (GGGGS)3 at the carboxyl terminal.180 Liu et al183 formulated the EGFR-targeted gene vectors (YPCs) by coupling YC21 with the PEI600-CD (PC) vectors composed of β-CD and low molecular weight polyethylenimine (PEI). Highly efficient gene delivery ability of EGFR-positive cells was proved in their in vitro study on SMMC-7721 cells. The in vivo investigation of gene transfection and antitumor activity also suggested its prominent gene transfection ability and therapeutic effects in the inhibition of tumor growth.
Fibroblast growth factors (FGFs) are another family of growth factors, and their receptors (FGFRs) are transmembrane proteins which are overexpressed in various human solid cancers and relative to angiogenesis.184 A study demonstrated that a heptapeptide sequence (MQLPLAT) is binding to FGFR with high affinity,185 suggesting that peptide is also a ligand to FGFR. Hu et al186 developed CY11 (CGMQLPLATWY)-coupled gene delivery to specifically transfect plasmid DNA into FGFR+ HepG2 cells. The results indicated that the CY11 could enhance the gene delivery efficiency due to the specific binding between the FGFR on the surface of hepatoma cells and the peptide on vectors.
SP94 (SFSIIHTPILPL) is a peptide that can selectively target HCC cells both in vitro and in vivo through an unknown target molecule.187 Lo et al187 synthesized a targeted drug delivery system by coupling SP94 with PEGylated liposomal DOX (SP94-LD). Studies that they implemented on mice bearing HCC xenografts confirmed the targeting efficiency of SP94 and demonstrated the superior targeting ability of SP94 over the phage clone PC94 and the enhanced therapeutic effect of SP94-LD compared with control peptide conjugated LD (CP-LD) and LD. Toita et al188 formulated HspG41C-SM(PEG)n-SP94 peptide conjugates by modifying a naturally occurred heat shock protein cage (HspG41C cage) with SP94 through the heterobifunctional linker (SM(PEG)n). The cellular binding investigation demonstrated the binding selectivity of this conjugate to human HCC-derived cell lines, while suggesting that the binding efficiency could be influenced by the amount of SP94 peptides on Hsp cages, conjugation site of SP94 peptide, and linker length between a Hsp cage and a SP94 peptide. They further found that the optimized strategy was conjugating the Hsp cage with high levels of SP94 at the N-terminus of SP94 through a longer linker, which might as well guide the usage of other peptide ligands. SP94 was also utilized by Moon et al189 to modify a protein cage, encapsulin, for HCC cells targeting, and SP94 was displayed on the surface of encapsulin through conjugation or genetic insertion. The studies they conducted proved the effective and specific targeting capability of SP94.
T7 (HAIYPRH), a Tf receptor-specific peptide, binds to a different site of Tf receptor compared with Tf and can exhibit enhanced uptake due to the help of endogenous Tf.190 Han et al conjugated PEG-modified polyamidoamine dendrimer (PAMAM) with T7 to deliver DOX to Tf receptor-overexpressed tumors.191 Enhanced cellular uptake and in vitro antitumor effect were detected in BEL-7402 cells. In the investigation of in vivo antitumor effect, PAMAM-PEG-T7/DOX NPs showed higher accumulation in tumor tissues and led to more remarkable inhibition of tumor growth than PAMAM-PEG/DOX NPs and saline. It has been reported that the complex of DOX and DNA do not leak DOX in the bloodstream contributing to intercalation of DOX within the DNA strand.192,193 Accordingly, the PAMAM-PEG-T7 NPs loaded with therapeutic gene encoding human tumor necrosis factor-related apoptosis-inducing ligand (pORF-hTRAIL) and DOX were developed to improve the antitumor activity.194 This system yet exhibited improved cellular uptake and gene expression and consequently higher level of apoptosis in BEL-7402 cells compared with the unmodified system. Both the studies demonstrated the promising potential of T7 as a targeting ligand for liver cancer.
Aptamer-mediated active targeting
Aptamers are single-stranded DNA, RNA, or unnatural oligonucleotides with a molecular weight of 10–15 kDa and bind with high specificity and affinity for a wide range of target molecules including other nucleic acids, proteins, peptides, and small molecules.195 Although functionally similar to protein antibodies, aptamers also called as “chemical antibodies” offer several advantages over antibodies in terms of biomedical applications.196 Aptamer-conjugated NPs are thus empowered by these attributes to selective delivery vehicles for therapeutic applications to the target.197
CESN, SAL-loaded NPs conjugated with CD133 aptamers A15 and EGFR aptamers CL4, was developed using an emulsion/solvent evaporation method by Jiang et al to enhance the delivery of SAL and target not only HCC CSCs but also a large percent of non-CSCs, thus improving the therapeutic effect.198 The cytotoxicity investigation showed that A15-conjugated SAL-loaded NPs (CSN) and CL4-conjugated SAL-loaded NPs (ESN) could be 1.38- or 2.01-fold effective than SN against Huh7 cells, respectively, while the CESN was the most effective. Similar experiment using Hep3B cells corroborated this trend. Moreover, the in vivo antitumor assay further confirmed the superiority of CSN and ESN over nontargeting SAL-loaded NPs.
Epithelial cell adhesion molecule (EpCAM) is upregulated in HCC and could be selectively bound by DNA aptamers.199 For instance, a DNA-based EpCAM aptamer was utilized by Pilapong et al as a targeting moiety to develop a theranostic nanoprobe (called as EpCAM-MNPs) for HCC treatment and MRI.200 In vitro study indicated that the DOX-loaded EpCAM-MNPs had higher specificity in drug delivery for HBV-associated HCC cells and better intracellular uptake compared with free DOX. Moreover, they were located mainly in endosomes/lysosomes, where the relatively low pH enabled the ready release of DOX.
The aptamers in NTDDS for HCC were limited in previous literatures. However, current studies have established some targeted systems using aptamers to target molecules overexpressed on HCC cells, attaining higher cellular specificity and better therapeutic effect. The thiolated TLS11a aptamers were attached to AuNP-modified glassy carbon electrode surface by Sun et al,201 to enhance the specificity of cancer cell detection based on the hybrid nanoelectrocatalysts and enzyme for signal amplification. The specificity study using various types of cells revealed the excellent selectivity of the cytosensor for HepG2 cells. This aptamer was also used for ultrasensitive and selective electrochemical cytosensing for HCC cells.202 There are studies using the conjugates or adducts of aptamers and diverse kinds of therapeutic materials for treatment of HCC, as AS1411 (AGRO100)-DOX adduct,203 LY-1-conjugated QDs and magnetic particles,204 TLS11a-GC-guided DOX delivery,205 and so on. These aptamers in Table 5 are potential ligands for targeted therapy for HCC, and the specificity and efficiency would be investigated in further works.
Tf-based active targeting
Tf, an iron-transporting glycoprotein, has two homologous domains for ferric iron and can internalize them efficiently into cells with expression of Tf receptors (TfRs) by receptor-mediated endocytosis.206 When iron is removed from holo-Tf (iron-binding Tf), Tf becomes apotransferrin (apoTf).206 TfR1 and TfR2 are two chief TfRs responsible for cellular iron uptake, while TfR1 is more ubiquitously expressed and shows significantly higher affinity for Tf compared with TfR2.207 It is reported that the mRNA level of TfR1 is upregulated in human HCC but that of TfR2 is downregulated.208 Consistent conclusion has been drawn in a similar study on a rat model of HCC.209
The Tf-decorated core-shell NPs loading DOX in poly(vinyl alcohol) nanocore and sorafenib in albumin nanoshell were developed using sequential freeze–thaw/coacervation method by Malarvizhi et al.210 Upon conjugation of Tfs, the cellular uptake of the nanomedicine was significantly increased in HepG2 cells. Besides, notably improved synergistic cytotoxicity and cell death have been observed upon incubation of HepG2 and PLC/PRF/5 cells with the Tf-targeted NPs. In order to mimic in vivo system, studies of cellular uptake and cytotoxicity were performed in three dimensional HCC spheroids. The results suggested remarkable penetration and internalization of NPs into the HCC spheroids and promising synergistic cytotoxicity inflicted on HCC cells.
To enhance the transfection efficiency of negatively charged liposomes encapsulating miRNA-221 antisense oligonucleotide (anti-miR-221), Zhang et al211 synthesized anti-miR-221-encapsulated Tf-targeted liposomes (Tf-RL) by conjugating Tfs to the liposomes. In vitro study indicated that the Tf-RL delivered miRNA more efficiently to the hepatoma cells than nontargeted liposome containing anti-miR-221 (RL), showing higher silencing efficiency and expression of targeted downstream genes. Concordant results have been found in HepG2 tumor-bearing xenografted mice after intravenous injection of Tf-RL. Moreover, the biodistribution in tumor-bearing mice suggested that the Tf-RL could result in higher intensity of miRNA in tumors compared to RL and, as a consequence, induce more significant apoptosis in HepG2 cells through active targeting.
ApoTf, a form of iron-removed Tf, has a weaker binding affinity for TfR than diferric Tf212 but can also be used as targeting ligand and carrier for NTDDS. The conjugated and non-conjugated forms of Dox to apoTf NPs have been prepared by Krishna et al213 and the outcome suggested that the non-conjugated form (direct-nano) is superior to the conjugated form (conj-nano) in various aspects, including drug localization in nucleus, sustainability of half-life, efficiency of delivery and so on. Besides, lactoferrin (Lf), also known as lactotransferrin, is an 80 KDa glycoprotein in the Tf family and has notably high affinity for iron.214 Research has also been also conducted using apoTf or Lf as the targeting moiety and drug carriers for chemotherapy of HCC. Golla et al215 synthesized DOX-loaded apoTf NPs (Apodoxonano) and Lf NPs (Lactodoxonano), and administered the drug intravenously in rats, minimizing the cardiotoxicity caused by DOX while attaining relatively high drug accumulation in liver and extended bioavailability. To sustain optimal concentration levels of drug and simplify the modality of administration, oral formulation of DOX-protein NPs are developed and administered in rats though doubts remain about whether the oral adsorption would also be effective in humans and how to determine the dosage in human studies.216
Other ligands for HCC
Glycyrrhetinic acid (GA) is the hydrolysis product of glycyrrhizin which was extracted from the root of the traditional Chinese medicine Glycyrrhiza glabra (licorice).217 It is demonstrated that the GA and glycyrrhizin could mediate the active hepatic-targeting drug delivery system through GA receptor and glycyrrhizin receptor, respectively.218–220 In 1991, Negishi et al221 confirmed that the quantity of GA binding sites was higher than that of glycyrrhizin binding sites, that is to say, the GA could be more effective than glycyrrhizin to HCC. Thus, the GA has been developed in targeted therapy for HCC as a ligand, which was summarized in a review.218 In a recent work, the capacity of the GA to HCC was also proved through a drug and gene codelivery carrier modified with GA.219 The drug concentration and gene transfection efficiency of GA-modified carrier was remarkably higher than that of GA-modified carrier adding free GA. Moreover, the GA-modified carrier possessed much better antitumor efficacy on xenograft liver tumor. All results indicate that GA as a ligand plays an important role in enhancing HCC-targeting efficacy through active targeting.
In the aforementioned section, the EGFR was described as a targeting site to match antibodies for HCC. Epidermal growth factor (EGF), derived from the cleavage of prepro-EGF and generated primarily in kidney in humans, can bind with EGFR as well, which promotes embryonic development and stem cell regeneration and regulates ion transport.222 To resolve the problems of in vivo gene delivery, Wolschek et al223 synthesized PEGylated EGF-containing DNA/PEI complexes for HCC targeting. The study on 2 xenotransplantation models for human HCC showed that expression levels within the tumors were up to 97%–99% of the total transgene expression in vivo, which is two logs higher than levels in the major expressing organ, liver. However, the transfection levels were significantly lower upon systemic application of EGF-free complexes, indicating that specific delivery is mediated by the interaction between EGF and EGFR, which corroborated the conclusion of the in vitro competition study using free mEGF.
Low-density lipoprotein (LDL) particle, with an average diameter of ~22 nm (ranging from 18 to 25 nm), has an apolar core consisting of cholesteryl esters, triglycerides and free unesterified cholesterol, and a cholesterol-containing phospholipid monolayer shell wrapped by a single protein of apoB100.224 LDL is the main carrier of cholesterol to peripheral tissues and is internalized through interaction between its ApoB ligand and the LDL receptor (LDL-R).225 Increased expression of LDL-R was found in HCC compared with adjacent liver tissue.226 N-succinyl chitosan NPs were coupled with LDL for target co-delivery of cholesterol-conjugated siRNA and DOX by Zhu et al.227 The LDL-decorated delivery system exhibited superior cytotoxicity against HepG2 cells over non-targeted system and also manifested enhanced liver tumor-targeting effects and relatively lower systemic toxicity in mice bearing hepatoma cell tumor, suggesting their potential for HCC therapy. LDL was also utilized to modify osthole-loading N-succinyl chitosan NPs, attaining high targeting efficacy indicated by investigations in vitro and in vivo.228 Hepatic arterial infusion of LDL-based NPs was employed for docosahexaenoic acid (DHA) transporting.229 LDL-DHA showed selective cytotoxicity against rat (H4IIE), mouse (Hepa1C7, TIB-75) and human (SK-Hep1) HCC cells, and its hepatic artery injection generated improved therapeutic and biologic effects compared with LDL NPs loaded with triolein or sham surgery controls.
Hematoporphyrin (HP) can also bind to LDL-R on tumor cell membrane as a ligand.230 HP was conjugated to NPs by Chang et al for specific targeting of DOX to HCC cells.231 In vitro investigation on HepG2 cells showed that HP-modified DOX-loaded NPs (HP-NPs) led to enhanced cellular uptake and strongest cytotoxicity compared with untargeted DOX-loaded NPs and free DOX. HP-NPs also resulted in increased AUC ratio of DOX in the liver (target organ) to DOX in the heart (an organ of major side effects), improving targeting efficiency and minimizing side effects. To enhance anticancer efficacy of photodynamic therapy for HCC, this team also employed HP to modify NPs but HP here was not only a targeting ligand for LDL-R on hepatoma cells but also a photosensitizer.232 Their study found increased cellular phototoxicity in vitro and remarkably lower tumor growth in vivo caused by HP-NPs compared with free HP, which may result from the HCC specificity of HP-NPs and/or the cytotoxicity of DOX itself.
Recombinant high-density lipoprotein (rHDL) particle is composed of phosphatidylcholine, apolipoprotein A-1, cholesterol, and cholesteryl esters and can efficiently deliver drugs since cancer cells can acquire HDL core components through scavenger receptor type B-1 (SR-B1).233 The SR-B1 is broadly overexpressed among various cancer cells including HCC and SR-B1 receptor-mediated HDL uptake makes HDL a promising target delivery vehicle for therapeutics.234 The rHDL was utilized as a biomimetic nanovector by Ding et al,235 mediating HCC-targeted cholesterol-conjugated siRNA (Chol-siRNA) delivery. rHDL could enhance cellular uptake and in vitro cytotoxicity of the siRNA and specifically target it into cytoplasm. Besides, rHDL-targeted complexes could effectively accumulate in tumor with prolonged retention time compared with the non-targeted lipoplexes, leading to significant tumor growth suppression in vivo.
Heat-labile enterotoxin subunit B (LTB) is a subunit of heat-labile enterotoxin (LT) which is a heat-sensitive enterotoxigenic factor236 and is composed of a single A subunit (LTA) and a ring of five B subunits (LTB).237 LTB binds specifically to the monosialoganglioside GM1 which is the host receptor of LT.236 A novel NP was developed utilizing a mixture of LTB and BSA for HCC-targeted delivery of 5-FU.238 Investigation on SMMC-7721 cells showed enhanced cytotoxicity and cellular uptake of BSA-LTB NPs compared with BSA NPs, suggesting the promising targeting efficiency of LTB.
Epigallocatechin Gallate (EGCG) is the most abundant catechin in green tea infusions239 and has efficient antioxidant activity possibly due to a low reduction potential and its ability to chelate metal ions.240 EGCG binds with great affinity to 67 kDa laminin receptor (67LR) which is upregulated in HCC.241 EGCG-functionalized ruthenium NPs (RuNPs) loaded with luminescent ruthenium complexes were developed to improve the treatment of liver cancer.242 Significant internalization of EGCG-RuNPs was found and could be suppressed upon treatment with 67LR-blocking antibody or laminin before the addition of NPs, indicating that the uptake of RuBB-loaded EGCG-RuNPs was mediated by 67LR. Moreover, high antitumor efficacy was found on tumor-bearing nude mice.
Serotonin (5-HT), a famous neurotransmitter and vasoactive substance which also mediates many gastrointestinal functions,243 is involved in tumor growth of HCC244 and binds to 5-HT receptors. There are 7 receptor classes including 14 subtypes of 5-HT receptors, among which 5-HT receptors 1B and 2B were significantly increased in HCC compared with nontumoral tissue.245 Gopal et al246 utilized 5-HT as the targeting moiety for liposomes delivering DNA and observed enhanced targeted transfection in HepG2 cells compared with a non-targeted control liposome. Since there are several classes of 5-HT receptors located in places other than liver,245 the feasibility of employing 5-HT as a HCC-targeting ligand in vivo remains to be further investigated.
Somatostatin (SST) is a regulatory peptide produced by neuroendocrine, inflammatory, and immune cells and acts as an inhibitory regulator of diverse functions, including inhibiting hormone and growth factor hypersecretion from tumors which facilitates tumor growth and producing antiproliferative effect giving rise to variable tumor shrinkage.247 The functions of SST are achieved through high-affinity G-protein-coupled receptors which comprise five subtypes (SSTR1-5)247 and are expressed in cultured hepatoma cells and HCC.248 Octreotide, a somatostatin analog which binds primarily to SSTR2,249 was used to develop octreotide-polyethylene glycol(100) monostearate (OPMS) as a targeting moiety for the delivery of hydroxycamptothecine (HCPT) loaded in nanostructured lipid carrier (NLC).250 Enhanced cellular uptake by SMMC-7721 cells was observed and the results indicated that OPMS-modified NLC had remarkable targeting efficiency both in vitro and in vivo. Somatostatin was also attached to AuNPs to establish a targeted delivery system for antitumor agents.251 Investigation on HCC-1806 cell lines revealed that cellular uptake of AuNPs-SST was higher than citrate-AuNPs and can be suppressed upon addition of antagonist, suggesting the modification of AuNPs using SST could enhance drug internalization by HCC-1806 cells through the specific interaction between SST and SSTR.
Dual-ligand modification to further enhance active targeting
The efficiency and specificity of active targeting depends on direct point-to-point recognitions and interactions of the ligands on the surface of NPs and the matching receptors on tumor cell membrane.252,253 However, this receptor-mediated endocytosis is a saturated pathway due to the limited number of receptors and the recycling of receptors during endocytosis.254–257 On the other hand, most receptors such as ASGPR and FR are not only expressed on hepatoma cells but also on some normal cells, resulting in unfavorable uptake in these non-targeted cells.258 Recently, some studies have focused on further enhancement of targeting effect and reduction of non-specific uptake. As has been noted in this review, hepatoma cells overexpress multiple surface receptors. To overcome this obstacle, dual-ligand based NPs is a promising strategy.198,255–260 In our previous report, dual-ligand Gal moieties and GA-modified chitosan NPs were prepared as a drug or gene carrier for HCC.261 The cellular uptake in vitro and biodistribution in vivo demonstrates that this dual-ligand decorated nanosystem can enhance the specific recognition and internalization by hepatoma cells, compared with single-ligand decorated nanosystem. Besides, it seems to be another good alternative to modify HA with a small-molecular ligand to further enhance the targeting of HA vehicle. For example, GA-HA conjugate was developed by chemical reactions and loaded with chemotherapeutic agents.262,263 All results indicate that GA-HA NPs seem to be a potential drug carrier with “double target sites” for HCC intracellular delivery. Due to degradation of HA by HAase, HA has been applicable to compose smart delivery system as HAase-responded shell and active ligand. At the HAase-rich tumor microenvironment, HA shells are disintegrated and other ligand-decorated carriers are exposed to the interstitium in tumor tissue, then the active targeting is triggered.259
Conclusion
This study reviewed the ligands in NTDDS which recognize the membrane receptors and proteins on the surface of hepatoma cells. The further investigation should focus on the detection of targeting properties of these ligands for preclinical trials and explore new target sites on membrane of hepatoma cells. Apart from ligands mentioned earlier, there are ligands whose receptors were found to have increased expression on HCC cells or play a role in HCC development, such as adenosine A2b receptor,264 gamma-aminobutyric acid A receptor theta subunit,265 androgen receptor,266 E prostanoid receptor,267 mitogenic insulin receptor A,268 receptor of insulin-like growth factor 2,269 and so on. On the other hand, the spatial properties of ligands can affect the targeting performance.78,80 It is known that the targeting performance is enhanced as increasing the density of ligands on the NPs until the saturation of ligand-receptor interaction is occurred, whereas the space between ligand and vehicle is not definite. A recent study demonstrated that the space plays a key role in transfection efficiency,80 but the effect should be further investigated.
A great deal of studies have shown that immunotherapy is a potent therapeutic option for HCC.270–272 However, it is uncertain whether that combination with immunotherapy and NTDDS can induce synergistic effect of anti-HCC. Furthermore, the high expression of MDR results in the poor response of HCC to chemotherapy.273 Thus it is a potential approach to treat HCC cells by composite delivery system co-loaded with siRNA for MDR genes and chemotherapeutic agent or multiple complementary chemotherapeutic agents to improve the prognosis of HCC.274
Acknowledgments
This work was supported by National Natural Science Foundation of China (No 81372668, 81502527), Natural Science Foundation of Hubei Province, People’s Republic of China (No 2015CFB527) and Fundamental Research Funds for the Central Universities, People’s Republic of China (No 2014QN064). Dr Min Li thanks the support from the Research Grant of Union Hospital (No 02.03.2015-74).
Disclosure
The authors report no conflicts of interest in this work.
References
Verslype C, Rosmorduc O, Rougier P. Hepatocellular carcinoma: ESMO-ESDO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(Suppl 7):vii41–vii48. | ||
European Association for the Study of the Liver; European Organisation for Research and Treatment of Cancer. EASL-EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2012;56(4):908–943. | ||
Singh S, Singh PP, Roberts LR, Sanchez W. Chemopreventive strategies in hepatocellular carcinoma. Nat Rev. 2014;11(1):45–54. | ||
Gomaa A-I. Hepatocellular carcinoma: epidemiology, risk factors and pathogenesis. World J Gastroenterol. 2008;14(27):4300. | ||
Bruix J, Sherman M. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53(3):1020–1022. | ||
Gutierrez JA, Gish RG. Efficacy of combination treatment modalities for intermediate and advanced hepatocellular carcinoma: intra-arterial therapies, sorafenib and novel small molecules. Transl Cancer Res. 2013;2(6):460–471. | ||
Topp ZZ, Sigal DS. Beyond chemotherapy: systemic treatment options for hepatocellular carcinoma. Transl Cancer Res. 2013;2(6):482–491. | ||
Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099–7109. | ||
Chang YS, Adnane J, Trail PA, et al. Sorafenib (BAY 43-9006) inhibits tumor growth and vascularization and induces tumor apoptosis and hypoxia in RCC xenograft models. Cancer Chemother Pharmacol. 2007;59(5):561–574. | ||
Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. | ||
Gillet JP, Andersen JB, Madigan JP, et al. A gene expression signature associated with overall survival in patients with hepatocellular carcinoma suggests a new treatment strategy. Mol Pharmacol. 2016;89(2):263–272. | ||
Parveen S, Misra R, Sahoo SK. Nanoparticles: a boon to drug delivery, therapeutics, diagnostics and imaging. Nanomedicine. 2012;8(2):147–166. | ||
Ling D, Xia H, Park W, et al. pH-sensitive nanoformulated triptolide as a targeted therapeutic strategy for hepatocellular carcinoma. ACS Nano. 2014;8(8):8027–8039. | ||
Yuan Y, Feng G, Qin W, Tang BZ, Liu B. Targeted and image-guided photodynamic cancer therapy based on organic nanoparticles with aggregation-induced emission characteristics. Chem Commun (Camb). 2014;50(63):8757–8760. | ||
Ding D, Liu J, Feng G, Li K, Hu Y, Liu B. Bright far-red/near-infrared conjugated polymer nanoparticles for in vivo bioimaging. Small. 2013;9(18):3093–3102. | ||
Varshosaz J, Farzan M. Nanoparticles for targeted delivery of therapeutics and small interfering RNAs in hepatocellular carcinoma. World J Gastroenterol. 2015;21(42):12022–12041. | ||
Ye LY, Chen W, Bai XL, et al. Hypoxia-induced epithelial-to-mesenchymal transition in hepatocellular carcinoma induces an immunosuppressive tumor microenvironment to promote metastasis. Cancer Res. 2016;76(4):818–830. | ||
Ferrara N, Houck K, Jakeman L, Leung DW. Molecular and biological properties of the vascular endothelial growth factor family of proteins. Endocrine Rev. 1992;13(1):18–32. | ||
Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6(4):389–395. | ||
Compagni A, Wilgenbus P, Impagnatiello MA, Cotten M, Christofori G. Fibroblast growth factors are required for efficient tumor angiogenesis. Cancer Res. 2000;60(24):7163–7169. | ||
McDougall SR, Anderson AR, Chaplain MA. Mathematical modelling of dynamic adaptive tumour-induced angiogenesis: clinical implications and therapeutic targeting strategies. J Theor Biol. 2006;241(3):564–589. | ||
Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41(1):189–207. | ||
Kaminskas LM, McLeod VM, Kelly BD, et al. A comparison of changes to doxorubicin pharmacokinetics, antitumor activity, and toxicity mediated by PEGylated dendrimer and PEGylated liposome drug delivery systems. Nanomedicine. 2012;8(1):103–111. | ||
Gerlowski LE, Jain RK. Microvascular permeability of normal and neoplastic tissues. Microvasc Res. 1986;31(3):288–305. | ||
Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–6392. | ||
Danhier F, Feron O, Preat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148(2):135–146. | ||
Narang AS, Varia S. Role of tumor vascular architecture in drug delivery. Adv Drug Deliv Rev. 2011;63(8):640–658. | ||
Semela D, Dufour JF. Angiogenesis and hepatocellular carcinoma. J Hepatol. 2004;41(5):864–880. | ||
Wilhelm S, Tavares AJ, Dai Q, et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mater. 2016;1:16014. | ||
Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotech. 2015;33(9):941–951. | ||
Venturoli D, Rippe B. Ficoll and dextran vs globular proteins as probes for testing glomerular permselectivity: effects of molecular size, shape, charge, and deformability. Am J Physiol. 2005;288(4):F605–F613. | ||
Choi HS, Liu W, Misra P, et al. Renal clearance of quantum dots. Nat Biotechnol. 2007;25(10):1165–1170. | ||
Shubayev VI, Pisanic Ii TR, Jin S. Magnetic nanoparticles for theragnostics. Adv Drug Deliv Rev. 2009;61(6):467–477. | ||
Kievit FM, Zhang M. Cancer nanotheranostics: improving imaging and therapy by targeted delivery across biological barriers. Adv Mater. 2011;23(36):H217–H247. | ||
Warren A, Cogger VC, Arias IM, McCuskey RS, Le Couteur DG. Liver sinusoidal endothelial fenestrations in caveolin-1 knockout mice. Microcirculation. 2010;17(1):32–38. | ||
Cho K, Wang X, Nie S, Chen ZG, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14(5):1310–1316. | ||
Byrne JD, Betancourt T, Brannon-Peppas L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Deliv Rev. 2008;60(15):1615–1626. | ||
Brigger I, Dubernet C, Couvreur P. Nanoparticles in cancer therapy and diagnosis. Adv Drug Deliv Rev. 2002;54(5):631–651. | ||
Stylianopoulos T, Jain RK. Design considerations for nanotherapeutics in oncology. Nanomedicine. 2015;11(8):1893–1907. | ||
Gao H, Cheng T, Liu J, et al. Self-regulated multifunctional collaboration of targeted nanocarriers for enhanced tumor therapy. Biomacromolecules. 2014;15(10):3634–3642. | ||
Gaur U, Sahoo SK, De TK, Ghosh PC, Maitra A, Ghosh PK. Biodistribution of fluoresceinated dextran using novel nanoparticles evading reticuloendothelial system. Int J Pharm. 2000;202(1–2):1–10. | ||
Adams ML, Lavasanifar A, Kwon GS. Amphiphilic block copolymers for drug delivery. J Pharm Sci. 2003;92(7):1343–1355. | ||
Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nature Reviews Drug Discov. 2008;7(9):771–782. | ||
Upponi JR, Torchilin VP. Passive vs active targeting: an update of the EPR role in drug delivery to tumors. In: Alonso JM, Garcia-Fuentes M, editors. Nano-Oncologicals: New Targeting and Delivery Approaches. Cham: Springer International Publishing; 2014:3–45. | ||
Yameen B, Choi WI, Vilos C, Swami A, Shi J, Farokhzad OC. Insight into nanoparticle cellular uptake and intracellular targeting. J Control Release. 2014;190:485–499. | ||
Zhong Y, Meng F, Deng C, Zhong Z. Ligand-directed active tumor-targeting polymeric nanoparticles for cancer chemotherapy. Biomacromolecules. 2014;15(6):1955–1969. | ||
Jain K, Kesharwani P, Gupta U, Jain NK. A review of glycosylated carriers for drug delivery. Biomaterials. 2012;33(16):4166–4186. | ||
Baenziger JU, Maynard Y. Human hepatic lectin. Physiochemical properties and specificity. J Biol Chem. 1980;255(10):4607–4613. | ||
D’Souza AA, Devarajan PV. Asialoglycoprotein receptor mediated hepatocyte targeting – strategies and applications. J Control Release. 2015;203:126–139. | ||
Zhang X, Ng HL, Lu A, et al. Drug delivery system targeting advanced hepatocellular carcinoma: current and future. Nanomedicine. 2016;12(4):853–869. | ||
Wang B, Hu Q, Wan T, et al. Porous lactose-modified chitosan scaffold for liver tissue engineering: influence of galactose moieties on cell attachment and mechanical stability. Int J Polym Sci. 2016;2016:8. | ||
Li Y, Huang G, Diakur J, Wiebe LI. Targeted delivery of macromolecular drugs: asialoglycoprotein receptor (ASGPR) expression by selected hepatoma cell lines used in antiviral drug development. Curr Drug Deliv. 2008;5(4):299–302. | ||
Zhang X, Guo S, Fan R, et al. Dual-functional liposome for tumor targeting and overcoming multidrug resistance in hepatocellular carcinoma cells. Biomaterials. 2012;33(29):7103–7114. | ||
Frisch B, Carriere M, Largeau C, et al. A new triantennary galactose-targeted PEGylated gene carrier, characterization of its complex with DNA, and transfection of hepatoma cells. Bioconjug Chem. 2004;15(4):754–764. | ||
Liu H, Wang H, Xu Y, et al. Lactobionic acid-modified dendrimer-entrapped gold nanoparticles for targeted computed tomography imaging of human hepatocellular carcinoma. ACS Appl Mater Interfaces. 2014;6(9):6944–6953. | ||
Rajasekaran D, Srivastava J, Ebeid K, et al. Combination of nanoparticle-delivered siRNA for astrocyte elevated gene-1 (AEG-1) and all-trans retinoic acid (ATRA): an effective therapeutic strategy for hepatocellular carcinoma (HCC). Bioconjug Chem. 2015;26(8):1651–1661. | ||
Pan Q, Lv Y, Williams GR, et al. Lactobionic acid and carboxymethyl chitosan functionalized graphene oxide nanocomposites as targeted anticancer drug delivery systems. Carbohydr Polym. 2016;151:812–820. | ||
Wu DQ, Lu B, Chang C, et al. Galactosylated fluorescent labeled micelles as a liver targeting drug carrier. Biomaterials. 2009;30(7):1363–1371. | ||
Wei W, Yue ZG, Qu JB, Yue H, Su ZG, Ma GH. Galactosylated nanocrystallites of insoluble anticancer drug for liver-targeting therapy: an in vitro evaluation. Nanomedicine (Lond). 2010;5(4):589–596. | ||
Yang R, Meng F, Ma S, Huang F, Liu H, Zhong Z. Galactose-decorated cross-linked biodegradable poly(ethylene glycol)-b-poly(epsilon-caprolactone) block copolymer micelles for enhanced hepatoma-targeting delivery of paclitaxel. Biomacromolecules. 2011;12(8):3047–3055. | ||
Zhong Y, Yang W, Sun H, et al. Ligand-directed reduction-sensitive shell-sheddable biodegradable micelles actively deliver doxorubicin into the nuclei of target cancer cells. Biomacromolecules. 2013;14(10):3723–3730. | ||
Chen W, Zou Y, Meng F, et al. Glyco-nanoparticles with sheddable saccharide shells: a unique and potent platform for hepatoma-targeting delivery of anticancer drugs. Biomacromolecules. 2014;15(3):900–907. | ||
Zou Y, Song Y, Yang W, Meng F, Liu H, Zhong Z. Galactose-installed photo-crosslinked pH-sensitive degradable micelles for active targeting chemotherapy of hepatocellular carcinoma in mice. J Control Release. 2014;193:154–161. | ||
Duan C, Gao J, Zhang D, et al. Galactose-decorated pH-responsive nanogels for hepatoma-targeted delivery of oridonin. Biomacromolecules. 2011;12(12):4335–4343. | ||
Villa R, Cerroni B, Vigano L, et al. Targeted doxorubicin delivery by chitosan-galactosylated modified polymer microbubbles to hepatocarcinoma cells. Colloids Surf B Biointerfaces. 2013;110:434–442. | ||
Luo Z, Hu Y, Cai K, et al. Intracellular redox-activated anticancer drug delivery by functionalized hollow mesoporous silica nanoreservoirs with tumor specificity. Biomaterials. 2014;35(27):7951–7962. | ||
Xu Z, Chen L, Gu W, et al. The performance of docetaxel-loaded solid lipid nanoparticles targeted to hepatocellular carcinoma. Biomaterials. 2009;30(2):226–232. | ||
Kim TH, Park IK, Nah JW, Choi YJ, Cho CS. Galactosylated chitosan/DNA nanoparticles prepared using water-soluble chitosan as a gene carrier. Biomaterials. 2004;25(17):3783–3792. | ||
Kim EM, Jeong HJ, Park IK, et al. Asialoglycoprotein receptor targeted gene delivery using galactosylated polyethylenimine-graft-poly(ethylene glycol): in vitro and in vivo studies. J Control Release. 2005;108(2–3):557–567. | ||
Yang Y, Zhang Z, Chen L, Gu W, Li Y. Galactosylated poly(2-(2-aminoethyoxy)ethoxy)phosphazene/DNA complex nanoparticles: in vitro and in vivo evaluation for gene delivery. Biomacromolecules. 2010;11(4):927–933. | ||
Ma P, Liu S, Huang Y, Chen X, Zhang L, Jing X. Lactose mediated liver-targeting effect observed by ex vivo imaging technology. Biomaterials. 2010;31(9):2646–2654. | ||
Xue WJ, Feng Y, Wang F, et al. Asialoglycoprotein receptor-magnetic dual targeting nanoparticles for delivery of RASSF1A to hepatocellular carcinoma. Sci Rep. 2016;6:22149. | ||
Liang HF, Chen SC, Chen MC, Lee PW, Chen CT, Sung HW. Paclitaxel-loaded poly(gamma-glutamic acid)-poly(lactide) nanoparticles as a targeted drug delivery system against cultured HepG2 cells. Bioconjug Chem. 2006;17(2):291–299. | ||
Zhu D, Tao W, Zhang H, et al. Docetaxel (DTX)-loaded polydopamine-modified TPGS-PLA nanoparticles as a targeted drug delivery system for the treatment of liver cancer. Acta Biomater. 2016;30:144–154. | ||
Thapa B, Kumar P, Zeng H, Narain R. Asialoglycoprotein receptor-mediated gene delivery to hepatocytes using galactosylated polymers. Biomacromolecules. 2015;16(9):3008–3020. | ||
Quan S, Wang Y, Zhou A, Kumar P, Narain R. Galactose-based thermosensitive nanogels for targeted drug delivery of iodoazomycin arabinofuranoside (IAZA) for theranostic management of hypoxic hepatocellular carcinoma. Biomacromolecules. 2015;16(7):1978–1986. | ||
Lee YC, Townsend RR, Hardy MR, et al. Binding of synthetic oligosaccharides to the hepatic Gal/GalNAc lectin. Dependence on fine structural features. J Biol Chem. 1983;258(1):199–202. | ||
Managit C, Kawakami S, Yamashita F, Hashida M. Effect of galactose density on asialoglycoprotein receptor-mediated uptake of galactosylated liposomes. J Pharm Sci. 2005;94(10):2266–2275. | ||
Managit C, Kawakami S, Yamashita F, Hashida M. Uptake characteristics of galactosylated emulsion by HepG2 hepatoma cells. Int J Pharm. 2005;301(1–2):255–261. | ||
Fu F, Wu Y, Zhu J, Wen S, Shen M, Shi X. Multifunctional lactobionic acid-modified dendrimers for targeted drug delivery to liver cancer cells: investigating the role played by PEG spacer. ACS Appl Mater Interfaces. 2014;6(18):16416–16425. | ||
Craparo EF, Sardo C, Serio R, et al. Galactosylated polymeric carriers for liver targeting of sorafenib. Int J Pharm. 2014;466(1–2):172–180. | ||
Oh EJ, Park K, Kim KS, et al. Target specific and long-acting delivery of protein, peptide, and nucleotide therapeutics using hyaluronic acid derivatives. J Control Release. 2010;141(1):2–12. | ||
Rosso F, Quagliariello V, Tortora C, Di Lazzaro A, Barbarisi A, Iaffaioli RV. Cross-linked hyaluronic acid sub-micron particles: in vitro and in vivo biodistribution study in cancer xenograft model. J Mater Sci. 2013;24(6):1473–1481. | ||
Choi KY, Min KH, Yoon HY, et al. PEGylation of hyaluronic acid nanoparticles improves tumor targetability in vivo. Biomaterials. 2011;32(7):1880–1889. | ||
Rao NV, Yoon HY, Han HS, et al. Recent developments in hyaluronic acid-based nanomedicine for targeted cancer treatment. Exp Opin Drug Deliv. 2016;13(2):239–252. | ||
Xiao B, Han MK, Viennois E, et al. Hyaluronic acid-functionalized polymeric nanoparticles for colon cancer-targeted combination chemotherapy. Nanoscale. 2015;7(42):17745–17755. | ||
Yang RM, Fu CP, Li NN, et al. Glycosaminoglycan-targeted iron oxide nanoparticles for magnetic resonance imaging of liver carcinoma. Mater Sci Eng C. 2014;45:556–563. | ||
Kumar CS, Raja MD, Sundar DS, Gover Antoniraj M, Ruckmani K. Hyaluronic acid co-functionalized gold nanoparticle complex for the targeted delivery of metformin in the treatment of liver cancer (HepG2 cells). Carbohydr Polym. 2015;128:63–74. | ||
Opanasopit P, Apirakaramwong A, Ngawhirunpat T, Rojanarata T, Ruktanonchai U. Development and characterization of pectinate micro/nanoparticles for gene delivery. AAPS PharmSciTech. 2008;9(1):67–74. | ||
Chittasupho C, Jaturanpinyo M, Mangmool S. Pectin nanoparticle enhances cytotoxicity of methotrexate against HepG2 cells. Drug Deliv. 2013;20(1):1–9. | ||
Yu CY, Wang YM, Li NM, et al. In vitro and in vivo evaluation of pectin-based nanoparticles for hepatocellular carcinoma drug chemotherapy. Mol Pharm. 2014;11(2):638–644. | ||
Kaneo Y, Tanaka T, Nakano T, Yamaguchi Y. Evidence for receptor-mediated hepatic uptake of pullulan in rats. J Control Release. 2001;70(3):365–373. | ||
Zhang C, An T, Wang D, et al. Stepwise pH-responsive nanoparticles containing charge-reversible pullulan-based shells and poly(beta-amino ester)/poly(lactic-co-glycolic acid) cores as carriers of anticancer drugs for combination therapy on hepatocellular carcinoma. J Control Release. 2016;226:193–204. | ||
Leamon CP, Low PS. Folate-mediated targeting: from diagnostics to drug and gene delivery. Drug Discov Today. 2001;6(1):44–51. | ||
Lucock M. Folic acid: nutritional biochemistry, molecular biology, and role in disease processes. Mol Genet Metab. 2000;71(1–2):121–138. | ||
Kamen BA, Capdevila A. Receptor-mediated folate accumulation is regulated by the cellular folate content. Proc Natl Acad Sci U S A. 1986;83(16):5983–5987. | ||
Tengchaisri T, Chawengkirttikul R, Rachaphaew N, Reutrakul V, Sangsuwan R, Sirisinha S. Antitumor activity of triptolide against cholangiocarcinoma growth in vitro and in hamsters. Cancer Lett. 1998;133(2):169–175. | ||
Phillips PA, Dudeja V, McCarroll JA, et al. Triptolide induces pancreatic cancer cell death via inhibition of heat shock protein 70. Cancer Res. 2007;67(19):9407–9416. | ||
Alsaied OA, Sangwan V, Banerjee S, et al. Sorafenib and triptolide as combination therapy for hepatocellular carcinoma. Surgery. 2014;156(2):270–279. | ||
Niu C, Sun Q, Zhou J, Cheng D, Hong G. Folate-functionalized polymeric micelles based on biodegradable PEG-PDLLA as a hepatic carcinoma-targeting delivery system. Asian Pacific J Cancer Prev. 2011;12(8):1995–1999. | ||
Xu Z, Zhang Z, Chen Y, Chen L, Lin L, Li Y. The characteristics and performance of a multifunctional nanoassembly system for the co-delivery of docetaxel and iSur-pDNA in a mouse hepatocellular carcinoma model. Biomaterials. 2010;31(5):916–922. | ||
Lei XY, Zhong M, Feng LF, Zhu BY, Tang SS, Liao DF. siRNA-mediated Bcl-2 and Bcl-xl gene silencing sensitizes human hepatoblastoma cells to chemotherapeutic drugs. Clin Exp Pharm Physiol. 2007;34(5–6):450–456. | ||
Cao N, Cheng D, Zou S, Ai H, Gao J, Shuai X. The synergistic effect of hierarchical assemblies of siRNA and chemotherapeutic drugs co-delivered into hepatic cancer cells. Biomaterials. 2011;32(8):2222–2232. | ||
Hu BG, Liu LP, Chen GG, et al. Therapeutic efficacy of improved alpha-fetoprotein promoter-mediated tBid delivered by folate-PEI600-cyclodextrin nanopolymer vector in hepatocellular carcinoma. Exp Cell Res. 2014;324(2):183–191. | ||
Fan L, Li F, Zhang H, et al. Co-delivery of PDTC and doxorubicin by multifunctional micellar nanoparticles to achieve active targeted drug delivery and overcome multidrug resistance. Biomaterials. 2010;31(21):5634–5642. | ||
Tang ZY. Hepatocellular carcinoma – cause, treatment and metastasis. World J Gastroenterol. 2001;7(4):445–454. | ||
Ye QH, Qin LX, Forgues M, et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat Med. 2003;9(4):416–423. | ||
Uka K, Aikata H, Takaki S, et al. Clinical features and prognosis of patients with extrahepatic metastases from hepatocellular carcinoma. World J Gastroenterol. 2007;13(3):414–420. | ||
Zhang Y, Zhang H, Wu W, et al. Folate-targeted paclitaxel-conjugated polymeric micelles inhibits pulmonary metastatic hepatoma in experimental murine H22 metastasis models. Int J Nanomedicine. 2014;9:2019–2030. | ||
Wang W, Cheng D, Gong F, Miao X, Shuai X. Design of multifunctional micelle for tumor-targeted intracellular drug release and fluorescent imaging. Adv Mater. 2012;24(1):115–120. | ||
Li L, Tong R, Li M, Kohane DS. Self-assembled gemcitabine – gadolinium nanoparticles for magnetic resonance imaging and cancer therapy. Acta Biomater. 2016;33:34–39. | ||
Chen Y, Li M, Hong Y, Lam JW, Zheng Q, Tang BZ. Dual-modal MRI contrast agent with aggregation-induced emission characteristic for liver specific imaging with long circulation lifetime. ACS Appl Mater Interfaces. 2014;6(13):10783–10791. | ||
Maeng JH, Lee DH, Jung KH, et al. Multifunctional doxorubicin loaded superparamagnetic iron oxide nanoparticles for chemotherapy and magnetic resonance imaging in liver cancer. Biomaterials. 2010;31(18):4995–5006. | ||
Li X, Li H, Yi W, Chen J, Liang B. Acid-triggered core cross-linked nanomicelles for targeted drug delivery and magnetic resonance imaging in liver cancer cells. Int J Nanomedicine. 2013;8:3019–3031. | ||
Li YJ, Dong M, Kong FM, Zhou JP. Folate-decorated anticancer drug and magnetic nanoparticles encapsulated polymeric carrier for liver cancer therapeutics. Int J Pharm. 2015;489(1–2):83–90. | ||
Viota JL, Carazo A, Munoz-Gamez JA, et al. Functionalized magnetic nanoparticles as vehicles for the delivery of the antitumor drug gemcitabine to tumor cells. Physicochemical in vitro evaluation. Mater Sci Eng C. 2013;33(3):1183–1192. | ||
Guo Y, Wang J, Zhang L, et al. Theranostical nanosystem-mediated identification of an oncogene and highly effective therapy in hepatocellular carcinoma. Hepatology. 2016;63(4):1240–1255. | ||
Russell-Jones G, McTavish K, McEwan J, Rice J, Nowotnik D. Vitamin-mediated targeting as a potential mechanism to increase drug uptake by tumours. J Inorg Biochem. 2004;98(10):1625–1633. | ||
Chen S, Zhao X, Chen J, Kuznetsova L, Wong SS, Ojima I. Mechanism-based tumor-targeting drug delivery system. Validation of efficient vitamin receptor-mediated endocytosis and drug release. Bioconjug Chem. 2010;21(5):979–987. | ||
Mishra PR, Jain NK. Biotinylated methotrexate loaded erythrocytes for enhanced liver uptake. “A study on the rat.” Int J Pharm. 2002;231(2):145–153. | ||
Na K, Bum Lee T, Park K-H, Shin E-K, Lee Y-B, Choi H-K. Self-assembled nanoparticles of hydrophobically-modified polysaccharide bearing vitamin H as a targeted anti-cancer drug delivery system. Eur J Pharm Sci. 2003;18(2):165–173. | ||
Hong Y, Lam JWY, Tang BZ. Aggregation-induced emission: phenomenon, mechanism and applications. Chem Commun. 2009;(29):4332–4353. | ||
Hong Y, Lam JWY, Tang BZ. Aggregation-induced emission. Chem Soc Rev. 2011;40(11):5361–5388. | ||
Li M, Lam JWY, Mahtab F, et al. Biotin-decorated fluorescent silica nanoparticles with aggregation-induced emission characteristics: fabrication, cytotoxicity and biological applications. J Mater Chem B. 2013;1(5):676–684. | ||
Schug TT, Berry DC, Shaw NS, Travis SN, Noy N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell. 2007;129(4):723–733. | ||
Jinushi M, Takehara T, Tatsumi T, et al. Expression and role of MICA and MICB in human hepatocellular carcinomas and their regulation by retinoic acid. Int J Cancer. 2003;104(3):354–361. | ||
Sano K, Takayama T, Murakami K, Saiki I, Makuuchi M. Overexpression of retinoic acid receptor alpha in hepatocellular carcinoma. Clin Cancer Res. 2003;9(10 Pt 1):3679–3683. | ||
Varshosaz J, Hassanzadeh F, Sadeghi H, Ghelich Khan Z, Rostami M. Retinoic acid decorated albumin-chitosan nanoparticles for targeted delivery of doxorubicin hydrochloride in hepatocellular carcinoma. J Nanomater. 2013;2013:1–12. | ||
Michel V, Yuan Z, Ramsubir S, Bakovic M. Choline transport for phospholipid synthesis. Exp Biol Med (Maywood). 2006;231(5):490–504. | ||
Airley RE, Mobasheri A. Hypoxic regulation of glucose transport, anaerobic metabolism and angiogenesis in cancer: novel pathways and targets for anticancer therapeutics. Chemotherapy. 2007;53(4):233–256. | ||
Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006;66(18):8927–8930. | ||
Wood IS, Trayhurn P. Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins. Br J Nutr. 2003;89(1):3–9. | ||
Rumsey SC, Kwon O, Xu GW, Burant CF, Simpson I, Levine M. Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J Biol Chem. 1997;272(30):18982–18989. | ||
Vera JC, Rivas CI, Zhang RH, Farber CM, Golde DW. Human HL-60 myeloid leukemia cells transport dehydroascorbic acid via the glucose transporters and accumulate reduced ascorbic acid. Blood. 1994;84(5):1628–1634. | ||
Guo Y, Zhang Y, Li J, et al. Cell microenvironment-controlled antitumor drug releasing-nanomicelles for GLUT1-targeting hepatocellular carcinoma therapy. ACS Appl Mater Interfaces. 2015;7(9):5444–5453. | ||
Arruebo M, Valladares M, González-Fernández Á. Antibody-conjugated nanoparticles for biomedical applications. J Nanomater. 2009;2009:1–24. | ||
Goodall S, Jones ML, Mahler S. Monoclonal antibody-targeted polymeric nanoparticles for cancer therapy – future prospects. J Chem Technol Biotechnol. 2015;90(7):1169–1176. | ||
He Q, Lu WS, Liu Y, Guan YS, Kuang AR. 131I-labeled metuximab combined with chemoembolization for unresectable hepatocellular carcinoma. World J Gastroenterol. 2013;19(47):9104–9110. | ||
Xu J, Xu HY, Zhang Q, et al. HAb18G/CD147 functions in invasion and metastasis of hepatocellular carcinoma. Mol Cancer Res. 2007;5(6):605–614. | ||
Jin C, Qian N, Zhao W, et al. Improved therapeutic effect of DOX-PLGA-PEG micelles decorated with bivalent fragment HAb18 F(ab’)(2) for hepatocellular carcinoma. Biomacromolecules. 2010;11(9):2422–2431. | ||
Zhu R, Zhang CG, Liu Y, et al. CD147 monoclonal antibody mediated by chitosan nanoparticles loaded with alpha-hederin enhances antineoplastic activity and cellular uptake in liver cancer cells. Sci Rep. 2015;5:17904. | ||
Li B, Wang H, Zhang D, et al. Construction and characterization of a high-affinity humanized SM5-1 monoclonal antibody. Biochem Biophys Res Commun. 2007;357(4):951–956. | ||
Wang H, Song S, Kou G, et al. Treatment of hepatocellular carcinoma in a mouse xenograft model with an immunotoxin which is engineered to eliminate vascular leak syndrome. Cancer Immunol Immunother. 2007;56(11):1775–1783. | ||
Kou G, Wang S, Cheng C, et al. Development of SM5-1-conjugated ultrasmall superparamagnetic iron oxide nanoparticles for hepatoma detection. Biochem Biophys Res Commun. 2008;374(2):192–197. | ||
Ma X, Hui H, Jin Y, et al. Enhanced immunotherapy of SM5-1 in hepatocellular carcinoma by conjugating with gold nanoparticles and its in vivo bioluminescence tomographic evaluation. Biomaterials. 2016;87:46–56. | ||
Kou G, Gao J, Wang H, et al. Preparation and characterization of paclitaxel-loaded PLGA nanoparticles coated with cationic SM5-1 single-chain antibody. J Biochem Mol Biol. 2007;40(5):731–739. | ||
Gao J, Kou G, Chen H, et al. Treatment of hepatocellular carcinoma in mice with PE38KDEL type I mutant-loaded poly(lactic-co-glycolic acid) nanoparticles conjugated with humanized SM5-1 F(ab’) fragments. Mol Cancer Therap. 2008;7(10):3399–3407. | ||
Kiss A, Wang NJ, Xie JP, Thorgeirsson SS. Analysis of transforming growth factor (TGF)-alpha/epidermal growth factor receptor, hepatocyte growth Factor/c-met, TGF-beta receptor type II, and p53 expression in human hepatocellular carcinomas. Clin Cancer Res. 1997;3(7):1059–1066. | ||
Sibilia M, Kroismayr R, Lichtenberger BM, Natarajan A, Hecking M, Holcmann M. The epidermal growth factor receptor: from development to tumorigenesis. Differentiation. 2007;75(9):770–787. | ||
Liu P, Li Z, Zhu M, et al. Preparation of EGFR monoclonal antibody conjugated nanoparticles and targeting to hepatocellular carcinoma. J Mater Sci. 2010;21(2):551–556. | ||
Wang JL, Tang GP, Shen J, et al. A gene nanocomplex conjugated with monoclonal antibodies for targeted therapy of hepatocellular carcinoma. Biomaterials. 2012;33(18):4597–4607. | ||
Chen J, Wu H, Han D, Xie C. Using anti-VEGF McAb and magnetic nanoparticles as double-targeting vector for the radioimmunotherapy of liver cancer. Cancer Lett. 2006;231(2):169–175. | ||
Mathew J, Hines JE, Obafunwa JO, Burr AW, Toole K, Burt AD. CD44 is expressed in hepatocellular carcinomas showing vascular invasion. J Pathol. 1996;179(1):74–79. | ||
Wang L, Su W, Liu Z, et al. CD44 antibody-targeted liposomal nanoparticles for molecular imaging and therapy of hepatocellular carcinoma. Biomaterials. 2012;33(20):5107–5114. | ||
Zhang X-X, Li H-D, Zhao S, et al. The cell surface GRP78 facilitates the invasion of hepatocellular carcinoma cells. Biomed Res Int. 2013;2013:1–8. | ||
Li Z, Li Z. Glucose regulated protein 78: A critical link between tumor microenvironment and cancer hallmarks. Biochim Biophys Acta. 2012;1826(1):13–22. | ||
Zhao L, Li H, Shi Y, et al. Nanoparticles inhibit cancer cell invasion and enhance antitumor efficiency by targeted drug delivery via cell surface-related GRP78. Int J Nanomedicine. 2015;10:245–256. | ||
Takahashi H, Ozturk M, Wilson B, et al. In vivo expression of two novel tumor-associated antigens and their use in immunolocalization of human hepatocellular carcinoma. Hepatology. 1989;9(4):625–634. | ||
Moradpour D, Compagnon B, Wilson BE, Nicolau C, Wands JR. Specific targeting of human hepatocellular carcinoma cells by immunoliposomes in vitro. Hepatology. 1995;22(5):1527–1537. | ||
Filmus J, Selleck SB. Glypicans: proteoglycans with a surprise. J Clin Invest. 2001;108(4):497–501. | ||
Capurro M, Wanless IR, Sherman M, et al. Glypican-3: a novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology. 2003;125(1):89–97. | ||
Ishiguro T, Sugimoto M, Kinoshita Y, et al. Anti-glypican 3 antibody as a potential antitumor agent for human liver cancer. Cancer Res. 2008;68(23):9832–9838. | ||
Phung Y, Gao W, Man YG, Nagata S, Ho M. High-affinity monoclonal antibodies to cell surface tumor antigen glypican-3 generated through a combination of peptide immunization and flow cytometry screening. MAbs. 2012;4(5):592–599. | ||
Nakano K, Orita T, Nezu J, et al. Anti-glypican 3 antibodies cause ADCC against human hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2009;378(2):279–284. | ||
Feng M, Gao W, Wang R, et al. Therapeutically targeting glypican-3 via a conformation-specific single-domain antibody in hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2013;110(12):E1083–E1091. | ||
Hanaoka H, Nagaya T, Sato K, et al. Glypican-3 targeted human heavy chain antibody as a drug carrier for hepatocellular carcinoma therapy. Mol Pharm. 2015;12(6):2151–2157. | ||
Wang K, Kievit FM, Sham JG, et al. Iron-oxide-based nanovector for tumor targeted siRNA delivery in an orthotopic hepatocellular carcinoma xenograft mouse model. Small. 2016;12(4):477–487. | ||
Zitzmann S, Ehemann V, Schwab M. Arginine-glycine-aspartic acid (RGD)-peptide binds to both tumor and tumor-endothelial cells in vivo. Cancer Res. 2002;62(18):5139–5143. | ||
Danhier F, Le Breton A, Preat V. RGD-based strategies to target alpha(v) beta(3) integrin in cancer therapy and diagnosis. Mol Pharm. 2012;9(11):2961–2973. | ||
Bellis SL. Advantages of RGD peptides for directing cell association with biomaterials. Biomaterials. 2011;32(18):4205–4210. | ||
Chen L, Liu Y, Wang W, Liu K. Effect of integrin receptor-targeted liposomal paclitaxel for hepatocellular carcinoma targeting and therapy. Oncol Lett. 2015;10(1):77–84. | ||
Marelli UK, Rechenmacher F, Sobahi TR, Mas-Moruno C, Kessler H. Tumor targeting via integrin ligands. Front Oncol. 2013;3:222. | ||
Mitra A, Coleman T, Borgman M, Nan A, Ghandehari H, Line BR. Polymeric conjugates of mono- and bi-cyclic alphaVbeta3 binding peptides for tumor targeting. J Control Release. 2006;114(2):175–183. | ||
Shen JM, Gao FY, Yin T, et al. cRGD-functionalized polymeric magnetic nanoparticles as a dual-drug delivery system for safe targeted cancer therapy. Pharmacol Res. 2013;70(1):102–115. | ||
Sugahara KN, Teesalu T, Karmali PP, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16(6):510–520. | ||
Zhu Z, Xie C, Liu Q, et al. The effect of hydrophilic chain length and iRGD on drug delivery from poly(epsilon-caprolactone)-poly(N-vinylpyrrolidone) nanoparticles. Biomaterials. 2011;32(35):9525–9535. | ||
Mao X, Liu J, Gong Z, et al. iRGD-conjugated DSPE-PEG2000 nanomicelles for targeted delivery of salinomycin for treatment of both liver cancer cells and cancer stem cells. Nanomedicine (Lond). 2015;10(17):2677–2695. | ||
Zhang J, Hu J, Chan HF, Skibba M, Liang G, Chen M. iRGD decorated lipid-polymer hybrid nanoparticles for targeted co-delivery of doxorubicin and sorafenib to enhance anti-hepatocellular carcinoma efficacy. Nanomedicine. 2016;12(5):1303–1311. | ||
Walker PS, Reid ME. The Gerbich blood group system: a review. Immunohematology. 2010;26(2):60–65. | ||
Li Z, Zhao R, Wu X, et al. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J. 2005;19(14):1978–1985. | ||
Tang H, Chen X, Rui M, et al. Effects of surface displayed targeting ligand GE11 on liposome distribution and extravasation in tumor. Mol Pharm. 2014;11(10):3242–3250. | ||
Klutz K, Schaffert D, Willhauck MJ, et al. Epidermal growth factor receptor-targeted (131)I-therapy of liver cancer following systemic delivery of the sodium iodide symporter gene. Mol Ther. 2011;19(4):676–685. | ||
Liu M, Li ZH, Xu FJ, et al. An oligopeptide ligand-mediated therapeutic gene nanocomplex for liver cancer-targeted therapy. Biomaterials. 2012;33(7):2240–2250. | ||
Zheng N, Wei W, Wang Z. Emerging roles of FGF signaling in hepatocellular carcinoma. Transl Cancer Res. 2016;5(1):1–6. | ||
Maruta F, Parker AL, Fisher KD, et al. Identification of FGF receptor-binding peptides for cancer gene therapy. Cancer Gene therapy. 2002;9(6):543–552. | ||
Hu Y, Tang G, Liu J, et al. FGF receptor-mediated gene delivery using ligands coupled to PEI-beta-CyD. J Biomed Biotechnol. 2012;2012:989235. | ||
Lo A, Lin CT, Wu HC. Hepatocellular carcinoma cell-specific peptide ligand for targeted drug delivery. Mol Cancer Ther. 2008;7(3):579–589. | ||
Toita R, Murata M, Tabata S, et al. Development of human hepatocellular carcinoma cell-targeted protein cages. Bioconjug Chem. 2012;23(7):1494–1501. | ||
Moon H, Lee J, Min J, Kang S. Developing genetically engineered encapsulin protein cage nanoparticles as a targeted delivery nanoplatform. Biomacromolecules. 2014;15(10):3794–3801. | ||
Oh S, Kim BJ, Singh NP, Lai H, Sasaki T. Synthesis and anti-cancer activity of covalent conjugates of artemisinin and a transferrin-receptor targeting peptide. Cancer Lett. 2009;274(1):33–39. | ||
Han L, Huang R, Liu S, Huang S, Jiang C. Peptide-conjugated PAMAM for targeted doxorubicin delivery to transferrin receptor overexpressed tumors. Mol Pharm. 2010;7(6):2156–2165. | ||
Trouet A, Deprez-De Campeneere D. Daunorubicin-DNA and doxorubicin-DNA. A review of experimental and clinical data. Cancer Chemother Pharmacol. 1979;2(1):77–79. | ||
Bagalkot V, Lee IH, Yu MK, et al. A combined chemoimmunotherapy approach using a plasmid-doxorubicin complex. Mol Pharm. 2009;6(3):1019–1028. | ||
Han L, Huang R, Li J, Liu S, Huang S, Jiang C. Plasmid pORF-hTRAIL and doxorubicin co-delivery targeting to tumor using peptide-conjugated polyamidoamine dendrimer. Biomaterials. 2011;32(4):1242–1252. | ||
Farokhzad OC, Cheng J, Teply BA, et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci U S A. 2006;103(16):6315–6320. | ||
Sun H, Zu Y. Aptamers and their applications in nanomedicine. Small. 2015;11(20):2352–2364. | ||
Li X, Zhao Q, Qiu L. Smart ligand: aptamer-mediated targeted delivery of chemotherapeutic drugs and siRNA for cancer therapy. J Control Release. 2013;171(2):152–162. | ||
Jiang J, Chen H, Yu C, et al. The promotion of salinomycin delivery to hepatocellular carcinoma cells through EGFR and CD133 aptamers conjugation by PLGA nanoparticles. Nanomedicine (Lond). 2015;10(12):1863–1879. | ||
Song Y, Zhu Z, An Y, et al. Selection of DNA aptamers against epithelial cell adhesion molecule for cancer cell imaging and circulating tumor cell capture. Anal Chem. 2013;85(8):4141–4149. | ||
Pilapong C, Sitthichai S, Thongtem S, Thongtem T. Smart magnetic nanoparticle-aptamer probe for targeted imaging and treatment of hepatocellular carcinoma. Int J Pharm. 2014;473(1–2):469–474. | ||
Sun D, Lu J, Zhong Y, et al. Sensitive electrochemical aptamer cytosensor for highly specific detection of cancer cells based on the hybrid nanoelectrocatalysts and enzyme for signal amplification. Biosens Bioelectron. 2016;75:301–307. | ||
Sun D, Lu J, Chen Z, Yu Y, Mo M. A repeatable assembling and disassembling electrochemical aptamer cytosensor for ultrasensitive and highly selective detection of human liver cancer cells. Anal Chim Acta. 2015;885:166–173. | ||
Trinh TL, Zhu G, Xiao X, et al. A synthetic aptamer-drug adduct for targeted liver cancer therapy. PLoS One. 2015;10(11):e0136673. | ||
Wang FB, Rong Y, Fang M, et al. Recognition and capture of metastatic hepatocellular carcinoma cells using aptamer-conjugated quantum dots and magnetic particles. Biomaterials. 2013;34(15):3816–3827. | ||
Meng L, Yang L, Zhao X, et al. Targeted delivery of chemotherapy agents using a liver cancer-specific aptamer. PLoS One. 2012;7(4):e33434. | ||
Tros de Ilarduya C, Duzgunes N. Delivery of therapeutic nucleic acids via transferrin and transferrin receptors: lipoplexes and other carriers. Exp Opin Drug Deliv. 2013;10(11):1583–1591. | ||
Tortorella S, Karagiannis TC. Transferrin receptor-mediated endocytosis: a useful target for cancer therapy. J Membrane Biol. 2014;247(4):291–307. | ||
Tseng HH, Chang JG, Hwang YH, Yeh KT, Chen YL, Yu HS. Expression of hepcidin and other iron-regulatory genes in human hepatocellular carcinoma and its clinical implications. J Cancer Res Clin Oncol. 2009;135(10):1413–1420. | ||
Holmstrom P, Gafvels M, Eriksson LC, et al. Expression of iron regulatory genes in a rat model of hepatocellular carcinoma. Liver Int. 2006;26(8):976–985. | ||
Malarvizhi GL, Retnakumari AP, Nair S, Koyakutty M. Transferrin targeted core-shell nanomedicine for combinatorial delivery of doxorubicin and sorafenib against hepatocellular carcinoma. Nanomedicine. 2014;10(8):1649–1659. | ||
Zhang W, Peng F, Zhou T, et al. Targeted delivery of chemically modified anti-miR-221 to hepatocellular carcinoma with negatively charged liposomes. Int J Nanomedicine. 2015;10:4825–4836. | ||
Richardson DR, Ponka P. The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochim Biophys Acta. 1997;1331(1):1–40. | ||
Krishna AD, Mandraju RK, Kishore G, Kondapi AK. An efficient targeted drug delivery through apotransferrin loaded nanoparticles. PLoS One. 2009;4(10):e7240. | ||
Baker EN, Baker HM. Molecular structure, binding properties and dynamics of lactoferrin. Cell Mol Life Sci. 2005;62(22):2531–2539. | ||
Golla K, Cherukuvada B, Ahmed F, Kondapi AK. Efficacy, safety and anticancer activity of protein nanoparticle-based delivery of doxorubicin through intravenous administration in rats. PLoS One. 2012;7(12):e51960. | ||
Golla K, Bhaskar C, Ahmed F, Kondapi AK. A target-specific oral formulation of doxorubicin-protein nanoparticles: efficacy and safety in hepatocellular cancer. J Cancer. 2013;4(8):644–652. | ||
Chintharlapalli S, Papineni S, Jutooru I, McAlees A, Safe S. Structure-dependent activity of glycyrrhetinic acid derivatives as peroxisome proliferator–activated receptor γ agonists in colon cancer cells. Mol Cancer Ther. 2007;6(5):1588–1598. | ||
Cai Y, Xu Y, Chan HF, Fang X, He C, Chen M. Glycyrrhetinic acid mediated drug delivery carriers for hepatocellular carcinoma therapy. Mol Pharm. 2016;13(3):699–709. | ||
Wang FZ, Xing L, Tang ZH, et al. Codelivery of Doxorubicin and shAkt1 by poly(ethylenimine)-glycyrrhetinic acid nanoparticles to induce autophagy-mediated liver cancer combination therapy. Mol Pharm. 2016;13(4):1298–1307. | ||
Anirudhan TS, Binusreejayan. Dextran based nanosized carrier for the controlled and targeted delivery of curcumin to liver cancer cells. Int J Biol Macromol. 2016;88:222–235. | ||
Negishi M, Irie A, Nagata N, Ichikawa A. Specific binding of glycyrrhetinic acid to the rat liver membrane. Biochim Biophys Acta. 1991;1066(1):77–82. | ||
Zeng F, Harris RC. Epidermal growth factor, from gene organization to bedside. Semin Cell Dev Biol. 2014;28:2–11. | ||
Wolschek MF, Thallinger C, Kursa M, et al. Specific systemic nonviral gene delivery to human hepatocellular carcinoma xenografts in SCID mice. Hepatology. 2002;36(5):1106–1114. | ||
Prassl R, Laggner P. Molecular structure of low density lipoprotein: current status and future challenges. Eur Biophys J. 2009;38(2):145–158. | ||
Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29(4):431–438. | ||
Bhat M, Skill N, Marcus V, et al. Decreased PCSK9 expression in human hepatocellular carcinoma. BMC Gastroenterol. 2015;15:176. | ||
Zhu QL, Zhou Y, Guan M, et al. Low-density lipoprotein-coupled N-succinyl chitosan nanoparticles co-delivering siRNA and doxorubicin for hepatocyte-targeted therapy. Biomaterials. 2014;35(22):5965–5976. | ||
Zhang CG, Zhu QL, Zhou Y, et al. N-Succinyl-chitosan nanoparticles coupled with low-density lipoprotein for targeted osthole-loaded delivery to low-density lipoprotein receptor-rich tumors. Int J Nanomedicine. 2014;9:2919–2932. | ||
Wen X, Reynolds L, Mulik RS, et al. Hepatic arterial infusion of low-density lipoprotein docosahexaenoic acid nanoparticles selectively disrupts redox balance in hepatoma cells and reduces growth of orthotopic liver tumors in rats. Gastroenterology. 2016;150(2):488–498. | ||
Berg K, Selbo PK, Weyergang A, et al. Porphyrin-related photosensitizers for cancer imaging and therapeutic applications. J Microsc. 2005;218(Pt 2):133–147. | ||
Chang JE, Shim WS, Yang SG, et al. Liver cancer targeting of doxorubicin with reduced distribution to the heart using hematoporphyrin-modified albumin nanoparticles in rats. Pharm Res. 2012;29(3):795–805. | ||
Chang JE, Yoon IS, Sun PL, Yi E, Jheon S, Shim CK. Anticancer efficacy of photodynamic therapy with hematoporphyrin-modified, doxorubicin-loaded nanoparticles in liver cancer. J Photochem Photobiol B. 2014;140:49–56. | ||
Lacko AG, Nair M, Paranjape S, Johnso S, McConathy WJ. High density lipoprotein complexes as delivery vehicles for anticancer drugs. Anticancer Res. 2002;22(4):2045–2049. | ||
Damiano MG, Mutharasan RK, Tripathy S, McMahon KM, Thaxton CS. Templated high density lipoprotein nanoparticles as potential therapies and for molecular delivery. Adv Drug Deliv Rev. 2013;65(5):649–662. | ||
Ding Y, Wang W, Feng M, et al. A biomimetic nanovector-mediated targeted cholesterol-conjugated siRNA delivery for tumor gene therapy. Biomaterials. 2012;33(34):8893–8905. | ||
Mudrak B, Kuehn MJ. Heat-labile enterotoxin: beyond G(m1) binding. Toxins. 2010;2(6):1445–1470. | ||
Hardy SJ, Holmgren J, Johansson S, Sanchez J, Hirst TR. Coordinated assembly of multisubunit proteins: oligomerization of bacterial enterotoxins in vivo and in vitro. Proc Natl Acad Sci U S A. 1988;85(19):7109–7113. | ||
Zhao L, Su R, Cui W, Shi Y, Liu L, Su C. Preparation of biocompatible heat-labile enterotoxin subunit B-bovine serum albumin nanoparticles for improving tumor-targeted drug delivery via heat-labile enterotoxin subunit B mediation. Int J Nanomedicine. 2014;9:2149–2156. | ||
Del Rio D, Stewart AJ, Mullen W, et al. HPLC-MSn analysis of phenolic compounds and purine alkaloids in green and black tea. J Agric Food Chem. 2004;52(10):2807–2815. | ||
Legeay S, Rodier M, Fillon L, Faure S, Clere N. Epigallocatechin gallate: a review of its beneficial properties to prevent metabolic syndrome. Nutrients. 2015;7(7):5443–5468. | ||
Ozaki I, Yamamoto K, Mizuta T, et al. Differential expression of laminin receptors in human hepatocellular carcinoma. Gut. 1998;43(6):837–842. | ||
Zhou Y, Yu Q, Qin X, et al. Improving the anticancer efficacy of laminin receptor-specific therapeutic ruthenium nanoparticles (RuBB-Loaded EGCG-RuNPs) via ROS-dependent apoptosis in SMMC-7721 cells. ACS Appl Mater Interfaces. 2015;8(24):15000–15012. | ||
Lesurtel M, Soll C, Graf R, Clavien PA. Role of serotonin in the hepato-gastroIntestinal tract: an old molecule for new perspectives. Cell Mol Life Sci. 2008;65(6):940–952. | ||
Soll C, Jang JH, Riener MO, et al. Serotonin promotes tumor growth in human hepatocellular cancer. Hepatology. 2010;51(4):1244–1254. | ||
Soll C, Riener MO, Oberkofler CE, et al. Expression of serotonin receptors in human hepatocellular cancer. Clin Cancer Res. 2012;18(21):5902–5910. | ||
Gopal V, Xavier J, Dar GH, Jafurulla M, Chattopadhyay A, Rao NM. Targeted liposomes to deliver DNA to cells expressing 5-HT receptors. Int J Pharm. 2011;419(1–2):347–354. | ||
Patel YC. Somatostatin and its receptor family. Front Neuroendocrinol. 1999;20(3):157–198. | ||
Reynaert H, Rombouts K, Vandermonde A, et al. Expression of somatostatin receptors in normal and cirrhotic human liver and in hepatocellular carcinoma. Gut. 2004;53(8):1180–1189. | ||
Lamberts SW, de Herder WW, Hofland LJ. Somatostatin analogs in the diagnosis and treatment of cancer. Trends Endocrinol Metab. 2002;13(10):451–457. | ||
Su Z, Niu J, Xiao Y, et al. Effect of octreotide-polyethylene glycol(100) monostearate modification on the pharmacokinetics and cellular uptake of nanostructured lipid carrier loaded with hydroxycamptothecine. Mol Pharm. 2011;8(5):1641–1651. | ||
Abdellatif AA, Zayed G, El-Bakry A, Zaky A, Saleem IY, Tawfeek HM. Novel gold nanoparticles coated with somatostatin as a potential delivery system for targeting somatostatin receptors. Drug Dev Ind Pharm. 2016;42(11):1782–1791. | ||
Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. | ||
van der Meel R, Vehmeijer LJ, Kok RJ, Storm G, van Gaal EV. Ligand-targeted particulate nanomedicines undergoing clinical evaluation: current status. Adv Drug Deliv Rev. 2013;65(10):1284–1298. | ||
Ciechanover A, Schwartz AL, Lodish HF. Sorting and recycling of cell surface receptors and endocytosed ligands: the asialoglycoprotein and transferrin receptors. J Cell Biochem. 1983;23(1–4):107–130. | ||
Kibria G, Hatakeyama H, Ohga N, Hida K, Harashima H. Dual-ligand modification of PEGylated liposomes shows better cell selectivity and efficient gene delivery. J Control Release. 2011;153(2):141–148. | ||
Takara K, Hatakeyama H, Kibria G, Ohga N, Hida K, Harashima H. Size-controlled, dual-ligand modified liposomes that target the tumor vasculature show promise for use in drug-resistant cancer therapy. J Control Release. 2012;162(1):225–232. | ||
Mei L, Fu L, Shi K, et al. Increased tumor targeted delivery using a multistage liposome system functionalized with RGD, TAT and cleavable PEG. Int J Pharm. 2014;468(1–2):26–38. | ||
Li X, Zhou H, Yang L, et al. Enhancement of cell recognition in vitro by dual-ligand cancer targeting gold nanoparticles. Biomaterials. 2011;32(10):2540–2545. | ||
Jiang T, Zhang Z, Zhang Y, et al. Dual-functional liposomes based on pH-responsive cell-penetrating peptide and hyaluronic acid for tumor-targeted anticancer drug delivery. Biomaterials. 2012;33(36):9246–9258. | ||
Xiao B, Wang X, Qiu Z, et al. A dual-functionally modified chitosan derivative for efficient liver-targeted gene delivery. J Biomed Mater Res A. 2013;101A(7):1888–1897. | ||
Chen H, Li M, Wan T, et al. Design and synthesis of dual-ligand modified chitosan as a liver targeting vector. J Mater Sci. 2012;23(2):431–441. | ||
Zhang L, Yao J, Zhou J, Wang T, Zhang Q. Glycyrrhetinic acid-graft-hyaluronic acid conjugate as a carrier for synergistic targeted delivery of antitumor drugs. Int J Pharm. 2013;441(1–2):654–664. | ||
Mezghrani O, Tang Y, Ke X, et al. Hepatocellular carcinoma dually-targeted nanoparticles for reduction triggered intracellular delivery of doxorubicin. Int J Pharm. 2015;478(2):553–568. | ||
Xiang HJ, Liu ZC, Wang DS, Chen Y, Yang YL, Dou KF. Adenosine A(2b) receptor is highly expressed in human hepatocellular carcinoma. Hepatol Res. 2006;36(1):56–60. | ||
Li YH, Liu Y, Li YD, et al. GABA stimulates human hepatocellular carcinoma growth through overexpressed GABAA receptor theta subunit. World J Gastroenterol. 2012;18(21):2704–2711. | ||
Ma WL, Lai HC, Yeh S, Cai X, Chang C. Androgen receptor roles in hepatocellular carcinoma, fatty liver, cirrhosis and hepatitis. Endocr Relat Cancer. 2014;21(3):R165–R182. | ||
Bai X, Wang J, Guo Y, et al. Prostaglandin E2 stimulates beta1-integrin expression in hepatocellular carcinoma through the EP1 receptor/PKC/NF-kappaB pathway. Sci Rep. 2014;4:6538. | ||
Chettouh H, Fartoux L, Aoudjehane L, et al. Mitogenic insulin receptor-A is overexpressed in human hepatocellular carcinoma due to EGFR-mediated dysregulation of RNA splicing factors. Cancer Res. 2013;73(13):3974–3986. | ||
Chun YS, Huang M, Rink L, Von Mehren M. Expression levels of insulin-like growth factors and receptors in hepatocellular carcinoma: a retrospective study. World J Surg Oncol. 2014;12:231. | ||
Takayama T, Sekine T, Makuuchi M, et al. Adoptive immunotherapy to lower postsurgical recurrence rates of hepatocellular carcinoma: a randomised trial. Lancet. 2000;356(9232):802–807. | ||
Greten TF, Manns MP, Korangy F. Immunotherapy of HCC. Rev Recent Clin Trials. 2008;3(1):31–39. | ||
Shi L, Lin H, Li G, et al. Cisplatin enhances NK cells immunotherapy efficacy to suppress HCC progression via altering the androgen receptor (AR)-ULBP2 signals. Cancer Lett. 2016;373(1):45–56. | ||
Chenivesse X, Franco D, Bréchot C. MDR1 (multidrug resistance) gene expression in human primary liver cancer and cirrhosis. J Hepatol. 1993;18(2):168–172. | ||
Jin G, Feng G, Qin W, Tang BZ, Liu B, Li K. Multifunctional organic nanoparticles with aggregation-induced emission (AIE) characteristics for targeted photodynamic therapy and RNA interference therapy. Chem Commun. 2016;52(13):2752–2755. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.