Back to Journals » Clinical Ophthalmology » Volume 11
Lifitegrast ophthalmic solution in the treatment of signs and symptoms of dry eye disease: design, development, and place in therapy
Received 6 February 2017
Accepted for publication 20 April 2017
Published 22 May 2017 Volume 2017:11 Pages 951—957
DOI https://doi.org/10.2147/OPTH.S117188
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Morgan R Godin, Preeya K Gupta
Department of Ophthalmology, Division of Cornea and Refractive Surgery, Duke University Eye Center, Durham, NC, USA
Abstract: A new topical ophthalmic medication, lifitegrast 5%, was recently approved by the US Food and Drug Administration for use in dry eye patients. Lifitegrast is an integrin antagonist that decreases inflammation on the ocular surface, thereby improving dry eye disease (DED). Through a series of prospective, multicenter, randomized, masked, placebo-controlled studies in >2,000 patients total, lifitegrast was shown to be effective for improvement in both the signs and symptoms of DED. A subsequent study focused on the safety profile of lifitegrast and demonstrated that the majority of adverse events were mild and resolved over time. Lifitegrast is now available for use in clinical practice.
Keywords: OPUS-1, OPUS-2, OPUS-3, SONATA, LFA-1, ICAM-1, OSDI, ocular surface
Introduction
Dry eye is defined by the International Dry Eye Workshop as “a multifactorial disease of the tears and ocular surface that results in symptoms of discomfort, visual disturbance, and tear instability with potential damage to the ocular surface. It is accompanied by increased osmolarity of the tear film and inflammation of the ocular surface.”1 Stress of the ocular surface can lead to activation of the mitogen activated protein kinase and nuclear factor-kappa B signaling pathways.2 Chronic inflammation is a key component in the pathogenesis of dry eye as increased levels of cytokines expressed by T cells have been found in tears of patients with both evaporative and aqueous-deficient types of dry eye disease (DED). These inflammatory cascades can then lead to apoptosis of ocular surface cells.3 The treatment of DED with topical anti-inflammatory pharmaceutical medications has been limited to topical cyclosporine 0.05% for the past few years, until the recent Food and Drug Administration (FDA) approval of lifitegrast 5% in July 2016. Lifitegrast is approved for the treatment of both the signs and symptoms of DED. It has been studied in >2,000 dry eye patients, ranging in severity from mild to severe, and appears to have a rapid onset of action. The purpose of this review is to discuss the design and clinical data supporting lifitegrast 5% ophthalmic solution.
Lifitegrast: mechanism of action
The etiology of dry eye is complex. However, chronic inflammation is at the root of the problem with CD4 T-helper cells intimately involved. Therefore, therapies targeting pro-inflammatory T cells are key in the treatment of DED.4 Intercellular adhesion molecule-1 (ICAM-1) has been previously shown to be upregulated in the conjunctiva of dry eye patients.5 Lymphocyte function-associated antigen-1 (LFA-1) is an integrin protein on CD4 lymphocytes. ICAM-1 is the ligand to LFA-1 and is found on the surface of inflamed endothelial and epithelial cells as well as immune cells. When LFA-1 binds to ICAM-1, T cells are recruited to the inflamed tissues; specifically, T cells bind to the vascular endothelium and then this interaction further modulates T-cell migration out of the vasculature into the ocular surface tissues. Cytokine release stimulates more lymphocytes to the site of inflammation, thereby propagating a chronic inflammatory process.4,6–8
Lifitegrast is a T-cell inhibitor, previously referred to as SAR 1118. Specifically, lifitegrast is an LFA-1 direct competitive antagonist that mimics the binding epitope of ICAM-1.4,9 As a result, inflammation is decreased by preventing binding of LFA-1 to ICAM-1, which in turn reduces the pathogenicity of dry eye.6,7 Key mechanisms of lifitegrast are inhibition of adhesion, migration, activation, and recruitment of T cells.7 Lifitegrast has high aqueous solubility and demonstrates rapid absorption into the conjunctiva and cornea in animal models.10 It is also rapidly cleared out of the systemic circulation, giving it an overall favorable pharmacokinetic profile.4
Clinical trial data
For approval, the FDA requires two adequate and well-controlled trials that demonstrate statistically significant improvement in both a sign and a symptom of DED. Discussed in more detail below, the Phase II trial on lifitegrast achieved statistical significance for corneal staining (sign), and the OPUS-1 trial replicated this result. However, efficacy with regard to symptom(s) was not demonstrated in these trials. Subsequently, OPUS-2 showed statistical significance for improvement in eye dryness (symptom), which was confirmed by OPUS-3. At the completion of OPUS-3, both FDA criteria for drug approval had been met (Figure 1).6
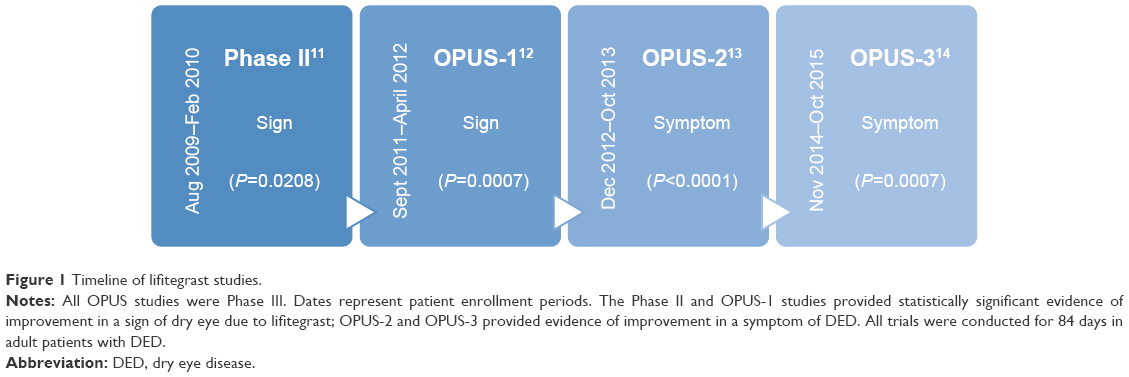 | Figure 1 Timeline of lifitegrast studies. |
The Phase II lifitegrast study was a prospective, multicenter, randomized, double-masked, placebo-controlled trial of lifitegrast 0.1%, 1%, and 5% versus vehicle twice daily for 84 days with controlled adverse environment (CAE) used for patient selection.11 The CAE worsens dry eye signs and symptoms due to creation of a negative environment for dry eye with respect to humidity, temperature, airflow, and lighting conditions over a period of 90 minutes. Key eligibility criteria included being an adult dry eye patient with unanesthetized Schirmer tear test result >1 and <10 mm/5 min, no active lid margin disease, corneal staining score ≥2, and CAE-induced increase in inferior corneal staining score (ICSS) as well as increase in ocular discomfort score (ODS) after exposure. Furthermore, patients should not have used topical cyclosporine within 6 weeks of visit 1, should not have had ocular surgery in the preceding 12 months, and were not allowed to wear contact lenses during the study. Subjects using artificial tears had to discontinue 72 hours prior to the first visit. Enrollment occurred between August 2009 and February 2010 at 5 sites in the United States. The study consisted of 3 periods: day −14 to 0 (screening), day 0–84 (treatment), and day 85–86 (follow-up phone interview). There were a total of 5 study visits: 2 during screening, and 3 during treatment at days 14, 42, and 84. The ocular surface disease index (OSDI) was completed at each of these visits. No supplemental artificial tears were allowed. The study eye had to have a Schirmer tear test result >1 and <10 mm and >1 point increase in ICSS and ODS from baseline at visit 1 post-CAE. Of note, patients with pre-CAE ICSS and/or ODS of 4 (maximum score) were excluded. All patients meeting criteria after visit 1 were started on a 2-week vehicle run-in period until visit 2 (day 0). Vehicle was composed of inactive ingredients which included sodium chloride, sodium phosphate dibasic anhydrous, sodium thiosulfate pentahydrate, sodium hydroxide and/or hydrochloric acid (for pH adjustment), and water. At visit 2, assessments including CAE were repeated; all patients who still met the criteria were randomized to either lifitegrast 0.1%, 1%, 5%, or vehicle twice daily for 84 days. The primary endpoint of the Phase II study was ICSS. Secondary objective endpoints included Schirmer tear test, conjunctival staining score, tear film break up time, and blink rate. Secondary subjective endpoints included OSDI, ODS, and visual analog scale (VAS, 7 item symptom inventory with each on a 0–100 scale). ICSS was analyzed across all time points by analysis of covariance. Primary and secondary analyses were intent to treat. In all, 546 patients were screened to achieve 230 enrolled patients (51 men, 179 women).
The average age was 62.3 years (range 26–91 years), 78% were females, and 93% were white. Two hundred and one patients completed the post-study survey, and a total of 29 discontinued. Baseline ICSS ranged from 1.65 to 1.78 with no differences between groups. A dose response was observed for ICSS at day 84 favoring lifitegrast 5.0% (P=0.0566). Confirmatory analyses showed dose response for lifitegrast 1.0% (P=0.0433) and 5.0% (P=0.0208). An increase in tear production was seen at day 14 for lifitegrast 5.0% (P=0.0392). No statistical trends were seen for conjunctival lissamine green staining. Baseline OSDI scores ranged from 28.84 to 32.76 with no differences between groups. Improvements in OSDI scores from baseline to day 14 were observed in all lifitegrast groups (0.1%, P=0.0390; 1%, P=0.0137; 5%, P=0.0341). The visual-related (VR) component of the OSDI (OSDI-VR) showed improvements from baseline to day 14 and to day 84 for the lifitegrast 1% (P=0.0231 and 0.0342, respectively) and 5% groups (P=0.0465 and 0.0394, respectively). Baseline ODS showed trends toward improvement for all groups including vehicle.
Adverse events were mild and transient, and no severe ocular adverse events occurred. The most commonly reported event was irritation after instillation of the eye drop; 93% of these events were reported on day 0. Previous studies had allowed unlimited artificial tear use, which may affect corneal staining results; thus, tear use was not permitted in this study. Also noted is the importance of the run-in period to eliminate patients with a robust placebo effect. In summary, this Phase II study demonstrated a dose-dependent reduction in corneal staining for lifitegrast compared to vehicle. This finding provided statistically significant evidence that lifitegrast effectively improves inferior corneal staining, a sign of DED, but investigators needed to demonstrate replication of this result.
The OPUS-1 study was a multicenter, randomized, double-masked, placebo-controlled trial of lifitegrast 5.0% versus vehicle twice daily for 84 days.12 The purpose was to confirm clinically meaningful responses from lifitegrast in a larger dry eye population. The study methods were very similar to the previously described Phase II study with several minor changes. First, the CAE was used for patient selection and conducted at visits 1 and 2 but not at visits 3–5 as in the prior study. Furthermore, participants with fixed fluorescein staining or ocular discomfort symptoms despite CAE were excluded. Lastly, stratification was used to provide balance in the study arms based on prior recent artificial tear use and severity of ICSS at baseline. In total, 1,016 patients were screened between September 2011 and April 2012 to obtain 588 participants at 13 sites in the United States. Mean participant age was 60.6 years (range 20–91 years), 76% of patients were females, and 93% were white. The primary objective outcome measure was ICSS change (sign) from baseline to day 84. Based on the results of the Phase II study, the co-primary subjective outcome measure was chosen to be VR-OSDI (vision-related subscale score of OSDI) change from baseline to day 84 (symptom). However, complete OSDI, VAS, and ODS inventories were obtained at every visit.
The OPUS-1 study met the primary objective endpoint, demonstrating that lifitegrast is superior to vehicle at day 84 (P=0.0007). Specifically, 22.2% of lifitegrast patients demonstrated ≥1.0 point reduction in ICSS versus 13.9% for vehicle. This was a replication and therefore confirmation that lifitegrast improves a sign of DED. Unfortunately, OPUS-1 did not meet the primary subjective efficacy endpoint of improvement in VR-OSDI for patients using lifitegrast compared to vehicle (P=0.7894). In fact, no significant differences in any OSDI parameter were seen at any follow-up visit. As for VAS scores, lifitegrast did significantly reduce patients’ eye dryness score (EDS) at day 42 (P=0.0441) and 84 (P=0.0291) compared to vehicle. On the ODS, lifitegrast significantly reduced mean ODS at day 84 (P=0.0273) but not at day 14 or 42. Therefore, symptom findings from the Phase II study were not replicated in this Phase III study.
There is no validated subjective outcome measure of dry eye symptoms, likely due to low correlation of dry eye signs and symptoms, placebo responses, transient nature of the disease, and issues with patient recall bias. However, based on the findings from OPUS-1, prescreening of patients using the VAS and only including patients with a threshold VAS score (at least 40) might help bring out symptomatic improvements due to lifitegrast. The above two controlled trials demonstrated a statistically significant improvement in a sign of dry eye, but investigators next set out to design a trial to demonstrate improvement in symptoms of dry eye in order to meet FDA requirements.
OPUS-2 was a Phase III multicenter, randomized, double-masked, placebo-controlled trial of lifitegrast 5.0% versus vehicle administered twice daily for 84 days.13 Based on a post hoc analysis of OPUS-1, the drug effect appeared to be increased for patients with recent artificial tear use and EDS ≥40. Therefore, eligibility criteria for the OPUS-2 study were: Schirmer tear test result ≥1 mm and ≤10 mm/5 min, no active lid margin disease, corneal staining score ≥2, artificial tear use within 30 days (prior to first screening), and EDS ≥40 on the VAS (scale from 0 to 100). Of note, eye dryness, and eye discomfort as a secondary outcome measure, were selected because they are the most consistently reported symptoms by dry eye patients. The primary objective endpoint was change in ICSS from baseline to day 84, and the primary subjective endpoint was change in EDS on the VAS from baseline to day 84. Totally, 1,450 patients were screened between December 2012 and October 2013 at 31 community clinics in the United States; 557 patients failed initial screening and an additional 178 patients failed post vehicle run-in screening. Therefore, 718 patients were enrolled with an average age of 58.8 years (range 19–97 years); 77% were females, 85% were white, 9% were black, 5% were Asian. Sites randomized patients using stratification by baseline ICSS (either ≤1.5 or >1.5) and EDS (either <60 or ≥60). Patients still completed a 14-day vehicle run-in and were still prohibited from using artificial tears during the study (but must have used tears in 30 days prior to the study). Of note, CAE was not used in this study. Compliance was measured by the amount of used/unused study product in collected vials with <80% of expected use indicating noncompliance and >120% of expected use indicating overuse.
In this study, lifitegrast patients had a significant improvement in EDS compared to vehicle patients (P<0.0001, 95% confidence interval 8.51–16.70). Specifically, the average decrease in EDS for lifitegrast patients was −35.30 compared to −22.75 for vehicle. In addition, post hoc analysis of OPUS-2 showed that the improvement in EDS occurred as early as the first treatment visit (visit 3, day 14). However, no significant difference between groups’ posttreatment ICSS was observed (P=0.6186). Thus, OPUS-2 was the first Phase III study to demonstrate a significant decrease in dry eye symptoms (eye dryness) due to lifitegrast but did not replicate the previous statistically significant ICSS (sign) data. As a result of the changes made to the eligibility criteria (namely, the requirement that patients must have an EDS ≥40 on the VAS and must have had recent artificial tear use), OPUS-2 likely enrolled patients with more severe dry eye symptoms. The authors hypothesize that the reason the sign was not replicated in this study may have been related to patients’ recent artificial tear use which could have made the drug effect more difficult to detect; alternatively, perhaps, more symptomatic patients have more advanced epithelial disease which makes it more difficult to demonstrate a treatment response.
Since only one of the co-primary outcome measures was statistically significant, nominal P-values had to be reported for the secondary endpoints. Change in eye discomfort score on the VAS was statistically significant with −26.46 for lifitegrast patients and −16.73 for vehicle patients (nominal P<0.0001); also, average change in ODS from baseline to day 84 was −0.91 for lifitegrast compared to −0.57 for vehicle (nominal P=0.0005). Of note, 29 patients (4.0% of the study population) had treatment-emergent adverse events (TEAEs), resulting in discontinuation from the study; 26 (3.6%) of these were in the lifitegrast group. TEAEs were further addressed in the Safety Of a 5.0% coNcentrATion of lifitegrAst ophthalmic solution (SONATA) safety study, discussed below. At this point, after completion of the Phase II as well as the Phase III OPUS-1 and OPUS-2 trials, investigators had shown replicated evidence of significant improvement in signs of dry eye due to lifitegrast (Phase II and OPUS-1) as well as one trial providing evidence of significant improvement in a symptom of dry eye (OPUS-2).
OPUS-3 was a multicenter, randomized, double-masked, placebo-controlled trial of lifitegrast 5.0% versus vehicle twice daily for 84 days.14 Patients were recruited from 41 centers in the United States between November 2014 and October 2015. In all, 1,542 participants were screened, and 711 were randomized; 637 completed the trial. Mean age was 58.7 years (range 18–93 years); 76% were females, 77% were white, 13% were black, 7% were Asian. Overall, 97.6% were compliant with placebo treatment, and 96.5% were compliant with lifitegrast treatment. To achieve totality of evidence, OPUS-3 was designed to replicate symptom data. Patients had to meet the following criteria in at least one eye: ICSS ≥0.5 and Schirmer tear test result ≥1 mm but ≤10 mm, artificial tear use within 30 days, and EDS ≥40 on VAS. The eye with the worse ICSS was designated as the study eye. Otherwise, this study was conducted identically to OPUS-2 with a 14-day run-in period followed by random assignment to treatment with lifitegrast 5.0% versus vehicle. Randomization was again stratified by baseline ICSS score (≤1.5 or >1.5) and EDS (<60 or ≥60) to provide balance. The primary endpoint of this study was only EDS from the VAS (symptom). Of note, this was the only study with a single primary endpoint design. Key secondary endpoints were changes in EDS from baseline to days 14 and 42 in order to evaluate timing of symptom improvement. The VAS was completed at all study visits.
In this study, lifitegrast participants had significantly greater improvement in EDS from baseline to day 84 compared to placebo participants (P=0.0007); results were also significant at day 14 (P<0.0001) and day 42 (P<0.0001). Improvements were also observed in symptoms of itching (nominal P=0.0318), foreign body sensation (nominal P=0.0418), and eye discomfort (nominal P=0.0048). ODS decreased in both treatment groups, but no significant differences in change from baseline were seen at day 84 (P=0.6655), day 42 (P=0.1293), or day 14 (P=0.8893). Safety assessments in OPUS-3 included conjunctival redness score, corneal and conjunctival staining scores, Schirmer tear test, best corrected visual acuity (BCVA), slit-lamp biomicroscopy, and dilated fundoscopy. Also, assessments of drop comfort score (DCS) were obtained at baseline and subsequent visits for each eye at instillation and 1, 2, and 3 minutes after instillation. If DCS was not ≤3 at 3 minutes, assessment was repeated at 5, 10, and 15 minutes and if still >3 at 15 minutes, recorded as an adverse event. More participants in the lifitegrast group (48.2%) compared to the placebo group (24.6%) had TEAEs. Also, there were more treatment-related TEAEs in the lifitegrast group (35.3% ocular, 14.6% non-ocular) than in the placebo group (13.3% ocular, 0.8% non-ocular). The most common ocular side effects were instillation site irritation and instillation site reaction, and all cases were mild to moderate in severity. The most common non-ocular side effect was dysgeusia (n=46 lifitegrast, n=1 vehicle). Twenty one (5.9%) lifitegrast patients and 9 (2.5%) placebo patients discontinued the study due to TEAEs, most commonly instillation site reaction or irritation. Eight serious TEAEs were reported but all were non-ocular and not related to the study drug. With regard to other safety parameters, conjunctival redness score, corneal and conjunctival staining scores, and Schirmer tear test, all showed improvement over time in the lifitegrast group compared to vehicle. Finally, improvements in DCS at instillation were reported over time; on days 14, 42, 84, the majority of patients reported DCS <3 at 3 minutes. For participants with DCS >3 at 3 minutes, average DCS in the lifitegrast group was similar to or better than vehicle at 5, 10, 15 minutes.
In summary, in OPUS-3 lifitegrast met the primary endpoint of change from baseline in dry eye symptoms as measured by EDS, and benefit was observed as early as 2 weeks. Therefore, statistically significant symptom data were replicated and confirmed by this study. Again, the authors noted that, based on the prior 4 studies, improvements in signs of dry eye due to lifitegrast were most notable in patients with mild to moderate DED whereas improvements in symptoms were most dramatic in patients with moderate to severe DED.
Safety data
Alongside the clinical trial data outlined above, a safety study was completed to provide further long-term evidence regarding safety of lifitegrast use. SONATA was a multicenter, randomized (2:1 lifitegrast:vehicle), double-masked, placebo-controlled trial of lifitegrast 5.0% versus vehicle twice daily for 1 year.15 Eligibility criteria included Schirmer tear test result ≥1 and ≤10 mm/5 min, no active lid margin disease, corneal staining score ≥2.0, EDS ≥40, and use and/or desire to use artificial tears in the past 6 months. Totally, 504 patients were screened at 22 sites in the United States between October 2012 and March 2014; 331 patients were randomized and followed at 7 visits over 1 year (day −7, day 0, day 14, day 90, day 180, day 270, and day 360). Of note, after day 14 (visit 3), patients were allowed to resume use of artificial tears (≤4 times daily prn), contact lenses (daily disposable only), loteprednol (for ≤4 weeks at a time), antihistamines, and mast cell stabilizers; use of these medications was tracked. DCS was evaluated in a similar fashion to the OPUS-3 study. The primary endpoint was percentage and severity of TEAEs, which were any adverse events that occurred after the first dose of lifitegrast. Secondary endpoints were corneal staining, drop comfort, BCVA, slit-lamp biomicroscopy, and intraocular pressure (IOP). This study was not powered to determine statistically significant improvement in signs/symptoms of dry eye due to lifitegrast.
A higher percentage of patients in the lifitegrast group (53.6%) had TEAEs compared to the placebo group (34.2%). These were categorized by the investigator as mild, moderate, or severe; most TEAEs were mild to moderate in severity. TEAEs included instillation site irritation (15.0% lifitegrast vs 4.5% vehicle), instillation site reaction (13.2% lifitegrast vs 1.8% vehicle), reduction in visual acuity (11.4% lifitegrast vs 6.3% vehicle), dry eye (1.8% lifitegrast vs 5.4% vehicle), and dysgeusia (16.4% lifitegrast vs 1.8% vehicle) (Table 1). Of note, the lifitegrast group had nearly twice the frequency of participants with reduced visual acuity than in the placebo group, but mean changes in BCVA from baseline to day 360 were minimal in both groups. This visual acuity reduction may have been due to transient alterations in the tear film, which contributes to the refractive properties of the eye. Also, it is hypothesized that dysgeusia was likely due to tear drainage through the lacrimal system into the oropharynx and was usually short in duration.
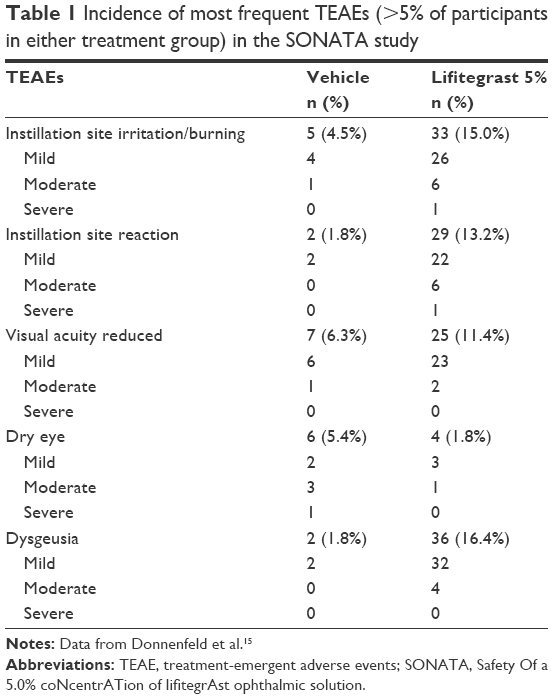 | Table 1 Incidence of most frequent TEAEs (>5% of participants in either treatment group) in the SONATA study |
A total of 24 participants (18 lifitegrast, 6 vehicle) had ≥1 ocular TEAE and 13 (9 lifitegrast, 4 vehicle) had ≥1 non-ocular TEAE that resulted in discontinuation. Discontinuation due to burning occurred in only 2 lifitegrast patients. No serious ocular adverse events occurred. Fifteen participants had serious non-ocular TEAEs (including cardiac arrhythmia resulting in death in one placebo group patient), and all non-ocular TEAEs were considered not related to the investigational drug. Overall, drop comfort improved at each visit in the lifitegrast patients. The mean IOPs of both eyes were similar between treatments groups, and no increased incidence of cataract was seen in the lifitegrast group. Interestingly, a lower percentage (32.8%) of patients in the lifitegrast group used artificial tears after day 14 compared to vehicle (43.9%), but patients in both groups who used artificial tears had higher rates of TEAEs (ocular: lifitegrast 67.2% vs 45.0%; vehicle 44.2% vs 25.5%; non-ocular: lifitegrast 60.9% vs 42.7%; vehicle 44.2% vs 32.7%). With regard to systemic safety, no immunosuppression was seen in lifitegrast patients based on serum CD3, CD4, and CD8 levels, and no opportunistic infections were reported. Average plasma lifitegrast concentration at 360 days was undetectable (<0.500 ng/mL).
Summary
Lifitegrast 5% is the first ophthalmic medication to be approved for the treatment of signs and symptoms of dry eye. Based on the Phase II and three Phase III OPUS studies discussed above, there are reproducible statistically significant data that demonstrate lifitegrast improves ICSS (sign) especially in mild to moderate dry eye patients as well as improves EDS (symptom) especially in moderate to severe dry eye patients. Furthermore, lifitegrast has a favorable safety profile, as demonstrated by the SONATA study, with the majority of adverse events being mild instillation site reaction usually early on in therapy. Lifitegrast may be used in addition to other conventional dry eye therapies and will hopefully provide clinically meaningful improvement in this disease for many patients in the future.
Disclosure
Dr Gupta is a consultant to Shire. Dr Godin does not have any conflicts of interest to report.
References
Foulks GN, Jester JV, Murube J, et al. 2007 Report of the international dry eye workshop (DEWS). Ocul Surf. 2007;5(2):65–204. | ||
Li DQ, Chen Z, Song XJ, Luo L, Pflugfelder SC. Stimulation of matrix metalloproteinases by hyperosmolarity via a JNK pathway in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2004;45:4302–4311. | ||
Yeh S, Song XJ, Farley W, Li DQ, Stern ME, Pflugfelder SC. Apoptosis of ocular surface cells in experimentally induced dry eye. Invest Ophthalmol Vis Sci. 2003;44:124–129. | ||
Perez VL, Pflugfelder SC, Zhang S, Shojaei A, Haque R. Lifitegrast, a novel integrin antagonist for treatment of dry eye disease. Ocul Surf. 2016;14(2):207–215. | ||
Gao J, Morgan G, Tieu D, et al. ICAM-1 expression predisposes ocular tissues to immune-based inflammation in dry eye patient and Sjögrens syndrome-like MRL/lpr mice. Exp Eye Res. 2004;78(4):823–835. | ||
Semba CP, Gadek TR. Development of lifitegrast: a novel T-cell inhibitor for the treatment of dry eye disease. Clin Ophthalmol. 2016;10:1083–1094. | ||
Zhong M, Gadek TR, Bui M, et al. Discovery and development of potent LFA-1/ICAM-1 antagonist SAR 1118 as ophthalmic solution for treating dry eye. ACS Med Chem Lett. 2012;3(3):203–206. | ||
Lam H, Bleiden L, de Paiva CS, Farley W, Stern ME, Pflugfelder SC. Tear cytokine profiles in dysfunctional tear syndrome. Am J Ophthalmol. 2009;147:198–205. | ||
Gadek TR, Burdick DJ, McDowell RS, et al. Generation of an LFA-1 antagonist by the transfer of the ICAM-1 immunoregulatory epitope to a small molecule. Science. 2002;295:1086–1090. | ||
Rao VR, Prescott E, Shelke NB, et al. Delivery of SAR 1118 to the retina via ophthalmic drops and its effectiveness in a rat streptozotocin (STZ) model of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2010;51:5198–5204. | ||
Semba CP, Torkildsen GL, Lonsdale JD, et al. A phase 2 randomized, double-masked, placebo-controlled study of a novel integrin antagonist (SAR 1118) for the treatment of dry eye. Am J Ophthalmol. 2012;153(6):1050–1060. | ||
Sheppard JD, Torkildsen GL, Lonsdale JD, et al. Lifitegrast ophthalmic solution 5.0% for treatment of dry eye disease: results of the OPUS-1 phase 3 study. Ophthalmology. 2014;121(2):475–483. | ||
Tauber J, Karpecki P, Latkany R, et al. Lifitegrast ophthalmic solution 5.0% versus placebo for treatment of dry eye disease: results of the randomized phase III OPUS-2 study. Ophthalmology. 2015;122(12):2423–2431. | ||
Holland EJ, Luchs J, Karpecki PM, et al. Lifitegrast for the treatment of dry eye disease: results of a phase III, randomized, double-masked, placebo-controlled trial (OPUS-3). Ophthalmology. 2017;124(1):53–59. | ||
Donnenfeld ED, Karpecki PM, Majmudar PA, et al. Safety of lifitegrast ophthalmic solution 5.0% in patients with dry eye disease: a 1-year, multicenter, randomized, placebo-controlled study. Cornea. 2016;35(6):741–748. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.