Back to Journals » Clinical Ophthalmology » Volume 9
Leber hereditary optic neuropathy: current perspectives
Authors Meyerson C, Van Stavern G, McClelland C
Received 17 March 2015
Accepted for publication 1 May 2015
Published 26 June 2015 Volume 2015:9 Pages 1165—1176
DOI https://doi.org/10.2147/OPTH.S62021
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Cherise Meyerson, Greg Van Stavern, Collin McClelland
Department of Ophthalmology and Visual Sciences, Washington University School of Medicine, St Louis, MO, USA
Abstract: Leber hereditary optic neuropathy (LHON) is one of the most common inherited optic neuropathies causing bilateral central vision loss. The disorder results from point mutations in mitochondrial DNA and subsequent mitochondrial dysfunction. The primary cell type that is lost in LHON is the retinal ganglion cell, which is highly susceptible to disrupted ATP production and oxidative stress. Inheritance of LHON follows that of mitochondrial genetics, and it has a highly variable clinical phenotype, as other genetic and environmental factors also play a role. Although LHON usually presents with isolated vision loss, some patients suffer other neurological sequelae. For ill-defined reasons, male LHON mutation carriers are more affected than females. Most LHON patients remain legally blind, but a small proportion can experience spontaneous partial recovery, often within the first year of symptom onset. Unfortunately, at this time there are no established curative interventions and treatment is largely supportive. Patients should be offered low vision services and counseled on mitigating risk factors for additional vision loss, such as smoking and consuming alcohol. Encouraging treatments currently undergoing investigation includes ubiquinone analogs, such as idebenone, as well as gene therapy and stem cells to restore ATP synthesis and provide neuroprotection to surviving retinal ganglion cells.
Keywords: Leber hereditary optic neuropathy, mitochondria, neuro-ophthalmology, mitochondrial DNA
Introduction
Mitochondrial diseases were once thought of as rare disorders, in part due to failure to recognize the diagnosis because of the wide variability among phenotypes. They are increasingly recognized as a common cause of neurologic and visual dysfunction. Ophthalmic manifestations are frequent among mitochondrial disorders and can result in retinopathy, ocular motility disorders, or optic neuropathy. It is important for ophthalmologists to remain cognizant of mitochondrial disease when a patient presents with vision loss. Among the mitochondrial diseases, Leber hereditary optic neuropathy (LHON) is often considered a prototypical disorder. It was the first mitochondrial disease to be recognized by Dr Albrecht von Graefe in 1858, but it was named after Dr Theodore Leber who described 15 patients with the disease among four families.1 LHON was the first disorder recognized to be maternally inherited and the first to be attributed to a point mutation in mitochondrial DNA (mtDNA).2,3 Vision loss from LHON results from selective degeneration of retinal ganglion cells (RGCs), which are highly sensitive to mitochondrial dysfunction and metabolic insult.4 The mechanisms involved in the pathogenesis of LHON continue to be elucidated, paving the way for research into potential therapeutic interventions.
Epidemiology
LHON is the most common optic neuropathy caused by a primary mutation in mtDNA.5–7 The minimum prevalence of vision loss due to the three most common pathogenic point mutations in LHON is one in 31,000 in the northern UK.8 Other epidemiological studies report a prevalence of one in 39,000 and one in 50,000 in the Netherlands and Finland, respectively.9,10 LHON affects predominantly males (in 80%–90% of cases).5 Symptom onset typically occurs in the second and third decades of life. LHON carriers rarely lose vision after the age of 50 years, but there have been reports of LHON onset from 2 to 87 years of age.5,7,11,12 Most LHON patients are aware of a family member with LHON-compatible vision loss, although 40% deny a known family history. Given that de novo mutations are rare, this relatively high percentage without a family history could be attributable to unrecognized mutation carriers in the family who had never lost vision or to the innate difficulty in accurately tracing family history.8,13
Clinical features of vision loss
LHON usually presents as painless, subacute, central visual loss in one eye. Weeks to months later, the second eye becomes involved, with a median delay of 6–8 weeks.7,14 Within 1 year, 97% of those affected have involvement of the second eye, such that a patient presenting with a unilateral optic neuropathy for longer than 1 year is highly unlikely to suffer from LHON-related vision loss.5,12,15 Approximately 25% have bilateral simultaneous vision loss, but in some individuals it may be that vision loss in the first eye is not noticed before the second eye becomes involved.5,12,14
The majority of individuals progress to a visual acuity of 20/200 or worse.16 Due to preferential involvement of the papillomacular bundle, the earliest visual field abnormality is a cecocentral scotoma (Figure 1), which can enlarge to become a larger central defect.16 Dyschromatopsia is common and usually parallels the degree of visual acuity loss.5,12,16 Pupillary light reflexes usually remain intact due to relative sparing of melanopsin-containing RGCs. This subset of RGCs are thought to be more resistant to metabolic insult from mitochondrial dysfunction when compared with all RGCs.4,17,18 In cases of asymmetric bilateral or monocular vision loss, however, a relative afferent pupillary defect can occur.19
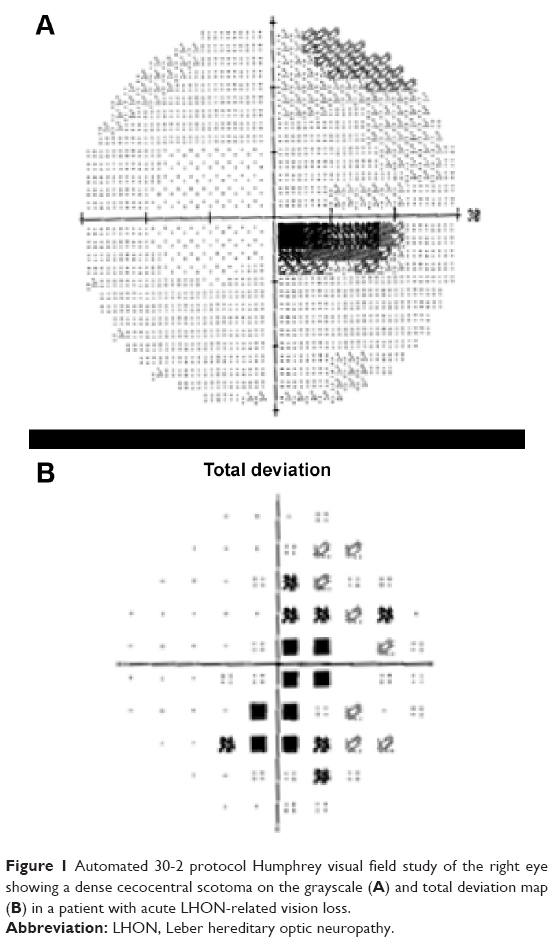 | Figure 1 Automated 30-2 protocol Humphrey visual field study of the right eye showing a dense cecocentral scotoma on the grayscale (A) and total deviation map (B) in a patient with acute LHON-related vision loss. |
On fundus examination preceding or during the acute stage of vision loss, there can be characteristic findings, including optic disc hyperemia, peripapillary telangiectatic blood vessels, vascular tortuosity, and swelling of the retinal nerve fiber layer (RNFL) around the optic disc (Figure 2A) without corresponding leakage on fluorescein angiography (sometimes termed “pseudoedema”). The fundus can look normal in 20%–40% of those in the active stage of vision loss, which can delay diagnosis.12,20,21 Eventually, as the disease progresses, disc hyperemia, peripapillary telangiectasias, and pseudoedema resolve. Approximately 6 weeks after onset of vision loss, optic disc pallor develops and can be accompanied by cupping of the optic disc, reflecting the death of RGCs that occurs in the chronic atrophic phase (Figure 2B).5,22
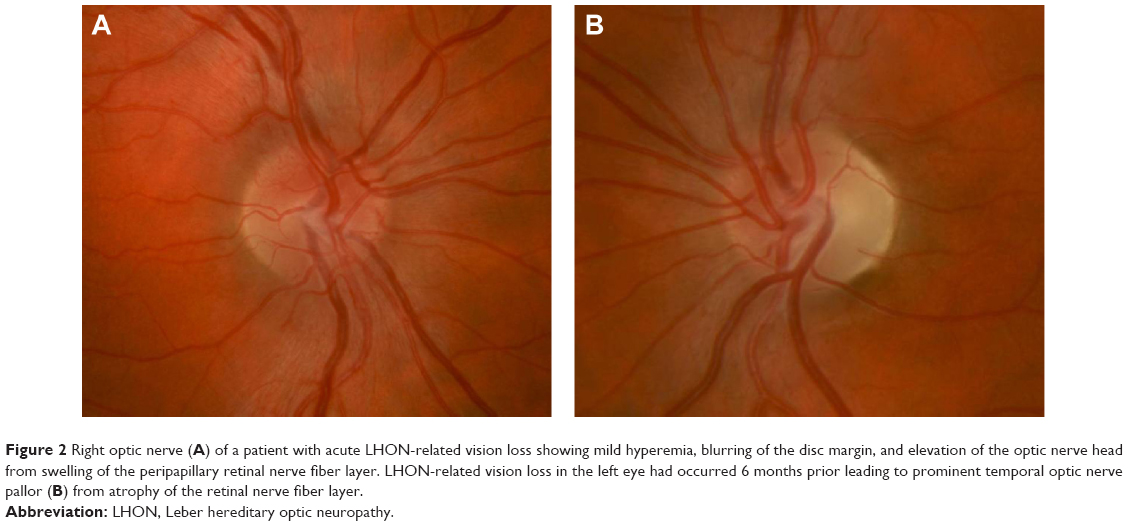 | Figure 2 Right optic nerve (A) of a patient with acute LHON-related vision loss showing mild hyperemia, blurring of the disc margin, and elevation of the optic nerve head from swelling of the peripapillary retinal nerve fiber layer. LHON-related vision loss in the left eye had occurred 6 months prior leading to prominent temporal optic nerve pallor (B) from atrophy of the retinal nerve fiber layer. |
Fundus changes seen in LHON can be further characterized and quantified through optical coherence tomography. In the acute phase, the RNFL thickens first in the temporal and inferior quadrants, then the superior and nasal quadrants.23 This is consistent with early preferential involvement of the papillomacular bundle of RGCs. RNFL thickening is related to axonal swelling from impaired mitochondrial function and axonal transport.4 The RNFL then becomes thinned during the chronic phase in the months following the onset of vision loss.24 Macular thickness may thin before the RNFL, suggesting that disease progression could be better monitored with both macular thickness and RNFL measurements by optical coherence tomography.25
Visual evoked potentials (VEPs) and electroretinograms (ERGs) are often abnormal in affected LHON patients, as they reflect optic nerve fiber degeneration and RGC loss, respectively. In a study of two brothers with LHON who had normal VEP latencies and configurations prior to the onset of symptoms, earliest abnormalities included prolonged VEP latency and abnormal VEP morphology. With symptom progression, there was a progressive prolongation of VEP latency.26 In a Brazilian family with a strong history of LHON, three affected members had increased latencies, decreased amplitudes, or absent responses for pattern-reversal VEP. Two family members also had reduced ERG cone responses.27 Another study found that affected LHON patients had increased VEP latency and decreased VEP amplitude that was associated with a decreased pattern ERG N50–N95 amplitude.28 Notably, VEPs were normal in unaffected carriers in these studies. VEPs and ERGs can therefore be useful in diagnosing and monitoring disease progression in affected patients.
Magnetic resonance images of the brain and orbits are usually normal in affected LHON patients.29 There are, however, reports of patients with non-specific increased signal in the retrobulbar optic nerve on T2-weighted fast spin echo and short time inversion recovery sequences months after vision loss.30,31 Intravenous gadolinium magnetic resonance imaging contrast leads to optic nerve enhancement in pathologic states in which there is disruption of the blood–brain barrier within the optic nerve.32 Patients with LHON have rarely demonstrated optic nerve enhancement on post-contrast images, suggesting that in some cases there may be an inflammatory component to the pathogenesis of LHON.32
Interestingly, unaffected LHON point mutation carriers can display subclinical signs of disease on fundus examination, including peripapillary microangiopathy, zones of mild disc pseudoedema, and telangiectasias.33 Some seemingly asymptomatic carriers do exhibit subclinical dyschromatopsia.34 Among asymptomatic carriers, temporal RNFL thickening on optical coherence tomography may be present, further highlighting early involvement of the papillomacular bundle in subclinical LHON.35 One study that recorded the pattern ERG in asymptomatic LHON mutation carriers showed a significant decline in N95 amplitude by ~40% from baseline over 36 months, suggesting that subclinical RGC loss is progressive in unaffected carriers and precedes the onset of visual field defects.36 Pattern ERG may therefore be a useful tool to monitor subclinical disease progression in LHON mutation carriers.
Prognosis
The visual prognosis in LHON is poor. Most individuals have permanent vision loss and are legally registered as visually impaired.37 Although vision loss is unlikely to progress after the active phase of the disease, patients report that the acute phase vision loss has a strong negative impact on quality of life.37 Spontaneous visual improvement is a well described phenomenon in LHON, usually occurring within the first year following vision loss. Recovery can also occur decades later.5 Recovery typically presents first with the appearance of small islands of vision within the central defect bilaterally.20,21 Spontaneous visual recovery is more common in patients with the 14484 mutation, with a partial recovery rate of 37%–58%, while the 11778 mutation has the lowest partial recovery rate of 4%.7,8,14,38 Patients with the 3460 mutation have an intermediate prognosis, with an approximate 20% partial recovery rate.39 Earlier age of onset (younger than 20 years), a subacute time course of vision loss, and a larger optic disc are all associated with a better visual prognosis.24,40,41
Mitochondrial function and oxidative phosphorylation
Mitochondria are double-membraned intracellular organelles present in nucleated eukaryotic cells that play a vital role in energy generation through production of ATP used for cell growth and function.42,43 The number of mitochondria present in a particular cell type depends on the energy demands of the cell. Cells in highly metabolically active tissues, including RGCs within the papillomacular bundle, the cardiac conduction system, pancreas, kidney, liver, and muscle, have high ATP requirements and high numbers of mitochondria. Therefore, mitochondrial diseases predominantly affect these tissues, as they are most susceptible to disrupted ATP production.4,44,45
ATP generation occurs via oxidative phosphorylation (OXPHOS) through the mitochondrial respiratory chain. The respiratory chain is made of five complexes (I–V) embedded in the inner membrane. Complexes I–IV oxidize NADH and FADH2 generated from glycolysis and beta-oxidation of fatty acids. Complex V, also known as ATP synthase, then pumps protons into the intermembrane space and generates an electrochemical gradient that facilitates phosphorylation of ADP to ATP. Several cofactors, including ubiquinone, play an important role in shuttling electrons between respiratory chain complexes.4,15,45–47
While ATP production is an essential task of mitochondria, the organelles also play other vital roles for the cell, including detoxification of reactive oxygen species (ROS), iron metabolism, fatty acid oxidation, amino acid biosynthesis, and regulation of cellular apoptosis.4,48 Although dysfunction of the OXPHOS pathway is one of the most important factors in the pathogenesis of mitochondrial diseases, defects of other cofactors or in the cell machinery that assembles mitochondrial components can also disrupt the ATP supply and increase production of ROS, which can have devastating effects on cells.
Mitochondrial genetics
The genetics of the mitochondria reflect the organelle’s evolutionary origins; autonomous proto-mitochondrial prokaryotic organisms were thought to be phagocytosed by larger eukaryotic cells, forming an endosymbiotic relationship in which the proto-mitochondria supplied ATP and detoxified ROS for the eukaryotic cell.48,49 Similar to their proposed prokaryotic origin, mitochondria also have a genome in the form of circular double-stranded mtDNA. Importantly, mitochondria rely heavily on nuclear DNA to encode proteins required for mitochondrial function. More than 80 subunits required for the OXPHOS system are encoded in the nuclear genome and are imported into mitochondria.4 Diseases with mutations in the nuclear genome often follow Mendelian inheritance (eg, autosomal dominant, autosomal recessive, or X-linked inheritance). In contrast, those with primary mutations in mtDNA have unique patterns of inheritance and penetrance governed by the principles of maternal inheritance, heteroplasmy, replicative segregation, and the critical threshold.50
Transmission of the mitochondrial genome occurs from a mother to her progeny. There is no paternal contribution, as the few mitochondria present in sperm are proteolytically destroyed by the zygote.51 Therefore, LHON results when a mother carrying the mutation transmits it to her children, while fathers cannot transmit the mutation to offspring.
Unlike nuclear DNA, mtDNA replicates continuously and independently of the cell cycle in both dividing and non-dividing cells. Often there are several thousands of mtDNA molecules present, and all copies are identical. This is called homoplasmy. However, a mutation can arise in one copy of mtDNA, resulting in coexistence of both mutant and wild-type mtDNA within a mitochondrion, termed heteroplasmy.45 The principle of heteroplasmy is critical when host cells divide and the mitochondria are separated between daughter cells. This process is termed replicative segregation and occurs stochastically. The proportion of mutant and wild-type mtDNA between daughter cells is subject to random segregation and therefore unequal. Heteroplasmy and replicative segregation contribute to the heterogeneity of mitochondrial disease phenotypes, even among related individuals.
In the case of heteroplasmy, a certain amount of wild-type mtDNA can compensate for the mutant mtDNA in a cell or tissue. If the effects of the mutant mtDNA exceed a certain threshold, however, the wild-type mtDNA can no longer compensate. This critical threshold level is often tissue-specific; metabolically active tissues, particularly the RGCs, which constitute the optic nerve and the cells of the retinal pigment epithelium, have a lower threshold than less metabolically active tissues.52 In addition, post-mitotic cells like neurons accumulate more mutant mtDNA copies over time, which eventually exceed the critical threshold and account for the age-dependent expression of many mitochondrial diseases.44
Over 200 point mutations in mtDNA account for mitochondrial diseases. Most point mutations that cause disease are heteroplasmic. However, there are several exceptions, including the common mutations in LHON, which are usually homoplasmic. Deletions and duplications in mtDNA can also lead to mitochondrial diseases including mitochondrial myopathies, which are often sporadic.15,45,53
Genetics and incomplete penetrance of LHON
Ninety percent of all cases of LHON are due to one of three point mutations in mtDNA, which are located at nucleotide positions 3460, 11778, and 14484.5 The most common point mutation is 11778, which accounts for 70% of all cases.5 The 14484 and 3460 mutations account for ~14% and ~13%, respectively.20,50 An exception is among those of French Canadian descent, in which the 14484 mutation accounts for about 90% of cases within that population due to a founder effect.54 Other relatively rare mutations have also been discovered but occur in single families.20,21 All of the mutations occur in genes encoding subunits for complex I in the respiratory chain, particularly in those encoding the ND1 and ND6 subunits.55,56
LHON is notable for incomplete penetrance, meaning that not all mutation carriers will develop vision loss. While the mechanisms involved have not been fully elucidated, it appears that complex genetic and environmental factors play a role.
Like several other mitochondrial disorders, heteroplasmy is thought to influence the penetrance of LHON. For instance, it has been observed that offspring born to mothers with less than 80% mutated mtDNA present in their blood are less likely to be symptomatic than offspring born to homoplasmic mothers.57 Even more variability is present because tissue-specific segregation of mutant mtDNA is stochastic during embryogenesis. Considering that 80%–90% of all LHON individuals carry homoplasmic mutations, heteroplasmy alone does not explain the incomplete penetrance of LHON.8,14
Another risk factor for vision loss is the set of inherited genes that commonly segregate with the pathogenic mtDNA mutation. Over the course of evolution, mtDNA polymorphisms have clustered together in groups called haplogroups, which tend to be inherited together. The other polymorphisms present within a haplogroup can have deleterious effects on the OXPHOS pathway, including decreased levels of protein synthesis and ATP production, which could then synergize with the deleterious effect of the primary LHON mutation.58,59 For instance, among Caucasians, haplogroup J is associated with increased penetrance for the 11778 and 14484 mutation carriers, while haplogroup K is associated with increased penetrance for the 3460 mutation carriers.60 Chinese carriers of the 11778 mutation had an increased risk of vision loss when inherited with haplogroup M7b1′2 and a decreased risk with haplogroup M8a.61
Differences in mitochondrial mass have also been postulated to play a role in the incomplete penetrance of LHON. Unaffected LHON mutation carriers have significantly higher mtDNA copy numbers in leukocytes than affected mutation carriers. Comparison of fibroblasts from unaffected, affected, and control patients has shown that cells from unaffected carriers have increases in mitochondrial transcripts, respiratory chain proteins, enzyme activity, and mitochondrial biogenesis factors relative to controls and affected carriers. Higher levels of mtDNA in leukocytes in unaffected carriers were also associated with milder signs of ocular pathology. Thus, mitochondrial mass increases in unaffected carriers may be protective, whereby the increased amount of mitochondria can compensate for complex I dysfunction. Although the specific factors that activate mitochondrial biogenesis remains to be discovered, these observations could have implications for the treatment of LHON.62
LHON is maternally inherited. Men cannot transmit the mutation. Both male and female offspring can, however, inherit the mutation, yet 50% of males and only 10% of females experience vision loss; this sex predilection cannot be explained solely by the principles of mitochondrial inheritance.7 It has been hypothesized that there is a recessive X-linked susceptibility gene that works in concert with the mitochondrial mutation, which could explain the male predominance among carriers who lose vision. Those female carriers who do lose vision may either have homozygosity at the X-linked locus or may have experienced an unfortunate X chromosome inactivation.63 Recent linkage analyses have discovered a region on the long arm of the X chromosome that may contain the susceptibility gene, but the actual gene responsible has not yet been identified.64–66
Hormonal differences between males and females have also been hypothesized to influence the male predominance in LHON. In females, estrogens are thought to modify the severity of mitochondrial dysfunction, including defective ATP synthesis, oxidative stress, and apoptosis. In cybrid cell lines containing homoplasmic LHON mutations, addition of 17β-estradiol led to activation of mitochondrial biogenesis, increased superoxide dismutase 2 (SOD2) activity, decreased production of ROS, and reduction of apoptosis. Estrogen receptor β was also shown to be particularly abundant in RGCs and their axons within the RNFL.67 The data obtained from the cybrid cell model may approximate the effects of estrogen on the in vivo target tissue affected in LHON.
Environmental factors have been implicated as contributing to vision loss in LHON mutation carriers. Alcohol consumption and tobacco smoking have been associated with the onset of vision loss in those genetically predisposed to LHON. A retrospective study of 125 European LHON pedigrees showed an association between smoking and vision loss, in which heavy smokers were more likely to experience vision loss than light smokers.68 The study showed a 93% penetrance of vision loss in male smokers versus a 66% penetrance in male non-smokers. Cigarette smoke can compromise complex I activity, reduce cytochrome c oxidase activity, and increase production of ROS. Heavy alcohol intake was also associated with a greater likelihood of vision loss, but this relationship was less significant than that of smoking. Other associated environmental triggers include head trauma, industrial toxins, and drugs that have mitochondrial toxicity, such as antiretrovirals and ethambutol.20 Nutritional deficiencies have also been implicated as a trigger for vision loss in LHON carriers, including low vitamin B12 levels.69
Optic nerve vulnerability
In LHON, RGCs appear to be selectively vulnerable to mitochondrial dysfunction. The papillomacular bundle of RGCs responsible for central vision is affected first and more severely because of its small-diameter fibers and heavy dependence on mitochondria.70 Studies of RGC axons show mitochondrial clustering in the prelaminar nerve fiber layer, and mitochondria significantly decrease in number posterior to the lamina cribrosa where the axons are myelinated. Such an uneven distribution of mitochondria partly explains why the unmyelinated portion of RGC axons is particularly affected.71–73 It is unclear whether the precipitation of RGC loss is due to low ATP production, increased production of ROS, or both, and there are data that support both mechanisms.74 A mouse mtDNA mutant model was developed that expressed key features of LHON, including degeneration of the papillomacular bundle, axonal swelling, and dysmorphic mitochondria in unmyelinated segments. There was no reduction in ATP synthesis, but there was increased ROS production; this research suggests that oxidative stress plays a larger role in the pathogenesis of LHON than defective ATP production.75
Of note, optic neuropathy can occur in other mitochondrial disorders that have prominent systemic manifestations, such as myoclonic epilepsy with ragged red fibers, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), Leigh syndrome, Friedreich ataxia, Mohr–Tranebjerg syndrome, and complicated hereditary spastic paraplegia. It remains unclear why mitochondrial mutations in other disorders lead to a phenotype with systemic disease. With time, it has become more apparent that LHON can be associated with other non-visual neurologic sequelae.76
LHON plus
While the typical manifestation of LHON is vision loss, there have been other manifestations reported within select pedigrees. Cardiac arrhythmias, peripheral neuropathies, dystonia, and myoclonus can occur in LHON carriers.20 Some families from Holland, Australia, and North America have particularly severe manifestations, including ataxia, juvenile onset encephalopathy, spastic dystonia, and psychiatric disturbances. These phenotypes have been called “LHON plus syndromes” and have been linked to other mtDNA point mutations that affect OXPHOS complex I activity but differ from the three commonly seen in LHON.20,77,78
An association between the 11778 mutation in LHON and a demyelinating syndrome that is radiologically and clinically identical to multiple sclerosis was described in 1992 and named Harding syndrome after the reporting author.79 This posed an interesting hypothesis that vision loss in LHON may be caused by an abnormal immune response against the optic nerve because of mtDNA mutations. In females with the 11778 mutation, this immune response may involve other myelinated axons and result in a disorder indistinguishable from multiple sclerosis. It also poses the question of whether mitochondrial genes contribute to susceptibility to multiple sclerosis. Some report that the co-occurrence of LHON mutations and multiple sclerosis is likely due to chance and note that those patients present with atypical symptoms of LHON, in which there is a higher female predominance and persisting unilateral vision loss.80 Others report a risk 50 times greater than expected for developing both multiple sclerosis and LHON, but given the rarity of the syndrome, much more remains to be elucidated.81
Treatment
Symptomatic and supportive treatments
The treatment of mitochondrial disorders is still in its infancy. Even though there is promising research underway, the mainstay of clinical treatment for LHON remains supportive rather than curative. Low vision aids can be of substantial benefit for those with central vision loss from LHON.82 In addition, low vision rehabilitation is a viable option because most LHON patients are young adults with intact peripheral vision. Patients should also be counselled to optimize environmental risk factors for vision loss by avoiding tobacco use, heavy alcohol consumption, medications with mitochondrial toxicity, and exposure to environmental toxins.20,68 Other supportive measures, particularly for the LHON plus syndromes, includes screening for extraocular manifestations of LHON, such as a screening electrocardiogram and neurological surveillance.83
Nutritional supplements
Several combinations of vitamins (eg, B2, B3, B12, C, E, and folic acid) and other supplements such as alpha-lipoic acid, carnitine, creatine, L-arginine, and dichloroacetate have been tried to treat mitochondrial disorders, including LHON. However, analyses have shown little proof of efficacy with these interventions.83,84 The rarity of mitochondrial disorders makes it particularly challenging to determine efficacy. One review noted that only 35 of 1,039 publications had trials with more than five patients.85
Brimonidine
Brimonidine is a topical α2-agonist that is used to manage patients with glaucoma. Brimonidine has been shown to be antiapoptotic, and it can protect RGCs from oxidative damage in animal models.86 Nine patients with monocular vision loss were treated with topical brimonidine in an open-label trial to prevent vision loss in the other eye. However, the study was stopped prematurely because all of the patients experienced vision loss in their second eye, and brimonidine did not appear to mitigate the amount of vision loss.87 Nonetheless, there are reports of patients who had both glaucoma and LHON in which vision loss was accelerated or worse than expected with either disorder alone. It has been proposed that increased intraocular pressure may further exacerbate axoplasmic stasis in RGCs already under high metabolic stress.88 Brimonidine could therefore be potentially useful in asymptomatic LHON carriers with glaucoma due to its pressure-lowering effects and putative neuroprotective effects.
Ubiquinone analogs
Ubiquinone is a molecule in the mitochondrial membrane that carries electrons from complexes I and II to complex III. LHON mtDNA mutations affect complex I, so the ubiquinone analog coenzyme Q10 is thought to help facilitate the flow of electrons and maximize the electrons shuttled to complex III. It has long been used in attempts to treat mitochondrial disorders, yet recent analyses have not shown significant benefit.83–85 Given that coenzyme Q10 is highly lipophilic, it is unlikely that oral administration allows for delivery to mitochondria.
Idebenone and EPI-743 are short-chain ubiquinone analogs that easily enter the brain and reach mitochondria, unlike coenzyme Q10. Idebenone has been shown to bypass complex I and maintain ATP production, and it inhibits lipid peroxidation to protect mitochondria from oxidative damage.89,90 Idebenone was initially successful in 1992 with a 10-year-old boy who had the 11778 mutation, but this is confounded by the fact that childhood-onset LHON has higher rates of spontaneous recovery.40,91 A randomized, double-blind, placebo-controlled trial called RHODOS (Rescue of Hereditary Optic Disease Outpatient Study) enrolled 85 LHON patients who had a disease duration of up to 5 years. After 24 weeks of idebenone 900 mg/day, the study did not show any benefit for the primary outcome of best recovery of visual acuity after 24 weeks. However, the data showed a positive trend for the secondary end points, including change from baseline in best visual acuity and change in visual acuity for both eyes, particularly for patients with discordant visual acuities.92 The study also found the largest treatment effect for patients with the 11778 or 3460 mutation, while those with the 14484 mutation had a high spontaneous recovery rate in the placebo group, thought to abolish any treatment effect. In the observational follow-up study (RHODOS-OFU), 59 of the original patients were followed up 30 months later, and those who were treated with idebenone still showed beneficial effects, especially those who had a short disease history.93 In a retrospective study of 103 LHON patients, 44 patients with disease onset within one year were treated with idebenone and followed for 5 years or more. Those treated with idebenone recovered vision more than the control cohort, and the improvement was more prominent with early initiation of treatment and longer duration of treatment.94 Another randomized, double-blind, placebo-controlled trial of 39 LHON patients with a disease duration of less than 5 years treated patients with idebenone 900 mg/day for 24 weeks. The treatment group showed improvement in blue-yellow color vision and less impairment of red-green color vision, and this effect was more prominent in those younger than 30 years of age and with less than 1 year of symptoms.95 These trials show promise, but more research is needed to determine the optimal idebenone dose and treatment duration, as well as whether asymptomatic carriers and those with long disease duration can be treated. While not as widely studied as idebenone, EPI-743 was used in a trial including five patients with a disease duration of 90 days, and four of the five patients showed disease arrest and reversal after treatment for a minimum of 1 year.96
Pharmacologic activators of mitochondrial biogenesis
As noted previously, increased mitochondrial biogenesis is significantly associated with unaffected LHON mutation carriers and may have a protective effect.62 Mitochondrial biogenesis is partly regulated by the transcriptional activator PGC-1α, which is controlled by peroxisome proliferator-activated receptors and AMP-activated protein kinase.97 Pharmacologic activators of these proteins include fibrates, rosiglitazone, metformin, and AICAR (5-aminoimidazole-4-carboxamide ribonucleoside).98 In a mouse model of mitochondrial myopathy treated with bezafibrate, there was increased mitochondrial biogenesis, a delay in onset of the myopathy, and a longer life span.99 However, it remains to be seen whether pharmaceutical activation of PGC-1α will be beneficial for affected LHON patients or mutation carriers.
Gene therapy
Gene therapy has shown some promise for mitochondrial diseases. For LHON in particular, it is an even more appealing treatment option because the RGC layer in the retina can be easily accessed. However, the ability to introduce genes directly into the mitochondrial genome has not been well developed, and a large obstacle is the double membrane of the mitochondrion. Thus, researchers have utilized allotopic expression by transfecting the desired gene into the nuclear genome instead.100–102 For this technique, the desired wild-type gene is engineered with a mitochondria-specific targeting sequence so that the wild-type protein product is imported into the mitochondria. The gene is then transfected into the nucleus with an adeno-associated virus (AAV) vector. One of the first successful attempts in vitro was on a cybrid cell line that was homoplasmic for the 11778 mutation, in which researchers used allotopic expression to complement the mutated ND4 subunit with a wild-type one, and rescued ATP synthesis.103 Two animal models expressing the mutated ND4 subunit also showed an improvement in vision and rescue of RGCs following intravitreal injection and allotopic expression of the wild-type gene for ND4.104,105 However, limitations remain due to inefficient mitochondrial import of the hydrophobic ND4 subunit, and allotopic expression can be optimized by targeting the mRNAs to the mitochondrial surface to couple translation and translocation for a longer duration of gene expression.106 Clinical trials associated with the University of Miami, Huazhong University of Science and Technology, and Gensight Biologics are currently recruiting patients with the 11778 mutation to assess the safety of gene therapy with an AAV vector carrying the wild-type ND4 gene.107–109
Another gene that has been allotopically expressed in cells harboring the 11778 mutation is the SOD2 gene, which detoxifies free radicals in mitochondria. Even though the SOD2 gene is encoded in the nuclear genome and expressed in LHON carriers, SOD2 activity is attenuated in cells homoplasmic for an LHON mutation.110 In LHON cybrids, overexpression of SOD2 through transfection with an AAV vector led to a decreased rate of apoptosis, which highlights how antioxidant mechanisms could improve RGC survival in LHON.111
More recent approaches have shown that it is possible to introduce genes into mitochondria, although it remains technically difficult. One study isolated whole mtDNA from healthy human donors and developed a construct with recombinant mitochondrial transcription factor A protein that could gain direct entry into mitochondria. This construct was then applied to LHON cybrids that were homoplasmic for the 11778 mutation, and it stimulated mitochondrial biogenesis.112 Another study used a mitochondrial targeting sequence on the AAV vector capsid that contained the wild-type ND4 gene. The mitochondrial targeting sequence led to direct introduction of the wild-type ND4 gene into mitochondria in vitro and in a mouse model, in which there was restoration of ATP synthesis.113 The same researchers then discovered that the mitochondrial targeting sequence-guided gene construct did not recombine with the host mitochondrial genes but rather remained episomal in the mitochondria.114 This discovery is encouraging, as it appears unlikely that the gene construct recombines with endogenous genes, which could further disrupt the OXPHOS pathway. These studies show immense therapeutic potential, but research remains preliminary and further studies are required to evaluate adverse effects and determine how long the beneficial effects would last.
Stem cells
The use of stem cells to treat mitochondrial optic neuropathies is still in the preliminary stages, but two techniques are currently under investigation. One technique involves transplantation of RGCs. However, harvesting mature and fully functioning RGCs from embryonic stem cells or induced pluripotent stem cells remains challenging, and is further compounded by the difficulty in ensuring that transplanted RGCs make the correct neuronal connections when integrated into the retina.115 For now, it appears that mature RGCs derived from stem cells can be used for further in vitro research of LHON pathogenesis and potential treatments.
Also under current investigation is the use of stem cells to provide neurotrophic factors that protect RGCs.116 Because some patients with LHON experience visual recovery, they have some RGCs that survive despite mitochondrial dysfunction and oxidative stress. To protect these surviving RGCs, use of mesenchymal stem cells that secrete neurotrophic factors and anti-inflammatory cytokines appears promising. Intravitreal injection of autologous mesenchymal stem cells in a rat model with glaucoma showed a neuroprotective effect with increased RGC axon survival.117 Another study used intravenous infusions of autologous bone-marrow derived mesenchymal stem cells in ten patients with secondary progressive multiple sclerosis, who experienced an improvement in visual acuity and an increase in optic nerve area without significant adverse effects.118 Given this proof-of-concept study, the same strategy could be employed in the future to slow the loss of RGCs in LHON. Patients should be cautioned that lucrative, unregulated stem cell treatments offered at clinics not affiliated with academic research centers may make inflated promises of efficacy and could be dangerous.
Conclusion
LHON remains the prototype mitochondrial optic neuropathy and is marked by acute onset of sequential bilateral optic neuropathy due to point mutations in mtDNA. Symptom onset typically begins in the second or third decade of life as a painless and subacute monocular loss of central vision that then progresses to involve the other eye. While partial visual recovery may occur, most patients remain legally blind and require low vision services. Optic nerve degeneration results from dysfunctional OXPHOS and increased production of ROS within mitochondria, and RGCs within the papillomacular bundle serving central vision are particularly susceptible to oxidative stress. The LHON inheritance pattern follows that of mitochondrial genetics and is maternally inherited. Incomplete penetrance occurs as a result of both genetic and environmental factors, leading to wide symptom variability among mutation carriers.
Although research for LHON is ongoing and new discoveries are continuously made, many questions remain to be answered before a curative treatment becomes first-line therapy for patients with this debilitating disease. In vitro cell studies and animal models have shown promise, and now the field is progressing to early phase clinical trials. At this point, however, the efficacy, adverse effects, and duration of treatment benefit have yet to be determined.
Disclosure
The authors report no conflicts of interest in this work.
References
Leber T. Ueber hereditäre und congenital-angelegte Sehnervenleiden [About hereditary and congenital optic nerve disorders]. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1871;17:249–291. German. | ||
Wallace D. A new manifestation of Leber’s disease and a new explanation for the agency responsible for its unusual pattern of inheritance. Brain. 1970;93:121–132. | ||
Wallace D, Singh G, Lott M, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242:1427–1430. | ||
Carelli V, Ross-Cisneros FN, Sadun A. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res. 2004;23:53–89. | ||
Newman NJ. Hereditary optic neuropathies: from the mitochondria to the optic nerve. Am J Ophthalmol. 2005;140:517–523. | ||
Riordan-Eva P. Neuro-ophthalmology of mitochondrial diseases. Curr Opin Opthalmol. 2000;11:408–412. | ||
Yu-Wai-Man P, Griffiths PG, Hudson G, Chinnery PF. Inherited mitochondrial optic neuropathies. J Med Genet. 2009;46:145–158. | ||
Man P, Griffiths P, Brown D. The epidemiology of Leber hereditary optic neuropathy in the North East of England. Am J Hum Genet. 2003;72:333–339. | ||
Spruijt L, Kolbach DN, de Coo RF, et al. Influence of mutation type on clinical expression of Leber hereditary optic neuropathy. Am J Ophthalmol.2006;141:676–682. | ||
Puomila A, Hämäläinen P, Kivioja S, et al. Epidemiology and penetrance of Leber hereditary optic neuropathy in Finland. Eur J Hum Genet.2007;15:1079–1089. | ||
Giraudet S, Lamirel C, Amati-Bonneau P, et al. Never too old to harbour a young man’s disease? Br J Ophthalmol. 2011;95:887–896. | ||
Riordan-Eva P, Harding A, Da Costa J, Sanders M, Govan G, Sweeney M. The clinical features of Leber’s hereditary optic neuropathy defined by the presence of a pathogenic mitochondrial DNA mutation. Brain. 1995;118 Pt 2:319–337. | ||
Biousse V, Brown M, Newman N, et al. De novo 14484 mitochondrial DNA mutation in monozygotic twins discordant for Leber’s hereditary optic neuropathy. Neurology. 1997;49:1136–1138. | ||
Harding A, Sweeney M. Pedigree analysis in Leber hereditary optic neuropathy families with a pathogenic mtDNA mutation. Am J Hum Genet. 1995;57:77–86. | ||
DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci. 2008;31:91–123. | ||
Newman NJ, Biousse V, Newman SA, et al. Progression of visual field defects in Leber hereditary optic neuropathy: experience of the LHON treatment trial. Am J Ophthalmol. 2006;141:1061–1067. | ||
La Morgia C, Ross-Cisneros FN, Sadun AA, et al. Melanopsin retinal ganglion cells are resistant to neurodegeneration in mitochondrial optic neuropathies. Brain. 2010;133 Pt 8:2426–2438. | ||
Moura ALA, Nagy BV, La Morgia C, et al. The pupil light reflex in Leber’s hereditary optic neuropathy: evidence for preservation of melanopsin-expressing retinal ganglion cells. Invest Ophthalmol Vis Sci. 2013;54:4471–4477. | ||
Jacobson DM, Stone EM, Miller NR, et al. Relative afferent pupillary defects in patients with Leber hereditary optic neuropathy and unilateral visual loss. Am J Ophthalmol. 1998;126:291–295. | ||
Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies – disease mechanisms and therapeutic strategies. Prog Retin Eye Res. 2011;30:81–114. | ||
Yu-Wai-Man P, Votruba M, Moore AT, Chinnery PF. Treatment strategies for inherited optic neuropathies: past, present and future. Eye. 2014;28:521–537. | ||
Mashima Y, Kimura I, Yamamoto Y, et al. Optic disc excavation in the atrophic stage of Leber’s hereditary optic neuropathy: comparison with normal tension glaucoma. Graefes Arch Clin Exp Ophthalmol. 2003;241:75–80. | ||
Barboni P, Carbonelli M, Savini G, et al. Natural history of Leber’s hereditary optic neuropathy: longitudinal analysis of the retinal nerve fiber layer by optical coherence tomography. Ophthalmology. 2010;117:623–627. | ||
Barboni P, Savini G, Valentino ML, et al. Retinal nerve fiber layer evaluation by optical coherence tomography in Leber’s hereditary optic neuropathy. Ophthalmology. 2005;112:120–126. | ||
Zhang Y, Huang H, Wei S, et al. Characterization of macular thickness changes in Leber’s hereditary optic neuropathy by optical coherence tomography. BMC Ophthalmol. 2014;14:105. | ||
Dorfman LJ, Nikoskelainen E, Rosenthal AR, Sogg RL. Visual evoked potentials in Leber’s hereditary optic neuropathy. Ann Neurol. 1977;1:565–568. | ||
Salomão SR, Berezovsky A, Andrade RE, Belfort R, Carelli V, Sadun AA. Visual electrophysiologic findings in patients from an extensive Brazilian family with Leber’s hereditary optic neuropathy. Doc Ophthalmol. 2004;108:147–155. | ||
Ziccardi L, Sadun F, De Negri AM, et al. Retinal function and neural conduction along the visual pathways in affected and unaffected carriers with Leber’s hereditary optic neuropathy. Invest Ophthalmol Vis Sci. 2013;54:6893–6901. | ||
Newman NJ. Leber’s hereditary optic neuropathy: new genetic considerations. Arch Neurol. 1993;50:540–548. | ||
Mashima Y, Oshitari K, Imamura Y, Momoshima S, Shiga H, Oguchi Y. Orbital high resolution magnetic resonance imaging with fast spin echo in the acute stage of Leber’s hereditary optic neuropathy. J Neurol Neurosurg Psychiatry. 1998;64:124–127. | ||
Kermode AG, Moseley IF, Kendall BE, Miller DH, MacManus DG, McDonald WI. Magnetic resonance imaging in Leber’s optic neuropathy. J Neurol Neurosurg Psychiatry. 1989;52:671–674. | ||
Vaphiades MS, Phillips PH, Turbin RE. Optic nerve and chiasmal enhancement in Leber hereditary optic neuropathy. J Neuroophthalmol. 2003;23:104–105. | ||
Sadun F, De Negri AM, Carelli V, et al. Ophthalmologic findings in a large pedigree of 11778/Haplogroup J Leber hereditary optic neuropathy. Am J Ophthalmol. 2004;137:271–277. | ||
Ventura DF, Quiros P, Carelli V, et al. Chromatic and luminance contrast sensitivities in asymptomatic carriers from a large Brazilian pedigree of 11778 Leber hereditary optic neuropathy. Invest Ophthalmol Vis Sci. 2005;46:4809–4814. | ||
Savini G, Barboni P, Valentino ML, et al. Retinal nerve fiber layer evaluation by optical coherence tomography in unaffected carriers with Leber’s hereditary optic neuropathy mutations. Ophthalmology. 2005;112:127–131. | ||
Guy J, Feuer WJ, Porciatti V, et al. Retinal ganglion cell dysfunction in asymptomatic G11778A: Leber hereditary optic neuropathy. Invest Ophthalmol Vis Sci. 2014;55:841–848. | ||
Kirkman MA, Korsten A, Leonhardt M, et al. Quality of life in patients with Leber hereditary optic neuropathy. Invest Ophthalmol Vis Sci. 2009;50:3112–3115. | ||
Johns DR, Heher KL, Miller NR, Smith KH. Leber’s hereditary optic neuropathy: clinical manifestations of the 14484 mutation. Arch Ophthalmol.1993;111:495–498. | ||
Johns DR, Smith KH, Miller NR. Leber’s hereditary optic neuropathy: clinical manifestations of the 3460 mutation. Arch Ophthalmol. 1992;110:1577–1581. | ||
Barboni P, Savini G, Valentino ML, et al. Leber’s hereditary optic neuropathy with childhood onset. Invest Ophthalmol Vis Sci. 2006;47:5303–5309. | ||
Ramos Cdo V, Bellusci C, Savini G, et al. Association of optic disc size with development and prognosis of Leber’s hereditary optic neuropathy. Invest Ophthalmol Vis Sci. 2009;50:1666–1674. | ||
DiMauro S, Schon E. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–2668. | ||
Schapira AH. Mitochondrial diseases. Lancet. 2012;379:1825–1834. | ||
McFarland R, Turnbull DM. Batteries not included: diagnosis and management of mitochondrial disease. J Intern Med. 2009;265:210–228. | ||
Zeviani M, Di Donato S. Mitochondrial disorders. Brain. 2004;127 Pt 10:2153–2172. | ||
Koene S, Smeitink J. Mitochondrial medicine: entering the era of treatment. J Intern Med. 2009;265:193–209. | ||
Wallace D. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. | ||
Sproule DM, Kaufmann P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann N Y Acad Sci. 2008;1142:133–158. | ||
Margulis L. Genetic and evolutionary consequences of symbiosis. Exp Parasitol. 1976;39:277–349. | ||
Fraser JA, Biousse V, Newman NJ. The neuro-ophthalmology of mitochondrial disease. Surv Ophthalmol. 2010;55:299–334. | ||
Sutovsky P, Moreno RD, Ramalho-Santos J, Dominko T, Simerly C, Schatten G. Ubiquitin tag for sperm mitochondria. Nature. 2005;2005:1998–1999. | ||
Jenuth J, Peterson A, Shoubridge E. Tissue-specific selection for different mtDNA genotypes in heteroplasmic mice. Nat Genet. 1997;16:93–95. | ||
Chinnery PF, DiMauro S, Shanske S, et al. Risk of developing a mitochondrial DNA deletion disorder. Lancet. 2004;364:592–596. | ||
Macmillan C, Kirkham T, Fu K, et al. Pedigree analysis of French Canadian families with T14484C Leber’s hereditary optic neuropathy. Neurology.1998;50:417–422. | ||
Valentino ML, Barboni P, Ghelli A, et al. The ND1 gene of complex I is a mutational hot spot for Leber’s hereditary optic neuropathy. Ann Neurol.2004;56:631–641. | ||
Chinnery PF, Brown DT, Andrews RM, et al. The mitochondrial ND6 gene is a hot spot for mutations that cause Leber’s hereditary optic neuropathy. Brain. 2001;124:209–218. | ||
Chinnery P, Andrews R, Turnbull D, Howell N. Leber hereditary optic neuropathy: does heteroplasmy influence the inheritance and expression of the G11778A mitochondrial DNA mutation? Am J Med Genet. 2001;98:235–243. | ||
Gómez-Durán A, Pacheu-Grau D, López-Gallardo E, et al. Unmasking the causes of multifactorial disorders: OXPHOS differences between mitochondrial haplogroups. Hum Mol Genet. 2010;19:3343–3353. | ||
Gómez-Durán A, Pacheu-Grau D, Martínez-Romero I, et al. Oxidative phosphorylation differences between mitochondrial DNA haplogroups modify the risk of Leber’s hereditary optic neuropathy. Biochim Biophys Acta. 2012;1822:1216–1222. | ||
Hudson G, Carelli V, Spruijt L, et al. Clinical expression of Leber hereditary optic neuropathy is affected by the mitochondrial DNA-haplogroup background. Am J Hum Genet. 2007;81:228–233. | ||
Ji Y, Zhang A-M, Jia X, et al. Mitochondrial DNA haplogroups M7b1’2 and M8a affect clinical expression of Leber hereditary optic neuropathy in Chinese families with the m.11778G – >A mutation. Am J Hum Genet.2008;83:760–768. | ||
Giordano C, Iommarini L, Giordano L, et al. Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy.Brain. 2014;137 Pt 2:335–353. | ||
Bu X, Rotter J. X chromosome-linked and mitochondrial gene control of Leber hereditary optic neuropathy: evidence from segregation analysis for dependence on X chromosome. Proc Natl Acad Sci U S A. 1991;88:8198–8202. | ||
Hudson G, Keers S, Man P, et al. Identification of an X-chromosomal locus and haplotype modulating the phenotype of a mitochondrial DNA disorder. Am J Hum Genet. 2005;77:1086–1091. | ||
Shankar SP, Fingert JH, Carelli V, et al. Evidence for a novel x-linked modifier locus for Leber hereditary optic neuropathy. Ophthalmic Genet. 2008;29:17–24. | ||
Ji Y, Jia X, Li S, Xiao X, Guo X, Zhang Q. Evaluation of the X-linked modifier loci for Leber hereditary optic neuropathy with the G11778A mutationin Chinese. Mol Vis. 2010;8090:416–424. | ||
Giordano C, Montopoli M, Perli E, et al. Oestrogens ameliorate mitochondrial dysfunction in Leber’s hereditary optic neuropathy. Brain. 2011;134 Pt 1:220–234. | ||
Kirkman MA, Yu-Wai-Man P, Korsten A, et al. Gene-environment interactions in Leber hereditary optic neuropathy. Brain. 2009;132 Pt 9:2317–2326. | ||
Pott JW, Wong KH. Leber’s hereditary optic neuropathy and vitamin B12 deficiency. Graefes Arch Clin Exp Ophthalmol. 2006;244:1357–1359. | ||
Sadun A, Win P, Ross-Cisneros F, Walker S, Carelli V. Leber’s hereditary optic neuropathy differentially affects smaller axons in the optic nerve. Trans Am Ophthalmol Soc. 2000;98:223–235. | ||
Andrews RM, Griffiths PG, Johnson MA, Turnbull DM. Histochemical localisation of mitochondrial enzyme activity in human optic nerve and retina. Br J Ophthalmol. 1999;83:231–235. | ||
Bristow E, Griffiths P, Andrews R, Johnson M, Turnbull D. The distribution of mitochondrial activity in relation to optic nerve structure. Arch Ophthalmol. 2002;120:791–796. | ||
Yu Wai Man CY, Chinnery PF, Griffiths PG. Optic neuropathies – importance of spatial distribution of mitochondria as well as function. Med Hypotheses. 2005;65:1038–1042. | ||
Carelli V, Rugolo M, Sgarbi G, et al. Bioenergetics shapes cellular death pathways in Leber’s hereditary optic neuropathy: a model of mitochondrial neurodegeneration. Biochim Biophys Acta. 2004;1658:172–179. | ||
Lin CS, Sharpley MS, Fan W, et al. Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc Natl Acad Sci U S A. 2012;109:20065–20070. | ||
Carelli V, La Morgia C, Valentino ML, Barboni P, Ross-Cisneros FN, Sadun AA. Retinal ganglion cell neurodegeneration in mitochondrial inherited disorders. Biochim Biophys Acta. 2009;1787:518–528. | ||
Gropman A, Chen T-J, Perng C-L, et al. Variable clinical manifestation of homoplasmic G14459A mitochondrial DNA mutation. Am J Med Genet. 2004;124A:377–382. | ||
Howell N, Kubacka I, McCullough D. Leber hereditary optic neuropathy: involvement of the mitochondrial ND1 gene and evidence for an intragenic suppressor mutation. Am J Hum Genet. 1991;48:935–942. | ||
Harding A, Sweeney M, Miller D, et al. Occurrence of a multiple sclerosis-like illness in women who have a Leber’s hereditary optic neuropathy mitochondrial DNA mutation. Brain. 1992;115 Pt 4:979–989. | ||
Pfeffer G, Burke A, Yu-Wai-Man P, Compston DA, Chinnery PF. Clinical features of MS associated with Leber hereditary optic neuropathy mtDNA mutations. Neurology. 2013;81:2073–2081. | ||
Palace J. Multiple sclerosis associated with Leber’s hereditary optic neuropathy. J Neurol Sci. 2009;286:24–27. | ||
Stelmack J, Tang X, Reda D, Rine S, Mancil R, Massof R. Outcomes of the veterans affairs low vision intervention trial (LOVIT). Arch Ophthalmol. 2008;126:608–617. | ||
Newman NJ. Treatment of hereditary optic neuropathies. Nat Rev Neurol. 2012;8:545–556. | ||
Pfeffer G, Majamaa K, Turnbull D, Thorburn D, Chinnery P. Treatment for mitochondrial disorders. Cochrane Database Syst Rev. 2012;4:004426. | ||
Pfeffer G, Horvath R, Klopstock T, et al. New treatments for mitochondrial disease – no time to drop our standards. Nat Rev Neurol. 2013;9:474–481. | ||
Saylor M, McLoon LK, Harrison AR, Lee MS. Experimental and clinical evidence for brimonidine as an optic nerve and retinal neuroprotective agent. Arch Ophthalmol. 2009;127:402–406. | ||
Newman NJ, Biousse V, David R, et al. Prophylaxis for second eye involvement in Leber hereditary optic neuropathy: an open-labeled, nonrandomized multicenter trial of topical brimonidine purite. Am J Ophthalmol. 2005;140:407–415. | ||
Thouin A, Griffiths PG, Hudson G, Chinnery PF, Yu-Wai-Man P. Raised intraocular pressure as a potential risk factor for visual loss in Leber hereditary optic neuropathy. PloS One. 2013;8:63446. | ||
Giorgio V, Petronilli V, Ghelli A, et al. The effects of idebenone on mitochondrial bioenergetics. Biochim Biophys Acta. 2012;1817:363–369. | ||
Mordente A, Martorana G, Minotti G, Giardina B. Antioxidant properties of 2,3-dimethoxy-5-methyl-6-(10-hydroxydecyl)-1, 4-benzoquinone (idebenone). Chem Res Toxicol. 1998:54–63. | ||
Mashima Y, Hiida Y, Oguchi Y. Remission of Leber’s hereditary optic neuropathy with idebenone. Lancet. 1992;340:368–369. | ||
Klopstock T, Yu-Wai-Man P, Dimitriadis K, et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain. 2011;134 Pt 9:2677–2686. | ||
Klopstock T, Metz G, Yu-Wai-Man P, et al. Persistence of the treatment effect of idebenone in Leber’s hereditary optic neuropathy. Brain. 2013;136 Pt 2:e230. | ||
Carelli V, La Morgia C, Valentino ML, et al. Idebenone treatment in Leber’s hereditary optic neuropathy. Brain. 2011;134 Pt 9:e188. | ||
Rudolph G, Dimitriadis K, Büchner B, et al. Effects of idebenone on color vision in patients with Leber hereditary optic neuropathy. J Neuroophthalmol. 2013;33:30–36. | ||
Sadun A, Chicani C, Ross-Ciscernos F, et al. Effect of EPI-743 on the clinical course of the mitochondrial disease Leber hereditary optic neuropathy. Arch Neurol. 2012;69:331–338. | ||
Wenz T, Williams SL, Bacman SR, Moraes CT. Emerging therapeutic approaches to mitochondrial diseases. Dev Disabil Res Rev. 2010;16:219–229. | ||
La Morgia C, Carbonelli M, Barboni P, Sadun AA, Carelli V. Medical management of hereditary optic neuropathies. Front Neurol. 2014;5:141. | ||
Wenz T, Diaz F, Spiegelman BM, Moraes CT. Activation of the PPAR/PGC-1alpha pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab. 2008;8:249–256. | ||
Guy J. New therapies for optic neuropathies: development in experimental models. Curr Opin Ophthalmol. 2000;11:421–429. | ||
DiMauro S, Mancuso M. Mitochondrial diseases: therapeutic approaches. Biosci Rep. 2007;27:125–137. | ||
Manfredi G, Fu J, Ojaimi J, et al. Rescue of a deficiency in ATP synthesis by transfer of MTATP6, a mitochondrial DNA-encoded gene, to the nucleus. Nat Genet. 2002;30:394–399. | ||
Guy J, Qi X, Pallotti F, et al. Rescue of a mitochondrial deficiency causing Leber hereditary optic neuropathy. Ann Neurol. 2002;52:534–542. | ||
Ellouze S, Augustin S, Bouaita A, et al. Optimized allotopic expression of the human mitochondrial ND4 prevents blindness in a rat model of mitochondrial dysfunction. Am J Hum Genet. 2008;83:373–387. | ||
Guy J, Qi X, Koilkonda RD, et al. Efficiency and safety of AAV-mediated gene delivery of the human ND4 complex I subunit in the mouse visual system. Invest Ophthalmol Vis Sci. 2009;50:4205–4214. | ||
Cwerman-Thibault H, Augustin S, Ellouze S, Sahel J-A, Corral-Debrinski M. Gene therapy for mitochondrial diseases: Leber hereditary optic neuropathy as the first candidate for a clinical trial. C R Biol. 2014;337:193–206. | ||
Safety Study of an Adeno-associated Virus Vector for Gene Therapy of Leber’s Hereditary Optic Neuropathy (LHON) Caused by the G11778A Mutation. 2015. Available from: https://clinicaltrials.gov/ct2/show/NCT02161380. Accessed February 21, 2015. | ||
Safety and Efficacy Study of rAAV2-ND4 Treatment of Leber Hereditary Optic Neuropathy (LHON). 2015. Available from: https://clinicaltrials.gov/ct2/show/NCT01267422. Accessed February 21, 2015. | ||
Safety Evaluation of Gene Therapy in Leber Hereditary Optic Neuropathy (LHON) Patients. 2015. Available from: https://clinicaltrials.gov/ct2/show/NCT02064569. Accessed February 21, 2015. | ||
Floreani M, Napoli E, Martinuzzi A, et al. Antioxidant defences in cybrids harboring mtDNA mutations associated with Leber’s hereditary optic neuropathy. FEBS J. 2005;272:1124–1135. | ||
Qi X, Sun L, Hauswirth WW, Lewin AS, Guy J. Use of mitochondrial antioxidant defenses for rescue of cells with a Leber hereditary optic neuropathy-causing mutation. Arch Ophthalmol. 2007;125:268–272. | ||
Iyer S, Bergquist K, Young K, Gnaiger E, Rao RR, Bennett JP. Mitochondrial gene therapy improves respiration, biogenesis, and transcription in G11778A Leber’s hereditary optic neuropathy and T8993G Leigh’s syndrome cells. Hum Gene Ther. 2012;23:647–657. | ||
Yu H, Koilkonda R, Chou T, et al. Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber’s hereditary optic neuropathy in a mouse model. Proc Natl Acad Sci U S A. 2012;109:1238–1247. | ||
Yu H, Mehta A, Wang G, et al. Next-generation sequencing of mitochondrial targeted AAV transfer of human ND4 in mice. Mol Vis. 2013;19:1482–1491. | ||
Marchetti V, Krohne T, Friedlander D, Friedlander M. Stemming vision loss with stem cells. J Clin Investig. 2010;120:3012–3021. | ||
Dahlmann-Noor A, Vijay S, Jayaram H, Limb A, Khaw PT. Current approaches and future prospects for stem cell rescue and regeneration of the retina and optic nerve. Can J Ophthalmol. 2010;45:333–341. | ||
Johnson TV, Bull ND, Hunt DP, Marina N, Tomarev SI, Martin KR. Neuroprotective effects of intravitreal mesenchymal stem cell transplantation in experimental glaucoma. Invest Ophthalmol Vis Sci. 2010;51:2051–2059. | ||
Connick P, Kolappan M, Crawley C, et al. Autologous mesenchymal stem cells for the treatment of secondary progressive multiple sclerosis: an open-label phase 2a proof-of-concept study. Lancet Neurol. 2012;11:150–156. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.