Back to Journals » Research Reports in Clinical Cardiology » Volume 5
“Mitochondrial remodeling” in coronary heart disease
Authors Saotome M, Hajnóczky G, Katoh H, Satoh H, Hayashi H
Received 18 January 2014
Accepted for publication 3 April 2014
Published 12 June 2014 Volume 2014:5 Pages 111—122
DOI https://doi.org/10.2147/RRCC.S43364
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Masao Saotome,1 György Hajnóczky,2 Hideki Katoh,1 Hiroshi Satoh,1 Hideharu Hayashi1
1Internal Medicine III, Hamamatsu University School of Medicine, Hamamatsu, Japan; 2Department of Pathology, Anatomy, and Cell Biology, Thomas Jefferson University, Philadelphia, PA, USA
Abstract: Coronary heart disease is a major cause of morbidity and mortality in advanced countries. Despite remarkable developments and achievements in the field of coronary intervention, such as percutaneous catheter intervention and coronary bypass surgery, the mortality from coronary heart disease remains high because of lack of effective cardioprotective therapy against ischemia/reperfusion injury after coronary recanalization. The mitochondria play a crucial role in determination of cell death in ischemia/reperfusion injury, and furthermore provide myocardial protection against ischemia/reperfusion injury by ischemic preconditioning. Functional and structural alterations in the mitochondria help to decide cell death and survival, and many investigations have been conducted to explore the pathophysiological mechanisms of “mitochondrial remodeling” to gain clues regarding ischemia/reperfusion injury. In this review, we summarize the current state of knowledge concerning the pathophysiological role of bidirectional (detrimental and defensive) “mitochondrial remodeling” via which cell death or survival is determined in coronary heart disease. Further, we discuss clinical trials of mitochondria-targeted treatment in patients with coronary heart disease.
Keywords: coronary heart disease, mitochondrial remodeling, mitochondrial dynamics
Introduction
Coronary heart disease (CHD) is the most common form of heart disease, and is caused by disturbance of coronary flow due to atherosclerosis or spasm in the coronary vasculature. CHD is a significant cause of morbidity and mortality in advanced countries,1,2 and the World Health Organization estimates that 7.3 million people die from CHD each year. Despite the remarkable developments and achievements in the field of coronary intervention for CHD, such as percutaneous coronary intervention and coronary artery bypass graft surgery, morbidity and mortality in CHD patients remains high. One of the major reasons for this may be the lack of a significant effective cardioprotection strategy for ischemia/reperfusion (I/R) injury.3–5 It is well recognized that early coronary recanalization can improve the prognosis in patients with acute myocardial infarction by reducing myocardial infarct size.6 However, coronary reperfusion therapy paradoxically promotes the myocardial damage caused by I/R injury and limits the benefit of early coronary recanalization.7 Further efforts to establish therapeutic options for protecting the myocardium from I/R injury are required in order to achieve a better outcome in CHD patients. In addition to being a critical source of energy, the mitochondrion plays a pivotal role in the pathogenesis of I/R injury.5,8–12 A growing body of evidence suggests that structural and functional alterations in the mitochondria, known as “mitochondrial remodeling”, play an important role in the pathophysiology of I/R injury, not only as a critical determinant of cell death but also as a final effector of cardioprotection by ischemic preconditioning, and significant attention has been focused on the mitochondria as a potential therapeutic target in CHD. This review summarizes the current scientific knowledge regarding the pathophysiological role of bidirectional (detrimental and defensive) mitochondrial remodeling in CHD. In addition, we discuss the possible clinical application of treatments targeting the mitochondria.
Cardiac mitochondria under physiological conditions
Mitochondrial energy production in the normal heart
The heart is an organ with high energy requirements. In order to sustain continuous contractions of the heart, production of sufficient amounts of adenosine triphosphate (ATP, 3.5–5 kg/day) is required at a high rate (~0.5 mmol/per gram wet weight per second at rest).13 Under physiological conditions, almost all ATP (>95%) is produced by oxidative phosphorylation in the mitochondria.13 The mitochondria mainly supply the intracellular energy demands of the myocardium. Cardiac muscle contains a high number of efficiently distributed mitochondria (>50% of cardiac volume) located between the myofibrils (intermyofibrillar mitochondria) and below the sarcolemma (subsarcolemmal mitochondria) to supply intracellular ATP.14
Cardiac contraction takes place by excitation-contraction coupling, during which calcium flux plays an essential role (Figure 1). An action potential is conducted to the plasma membrane and transverse tubule, causing a small calcium influx from the voltage-dependent L-type calcium channel (known as the dihydropyridine receptor) located in the transverse tubule, which causes a large amount of calcium to be released from the sarcoplasmic reticulum via ryanodine receptors. This process, known as calcium-induced calcium release, triggers binding of calcium to troponin C in the myofilaments and initiates contraction. Release of calcium from the sarcoplasmic reticulum via ryanodine receptors also allows accumulation of calcium in the mitochondrial matrix through the mitochondrial calcium uniporter (MCU), which activates calcium-dependent matrix dehydrogenase and synthesizes ATP to support cardiac contraction.
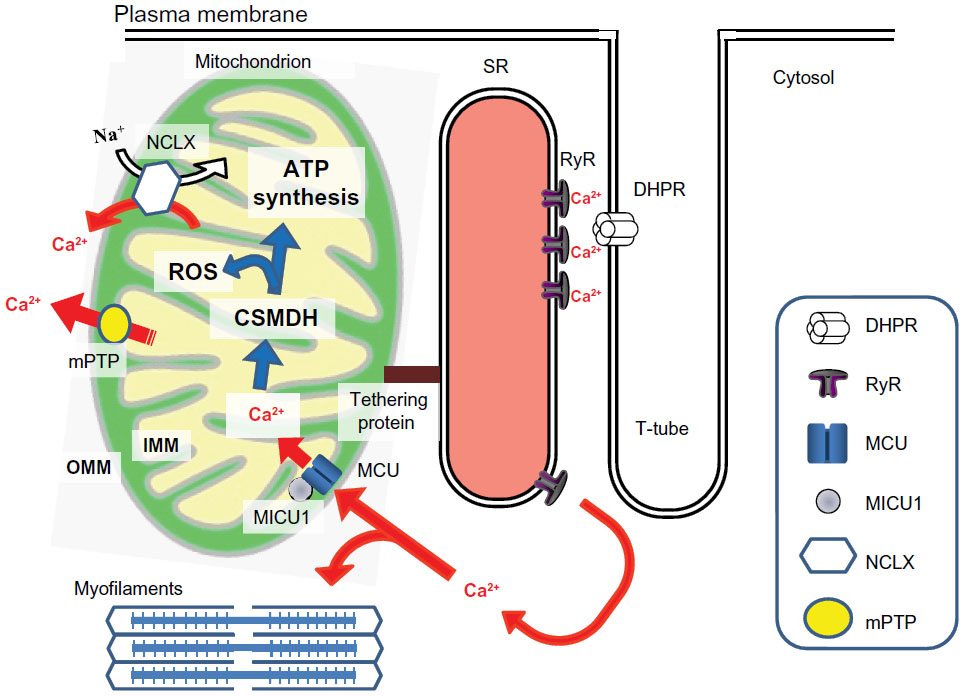 | Figure 1 Calcium-dependent physiological interactions between excitation-contraction coupling and mitochondrial energy production in cardiac myocytes. When an action potential is conducted, a small calcium influx from the voltage-dependent L-type calcium channel (known as the dihydropyridine receptor) located in the T-tube induces release of a large amount of calcium from the sarcoplasmic reticulum via RyR. This process, called “calcium-induced calcium release”, activates calcium-mediated myofilament contraction. The calcium release from the sarcoplasmic reticulum via RyR also induces accumulation of mitochondrial matrix calcium through the MCU, which is regulated by the MICU1, activates matrix calcium-dependent dehydrogenases, and then synthesis of intracellular ATP to support cardiac contraction. The calcium fluxes are indicated by a red arrow. |
Mitochondrial membrane potential, calcium, and ROS under normal conditions
The mitochondria produce ATP via redox reactions in the electron transport chain in the inner mitochondrial membrane. Electron transfer from donor to acceptor generates a potent electrical gradient across the mitochondrial membrane (ie, the mitochondrial membrane potential [ΔΨm] −180 to −200 mV). This electrochemical gradient is then effectively used for synthesis of ATP (F1-F0 ATPase), a process known as oxidative phosphorylation.
In spite of the high capacity of the mitochondria to accumulate calcium (as seen in isolated mitochondrial experiments), the mitochondrial matrix calcium concentration is relatively low under physiological conditions, and the contribution of mitochondria to bulk cytosolic calcium fluxes during cardiac excitation-contraction coupling is considered to be small (<5%).15 However, mitochondrial calcium regulation is important for various physiological processes in the cell, and production of ATP in the mitochondria is regulated by the mitochondrial matrix calcium concentration15 (Figure 1). Although it is still debated whether the mitochondria take up a small fraction of the calcium released during each cytosolic calcium spike or only respond to the changes of heart rate,16 mitochondrial calcium accumulates mainly in the mitochondrial matrix via the MCU. Classically, the driving force via which MCU accumulates calcium is the electrical gradient across the inner mitochondrial membrane, which is inhibited by the physiological cytosolic magnesium concentration, ruthenium red, and Ru360.17,18 Recent investigations have identified the molecular mechanisms of the MCU. The pore-forming protein is referred to as the MCU,19,20 and the MCU is regulated by mitochondrial calcium uptake 1 (MICU1), which is located in the inner mitochondrial membrane.21–23 The response of mitochondria to cardiac energy demand, which is changing in a beat-to-beat base cardiac energy demand may occur as a result of mobilization of calcium from the sarcoplasmic reticulum to the mitochondria,24 which is achieved by physical coupling (tethers) between the sarcoplasmic reticulum and the mitochondria25,26 (Figure 1). Tethers between the sarcoplasmic reticulum and mitochondria have been observed in cardiac myocytes,27 and although currently a matter of debate,28 mitofusin 2 is reported to be one of the molecules in cardiac muscle possibly contributing to tethering between these two organelles.26 The main mitochondrial efflux pathways in the heart are the mitochondrial sodium/calcium exchanger29 and the mitochondrial permeability transition pore (mPTP).30 The mitochondrial sodium/calcium exchanger is effectively inhibited by CGP37157, whereas mPTP opening can be attenuated by cyclosporine A, sanglifehrin A, and several related compounds. Genetic targeting of the specific inner mitochondrial membrane calcium transport pathway is expected to clarify the physiological role of mitochondrial calcium transport in the near future.
The mitochondria are major organelles producing reactive oxygen species (ROS) via mitochondrial electron transport chain activity, where 0.2%–2% of oxygen is converted to superoxide (O2−) by mitochondrial respiration.31,32 Under physiological conditions, myocardial ROS are present in relatively low numbers because mitochondria have effective detoxification systems, such as manganese superoxide dismutase, catalase, and gultathione peroxidase.11,33
Detrimental mitochondrial remodeling in CHD
Mitochondria are likely to be key players in the pathogenesis of CHD, given that they determine whether the cell dies or survives via necrosis or apoptosis, respectively. In this section, we discuss the detrimental mitochondrial remodeling determining ischemic myocardial damage during I/R injury and infarct size.
Pathophysiology of CHD
During myocardial ischemia, which is a situation of disrupted coronary artery blood flow as a result of atherosclerotic plaque rupture with thrombosis, myocardial contraction is rapidly impaired. During myocardial ischemia, the ATP supply to the mitochondria is disrupted because of impaired oxidative phosphorylation and loss of the ΔΨm by anoxia.34 Although cardiac myocytes embark on compensatory glycolytic ATP production, this results in intracellular acidification by accumulation of lactic acid and dysregulation of the intracellular ionic balance. Cytosolic sodium becomes elevated by accelerated sarcolemmal sodium/hydrogen ion exchange and/or by inhibition of Na+/K+-ATPase, with subsequent elevation of cytosolic calcium (Figure 2) due to reverse acceleration of the sarcolemmal sodium/calcium exchanger.35
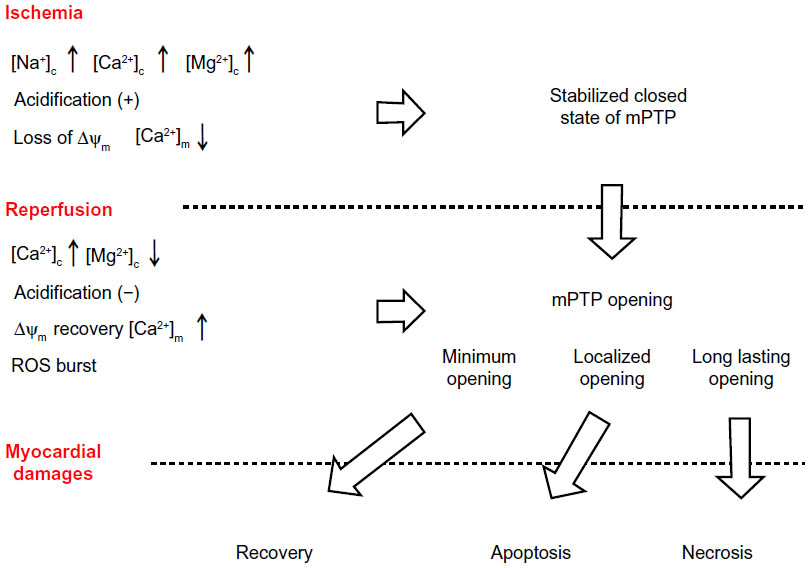 | Figure 2 Myocardial damage and mPTP opening during ischemia/reperfusion. During ischemia, alterations in intracellular circumstances, such as elevated [Ca2+]c, increase the likelihood of mPTP opening; however, elevated [Mg2+]c, intracellular acidification, and suppressed [Ca2+]m due to loss of ΔΨm remain the mPTP closed state. Upon reperfusion, recovery of ΔΨm by reoxygenation promotes mitochondrial calcium overload and a ROS burst, and these changes finally leads to myocardial damages by mPTP opening. |
Given that myocardial necrosis is mostly complete within 3 hours of the onset of coronary occlusion,36,37 early reperfusion by coronary intervention (using a thrombolytic agent and/or percutaneous coronary intervention) is required in order to salvage the myocardium.6 However, reperfusion therapy paradoxically exacerbates further myocardial damage due to I/R injury. After successful reperfusion by a thrombolytic agent or emergent percutaneous coronary intervention, reoxygenation enables the mitochondria to regenerate ΔΨm and to supply ATP by resumption of the electron transport chain. However, at the same time, the recovery process injures the mitochondria and cardiac myocytes. Recovery of ΔΨm induces mitochondrial calcium overload due to elevated cytosolic calcium concentration and massive release of ROS,38,39 resulting in increased susceptibility to mPTP opening (Figure 2), which is the final pathway leading to apoptosis and necrosis of the cell.8,10,12,34 Since I/R injury has a significant influence on the myocardial damage associated with acute myocardial infarction,40,41 the extent of mPTP opening is a critical determinant of the extent of irreversible myocardial damage and infarct size.8,42
Opening of mPTP and I/R injury
Opening of mPTP allows small molecules less than 1.5 kDa to cross the inner mitochondrial membrane.43–45 Sustained mPTP opening results in disruption of ΔΨm, mitochondrial swelling, and rupture of the outer mitochondrial membrane, leading to release of proapoptotic factors, such as cytochrome c, Smac/DIABLO, and apoptosis-inducing factor from the mitochondrial intermembrane space to the cytoplasm, and inducing apoptotic cell death.8,12,30 The extent of mPTP opening in cardiac myocytes determines the infarct size and prognosis in CHD patients, even if they have undergone successful recanalization by coronary intervention.
For a long time, the mPTP was considered to be comprised of a complex of the adenine nucleotide translocator in the inner mitochondrial membrane, cyclophilin D in the matrix, and the voltage-dependent anion channel in the outer mitochondrial membrane. However, recent investigations have suggested that the adenine nucleotide translocator and voltage-dependent anion channel are not necessary for mPTP opening,12,46 and genetic targeting investigations have confirmed that cyclophilin D is the main calcium sensor and regulator of the mPTP pore.47–49 Most recently, genetic evidence was provided for dimers of the ATP synthase forming the mPTP pore.50 Opening of mPTP is facilitated by binding of cyclophilin D to the inner mitochondrial membrane, which is regulated by calcium, Pi (inorganic phosphate), and ROS.30,51 Cyclosporine A and sanglifehrin A are recognized as specific inhibitors of mPTP; they inhibit mPTP by interfering with the binding of cyclophilin D to the inner mitochondrial membrane.51
The primary trigger for mPTP opening is calcium. Classical investigations conducted in isolated mitochondria showed that opening and closing of the mPTP is highly sensitive to calcium.52 Other factors, including pH, long-chain fatty acid accumulation, and ROS, can alter susceptibility to mPTP opening.8,10,12 In addition, certain proteins, including the benzodiazepine receptor, hexokinase, glycogen synthase kinase-3β, and creatine kinase, can regulate opening of the mPTP.53
Opening of the mPTP is more apparent in the reperfusion phase than in the ischemic phase.9 During ischemia, cytosolic acidification and elevated magnesium concentrations stabilize mPTP in the closed state. In contrast, reperfusion, where the cytosolic and matrix calcium concentrations, Pi, and ROS are elevated (at the same time cytosolic acidification and magnesium elevation are improved by recanalization) increases the susceptibility of mPTP to opening (Figure 2). Recovery of ΔΨm by reperfusion promotes mitochondrial calcium overload through recovered MCU, thereby opening the mPTP.8,10,11 Specific inhibitors of mPTP opening also protect the myocardium from I/R injury when they were applied during the reperfusion phase.54–56 Thus, mPTP opening upon reperfusion is a promising therapeutic target for cardioprotection.
The different contributions of the two mitochondrial subpopulations (subsarcolemmal mitochondria and intermyofibrillar mitochondria) during I/R injury remain unclear. The subsarcolemmal mitochondria were considered to be more susceptible to ischemic damage because calcium-induced mPTP opening and mitochondrial damage (cardiolipin and cytochrome c decrease) in these mitochondria are more apparent than in intermyofibrillar mitochondria.57 However, it is well known that opening of the mPTP is apparent after reperfusion, and several investigations have reported conflicting results regarding the differential sensitivity of these two subpopulations to calcium-induced mPTP opening.58,59
Mitochondrial calcium and mitochondrial ROS production during I/R
Overloading of mitochondrial calcium in the reperfusion phase is a critical trigger for opening of the mPTP. Classical investigations using ruthenium red, a potent MCU inhibitor, showed favorable effects on I/R injury.60,61 Inhibition of mitochondrial calcium uptake certainly seems to be effective in I/R injury by preventing calcium-induced mPTP opening. However, regarding the chemical inhibition of MCU, we have to bear in mind the fact that ruthenium red cannot pass readily through the plasma membrane because of its highly charged nature. A study by Hajnóczky et al found that ruthenium red failed to inhibit mitochondrial calcium uptake when it was applied to intact cells.18 Further, Griffiths et al showed that higher levels of ruthenium red were required to inhibit MCU in intact cardiac myocytes, and resulted in nonspecific damaging effects on the heart.62 Recently, Pan et al reported that mitochondria from MCU-knockout mice showed resistance to calcium-induced mPTP opening, with no evidence of protection against I/R injury, and also lacked cyclosporin A-dependent I/R injury.63 Recent investigations have revealed that MICU1 works as a gatekeeper for MCU-mediated mitochondrial calcium uptake.22,64 MICU1 locates in the inner mitochondrial membrane and exposes its two EF-hand domains (calcium-sensitive protein) toward the mitochondrial intermembrane space, enabling MICU1 to respond to changes in the cytosolic calcium concentration. MICU1 prevents mitochondrial calcium uptake when the cytosolic calcium concentration is low, and confers a cooperative activation of MCU at higher cytosolic calcium concentration.22 Although the pathophysiological role of MICU1 in I/R injury remains unclear, it is likely that dysregulation of MICU1 promotes mitochondrial calcium overload, underpinning the increased susceptibility to mPTP opening after reperfusion. Further investigations are required to clarify the involvement of MICU1 in the pathogenesis of I/R injury.
Oxidative stress is also relevant to opening of the mPTP during I/R injury. Elevation of mitochondrial ROS promotes a self-amplifying loop known as ROS-induced ROS release, ie, the initial elevation of mitochondrial ROS can induce a burst of mitochondrial ROS. Because ROS-induced ROS release is associated with mPTP opening and consequent dissipation of ΔΨm,65 the burst of myocardial ROS during the reperfusion phase might result from ROS-induced ROS release.
Dysregulation of mitochondrial morphology
Mitochondria are continuously altering their size, shape, and number as a result of mitochondrial dynamics (ie, fusion and fission events) in order to respond to changes in the intracellular environment (Figure 3). Mitochondrial dynamics are essential for cellular homeostasis, and misregulation of mitochondrial morphology is considered to be a pathogenetic trigger in many human diseases.66–68 Mitochondrial fusion is thought to be stimulated by energy demand and/or stress, and supports the exchange of proteins, substrates, and mitochondrial DNA between organelles to enhance the stability of the mitochondria.69 Mitochondrial fission enables an increase in the number and capability of mitochondria during cell division and facilitates control of mitochondrial quality by removing damaged mitochondria via lysosomal autophagy (so-called mitophagy).66,70 Major regulators of the mitochondrial fusion process include mitofusin 1, mitofusin 2, and optic atrophy-1, and the most relevant mitochondrial fission proteins are dynamin-related protein, mitochondrial fission factor,71 and fission 1 homolog protein.70
The evidence suggests that changes in mitochondrial morphology are correlated with the pathophysiology of CHD.67,68 Mitochondrial fragmentation (fission) is apparent in the failing myocardium after myocardial infarction, where both a decrease in fusion proteins and an increase in fission proteins have been observed.72 Questions remain concerning the pathophysiological role of mitochondrial dynamics in I/R injury, given that mitochondrial motility is of quite low amplitude in beating cardiac myocytes and little or no activity of fusion/fission is observed in adult myocytes under physiological conditions.66,73 Further, it is unclear if dysregulation of mitochondrial dynamics is a cause or a result of the pathogenesis of I/R injury. However, dysregulation of mitochondrial dynamics does seem to be associated with the various pathophysiologies of CHD, such as apoptotic cell death, mitophagy, and metabolic disorder.67
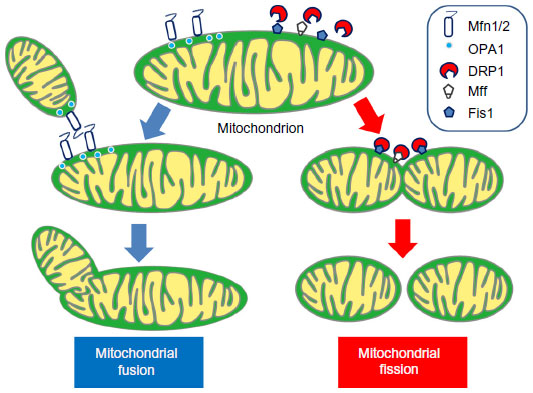 | Figure 3 Proposed mechanisms for mitochondrial fusion and fission. Under physiological conditions, the mitochondria dynamically alter their morphology through fusion (left) and fission (right) to maintain cellular homeostasis. Mfn1, Mfn2, and OPA1 proteins are the major regulators of mitochondrial fusion. Mfn1 and Mfn2 locate in the outer mitochondrial membrane with their GTPase site facing the cytosol to coordinate the fusion process with the outer membrane of opposing mitochondria, and OPA1 in the intermembrane space to coordinate inner mitochondrial membrane fusion. The mitochondrial fission process is mainly conducted by DRP1, Mff, and Fis1. DRP1 is normally located in the cytosolic space and is recruited to the outer mitochondrial membrane during the fission process. Fis1 and Mff are located in the outer mitochondrial membrane and work as the adaptor protein for DRP1. |
Defensive mitochondrial remodeling in CHD
Ischemic preconditioning is a potential way of reducing the cardiac damage resulting from I/R injury.5,74 Murry et al first reported that myocardium obtained a resistance against I/R injury when myocardium was exposed to repeated short episodes of ischemia before prolonged ischemia.75 Various factors, such as autacoids (eg, adenosine, bradykinin, and opioids), their plasma membrane receptors, signaling pathways, and mitochondrial modulation are involved in the cardioprotective mechanism of ischemic preconditioning. Although recent investigations have provided evidence of other cardioprotective therapeutic options against I/R injury, such as post conditioning and remote conditioning (reviewed elsewhere76,77), this section focuses on ischemic preconditioning and discusses the role of defensive mitochondrial remodeling during this process.
Remodeling of mitochondria by the ischemic preconditioning signal pathway
Previous investigations have suggested that various factors are involved in the ischemic preconditioning signal pathway.74,78 Many plasma membrane receptors, including G-protein-coupled receptors (adenosine A1, A3, opioids, and bradykinin-B2), cytokine receptors (erythropoietin and tumor necrosis factor-alpha receptor), tyrosine kinase receptors (epidermal growth factor receptor and insulin receptor), and alpha-adrenergic and beta-adrenergic receptors can act as triggers for ischemic preconditioning.3,53,74 Multiple signaling pathways are activated via these receptors, and their signaling cascades intricately stimulate each other. Accumulating evidence shows that the mitochondria are one of most important final effectors of ischemic preconditioning,8,53,74 which affords myocardial protection against I/R injury by inhibiting mPTP opening in the reperfusion phase.9,56,79
Brief and repetitive ischemia activates multiple signaling pathways in the cytosol, such as phosphatidylinositol 3-kinase/AKT, extracellular-regulated kinases, protein kinase C, and protein kinase G. One of the most important intracellular signals for cardioprotection afforded by ischemic preconditioning is protein kinase C, which generally requires second messengers including cytosolic calcium, diacylglycerol, and phospholipids for activation. A novel type of protein kinase C, which does not require cytosolic calcium for activation, is also involved in the cardiac protection induced by ischemic preconditioning, and has different actions according to subtype, ie, PKC-ε affords protection by activation whereas PKC-δ provides protection by inhibition. Phosphatidylinositol 3-kinase/AKT signaling, which is well recognized as a “reperfusion injury salvage kinase pathway”,80 stimulates the extracellular-regulated kinase/endothelial nitric oxide synthase/protein kinase G pathway, and then activates the mitochondrial ATP-sensitive potassium channel, which is a putative effector of ischemic preconditioning.81 The cardioprotection afforded by the mitochondrial ATP-sensitive potassium channel is considered to be regulated by stabilization of the inner mitochondrial membrane and prevention of membrane uncoupling,9 which can decrease susceptibility to mPTP opening after reperfusion.82
Certain protein kinases in the mitochondria, such as AKT,83 protein kinase C-ε, extracellular regulated kinases, glycogen synthase kinase-3β,84,85 and hexokinase I and II,78 are considered to confer the myocardial protection induced by ischemic preconditioning.53,74 Although the exact mechanism by which these protein kinases afford myocardial protection is still unclear, enhancement of hexokinase binding to the mitochondria78 and/or inactivation of glycogen synthase kinase-3β by phosphorylation53,74 seem to be the final effectors of the cardioprotective mechanism of ischemic preconditioning.86 Hexokinase binding with the voltage-dependent anion channel promotes cell survival by inhibiting mPTP opening,87 and inhibition of hexokinase detachment from the voltage-dependent anion channel decreases the likelihood of mitochondrial outer membrane permeability by competitive BCL-XL-voltage-dependent anion channel binding, which facilitates to interact Bax-Bak apoptotic proteins.86 Furthermore, Chiara et al have shown that mitochondrial hexokinases regulate mPTP opening via the adenine nucleotide translocator and cyclophilin D and not by interacting with the voltage-dependent anion channel.88 Inactivation of glycogen synthase kinase-3β by phosphorylation of serine also enables cell survival by inhibiting the detachment of hexokinase from the voltage-dependent anion channel.89 Since glycogen synthase kinase-3β phosphorylates threonine51 on the voltage-dependent anion channel and causes detachment of hexokinase, inactivation of glycogen synthase kinase-3β results in preservation of hexokinase binding to the voltage-dependent anion channel.89
Nitric oxide (NO) also plays an important role in cardioprotective signaling during ischemic preconditioning.90 In addition to the classical cGMP-dependent pathway, such as vasodilation and anti-inflammatory effects,91 recent investigations suggest that NO protects the myocardium through S-nitrosylation of protein, a reversible post transcriptional protein modification and inhibition of mPTP.90 Ngyuyen et al reported that S-nitrosylation in cysteine 203 of cyclophilin D, a critical mediator of mPTP opening, is associated with NO-induced cellular protection.92 However, NO is somewhat of a “double-edged sword” with regard to mPTP opening, in that the beneficial effects of NO are obtained at a relatively low concentration (close to the physiological concentration range), whereas higher NO concentrations increase the likelihood of mPTP opening.93
Remodeling of mitochondrial dynamics by ischemic preconditioning
As mentioned in the previous section, altered mitochondrial morphology (fragmentation) has been reported in ischemic cardiomyopathy.94 There have been reports suggesting that intervention on mitochondrial dynamics has a cardioprotective effect against I/R injury.95,96 Ong et al showed that expression of mitofusin 1/2 or suppression of dynamin-related protein (by DRP1-K38, the dominant negative form of dynamin-related protein) inhibited mPTP opening and consequent cell death after I/R injury. They also indicated that pharmacological inhibition of mitochondrial fission by mitochondrial division inhibitor-1 reduced myocardial infarct size in an in vitro mouse model.95
As described above, mitochondrial dynamics associate with the pathogenesis of I/R injury. However, currently there is no report indicating a clear association between ischemic preconditioning and altered mitochondrial dynamics. If an alteration of mitochondrial dynamics is involved in the mechanism of ischemic preconditioning, mitochondrial dynamics would be expected to cause mitochondrial fusion by activation of mitochondrial fusion protein and/or inactivation of fission protein during ischemic preconditioning, given that mitochondrial fusion is stimulated by energy demand and/or stress to respond to them by mixing the matrix components.69 Further investigations are needed to clarify the association between ischemic preconditioning and mitochondrial dynamics.
Transient mPTP opening by ischemic preconditioning
Previous investigations have shown that transient mPTP opening (a brief increase in mitochondrial permeability) can release mitochondrial calcium to avoid matrix calcium overload.97–99 In contrast with long-lasting mPTP opening, transient mPTP opening is considered to excrete excessive amounts of metabolites to preserve mitochondrial integrity. Therefore, transient mPTP opening may be a critical mediator of the cardioprotective mechanism in ischemic preconditioning. During ischemic preconditioning, brief and repetitive ischemia can activate transient mPTP opening, which enables a decrease in the matrix calcium concentration by direct activation of mitochondrial calcium extrusion and inhibition of mitochondrial calcium accumulation by temporal ΔΨm depolarization, and can confer resistance against subsequent long-lasting mPTP opening in the reperfusion phase.100 Thus, the mitochondria can perform “defensive remodeling” by transient mPTP opening.
ROS production during ischemic preconditioning may be a key trigger for transient mPTP opening. We have previously demonstrated that repetitive administration of a small amount of ROS (ie, hydrogen peroxide) had ischemic preconditioning-like cardioprotective effects, and this favorable effect was abolished by inhibition of mPTP.101 Hausenloy et al further revealed the involvement of transient mPTP opening in ischemic preconditioning in cyclophilin D-knockout mice. They suggested that transient mPTP opening during ischemic preconditioning promotes mitochondrial ROS, which stimulates prosurvival pathways such as AKT and extracellular-regulated kinases 1/2, thereby enabling cyclophilin D to resist long-lasting mPTP opening.102 Given that the contribution of transient mPTP opening to ischemic preconditioning is far from fully understood, further investigations are needed to identify the mechanism via which transient mPTP opening protects the myocardium from long-lasting mPTP opening in the reperfusion phase.
Translation to bedside: clinical investigations of mitochondria- targeting in CHD
It is important to translate basic research findings to clinical medicine (the so-called translation from bench to bedside). As already discussed, the mitochondria play an important role in the regulation of both cell death and survival, and have attracted considerable attention as a potential therapeutic target in CHD. Unfortunately, at present, very few treatments that target the mitochondria have successfully completed clinical trials.5,103 Regarding treatments targeting the ATP-sensitive mitochondrial potassium channel, two small clinical trials have indicated that diazoxide, an ATP-sensitive mitochondrial potassium channel agonist, protects against perioperative myocardial damage.104,105 However, J-WIND (Japan-Working Groups of Acute Myocardial Infarction for the Reduction of Necrotic Damage by Atrial Natriuretic Peptide or Nicorandil), a multicenter Phase III clinical trial in Japan, ie, indicates that nicorandil, a dual nitrate and mitochondrial ATP-sensitive potassium channel agonist, neither reduces infarct size nor improves left ventricular function in patients with acute myocardial infarction.106 Since the exact structure and molecular components of the mitochondrial ATP-sensitive potassium channel are not yet known, such drugs are substitutes which multiply affect to other ATP-sensitive potassium channels. Further exploration of the molecular components of the mitochondrial ATP-sensitive potassium channel is required to produce a new drug to act selectively on this channel. A pilot study has reported that intravenous administration of cyclosporin A decreased infarct size in patients with acute myocardial infarction,107 but further evidence from large-scale multicenter clinical trials may be needed before clinical application.
Currently, effective cardioprotection therapy against I/R injury is limited in the clinical setting, whereas numerous potential cardioprotective strategies, including mitochondria-targeting treatment,103 have been investigated at the basic medical research level. One of the major reasons for this problem is because of less successful translation research with an optimized clinical study design.4,5 To benefit CHD patients, further developments of translation research are needed.3–5
Summary and clinical implications
In this review, we have discussed the pathophysiological roles of bidirectional (detrimental and defensive) mitochondrial remodeling in CHD. Cardiac mitochondria are key organelles, since they provide not only high energy phosphate to maintain cardiac contraction but also cellular homeostasis through ion regulation. During I/R, the mitochondria start “detrimental remodeling” and myocardial cell death or survival is determined by mPTP opening (Figure 4, left). In contrast, the mitochondria can also provide “defensive remodeling” to protect the myocardium from I/R injury by ischemic preconditioning (Figure 4, right). Although it is apparent that the mitochondria are a promising therapeutic target for CHD at the basic research level, very few clinical trials have successfully translated this evidence to the clinical setting. Further efforts are required to promote successful translation of basic research to an optimal study design to improve the clinical outcome for CHD patients.
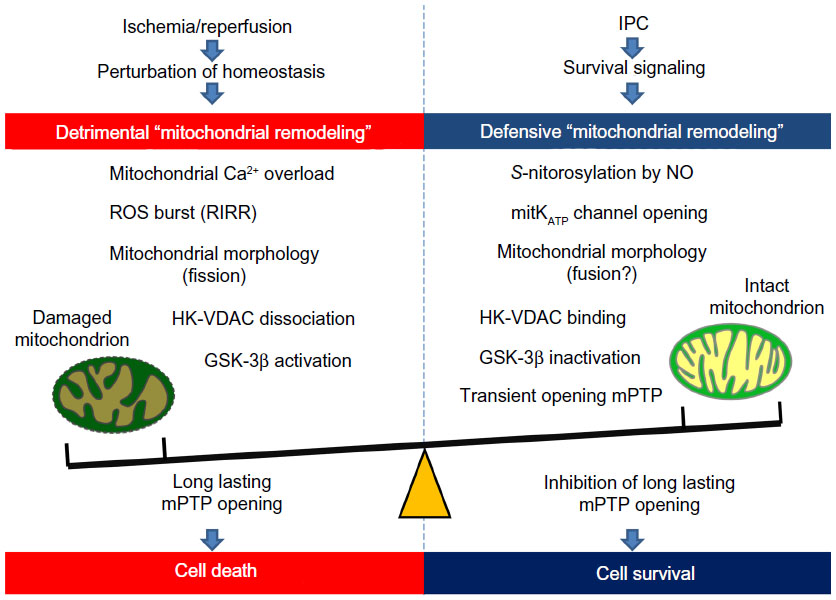 | Figure 4 Bidirectional (detrimental and defensive) “mitochondrial remodeling” in coronary heart disease. In ischemia/reperfusion injury, the mitochondria start their detrimental remodeling and play a crucial role in determination of cell death through mPTP opening. In contrast, the mitochondria can contribute to IPC-induced myocardial protection against ischemia/reperfusion injury. Defensive mitochondrial remodeling by IPC can decrease susceptibility of mPTP and thereby protects the myocardium from ischemia/reperfusion injury. |
Acknowledgments
This work was supported by Japan Grants-in-Aid (21590926 to MS, 23591036 to HK, 22590776 to HS, and 20590853 to HH), and by a grant-in-aid from the Ministry of Education, Culture, Sports, Science, and Technology.
Disclosure
The authors report no conflicts of interest in this work.
References
Takii T, Yasuda S, Takahashi J, et al. Trends in acute myocardial infarction incidence and mortality over 30 years in Japan: report from the MIYAGI-AMI Registry Study. Circ J. 2010;74(1):93–100. | |
Roger VL, Go AS, Lloyd-Jones DM, et al. Executive summary: heart disease and stroke statistics – 2012 update: a report from the American Heart Association. Circulation. 2012;125(1):188–197. | |
Minamino T. Cardioprotection from ischemia/reperfusion injury: basic and translational research. Circ J. 2012;76(5):1074–1082. | |
Heusch G. Cardioprotection: chances and challenges of its translation to the clinic. Lancet. 2013;381(9861):166–175. | |
Hausenloy DJ, Erik Botker H, Condorelli G, et al. Translating cardioprotection for patient benefit: position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res. 2013;98(1):7–27. | |
Ellis SG, Henschke CI, Sandor T, Wynne J, Braunwald E, Kloner RA. Time course of functional and biochemical recovery of myocardium salvaged by reperfusion. J Am Coll Cardiol. 1983;1(4):1047–1055. | |
Topol EJ, O’Neill WW, Langburd AB, et al. A randomized, placebo-controlled trial of intravenous recombinant tissue-type plasminogen activator and emergency coronary angioplasty in patients with acute myocardial infarction. Circulation. 1987;75(2):420–428. | |
Weiss JN, Korge P, Honda HM, Ping P. Role of the mitochondrial permeability transition in myocardial disease. Circ Res. 2003;93(4):292–301. | |
Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion – a target for cardioprotection. Cardiovasc Res. 2004;61(3):372–385. | |
Honda HM, Korge P, Weiss JN. Mitochondria and ischemia/reperfusion injury. Ann N Y Acad Sci. 2005;1047:248–258. | |
Gustafsson AB, Gottlieb RA. Heart mitochondria: gates of life and death. Cardiovasc Res. 2008;77(2):334–343. | |
Halestrap AP. A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochem Soc Trans. 2010;38(4):841–860. | |
Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90(1):207–258. | |
Andrienko T, Kuznetsov AV, Kaambre T, et al. Metabolic consequences of functional complexes of mitochondria, myofibrils and sarcoplasmic reticulum in muscle cells. J Exp Biol. 2003;206 Pt 12:2059–2072. | |
Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. 2nd ed. Kluwer Academic Publishers: Boston, MA, USA; 2001. | |
Dedkova EN, Blatter LA. Calcium signaling in cardiac mitochondria. J Mol Cell Cardiol. 2013;58:125–133. | |
Saotome M, Katoh H, Satoh H, et al. Mitochondrial membrane potential modulates regulation of mitochondrial Ca2+ in rat ventricular myocytes. Am J Physiol Heart Circ Physiol. 2005;288(4):H1820–H1828. | |
Hajnoczky G, Csordas G, Das S, et al. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium. 2006;40(5–6):553–560. | |
Baughman JM, Perocchi F, Girgis HS, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476(7360):341–345. | |
De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476(7360):336–340. | |
Perocchi F, Gohil VM, Girgis HS, et al. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. 2010;467(7313):291–296. | |
Csordas G, Golenar T, Seifert EL, et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab. 2013;17(6):976–987. | |
Drago I, Pizzo P, Pozzan T. After half a century mitochondrial calcium in- and efflux machineries reveal themselves. EMBO J. 2011;30(20):4119–4125. | |
Pacher P, Thomas AP, Hajnoczky G. Ca2+ marks: miniature calcium signals in single mitochondria driven by ryanodine receptors. Proc Natl Acad Sci U S A. 2002;99(4):2380–2385. | |
Eisner V, Csordas G, Hajnoczky G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle – pivotal roles in Ca(2)(+) and reactive oxygen species signaling. J Cell Sci. 2013;126 Pt 14:2965–2978. | |
Dorn GW 2nd, Maack C. SR and mitochondria: calcium cross-talk between kissing cousins. J Mol Cell Cardiol. 2013;55:42–49. | |
Hayashi T, Martone ME, Yu Z, et al. Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J Cell Sci. 2009;122 Pt 7:1005–1013. | |
Papanicolaou KN, Khairallah RJ, Ngoh GA, et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. 2011; 31(6):1309–1328. | |
Palty R, Silverman WF, Hershfinkel M, et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A. 2010;107(1):436–441. | |
Di Lisa F, Carpi A, Giorgio V, Bernardi P. The mitochondrial permeability transition pore and cyclophilin D in cardioprotection. Biochim Biophys Acta. 2011;1813(7):1316–1322. | |
Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552 Pt 2:335–344. | |
Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278(38):36027–36031. | |
Raedschelders K, Ansley DM, Chen DD. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol Ther. 2012;133(2):230–255. | |
Kadenbach B, Ramzan R, Moosdorf R, Vogt S. The role of mitochondrial membrane potential in ischemic heart failure. Mitochondrion. 2011;11(5):700–706. | |
Murphy E, Eisner DA. Regulation of intracellular and mitochondrial sodium in health and disease. Circ Res. 2009;104(3):292–303. | |
Basso C, Rizzo S, Thiene G. The metamorphosis of myocardial infarction following coronary recanalization. Cardiovasc Pathol. 2010;19(1):22–28. | |
Thiene G, Basso C. Myocardial infarction: a paradigm of success in modern medicine. Cardiovasc Pathol. 2010;19(1):1–5. | |
Kevin LG, Camara AK, Riess ML, Novalija E, Stowe DF. Ischemic preconditioning alters real-time measure of O2 radicals in intact hearts with ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2003;284(2):H566–H574. | |
Sack MN. Mitochondrial depolarization and the role of uncoupling proteins in ischemia tolerance. Cardiovasc Res. 2006;72(2):210–219. | |
Buja LM. Myocardial ischemia and reperfusion injury. Cardiovasc Pathol. 2005;14(4):170–175. | |
Buja LM. The pathobiology of acute coronary syndromes: clinical implications and central role of the mitochondria. Tex Heart Inst J. 2013;40(3):221–228. | |
Halestrap AP. Mitochondria and reperfusion injury of the heart – a holey death but not beyond salvation. J Bioenerg Biomembr. 2009;41(2):113–121. | |
Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys. 1979;195(2):460–467. | |
Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch Biochem Biophys. 1979;195(2):453–459. | |
Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. III. Transitional Ca2+ release. Arch Biochem Biophys. 1979;195(2):468–477. | |
Leung AW, Halestrap AP. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim Biophys Acta. 2008;1777(7–8):946–952. | |
Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of cyclophilin D. J Biol Chem. 2005;280(19):18558–18561. | |
Nakagawa T, Shimizu S, Watanabe T, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434(7033):652–658. | |
Baines CP, Kaiser RA, Purcell NH, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434(7033):658–662. | |
Giorgio V, von Stockum S, Antoniel M, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A. 2013;110(15):5887–5892. | |
Giorgio V, Soriano ME, Basso E, et al. Cyclophilin D in mitochondrial pathophysiology. Biochim Biophys Acta. 2010;1797(6–7):1113–1118. | |
Crompton M, Costi A, Hayat L. Evidence for the presence of a reversible Ca2+-dependent pore activated by oxidative stress in heart mitochondria. Biochem J. 1987;245(3):915–918. | |
Miura T, Tanno M, Sato T. Mitochondrial kinase signalling pathways in myocardial protection from ischaemia/reperfusion-induced necrosis. Cardiovasc Res. 2010;88(1):7–15. | |
Griffiths EJ, Halestrap AP. Protection by cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J Mol Cell Cardiol. 1993;25(12):1461–1469. | |
Clarke SJ, McStay GP, Halestrap AP. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J Biol Chem. 2002;277(38):34793–34799. | |
Hausenloy DJ, Duchen MR, Yellon DM. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovasc Res. 2003;60(3):617–625. | |
Hoppel CL, Tandler B, Fujioka H, Riva A. Dynamic organization of mitochondria in human heart and in myocardial disease. Int J Biochem Cell Biol. 2009;41(10):1949–1956. | |
Hofer T, Servais S, Seo AY, et al. Bioenergetics and permeability transition pore opening in heart subsarcolemmal and interfibrillar mitochondria: effects of aging and lifelong calorie restriction. Mech Ageing Dev. 2009;130(5):297–307. | |
Panasiuk O, Shysh A, Bondarenko A, Moibenko O. Omega-3 polyunsaturated fatty acid-enriched diet differentially protects two subpopulations of myocardial mitochondria against Ca(2+)-induced injury. Exp Clin Cardiol. 2013;18(1):e60–e64. | |
Miyamae M, Camacho SA, Weiner MW, Figueredo VM. Attenuation of postischemic reperfusion injury is related to prevention of [Ca2+]m overload in rat hearts. Am J Physiol. 1996;271(5 Pt 2):H2145–H2153. | |
Leperre A, Millart H, Prevost A, Trenque T, Kantelip JP, Keppler BK. Compared effects of ruthenium red and cis [Ru(NH3)4Cl2]Cl on the isolated ischaemic-reperfused rat heart. Fundam Clin Pharmacol. 1995;9(6):545–553. | |
Griffiths EJ. Use of ruthenium red as an inhibitor of mitochondrial Ca(2+) uptake in single rat cardiomyocytes. FEBS Lett. 2000; 486(3):257–260. | |
Pan X, Liu J, Nguyen T, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15(12):1464–1472. | |
Mallilankaraman K, Doonan P, Cardenas C, et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell. 2012;151(3):630–644. | |
Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192(7):1001–1014. | |
Beraud N, Pelloux S, Usson Y, et al. Mitochondrial dynamics in heart cells: very low amplitude high frequency fluctuations in adult cardiomyocytes and flow motion in non beating Hl-1 cells. J Bioenerg Biomembr. 2009;41(2):195–214. | |
Ong SB, Hausenloy DJ. Mitochondrial morphology and cardiovascular disease. Cardiovasc Res. 2010;88(1):16–29. | |
Archer SL. Mitochondrial dynamics – mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369(23):2236–2251. | |
Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337(6098):1062–1065. | |
Benard G, Karbowski M. Mitochondrial fusion and division: regulation and role in cell viability. Semin Cell Dev Biol. 2009;20(3):365–374. | |
Otera H, Wang C, Cleland MM, et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191(6):1141–1158. | |
Javadov S, Rajapurohitam V, Kilic A, et al. Expression of mitochondrial fusion-fission proteins during post-infarction remodeling: the effect of NHE-1 inhibition. Basic Res Cardiol. 2011;106(1):99–109. | |
Huang CH, Lazarou M, Youle RJ. Sequestration and autophagy of mitochondria do not cut proteins across the board. Proc Natl Acad Sci U S A. 2013;110(16):6252–6253. | |
Sanada S, Komuro I, Kitakaze M. Pathophysiology of myocardial reperfusion injury: preconditioning, postconditioning, and translational aspects of protective measures. Am J Physiol Heart Circ Physiol. 2011;301(5):H1723–H1741. | |
Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74(5):1124–1136. | |
Candilio L, Hausenloy DJ, Yellon DM. Remote ischemic conditioning: a clinical trial’s update. J Cardiovasc Pharmacol Ther. 2011;16(3–4):304–312. | |
Hausenloy DJ, Yellon DM. The therapeutic potential of ischemic conditioning: an update. Nat Rev Cardiol. 2011;8(11):619–629. | |
Zuurbier CJ, Eerbeek O, Meijer AJ. Ischemic preconditioning, insulin, and morphine all cause hexokinase redistribution. Am J Physiol Heart Circ Physiol. 2005;289(1):H496–H499. | |
Javadov SA, Clarke S, Das M, Griffiths EJ, Lim KH, Halestrap AP. Ischaemic preconditioning inhibits opening of mitochondrial permeability transition pores in the reperfused rat heart. J Physiol. 2003;549 Pt 2:513–524. | |
Hausenloy DJ, Tsang A, Yellon DM. The reperfusion injury salvage kinase pathway: a common target for both ischemic preconditioning and postconditioning. Trends Cardiovasc Med. 2005;15(2):69–75. | |
Downey JM, Davis AM, Cohen MV. Signaling pathways in ischemic preconditioning. Heart Fail Rev. 2007;12(3–4):181–188. | |
Costa AD, Garlid KD. Intramitochondrial signaling: interactions among mitoKATP, PKCepsilon, ROS, and MPT. Am J Physiol Heart Circ Physiol. 2008;295(2):H874–H882. | |
Kobayashi T, Sato S, Takamiya S, et al. Mitochondria and apicoplast of Plasmodium falciparum: behaviour on subcellular fractionation and the implication. Mitochondrion. 2007;7(1–2):125–132. | |
Tong H, Imahashi K, Steenbergen C, Murphy E. Phosphorylation of glycogen synthase kinase-3beta during preconditioning through a phosphatidylinositol-3-kinase-dependent pathway is cardioprotective. Circ Res. 2002;90(4):377–379. | |
Juhaszova M, Zorov DB, Kim SH, et al. Glycogen synthase kinase- 3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113(11):1535–1549. | |
Pastorino JG, Hoek JB. Regulation of hexokinase binding to VDAC. J Bioenerg Biomembr. 2008;40(3):171–182. | |
Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V. In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem J. 2004;377 Pt 2:347–355. | |
Chiara F, Castellaro D, Marin O, et al. Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS One. 2008;3(3):e1852. | |
Pastorino JG, Hoek JB, Shulga N. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005;65(22):10545–10554. | |
Sun J, Murphy E. Protein S-nitrosylation and cardioprotection. Circ Res. 2010;106(2):285–296. | |
Otani H. The role of nitric oxide in myocardial repair and remodeling. Antioxid Redox Signal. 2009;11(8):1913–1928. | |
Nguyen TT, Stevens MV, Kohr M, Steenbergen C, Sack MN, Murphy E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J Biol Chem. 2011;286(46):40184–40192. | |
Ohtani H, Katoh H, Tanaka T, et al. Effects of nitric oxide on mitochondrial permeability transition pore and thiol-mediated responses in cardiac myocytes. Nitric Oxide. 2012;26(2):95–101. | |
Chen L, Knowlton AA. Mitochondrial dynamics in heart failure. Congest Heart Fail. 2011;17(6):257–261. | |
Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121(18):2012–2022. | |
Sharp WW, Fang YH, Han M, et al. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 2014;28(1):316–326. | |
Ichas F, Mazat JP. From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim Biophys Acta. 1998; 1366(1–2):33–50. | |
Petronilli V, Miotto G, Canton M, et al. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys J. 1999;76(2):725–734. | |
Jacobson J, Duchen MR. Mitochondrial oxidative stress and cell death in astrocytes – requirement for stored Ca2+ and sustained opening of the permeability transition pore. J Cell Sci. 2002;115 Pt 6:1175–1188. | |
Korge P, Yang L, Yang JH, Wang Y, Qu Z, Weiss JN. Protective role of transient pore openings in calcium handling by cardiac mitochondria. J Biol Chem. 2011;286(40):34851–34857. | |
Saotome M, Katoh H, Yaguchi Y, et al. Transient opening of mitochondrial permeability transition pore by reactive oxygen species protects myocardium from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2009;296(4):H1125–H1132. | |
Hausenloy DJ, Lim SY, Ong SG, Davidson SM, Yellon DM. Mitochondrial cyclophilin-D as a critical mediator of ischaemic preconditioning. Cardiovasc Res. 2010;88(1):67–74. | |
Walters AM, Porter GA Jr, Brookes PS. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circ Res. 2012;111(9):1222–1236. | |
Wang X, Wei M, Kuukasjarvi P, et al. Novel pharmacological preconditioning with diazoxide attenuates myocardial stunning in coronary artery bypass grafting. Eur J Cardiothorac Surg. 2003;24(6):967–973. | |
Deja MA, Malinowski M, Golba KS, et al. Diazoxide protects myocardial mitochondria, metabolism, and function during cardiac surgery: a double-blind randomized feasibility study of diazoxide-supplemented cardioplegia. J Thorac Cardiovasc Surg. 2009;137(4):997–1004, 1004e1–2. | |
Kitakaze M, Asakura M, Kim J, et al. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): two randomised trials. Lancet. 2007;370(9597):1483–1493. | |
Piot C, Croisille P, Staat P, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008; 359(5):473–481. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.