Back to Journals » International Medical Case Reports Journal » Volume 19
Lamellar Ichthyosis in a Resource-Limited Setting: A Somaliland Case Report
Authors Aw Hashi MO, Derie AM ![]() , Ahmed SI
, Ahmed SI ![]()
Received 1 December 2025
Accepted for publication 25 March 2026
Published 30 March 2026 Volume 2026:19 583796
DOI https://doi.org/10.2147/IMCRJ.S583796
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Thomas E Hutson
Mohamed Osman Aw Hashi,1,2 Ahmed M Derie,3 Sadam Ismail Ahmed4
1College of medicine and surgery, University of Hargeisa, Hargeisa, Somaliland; 2Department of Dermatology and Venereology, Hargeisa Group Hospital, Hargeisa, Somaliland; 3Department of Pediatric, Gargaar Multispecialty and Teaching Hospital, Hargesia, Somaliland; 4Faculty of medicine and health science, University of Burao, Burao, Somaliland
Correspondence: Sadam Ismail Ahmed, Email [email protected]
Introduction: Lamellar Ichthyosis (LI) is a rare, severe autosomal recessive genodermatosis present at birth as a collodion membrane. Managing this condition in resource-limited settings like Somaliland presents significant challenges due to limited diagnostic capabilities and multidisciplinary care.
Case Presentation: A one-day-old, full-term male neonate was presented to the dermatology department with a history of generalized skin encasement since birth and incomplete eyelid closure. The patient was born to consanguineous parents, with a family history of similar skin disorders among relatives. Clinical examination revealed a classic collodion membrane with bilateral ectropion and eclabium. Systemic examination was otherwise unremarkable. A clinical diagnosis of Lamellar Ichthyosis was made based on the classic presentation. Management involved bland emollients, ophthalmology referral for ocular lubricants, and extensive parental counseling. The patient was lost to follow-up after discharge, highlighting a common barrier to care in this context.
Conclusion: This case underscores the diagnostic and management challenges of rare genodermatoses in resource-limited settings. It emphasizes the critical role of clinical diagnosis, basic supportive care, and the need for improved patient education and follow-up systems to ensure continuity of care.
Keywords: lamellar ichthyosis, collodion baby, genodermatosis, resource-limited setting, Gargaar Multispeciality and Teaching Hospital Hargeisa Somaliland
Introduction
Lamellar Ichthyosis (LI) is a rare autosomal recessive disorder of keratinization, with an estimated incidence of 1 in 200,000 to 300,000 births.1 It is caused by mutations in the transglutaminase-1 (TGM1) gene in most cases, leading to defective cornification and desquamation of the stratum corneum.2 The classic presentation is that of a “collodion baby,” where the neonate is born encased in a taut, cellophane-like membrane that sheds within the first few weeks of life, revealing underlying generalized scaling.3
Lamellar ichthyosis (LI) represents one of the most severe and clinically distinct phenotypes within the spectrum of autosomal recessive congenital ichthyoses (ARCI), a group of hereditary disorders of keratinization characterized by generalized hyperkeratosis and scaling.4,5 First described in the medical literature as early as the 19th century, this condition results from a fundamental defect in the formation of the stratum corneum, leading to a profoundly compromised epidermal barrier.6 The neonatal presentation of LI is often dramatic and immediately recognizable, presenting a unique set of diagnostic challenges and demanding intensive, multidisciplinary management from the moment of birth.
The initial neonatal period is critical due to high risks of transepidermal water loss, temperature instability, electrolyte imbalance, and septicemia. Furthermore, ectropion (eversion of the eyelids) and eclabium (eversion of the lips) are common associated features that require specialized care to prevent corneal damage and feeding difficulties.7
Managing LI in high-income settings involves a multidisciplinary approach including dermatology, ophthalmology, and genetic counseling. However, in resource-limited settings like Somaliland, the absence of genetic testing, limited specialist availability, and socioeconomic barriers pose significant challenges to optimal care. We present a case of a collodion baby diagnosed clinically with Lamellar Ichthyosis in Hargeisa, Somaliland, to highlight these challenges and discuss pragmatic management strategies.
Case Presentation
A one-day-old male neonate was brought to the dermatology department of Gargaar Multispeciality and Teaching Hospital, Hargeisa, Somaliland who presented with a shiny, tight membrane covering his entire body and had incomplete closure of the eyelids both while asleep and awake at birth.
The mother is a 50-year-old primigravida who conceived via in vitro fertilization (IVF). The father is 70 years old. The parents are consanguineous, and there was a reported history of a similar skin disorder among cousins and other relatives. The antenatal period was uncomplicated, with regular clinic attendance throughout all trimesters and reported normal obstetric ultrasounds. The mother took only vitamin and folic acid supplements during pregnancy. The family is of low socioeconomic status, residing in a rural village.
The baby was delivered via Cesarean section at term, with a birth weight of 2.4 kg. He cried spontaneously and required no resuscitation.
Clinical Findings
On examination, the neonate was alert with no signs of systemic distress or infection. Dermatological examination revealed a translucent, collodion-like membrane encasing the entire body, causing dryness and hardness of the skin. Significant bilateral ectropion and eclabium were present (Figure 1). There was no oozing, crusting, or erythema at this initial presentation. Systemic examinations of the cardiovascular, respiratory, and abdominal systems were unremarkable.
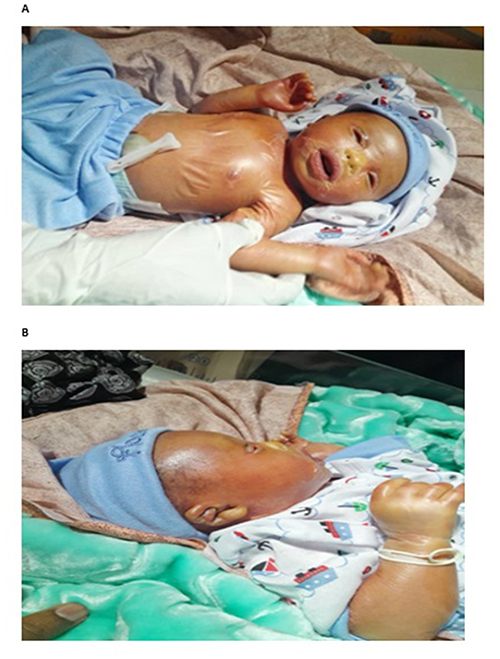 |
Figure 1 (A and B) The neonate at birth, showing encasement in a collodion membrane, bilateral ectropion, and eclabium. |
Diagnostic Focus and Assessment
A clinical diagnosis of Lamellar Ichthyosis was made based on the classic presentation of a collodion membrane with ectropion and a positive family history suggestive of an autosomal recessive pattern. Due to resource limitations, genetic testing was not available. Basic laboratory investigations, including a complete blood count (CBC), electrolytes, and erythrocyte sedimentation rate (ESR), were performed and were within normal limits, ruling out immediate electrolyte imbalance or infection.
Therapeutic Intervention and Follow-Up
The management plan focused on supportive care:
- Skin Care: Application of bland emollients (Vaseline and CeraVe cream) multiple times daily to promote membrane shedding and maintain skin hydration.
- Ophthalmological Care: Urgent referral to ophthalmology. The ophthalmologist prescribed ocular lubricants and prophylactic antibacterial eye drops to protect the cornea from exposure keratitis.
- Parental Counseling: The parents were extensively counseled on the chronic nature of the disease, its prognosis, and the critical importance of daily skin care and follow-up.
The patient was discharged on day 3 of life. At this point, the membrane had begun to shed, revealing underlying diffuse erythema and scaling, with some fissuring noted at the flexures (Figure 2).
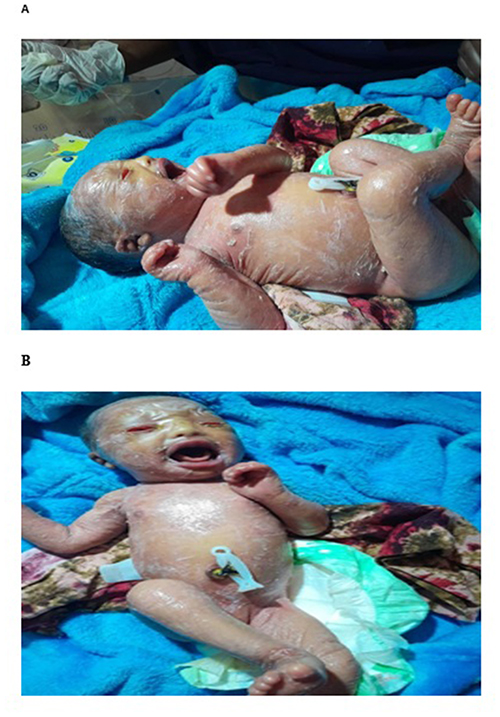 |
Figure 2 (A and B) Day 3, showing diffuse redness and scaling, with fissuring at the flexures. The eclabium has resolved, allowing for unrestricted crying. |
The family was advised to return for regular follow-up. However, they returned to their rural village and a follow-up visit at 3 weeks was achieved. At this visit (Figure 3), the skin showed thick, plate-like scales on the forehead, with persistent scaling on the face and abdomen.
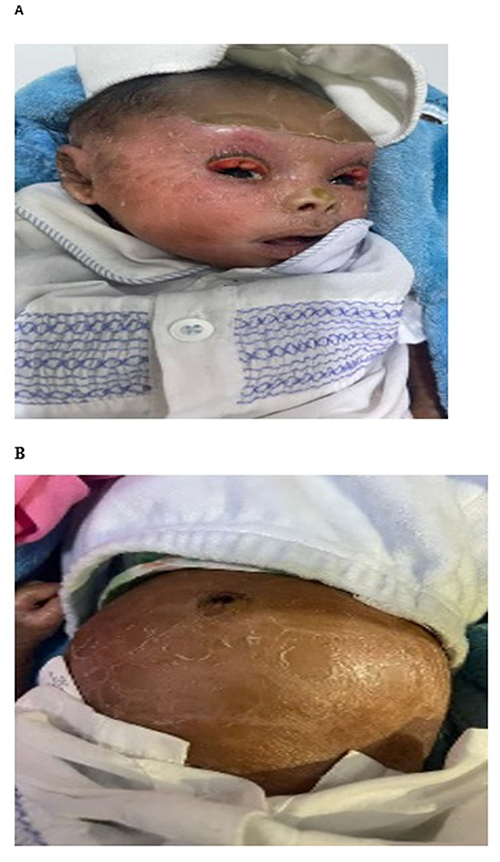 |
Figure 3 (A and B) Day 21, showing thick, plate-like scales on the forehead, face, and abdomen, with some improvement in eyelid closure. |
A partial improvement in the ectropion was noted, with some closure of the eyelids. The family reported applying emollients regularly but was subsequently lost to further follow-up.
Discussion
Lamellar Ichthyosis (LI) is a severe and rare autosomal recessive disorder of cornification, belonging to the heterogeneous group of congenital ichthyoses. Its estimated incidence ranges from 1 in 200,000 to 1 in 300,000 live births, though this may be higher in populations with high rates of consanguinity.8 The classic presentation is that of a “collodion baby,” where the neonate is born encased in a taut, translucent, cellophane-like membrane that predisposes them to critical complications. Genetically, LI is most commonly caused by mutations in the TGM1 gene, which encodes transglutaminase-1, a key enzyme responsible for the formation of the cornified cell envelope in the epidermis.2 This enzymatic defect leads to abnormal desquamation, impaired barrier function, and persistent hyperkeratosis. Other implicated genes include ABCA12, ALOXE3, ALOX12B, and CYP4F22, contributing to the genetic heterogeneity of the condition.4 The presence of consanguinity, as seen in our case from Somaliland, significantly increases the risk of autosomal recessive disorders like LI, and a positive family history among cousins is a strong corroborating factor. This case illustrates the classic presentation and natural history of Lamellar Ichthyosis, beginning with the collodion membrane phase and evolving into widespread scaling. The positive family history and parental consanguinity strongly support an autosomal recessive inheritance, which is well-documented in LI.1,2
The primary challenge in this setting was the reliance on clinical diagnosis alone. While genetic confirmation is the gold standard, it is inaccessible in Somaliland. This underscores the continued importance of skilled clinical assessment in dermatology. The initial management was successful in preventing life-threatening neonatal complications like hypernatremic dehydration or sepsis through basic but diligent emollient therapy and ophthalmological collaboration. The loss to follow-up is a significant concern and a common outcome in similar settings.9 Barriers include distance from the healthcare facility, low socioeconomic status, and potentially a lack of understanding of the long-term needs of a chronic condition. This highlights an area for crucial intervention: strengthening patient education and developing community-based follow-up systems to monitor these vulnerable patients.
Differential Diagnosis
Both NBCIE (nonbullous congenital ichthyosiform erythroderma) or LI (lamellar ichthyosis) have a similar presentation at birth and equally affects males and females.10 They can be differentiated only after infancy when LI presents with brown plate-like scales on non erythematous base while NBCIE is characterized by generalized erythroderma with overlying fine white scales. Although, both LI and NBCIE have a normal life span, symptoms of LI remain severe throughout life while NBCIE improves after puberty.11
Conclusion
This report describes the successful initial clinical diagnosis and management of a rare genodermatosis in a resource-limited setting. It demonstrates that even without advanced diagnostics, positive outcomes can be initiated through a structured clinical approach, basic supportive care, and effective interdisciplinary communication. However, the case also reveals the fragility of long-term care, emphasizing the urgent need for strategies to improve patient retention and follow-up to prevent long-term sequelae such as blindness and contractures.
Ethical Approval
In our institute, the ethical approval is not required for publication of case reports, so our hospital is waived for case reports.
Patient Perspective
The parents expressed profound anxiety about their child’s condition and future. They were grateful for the explanation and initial care but were concerned about the long-term management and costs associated with the disease, which contributed to their decision to return to their village.
Consent for Publication
Written informed consent was obtained from the patient’s parents for publication of this case report and accompanying images.
Informed Consent
Informed consent was obtained from the patient’s parents (Primary Care giver) for the publication of this case report.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work”.
Funding
No funding for this case.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Akiyama M. The pathogenesis of severe congenital ichthyosis of the neonate. J Dermatol Sci. 1999;21(2):96–6. doi:10.1016/S0923-1811(99)00024-9
2. Huber M, Rettler I, Bernasconi K, et al. Mutations of keratinocyte transglutaminase in lamellar ichthyosis. Science. 1995;267(5197):525–528. doi:10.1126/science.7824952
3. Vahlquist A, Gånemo A, Pigg M, et al. The clinical spectrum of congenital ichthyosis in Sweden: a review of 127 cases. Acta Derm Venereol Suppl. 2003;213:3–13.
4. Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the first ichthyosis consensus conference in sorèze 2009. J Am Acad Dermatol. 2010;63(4):607–641. doi:10.1016/j.jaad.2009.11.020
5. Williams ML, Elias PM. From basket weave to barrier. Unifying concepts for the pathogenesis of the disorders of cornification. Arch Dermatol. 1993;129(5):626–629. doi:10.1001/archderm.1993.01680260096015
6. Online Mendelian Inheritance in Man (OMIM). Ichthyosis, Lamellar, Autosomal Dominant; ADLI. Entry #146750. Available from:
7. Rodríguez-Pazos L, Zhang J, Dooley J, et al. Autosomal recessive congenital ichthyosis. Orphanet J Rare Dis. 2013;8:79. doi:10.1186/1750-1172-8-79
8. Vahlquist A, Bygum A, Gånemo A, et al. Genotypic and clinical spectrum of self-improving collodion ichthyosis: ALOX12B, ALOXE3, and TGM1 mutations in Scandinavian patients. J Invest Dermatol. 2010;130(2):438–443. doi:10.1038/jid.2009.346
9. Derseh D, et al. Challenges in the management of congenital ichthyosis in a low-resource setting: a case series from Ethiopia. Clin Case Rep. 2021;9;8:e04582.
10. Dyer JA, Spraker M, Williams M. Care of the newborn with ichthyosis. Dermatol Thera. 2013;26(1):1–5. doi:10.1111/j.1529-8019.2012.01555.x
11. Cortina WJ, Cruz MJ, Villalobos OA, Espinoza MA. Ictiosis congénita (feto arlequín). Bol Med Hosp Infant Mex. 1975;32:699–702.
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.