Back to Journals » International Journal of Nanomedicine » Volume 13
Lactoferrin- and RGD-comodified, temozolomide and vincristine-coloaded nanostructured lipid carriers for gliomatosis cerebri combination therapy
Authors Zhang J, Xiao X, Zhu J, Gao Z, Lai X, Zhu X, Mao G
Received 30 December 2017
Accepted for publication 22 March 2018
Published 22 May 2018 Volume 2018:13 Pages 3039—3051
DOI https://doi.org/10.2147/IJN.S161163
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Jicai Zhang, Xiang Xiao, Jianming Zhu, Ziyun Gao, Xianliang Lai, Xingen Zhu, Guohua Mao
Department of Neurosurgery, Second Affiliated Hospital, Nanchang University, Nanchang, Jiangxi, China
Purpose: Glioblastoma multiforme (GBM) is the most common malignant brain tumor originating in the central nervous system in adults. Based on nanotechnology such as liposomes, polymeric nanoparticles, and lipid nanoparticles, recent research efforts have been aimed to target drugs to the brain.
Methods: In this study, lactoferrin- and arginine–glycine–aspartic acid (RGD) dual-ligand-comodified, temozolomide and vincristine-coloaded nanostructured lipid carriers (L/R-T/V-NLCs) were introduced for GBM combination therapy. The physicochemical properties of L/R-T/V-NLCs such as particle size, zeta potential, and encapsulated efficiency are measured. The drug release profile, cellular uptake, cytotoxicity, tissue distribution, and antitumor activity of L/R-T/V-NLCs are further investigated in vitro and in vivo.
Results: L/R-T/V-NLCs were stable with nanosize and high drug encapsulation efficiency. L/R-T/V-NLCs exhibited sustained-release behavior, high cellular uptake, high cytotoxicity and synergy effects, increased drug accumulation in the tumor tissue, and obvious tumor inhibition efficiency with low systemic toxicity.
Conclusion: L/R-T/V-NLCs could be a promising drug delivery system for glioblastoma chemotherapy.
Keywords: gliomatosis cerebri, combination therapy, nanostructured lipid carriers, lactoferrin, arginine–glycine–aspartic acid peptide, vincristine, temozolomide
Introduction
Glioblastoma multiforme (GBM, World Health Organization grade IV astrocytoma) is the most common malignant brain tumor originating in the central nervous system in adults.1 The standard treatment includes resection followed by radiotherapy and temozolomide (TMZ) chemotherapy; a median survival of 14.6 months can be expected by GBM patients.2 However, due to the limited delivery of therapeutics across the blood–brain barrier (BBB) and therapeutic resistance, the poor prognosis can still be troublesome and there is a critical need for means to cross the BBB and improve the efficacy of current GBM therapies.3 On one hand, combined chemotherapy is emerging as an important strategy to defeat drug resistance, maximize the therapeutic effect, and reduce side effects.4 On the other hand, a novel targeted nanoparticulate delivery system as related to brain tumors is urgently needed to improve the solubility, prolong the circulation time, enhance the targeted effect, and then reduce the systemic toxicity.5
Based on nanotechnology such as liposome, polymeric nanoparticles (NPs), and lipid NPs, recent research efforts have been aimed to target drugs to the brain.6–8 Compared to conventional drug formulations, carefully designed nanoformulations offer significant advantages such as improved drug solubility, facilitation of drug delivery across the BBB, selective targeting, and reduced side effects.9 It has been reported that lipid NPs improve the drug absorption and bioavailability because of their diameter and absorption-enhancing effect of lipids.10 Nanostructured lipid carriers (NLCs) represent an improved generation of lipid NPs.11 They are obtained by controlled mixing of solid lipids with spatially incompatible liquid lipids, leading to a specific nanostructure to accommodate therapeutics, and thus achieve higher loading capacity.12 Our previous work has already proved that NLCs are better for GBM chemotherapy than solid lipid NPs and polymeric NPs.9
Lactoferrin (Lf), a member of the transferrin (Tf) family, is a double-lobe glycoprotein originally found in an iron-binding site to reversibly chelate and carry iron.13 It has been observed that GBM cells expressed Lf receptor.14 Hence, the conjugated Lf can be employed to modulate the transcytosis across the BBB and permeation to GBM through receptor-mediated signaling pathways. In fact, Lf is reported to cause an inhibition to the multiplication of malignant U87MG cells, one of the representative GBM cells, through downregulation of cyclin D1 and D4.15
The tripeptide arginine–glycine–aspartic acid (RGD) could specifically bind with αvβ3, which was overexpressed on neurovascular endothelial cells.16 RGD-linked nanocarriers have shown therapeutic efficacy in GBM.17 In our previous study, RGD-conjugated polyethylene glycol-b-distearoylphosphatidylethanolamine (PEG-DSPE) was synthesized and RGD-modified, TMZ-loaded NLCs were fabricated for the GBM chemotherapy.5
Combination chemotherapy is increasingly used as a primary cancer treatment regimen to avoid the poor response and resistance often associated with monotherapy.18 Our previous studies have established the antitumor efficacy on GBM by codelivery of TMZ and vincristine (VCR)4 or TMZ and gene10 by NPs. In this study, we would like to introduce Lf and RGD dual-ligand-comodified, TMZ and VCR–coloaded nanostructured lipid carriers (L/R-T/V-NLCs) for GBM combination therapy. The physicochemical properties of L/R-T/V-NLCs such as particle size, zeta potential, and encapsulated efficiency are measured. The drug release profile, cellular uptake, cytotoxicity, tissue distribution, and antitumor activity of L/R-T/V-NLCs are further investigated in vitro and in vivo.
Materials and methods
Materials
RGD peptide was supplied by the GL Biochem Ltd (Shanghai, China). Lf, 2-iminothiolane (Traut’s Reagent), stearic acid, soybean phosphatidylcholine (SPC), dimethyldioctadecylammonium bromide (DDAB), and MTT were purchased from Sigma Aldrich (St Louis, MO, USA). COMPRITOL® 888 ATO (888 ATO) was generously provided by Gattefossé (Paramus, NJ, USA). Polyoxyl castor oil (Cremophor ELP) was donated by BASF (Ludwigshafen, Germany). Injectable soya lecithin was obtained from Shanghai Taiwei Pharmaceutical Co, Ltd (Shanghai, China). Distearoylphosphatidylethanolamine-polyethylene glycol (MW~2 kDa)-maleimide (DSPE-PEG2000-MAL) and DSPE-PEG2000-NH2 were purchased from Xi’an ruixi Biological Technology Co., Ltd (Xi’an, China). VCR was purchased from Shaanxi Pioneer Biotech Co., Ltd (Xi’an, China). TMZ was kindly provided by Laimei Pharmaceutical Co., Ltd (Chongqing, China). All other chemicals were of analytical grade or high-performance liquid chromatography (HPLC) grade and used without further purification.
Cells and animals
U87 malignant glioma cells (U87 MG cells), T98G human malignant glioma cell line (T98G cells), and A549 lung adenocarcinoma cells (A549 cells) were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in DMEM (Sigma, St Louis, MO, USA) supplemented with 10% fetal bovine serum (Fisher Chemicals, Fairlawn, NJ, USA) in a 5% CO2 fully humidified atmosphere.
BALB/c nude mice (5–6 weeks old, 18–22 g) were purchased from the Shanghai Slack Laboratory Animal Co., Ltd. All animal experiments complied with the Guide of the National Institutes of Health for the care and use of laboratory animals and the Animal Care and Use Committee, Nanchang University approved the animal protocols used (No NCU001020170903001).
Synthesis of Lf-PEG-DSPE and RGD-PEG-DSPE
Lactoferrin-PEG-DSPE (Lf-PEG-DSPE) was synthesized by conjugating Lf to the distal MAL functional groups on DSPE-PEG2000-MAL (Figure 1). Lf was first thiolated with 1 equivalent of Traut’s reagent.19 The resulted thiolated Lf was added with 1 equivalent of DSPE-PEG2000-MAL and reacted at room temperature for 20 h to form a thioether bonding with the MAL functional group at the N-terminus of DSPE-PEG2000-MAL chain.20 The mixture was centrifuged at 10,000 g for 30 min at 4°C, and then resuspended in PBS (pH 7.4). Then, Lf-PEG-DSPE was obtained after freeze-drying, yielding a white, crystalline powder.
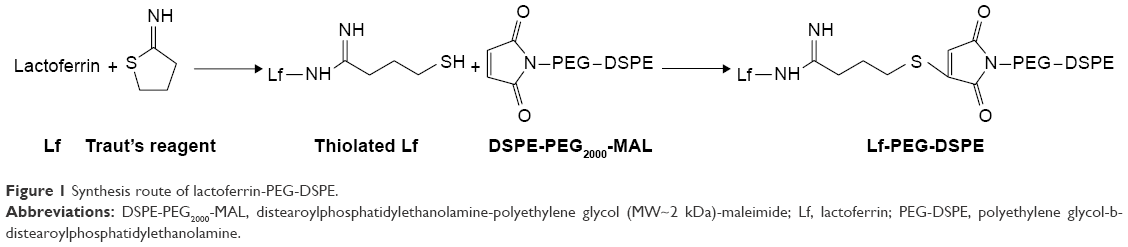 | Figure 1 Synthesis route of lactoferrin-PEG-DSPE. |
RGD-PEG-DSPE was synthesized as described in our previous study.5 DSPE-PEG2000-NH2 was dissolved in DMSO. RGD was added and stirred for 24 h. After completion of the reaction, the solution was dialyzed successively against Milli-Q water (membrane tubing, molecular weight cutoff 1,000 Da). Then, RGD-PEG-DSPE was obtained after freeze-drying.
Preparation of L/R-T/V-NLCs
L/R-T/V-NLCs (Figure 2) were prepared by solvent diffusion method.10 SPC (200 mg) and 888 ATO (200 mg) were dispersed in Cremophor ELP (1 mL) to form lipid dispersion. Injectable soya lecithin (100 mg), TMZ (50 mg), and VCR (50 mg) were dissolved in 1 mL of dimethyl formamide and added to the lipid dispersion with heating at the temperature of 80°C–85°C to form the lipid phase. Aqueous phase was prepared by dispersing Lf-PEG-DSPE (100 mg), RGD-PEG-DSPE (100 mg), and 0.5% DDAB weight/volume (w/v) in 10 mL of water. The lipid phase was rapidly injected into the stirred aqueous phase (800 rpm), and the resulting suspension was then dispersed with Milli-Q water and then dialyzed against Milli-Q water for 24 h to get the L/R-T/V-NLCs suspension. The L/R-T/V-NLCs suspension was washed using Milli-Q water twice and resuspended in PBS (pH 7.4), filtered through a membrane with 0.45 μm pore size (Phenomenex, 25 mm filter, CA) and stored at 2°C–8°C.
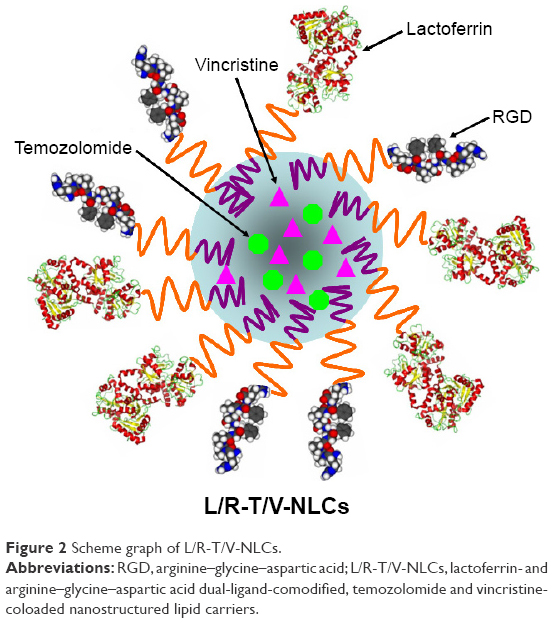 | Figure 2 Scheme graph of L/R-T/V-NLCs. |
Blank Lf and RGD dual-ligand-comodified NLCs (L/R-NLCs) were prepared using the same method without adding TMZ and VCR.
Lf and RGD dual-ligand-comodified, single-TMZ-loaded NLCs (L/R-T-NLCs) were prepared using the same method without adding VCR.
Lf and RGD dual-ligand-comodified, single-VCR-loaded NLCs (L/R-V-NLCs) were prepared using the same method without adding TMZ.
Single-Lf-ligand-modified, TMZ and VCR–coloaded NLCs (L-T/V-NLCs) were prepared using the same method without adding RGD-PEG-DSPE.
Single-RGD-ligand-modified, TMZ and VCR–coloaded NLCs (R-T/V-NLCs) were prepared using the same method without adding Lf-PEG-DSPE.
No-ligand-modified, TMZ and VCR–coloaded NLCs (T/V-NLCs) were prepared using the same method without adding Lf-PEG-DSPE and RGD-PEG-DSPE.
Characterization of particle size and surface charge
The suspensions were diluted with deionized water to an appropriate concentration for the measurement. The particle sizes and surface charges (zeta potentials) of the NPs were measured on a Nano-ZS Zetasizer dynamic light scattering detector (Malvern Instruments, Malvern, UK) at 25°C.21 The width of the size distributions was characterized by the polydispersity index (PDI).
Characterization of drug encapsulation and drug-loading efficiency
The drug encapsulation efficiency (EE) and drug-loading efficiency (DL) of NLCs were measured.4 For the TMZ measurement, the HITACHI P-4010 inductively coupled plasma mass spectrometry (ICP-MS) (Hitachi Ltd, Kyoto, Japan) was used. Briefly, 5 mL SLNs or NLCs was centrifuged (16,000 rpm and 4°C for 30 min) separately, and the supernatants were then determined using the ICP-MS. The EE was expressed as the percentage of the amount of TMZ encapsulated in the NLCs to the total amount of TMZ initially used. For the VCR measurement, HPLC was applied. Briefly, ethanol was added to disrupt the SLNs or NLCs, and 20 μL of the resulting transparent solution was injected into an HPLC system (Agilent 1260; Agilent Technologies, Santa Clara, CA, USA). A Kromasil C18 reverse phase column (150×4.6 mm, 5 μm; AkzoNobel, Separation Products, Bohus, Sweden) and the mobile phase consisting of acetonitrile and 0.01 M NaH2PO4 (55/45, volume/volume [v/v], pH 7.0 adjusted with triethylamine) were used to separate the targeted component. The samples were eluted by mobile phase at a flow rate of 1.0 mL/min at 35°C and monitored at 297 nm.
EE was calculated as follows: EE (%)=Weight of (total drug–free drug)/Weight of total drug×100.
DL was calculated as follows: DL (%)=Weight of (total drug–free drug)/Weight of total drug and carriers×100.
Stability of NLCs
NLC suspensions were maintained at 2°C–8°C and the stability of NLCs was evaluated by measuring the NLC sizes, PDI, and drug EE of samples for 90 days.22 Results are recorded at 0, 10, 20, 30, 60, and 90 days. Mean particle sizes and EE were determined by the methods as described in the sections Characterization of Particle Size and Surface Charge and Characterization of Drug Encapsulation and Drug-loading Efficiency.
In vitro drug release
For in vitro drug release study, NLCs were reconstituted in PBS (pH 7.4) and transferred into dialysis bags molecular weight cut-off (MWCO: 20 kDa) that were placed in the same PBS at 37°C and stirred at 100 rpm.23 At scheduled time intervals, 100 μL of the dialysate was collected and the dialysate replenished with the same amount of fresh PBS. The concentration of the released TMZ and VCR in the removed dialysate was determined by the methods as described in the section Characterization of Drug Encapsulation and Drug-loading Efficiency.
Cellular uptake of NLCs
To quantitatively determine the cellular uptake of the NLCs, intracellular accumulation assay was used on U87 MG cells and A549 cells.24 Coumarin 6 (C6) was applied as a model fluorescent molecule and was loaded into the NLCs by adding C6 to the lipid dispersion. The cells were equilibrated with Hank’s buffered salt solution at 37°C for 1 h before C6-loaded NLCs were added at a concentration of 200 mg/mL. The medium was removed after incubation for the determined time, and the wells were washed three times with cold PBS solution and detached with trypsin/EDTA. Then the cells were centrifuged at 1,500 rpm, 4°C for 5 min, the supernatant was discarded, 300 μL of PBS was added to resuspend the cells and directly introduced to a FACSCalibur flow cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ, USA).
In vitro cytotoxicity and synergistic effect
The cytotoxicity of NLCs was determined by MTT assay.25 U87 MG cells and T98G cells were seeded in 96-well plates at a density of 104 cells per well and grown overnight at 37°C in a 5% CO2 incubator, separately. All kinds of prepared NLCs, TMZ, and VCR mixed solution (T/V-solution), TMZ solution (T-solution), and VCR solutions (V-solution) at TMZ or VCR concentrations of 1, 5, 10, 50, and 100 μg/mL were added into each well and incubated for 72 h. After incubation, 10 μL MTT solution (5 mg/mL in PBS) aliquots were added to each well and incubated at 37°C for 4 h. Then, MTT formazan crystals were dissolved in 200 μL of DMSO. Cell viability was assessed according to the MTT manufacturer’s procedures and the absorbance at 570 nm was measured using a microplate reader (Model 680, Bio-Rad Laboratories Inc., Philadelphia, PA, USA). Cells without the addition of MTT reagents were used as a blank control. The drug concentration causing 50% inhibition (IC50) was calculated using the CalcuSyn software (Biosoft, Ferguson, MO, USA).
Combination index (CI) was measured to study the synergy in TMZ and VCR in the double drugs combination system on U87 MG cells. CI was calculated as follows: CI50=(D)1/(D50)1+(D)2/(D50)2. (D)1 and (D)2 represent the concentrations of TMZ and VCR in the combination system at the IC50 value. (D50)1 and (D50)2 represent the IC50 values of TMZ alone and VCR alone, respectively. CI50<1 and >1 represent synergism and antagonism, respectively.
In vivo tissue distribution of NLCs
In vivo tissue distribution of NLCs was conducted on U87 MG cells bearing BALB/c nude mice.26 U87 MG cells (106 cells in 200 μL of DMEM) were subcutaneously injected in the right flank regions of mice. When the tumor volumes reached around 100 mm3, the mice were randomly distributed into 10 groups with eight mice in each group, and intravenously injected with L/R-T/V-NLCs, R-T/V-NLCs, L-T/V-NLCs, T/V-NLCs, L/R-V-NLCs, L/R-T-NLCs, T/V-solution, T-solution, V-solution (each contains 10 μg of TMZ and/or VCR per g mice), and 0.9% saline. At 10 and 72 h after intravenous injection, mice were sacrificed and the tumor, heart, liver, spleen, lung, and kidney tissue samples were harvested, weighed, and homogenized with physiologic saline to determine the amount of drugs in each tissue. The amount of drugs was determined by the method mentioned in the section Characterization of Drug Encapsulation and Drug-loading Efficiency.
In vivo antitumor efficiency
To examine the in vivo antitumor efficiency of NLCs, U87 MG cells bearing BALB/c nude mice were utilized.27 The mice were randomly distributed into 11 groups with eight mice in each group, and intravenously injected with L/R-T/V-NLCs, R-T/V-NLCs, L-T/V-NLCs, T/V-NLCs, L/R-V-NLCs, L/R-T-NLCs, L/R-NLCs, T/V-solution, T-solution, V-solution, and 0.9% normal saline, and the intravenous injections were carried out every 3 days. Body weight and tumor measurements were also documented on the treatment day. Tumor sizes were measured by vernier calipers and the volume was calculated as follows: Tumor volume=(Width2×Length)/2.
The tumor inhibition rates were also determined as follows: Tumor growth inhibition (%)=(Mean tumor weight of a control group–Mean tumor weight of a treatment group)/Mean tumor weight of a control group×100.
Statistical analysis
Quantitative data were presented as means±SD. Statistical significance was analyzed using the Student’s t-test or one-way analysis of variance with the P-value <0.05 indicating significance.
Results
Structure confirmation of Lf-PEG-DSPE
The chemical structure of Lf-PEG-DSPE was determined by 1H-NMR spectroscopy, and the chemical structure shifts are marked by letters in Figure 3: (a) 1.26 (−CH2− of DSPE); (b) 2.53 (−CH2−NH−C=N); (c) 2.78 (−CH2−NH−C=O of Lf); (d) 3.47 (−CH2− of PEG); (e) 3.77 (−CH2−NH−C=O of DSPE); (f) 4.76, (g) 6.13, and (h) 8.08 belong to Lf. The chemical shifts of the thioether bonding, amido linkages, Lf, PEG, and DSPE could determine the formation of Lf-PEG-DSPE.
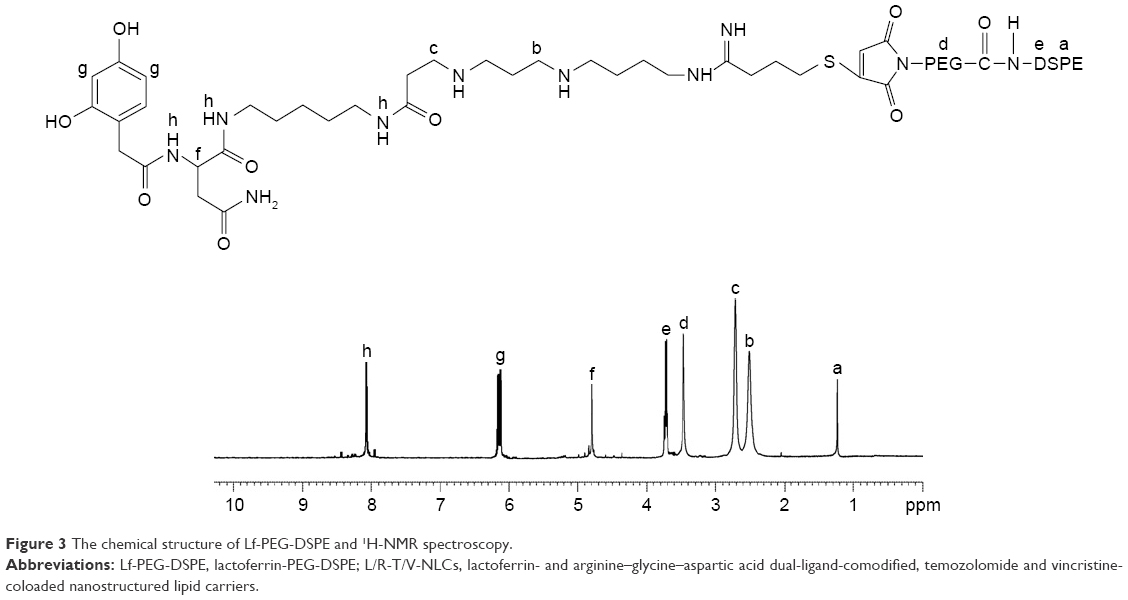 | Figure 3 The chemical structure of Lf-PEG-DSPE and 1H-NMR spectroscopy. |
Preparation and characterization of NLCs
Table 1 calculates the particle size and zeta potential changes from blank L/R-NLCs to L/R-T/V-NLCs. The average size of L/R-T/V-NLCs was 139 nm, almost the same as blank L/R-NLCs (P>0.05). However, the sizes of Lf- or RGD-modified NLCs are different, 136 nm for L-T/V-NLCs and 114 nm for R-T/V-NLCs, respectively. Furthermore, no-ligand-modified T/V-NLCs had the size of 96 nm, which is obviously smaller than the ligand(s)-modified NLCs. The zeta potentials of NLCs were positive, and L/R-T/V-NLCs had a zeta potential of +32 mV. The EE values of all the NLCs samples were over 80%. The DL of different formulations varies from 5.3% to 10.1% (Table 1).
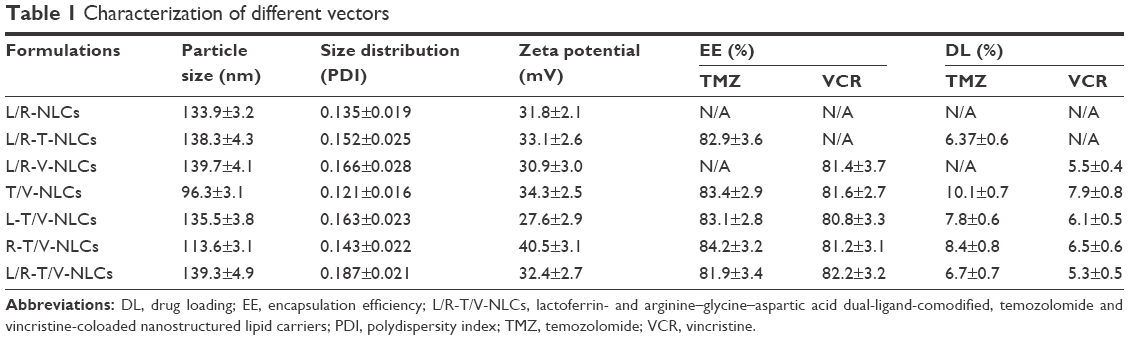 | Table 1 Characterization of different vectors |
Stability of NLCs
The stability of NLCs was evaluated over a period of 90 days (Figure 4). For all kinds of NLCs, the particle sizes remained almost constant throughout the period. There were also no significant changes with time of the PDIs of all the formulations tested. The TMZ and VCR EE of drug(s)-loaded NLCs remained stable at all the time points analyzed in the research.
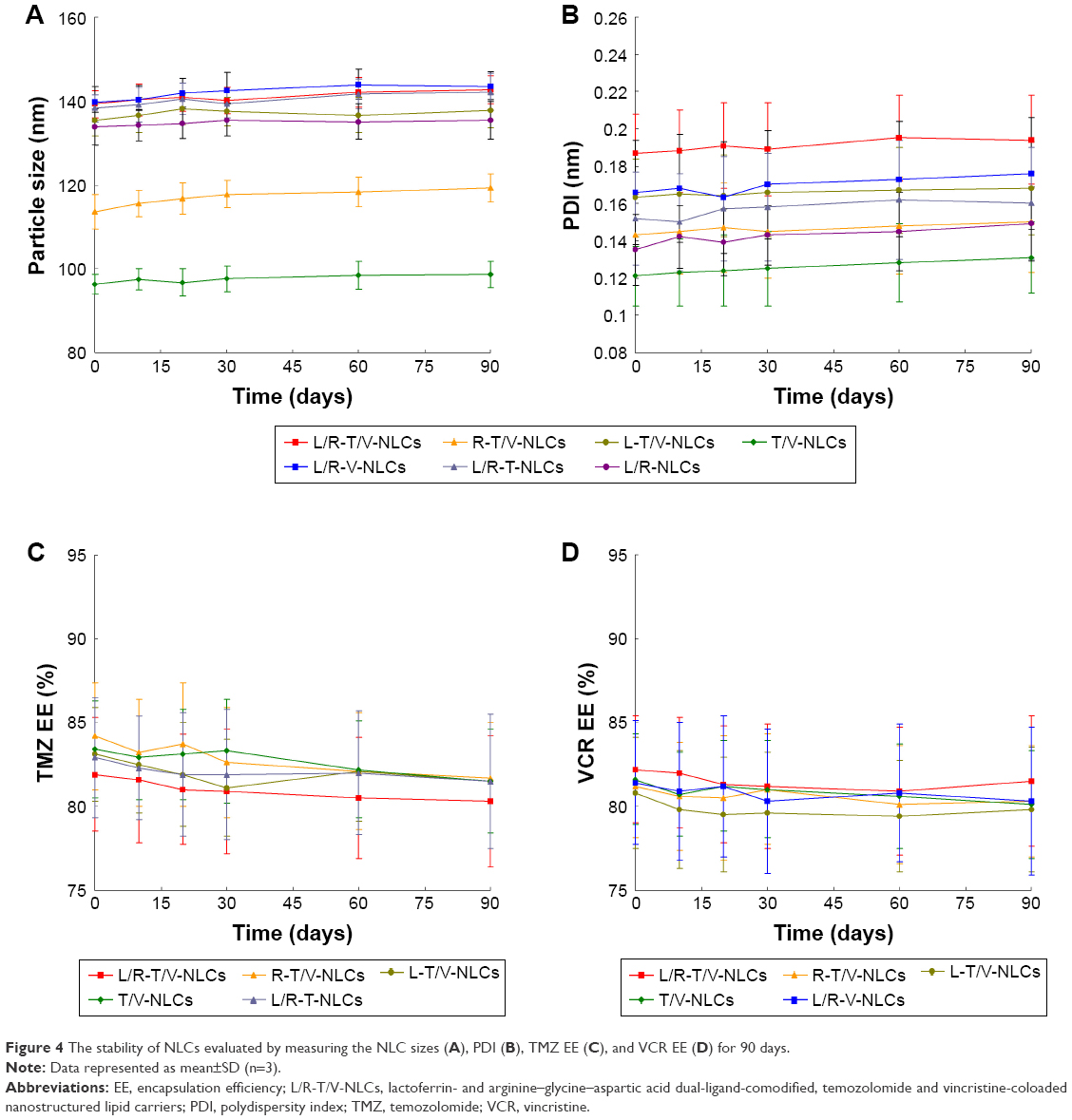 | Figure 4 The stability of NLCs evaluated by measuring the NLC sizes (A), PDI (B), TMZ EE (C), and VCR EE (D) for 90 days. |
In vitro drug release
In vitro drug release profiles of all kinds of NLCs are depicted in Figure 5. The drug release of all the samples showed the sustained-release behavior. The release of Lf and RGD dual-ligand-comodified NLCs was slower than that of single-ligand-modified NLCs. No-ligand-modified T/V-NLCs exhibited the fastest release among all the samples.
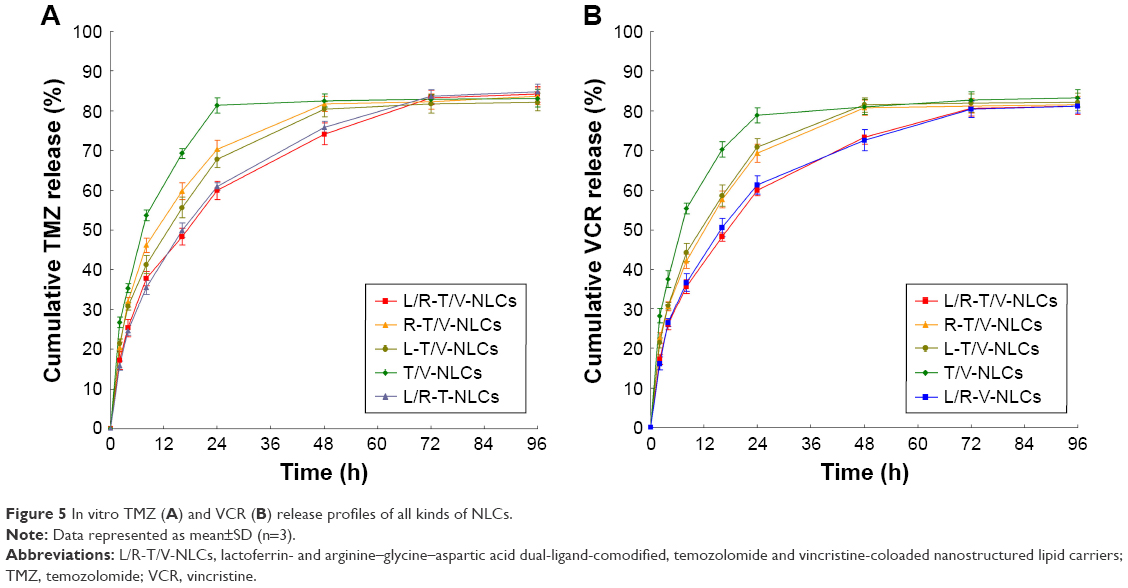 | Figure 5 In vitro TMZ (A) and VCR (B) release profiles of all kinds of NLCs. |
Cellular uptake of NLCs
Figure 6 illustrates the cellular uptake efficiency of the NLCs. Cellular uptake efficiency of Lf and RGD dual-ligand-comodified NLCs on U87 MG cells was significantly higher than single-ligand-modified NLCs (P<0.05). Single-ligand-modified NLCs showed obvious higher uptake on U87 MG cells compared with no-ligand-modified T/V-NLCs (P<0.05). On A549 cells, there were no obvious differences among modified and no-ligand-modified groups.
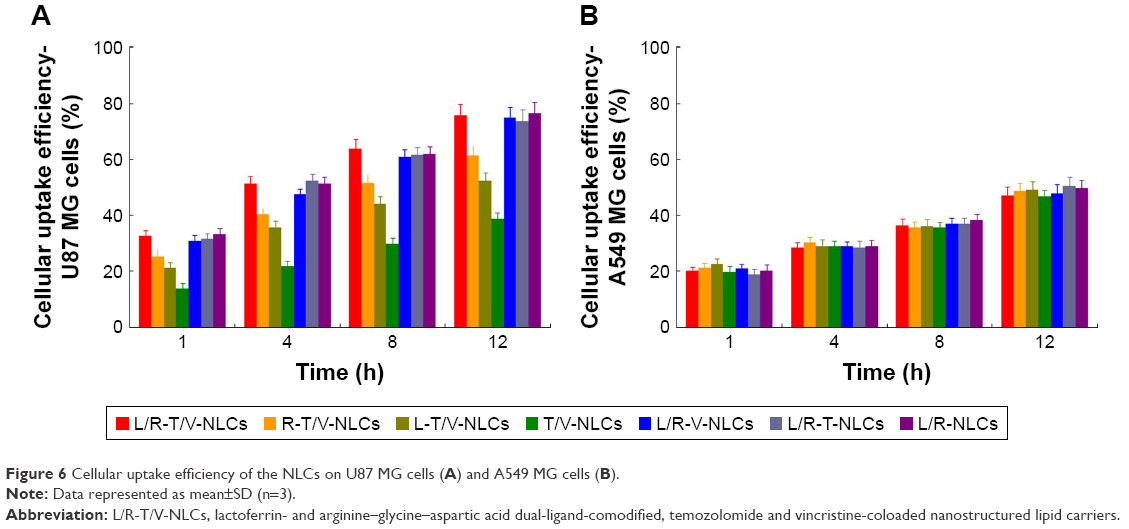 | Figure 6 Cellular uptake efficiency of the NLCs on U87 MG cells (A) and A549 MG cells (B). |
In vitro cytotoxicity and synergistic effect
In vitro cytotoxicity of NLCs was evaluated on U87 MG cells and T98G cells by MTT assay (Figure 7). At all the studied drug concentrations, the cytotoxicity of dual-drug-loaded NLCs was higher than single-drug-loaded NLCs, and drug-loaded NLCs were higher than free drugs (P<0.05). L/R-T/V-NLCs exhibited the highest cell inhibition effect. CI50 was measured to confirm the synergistic effect of TMZ and VCR in L/R-T/V-NLCs, R-T/V-NLCs, L-T/V-NLCs, and T/V-solution systems. TMZ and VCR–coloaded NLCs displayed an overall CI value <1 when fraction of affected cells (Fa) value was between 0.2 and 0.8, indicating the pronounced synergy effects of the NLC systems.
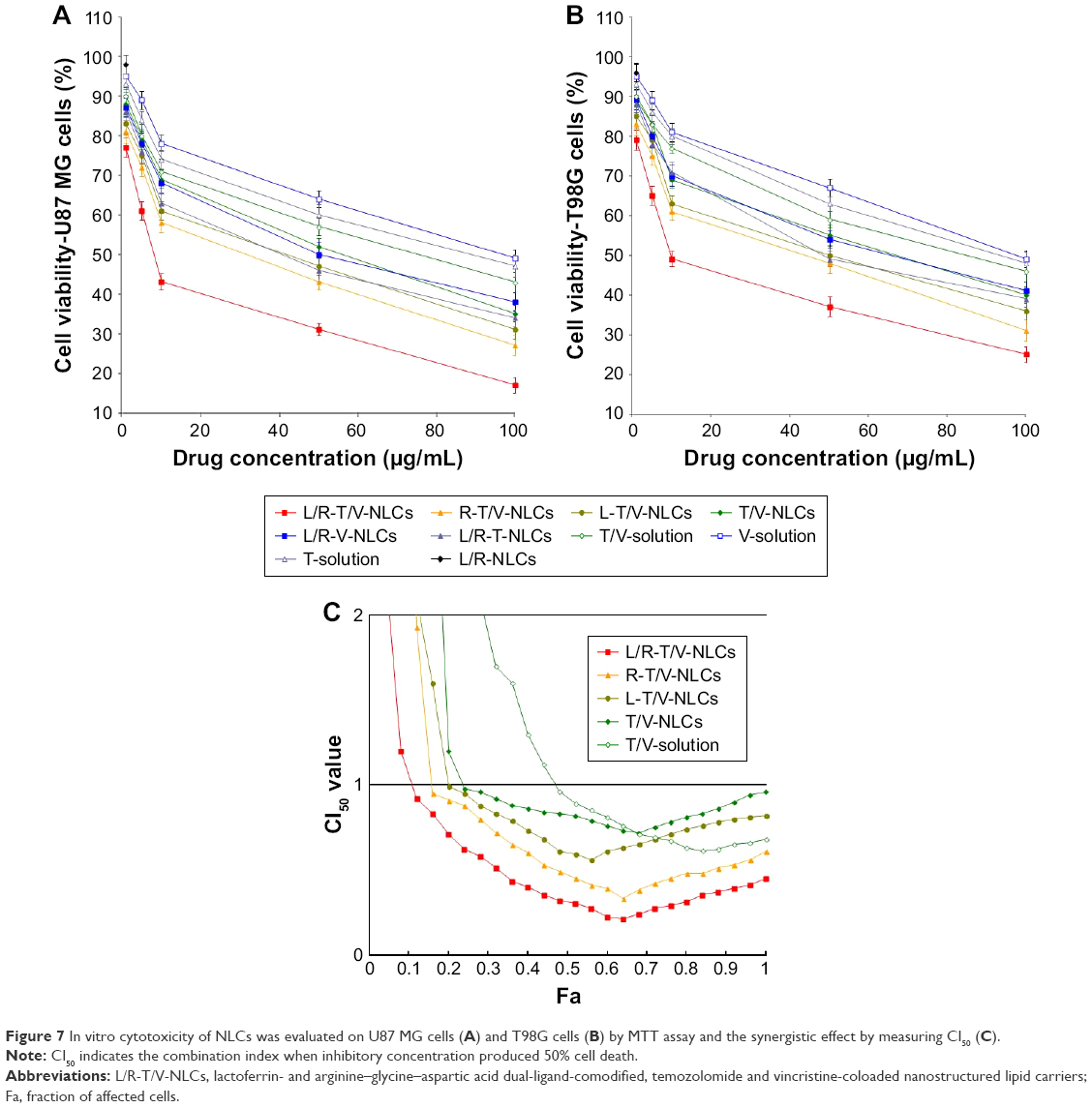 | Figure 7 In vitro cytotoxicity of NLCs was evaluated on U87 MG cells (A) and T98G cells (B) by MTT assay and the synergistic effect by measuring CI50 (C). |
In vivo tissue distribution
In vivo TMZ and VCR distribution of Lf and RGD dual-ligand-comodified NLCs was significantly higher than single-ligand-modified NLCs in tumor tissues (P<0.05; Figure 8). At 72 h, tumor tissue distribution of drug-loaded NLCs was higher than drug solutions (P<0.05). At 12 h, drug solutions’ distribution was higher in heart and kidney than the drug-loaded NLCs (P<0.05).
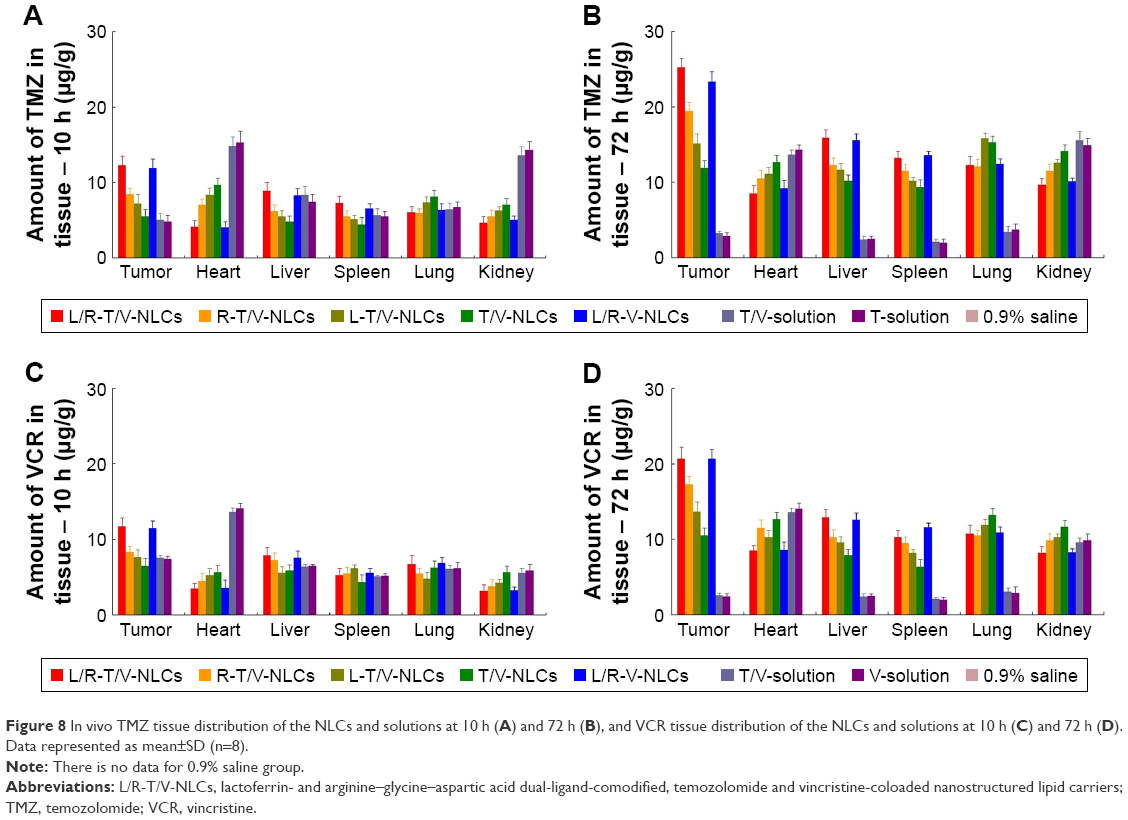 | Figure 8 In vivo TMZ tissue distribution of the NLCs and solutions at 10 h (A) and 72 h (B), and VCR tissue distribution of the NLCs and solutions at 10 h (C) and 72 h (D). Data represented as mean±SD (n=8). |
In vivo antitumor efficiency
In vivo antitumor efficiency of NLCs was evaluated in U87 MG cells–bearing BALB/c nude mice by measuring the tumor size after treatment (Figure 9). The tumor volumes of 0.9% saline control group and blank L/R-NLCs increased dramatically over time, while tumor volume of L/R-T/V-NLCs had the least rise. At the end of study, tumor inhibition rates of tumor-bearing mice treated with different formulations were calculated. L/R-T/V-NLCs inhibited the tumor growth better than that treated with single-ligand-modified NLCs, single-drug-loaded NLCs, and drug solutions. The body weight of the mice in any of the NLC treatment groups showed no obvious decrease in comparison with 0.9% saline control group. However, there was a significant decrease in body weight in drug solutions.
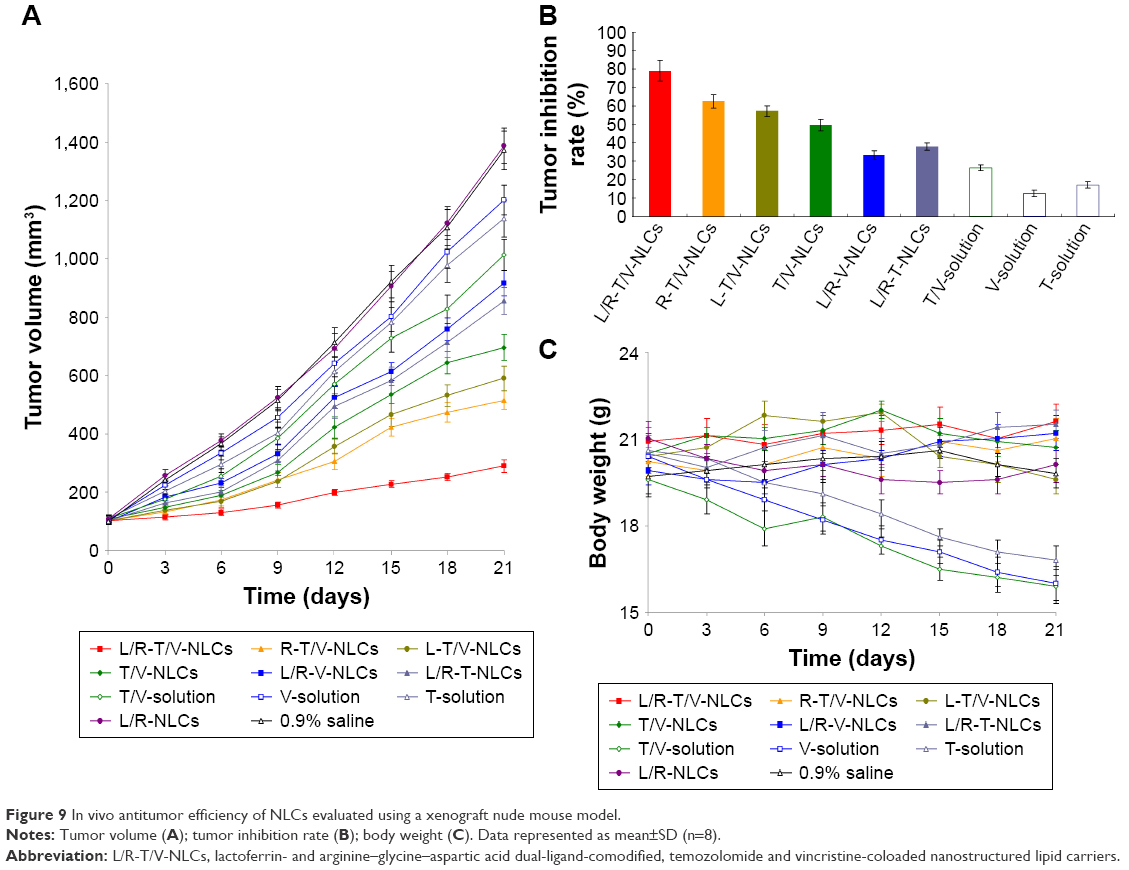 | Figure 9 In vivo antitumor efficiency of NLCs evaluated using a xenograft nude mouse model. |
Discussion
The present work aimed to introduce L/R-T/V-NLCs for GBM combination therapy. Lf, a single-chain iron-binding glycoprotein containing 690 amino acids folded into two globular lobes, is part of Tf family, which can penetrate the brain through receptor-mediated transcytosis.20 RGD is a peptide that was used for neovasculature targeting delivery because of its high binding efficiency with αvβ3 and αvβ5, which are overexpressed on the endothelial cells of tumor angiogenic vessels,28 as well as on GBM cells (eg, the U87 MG cell line). So, Lf- and RGD-containing ligands, Lf-PEG-DSPE and RGD-PEG-DSPE, were synthesized.
L/R-T/V-NLCs were prepared by solvent diffusion method. Table 1 calculated physicochemical characteristics of different NLC formulations. No statistical difference was observed between the average size of L/R-T/V-NLCs and blank L/R-NLCs (P>0.05). It could be the proof that the loading of TMZ and VCR did not expand the size of the NLC formulation. After modification of Lf or RGD, the size of L-T/V-NLCs and R-T/V-NLCs was 136 and 114 nm, respectively. This may be because the Lf modification gives larger size to the system. No-ligand-modified T/V-NLCs had the size of 96 nm, which is obviously smaller than the modified NLCs. Particle size can influence the distribution of nanocarriers.29 Particle size <150 nm is an advantage for NLCs because it decreases uptake by the liver, prolongs circulation time in the blood, and improves bioavailability.18 Small vectors are also minimally phagocytosed by macrophages, so destruction and clearance from the body are minimized. The zeta potentials of NLCs were positive, around +30 mV. Cationic surface charge of nanocarriers can affect the internalization process.30 Cationic vectors could exploit the negative charges present at the cell surface for increased residence time and penetration. Cationic NPs have been shown to keenly associate with negatively charged lipid bilayers, to be internalized rapidly, and enter cells through alternative mechanisms compared to anionic NPs.31 A more likely explanation is the increased affinity of cationic NPs for the negatively charged cell membrane. Besides uptake rates, surface charge also affects the intracellular trafficking. It is considered that cationic NPs present within a lysosome undergoing acidification may accumulate protons entering through the proton pump, therefore acting as a “proton sponge” and maintaining the pump active, resulting in osmotic swelling, lysis, and release of the lysosome content into the cytoplasm. The EE values of all the NLCs samples were greater than 80%. The ability of encapsulation of drugs is quite essential for the delivery system.32 The good drug EE of carriers along with the suitable size and surface charge of the system could bring about high antitumor activity.
Stability evaluation of the SLNs and NLCs was essential to confirm that the structural properties were preserved over storage time, because disruption of nanocarriers in the drug delivery systems could affect their therapeutic potential.33 The NLC samples were analyzed to verify their stability overtime. For all kinds of NLCs, no aggregate was found for the particle sizes that remained almost constant throughout the period.34 PDIs <0.2 are normally used to indicate the existence of a narrow particle size range. Large fluctuations with time could indicate aggregate formation, hence increasing the NP size range. There were no significant changes with time of the PDIs of all the samples tested, showing the stability of the NLCs. The TMZ and VCR EE of TAT-RVC/NCs and RVC/NCs also remained stable at all the time points tested in the research, indicating that the NLCs were stable and will not disassemble within 3 months.
For cancer therapy, a NP system for drug delivery is desired to improve the bioavailability of drugs to the tumor while reducing their side effects to normal tissues.23 The drug release may be controlled by diffusion, swelling, and/or erosion process of the carriers. Thus, a drug depot effect could be achieved by the carriers, which could act as a barrier against diffusion of hydrophobic drugs.35 Insoluble drugs loaded in the carriers may show a stable state and slow drug release over a prolonged period of time. The release rate of modified NLCs was slower than that of no-ligand-modified NLCs. This may be due to the sustained effect of the PEG coating on the surface of the NLCs.36 The sustained-release behavior of the carriers may give continuous protection of the drug and lead to the antitumoral therapeutic effect.
Cellular uptake research could provide some circumstantial evidence to display the advantages of the NP formulation.24 An in vitro cellular study was performed in order to investigate the efficacy of the NLCs as anticancer agent for GBM as well as the uptake of NLCs into the U87 MG cells.37 Cellular uptake efficiency of Lf and RGD dual-ligand-comodified NLCs on U87 MG cells was significantly higher than other NLCs. However, on A549 cells, there were no obvious differences among the modified and no-ligand-modified groups. This could be attributed to enhanced cancer cell–specific adherence of the Lf and RGD ligands. The improved activity and penetration of drugs delivered with Lf and RGD can be made use of to improve the efficacy of the standard drug dose, attenuate side effect, and overcome drug resistance.
In vitro cytotoxicity of NLCs was evaluated on U87 MG cells and T98G cells. At all the studied drug concentrations, the cytotoxicity of dual-drug-loaded NLCs was higher than single-drug-loaded NLCs, and drug-loaded NLCs were higher than free drugs (P<0.05). L/R-T/V-NLCs exhibited the highest cell inhibition effect. Significantly higher cytotoxicity was found for drug-loaded NLCs than free drugs. Constructions of NLC systems could target the Lf and/or RGD receptor, and may improve the ability of drugs thus gaining better efficiency than free drugs.25 When combination therapy is used in cancer chemotherapy, evaluation of the synergistic effect is important and CI analyses are one of the most reliable methods.38 CI values were determined using the Chou and Talalay method.39 This method is relatively simple and is not limited by whether the dose–effect relationships are hyperbolic or sigmoidal, whether the effects of the drugs are mutually exclusive or nonexclusive, whether the ligand interactions are competitive, noncompetitive, or uncompetitive, whether the drugs are agonists or antagonists, or the number of drugs involved. In this study, CI50 was measured to confirm the synergistic effect of TMZ and VCR in L/R-T/V-NLCs, R-T/V-NLCs, L-T/V-NLCs, and T/V-solution systems. TMZ and VCR–coloaded NLCs displayed an overall CI value <1 when Fa value was between 0.2 and 0.8, indicating the pronounced synergy effects of the NLCs systems.
In vivo tissue distribution results showed that the administration of drug-loaded NLCs led to a dramatic increase in drug accumulation in the tumor tissue than the free drug solutions. This can be explained by solid tumors having leakage microvasculatures. Nanosized particles may be delivered to the tumor tissue owing to the enhanced permeability and retention (EPR) effects.26 EPR effects at the same time selectively made the entry of the carriers into the tumor easier, and thus prevented the entry of the system in the normal tissue. Carriers of this size are typically found in liver and spleen, and with rather low signal from other organs. The high signal from the tumor in the first 72 h of the drug-loaded NPs may prove the long circulation time of the carriers, and the drug efficiency lasted until 72 h after injection. This behavior may assist with the performance of systems for the in vivo antitumor therapy.
In vivo antitumor efficiency of NLCs was assessed through measuring the mean tumor volume and the inhibition of tumor growth in tumor-bearing mice. NLC formulation was used to overcome the side effects of conventional chemotherapeutic treatment and achieve high anticancer efficiency. Mice in free NPs and 0.9% saline control groups shared the similar tumor growth pattern, suggesting that blank NPs were not capable of inhibiting tumor growth. In tumor xenograft mouse model, L/R-T/V-NLCs inhibited the tumor growth better than that treated with single-ligand-modified NLCs, single-drug-loaded NLCs, and drug solutions. No noticeable weight loss was observed in any of the NLC treatment groups, indicating that all the treatments were well tolerated. The mechanisms could be discussed by the lipid structure of NLCs having high affinity to the lipid cell surface, promoting the fusion of the carriers to the cells, and thus delivering the drug into them. These results are in agreement with the previous in vitro results, which indicate that L/R-T/V-NLCs have the best application in the treatment of GBM.
Conclusion
In this study, on the basis of our own studies, we introduced L/R-T/V-NLCs for GBM combination therapy. L/R-T/V-NLCs were stable with nanosize and high EE. L/R-T/V-NLCs exhibited sustained-release behavior, high cellular uptake, high cytotoxicity and synergy effects, increased drug accumulation in the tumor tissue, and obvious tumor inhibition efficiency with minimal systemic toxicity. L/R-T/V-NLCs could be a promising drug delivery system for glioblastoma chemotherapy.
Disclosure
The authors report no conflicts of interest in this work.
References
Higa M, Katagiri C, Shimizu-Okabe C, et al. Identification of a novel cell-penetrating peptide targeting human glioblastoma cell lines as a cancer-homing transporter. Biochem Biophys Res Commun. 2015;457:206–212. | ||
Hou LC, Veeravagu A, Hsu AR, Tse VC. Recurrent glioblastoma multiforme: a review of natural history and management options. Neurosurg Focus. 2006;20(4):E5. | ||
Kim SS, Harford JB, Pirollo KF, Chang EH. Effective treatment of glioblastoma requires crossing the blood-brain barrier and targeting tumors including cancer stem cells: the promise of nanomedicine. Biochem Biophys Res Commun. 2015;468:485–489. | ||
Wu M, Fan Y, Lv S, Xiao B, Ye M, Zhu X. Vincristine and temozolomide combined chemotherapy for the treatment of glioma: a comparison of solid lipid nanoparticles and nanostructured lipid carriers for dual drugs delivery. Drug Deliv. 2016;23(8):2720–2725. | ||
Song S, Mao G, Du J, Zhu X. Novel RGD containing, temozolomide-loading nanostructured lipid carriers for glioblastoma multiforme chemotherapy. Drug Deliv. 2016;23(4):1404–1408. | ||
Chekhonin VP, Baklaushev VP, Yusubalieva GM, et al. Targeted delivery of liposomal nanocontainers to the peritumoral zone of glioma by means of monoclonal antibodies against GFAP and the extracellular loop of Cx43. Nanomedicine. 2012;8:63–70. | ||
Zhang B, Sun X, Mei H, et al. LDLR-mediated peptide-22-conjugated nanoparticles for dual-targeting therapy of brain glioma. Biomaterials. 2013;34:9171–9182. | ||
Gastaldi L, Battaglia L, Peira E, et al. Solid lipid nanoparticles as vehicles of drugs to the brain: current state of the art. Eur J Pharm Biopharm. 2014;87:433–444. | ||
Qu J, Zhang L, Chen Z, et al. Nanostructured lipid carriers, solid lipid nanoparticles, and polymeric nanoparticles: which kind of drug delivery system is better for glioblastoma chemotherapy? Drug Deliv. 2016;23(9):3408–3416. | ||
Chen Z, Lai X, Song S, Zhu X, Zhu J. Nanostructured lipid carriers based temozolomide and gene co-encapsulated nanomedicine for gliomatosis cerebri combination therapy. Drug Deliv. 2016;23(4):1369–1373. | ||
Muller RH, Radtke M, Wissing SA. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv Drug Deliv Rev. 2002;54:S131–S155. | ||
Saupe A, Wissing SA, Lenk A, Schmidt C, Müller RH. Solid Lipid Nanoparticles (SLN) and Nanostructured Lipid Carriers (NLC)–structural investigations on two different carrier systems. Bio-Med Mater Eng. 2005;15:393–402. | ||
Kuo YC, Cheng SJ. Brain targeted delivery of carmustine using solid lipid nanoparticles modified with tamoxifen and lactoferrin for antitumor proliferation. Int J Pharm. 2016;499(1–2):10–19. | ||
Miao D, Jiang M, Liu Z, et al. Co-administration of dual-targeting nanoparticles with penetration enhancement peptide for antiglioblastoma therapy. Mol Pharm. 2014;11(1):90–101. | ||
Arcella A, Oliva MA, Staffieri S, et al. In vitro and in vivo effect of human lactoferrin on glioblastoma growth. J Neurosurg. 2015;123(4):1026–1035. | ||
Gao H, Xiong Y, Zhang S, Yang Z, Cao S, Jiang X. RGD and interleukin-13 peptide functionalized nanoparticles for enhanced glioblastoma cells and neovasculature dual targeting delivery and elevated tumor penetration. Mol Pharm. 2014;11(3):1042–1052. | ||
Miura Y, Takenaka T, Toh K, et al. Cyclic RGD-linked polymeric micelles for targeted delivery of platinum anticancer drugs to glioblastoma through the blood-brain tumor barrier. ACS Nano. 2013;7(10):8583–8592. | ||
Yan J, Wang Y, Jia Y, et al. Co-delivery of docetaxel and curcumin prodrug via dual-targeted nanoparticles with synergistic antitumor activity against prostate cancer. Biomed Pharmacother. 2017;88:374–383. | ||
Wang W, Zhou F, Ge L, Liu X, Kong F. Transferrin-PEG-PE modified dexamethasone conjugated cationic lipid carrier mediated gene delivery system for tumor-targeted transfection. Int J Nanomedicine. 2012;7:2513–2522. | ||
Huang FY, Chen WJ, Lee WY, Lo ST, Lee TW, Lo JM. In vitro and in vivo evaluation of lactoferrin-conjugated liposomes as a novel carrier to improve the brain delivery. Int J Mol Sci. 2013;14(2):2862–2874. | ||
Tian XH, Lin XN, Wei F, et al. Enhanced brain targeting of temozolomide in polysorbate-80 coated polybutylcyanoacrylate nanoparticles. Int J Nanomedicine. 2011;6:445–452. | ||
Chirio D, Gallarate M, Peira E, et al. Positive-charged solid lipid nanoparticles as paclitaxel drug delivery system in glioblastoma treatment. Eur J Pharm Biopharm. 2014;88(3):746–758. | ||
Wang H, Agarwal P, Zhao S, et al. Hyaluronic acid-decorated dual responsive nanoparticles of Pluronic F127, PLGA, and chitosan for targeted co-delivery of doxorubicin and irinotecan to eliminate cancer stem-like cells. Biomaterials. 2015;72:74–89. | ||
Wang H, Sun G, Zhang Z, Ou Y. Transcription activator, hyaluronic acid and tocopheryl succinate multi-functionalized novel lipid carriers encapsulating etoposide for lymphoma therapy. Biomed Pharmacother. 2017;91:241–250. | ||
Zhu B, Yu L, Yue Q. Co-delivery of vincristine and quercetin by nanocarriers for lymphoma combination chemotherapy. Biomed Pharmacother. 2017;91:287–294. | ||
Zhang R, Ru Y, Gao Y, Li J, Mao S. Layer-by-layer nanoparticles co-loading gemcitabine and platinum (IV) prodrugs for synergistic combination therapy of lung cancer. Drug Des Devel Ther. 2017;11:2631–2642. | ||
Zhu B, Zhang H, Yu L. Novel transferrin modified and doxorubicin loaded Pluronic 85/lipid-polymeric nanoparticles for the treatment of leukemia: in vitro and in vivo therapeutic effect evaluation. Biomed Pharmacother. 2017;86:547–554. | ||
Gao H, Yang Z, Cao S, et al. Tumor cells and neovasculature dual targeting delivery for glioblastoma treatment. Biomaterials. 2014;35:374–382. | ||
Shao Z, Shao J, Tan B, et al. Targeted lung cancer therapy: preparation and optimization of transferrin-decorated nanostructured lipid carriers as novel nanomedicine for co-delivery of anticancer drugs and DNA. Int J Nanomedicine. 2015;10:1223–1233. | ||
Harush-Frenkel O, Rozentur E, Benita S, Altschuler Y. Surface charge of nanoparticles determines their endocytic and transcytotic pathway in polarized MDCK cells. Biomacromolecules. 2008;9(2):435–443. | ||
Boyles MS, Kristl T, Andosch A, et al. Chitosan functionalisation of gold nanoparticles encourages particle uptake and induces cytotoxicity and pro-inflammatory conditions in phagocytic cells, as well as enhancing particle interactions with serum components. J Nanobiotechnol. 2015;13:84. | ||
Puglia C, Offerta A, Tirendi GG, et al. Design of solid lipid nanoparticles for caffeine topical administration. Drug Deliv. 2016;23:36–40. | ||
You P, Yuan R, Chen C. Design and evaluation of lidocaine- and prilocaine-coloaded nanoparticulate drug delivery systems for topical anesthetic analgesic therapy: a comparison between solid lipid nanoparticles and nanostructured lipid carriers. Drug Des Devel Ther. 2017;11:2743–2752. | ||
Chen C, You P. A novel local anesthetic system: transcriptional transactivator peptide-decorated nanocarriers for skin delivery of ropivacaine. Drug Des Devel Ther. 2017;11:1941–1949. | ||
Liu Y, Gao D, Zhang X, et al. Antitumor drug effect of betulinic acid mediated by polyethylene glycol modified liposomes. Mater Sci Eng C Mater Biol Appl. 2016;64:124–132. | ||
Peeters L, Sanders NN, Jones A, Demeester J, De Smedt SC. Post-pegylated lipoplexes are promising vehicles for gene delivery in RPE cells. J Control Release. 2007;121(3):208–217. | ||
Li H, Shi L, Wei J, et al. Cellular uptake and anticancer activity of salvianolic acid B phospholipid complex loaded nanoparticles in head and neck cancer and precancer cells. Colloids Surf B Biointerfaces. 2016;147:65–72. | ||
Li S, Wang L, Li N, Liu Y, Su H. Combination lung cancer chemotherapy: design of a pH-sensitive transferrin-PEG-Hz-lipid conjugate for the co-delivery of docetaxel and baicalin. Biomed Pharmacother. 2017;95:548–555. | ||
Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.