Back to Journals » Cancer Management and Research » Volume 10
Knockdown of PRL-3 increases mitochondrial superoxide anion production through transcriptional regulation of RAP1
Authors Yang Y, Lian S, Meng L, Tian Z, Feng Q, Wang Y, Wang P, Qu L, Shou C
Received 12 February 2018
Accepted for publication 10 August 2018
Published 30 October 2018 Volume 2018:10 Pages 5071—5081
DOI https://doi.org/10.2147/CMAR.S165344
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Kenan Onel
Yongyong Yang,1,* Shenyi Lian,2,* Lin Meng,1,* Zhihua Tian,3 Qin Feng,2 Yue Wang,2 Ping Wang,2 Like Qu,1 Chengchao Shou1
1Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Department of Biochemistry and Molecular Biology, Peking University Cancer Hospital & Institute, Beijing 100142, China; 2Department of Pathology, Peking University Cancer Hospital & Institute, Beijing 100142, China; 3Central Laboratory, Peking University Cancer Hospital & Institute, Beijing 100142, China
*These authors contributed equally to this work
Background: Phosphatase of regenerating liver-3 (PRL-3) has been shown to be highly expressed in various types of cancers and is related to poor prognosis. Our previous study showed that silencing of PRL-3 leads to increased reactive oxygen species (ROS). However, the mechanism of PRL-3 regulating ROS is not clear.
Materials and methods: PRL-3 or Repressor activator protein 1 (RAP1) was knockdown in human colorectal cancer cell lines HCT116 and SW480. The mRNA level was measured by quantitative real-time (qRT)-PCR and the protein level was measured by western blot. ROS was detected by specific oxidationsensitive fluorescent probes. Cell cycle was analyzed through flow cytometry. Luciferase assay and chromatin immunoprecipitation (ChIP) were performed to investigate the regulation of RAP1 by PRL-3. Gene expression correlation was analyzed through an interactive web server. Statistical analysis was performed with SPSS software.
Results: Knockdown of PRL-3 significantly increases mitochondrial superoxide anion, mitochondria membrane potential, and induces cell cycle arrest. Decreased PRL-3-induced mitochondrial superoxide anion accumulation is related to the downregulation of RAP1, which could also affect the level of mitochondria superoxide anion. PRL-3 regulates the expression of RAP1 through binding to the promoter of rap1 gene. PRL-3 could regulate the expression of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α) through the mediation of RAP1. Both PRL-3 and RAP1 could regulate the expression of manganese superoxide dismutase 2 (SOD2) and the uncoupling protein 2 (UCP2), which may be related to PRL-3 suppression induced mitochondria superoxide anion.
Conclusion: Our study presents the first evidence that PRL-3 is involved in the regulation of mitochondria superoxide anion as a transcriptional factor.
Keywords: PRL-3, cell cycle, mitochondria, RAP1, PGC-1α, superoxide anion
Introduction
The PTP superfamily of phosphatases (PTPs) consist of a large group of enzymes, which are important for the regulation of various cell mechanisms.1 The phosphatase of regenerating liver (PRL) family of PTPs includes PRL-1, PRL-2, and PRL-3, and all PRLs were discovered as potential biomarkers and therapeutic targets for diverse types of cancers.2 Among them, PRL-3 had been found to be highly expressed in a variety of cancer tissues, and high PRL-3 is related with disease progression and poor survival outcome.2–5 Several reports from different groups demonstrated PRL-3’s oncogenic role in promoting cancer metastasis via multiple oncogenic effector pathways, including PI3K/Akt,6 Src-ERK1/2.4,7–9 Besides, several proteomic analyses revealed that PRL-3 could widely increase intracellular protein phosphorylation.10,11 The previous study showed that maintenance of basal PRL-3 levels is important for proper cell cycle progression in normal cells.12 Our recent study showed that overexpression of PRL-3 could promote telomere deprotection through interaction with telomeric binding protein, repressor activator protein 1 (RAP1). Meanwhile, knockdown of PRL-3 could significantly inhibit cell proliferation and induce cell senescence,13 which is consistent with previous reports.14–17 Furthermore, knockdown of PRL-3-induced cell senescence is dependent on the accumulation of superoxide anion.13
RAP1 has been shown to bind to telomeric and extra-telomeric DNA and regulate metabolism and protect against obesity through regulating peroxisome proliferator-activated receptor alpha (PPARα) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α).18–20 RAP1 could also interact with IkappaB kinases and activates nuclear factor kappa B (NF-κB).21 The expression of RAP1 was significantly higher in tumor tissues than in adjacent non-tumor tissues,21,22 indicating that RAP1 plays a potential role in cancer progression. Our previous investigation showed that PRL-3 activates NF-κB signaling pathway through the interaction with RAP1.23
Here, in this study, we investigated the mechanism of PRL-3 knockdown increasing superoxide anion. We discovered that PRL-3 knockdown induces G1 phase arrest and knockdown of PRL-3-induced superoxide anion is mainly generated from mitochondria. Besides, we showed that PRL-3 transcriptionally regulates RAP1 expression through binding to the promoter region of rap1 gene and PRL-3 regulates the expression of PGC-1α through RAP1. Finally, our study suggested that PRL-3 knockdown increased mitochondria superoxide anion is related to decreased PGC-1α and its downstream genes. Our study revealed PRL-3’s novel function in regulating mitochondria superoxide anion as a potential transcriptional factor, which will contribute to further studying the role of PRL-3 in cancer progression.
Materials and methods
Cell culture and transfection
Human colorectal cancer cell lines HCT116 and SW480 were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and cultured in Roswell Park Memorial Institute 1640 supplemented with 10% FBS plus 1% penicillin–streptomycin. Cells were transfected with plasmid DNA using Lipofectamine 2000 Transfection Reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. For lentiviral infection, cells were seeded into six-well plates at a density of 50,000 cells per well and transduced with lentiviral particles at a multiplicity of infection of 20. The infection efficiency was determined by counting green fluorescent protein-expressing cells under fluorescence microscopy 72 hours after infection. Then, quantitative real-time (qRT)-PCR and Western blot (WB) were used to analyze the transfection or infection efficiency at various time points.
Plasmids, shRNAs and sgRNA
Full-length human PRL-3 gene was cloned from a LoVo cDNA library and inserted into the pcDNA3 vector with myc-tag as previously described.4 Full-length human RAP1 gene was amplified from an HCT116 cell cDNA library and PCR product was cloned into plasmid pcDNA3.0-HA verified by sequencing.22 Lentiviral vectors expressing human PRL-3-targeting shRNA (shPRL-3) and human RAP1-targeting shRNA (shRAP1) were designed and constructed by GenePharma (Shanghai, China).13 The target nucleotide sequences of the oligoduplexes were as follows: Lv-shPRL-3-3#: 5′- CCCAGCTCCTGTGTGGAGAAAG-3′; Lv-shPRL-3-4#: 5′- GACCAGATGCTCATGTGTTCC-3′; Lv-shRAP1-1#: 5′-GGAGAAGTTTAACTTGGATCT-3′; Lv-shRAP1-2#: 5′-GAATGTAGCTCGGAGGATTGA-3′; Control shRNA sequence was 5′-TTCTCCGAACGTGTCACGTTTC-3′. Generation of PRL-3 knockout cells through the clustered regularly interspaced short palindromic repeat (CRISPR)-associated endonuclease 9 (CRISPR/Cas9) system in SW480 cells was described previously.13 The PRL-3-specific sgRNA sequence was 5′-AGGACCTGAAGAAGTACGGGG-3′.
RNA extraction and quantitative reverse transcription PCR
Total RNA was extracted using Trizol reagent (Thermo Fisher Scientific) under RNase-free conditions. Qualified RNA samples were used to synthesize cDNA with GoScriptTM Reverse Transcription system (Promega, Madison, WI, USA). qRT-PCR was performed using a StepOne Real-time PCR system (Thermo Fisher Scientific) and SYBR Green PCR master mix reagents (Toyobo, Osaka, Osaka Prefecture, Japan). Expression data were normalized to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Primers used are listed in Table 1. Each sample was performed in triplicate. Relative expression quantification analysis relied on the classical delta-delta-Ct method.
 | Table 1 The primers of real-time PCR |
Western blotting
Total protein from cells was extracted with RIPA lysis buffer, quantified by a BCA kit (23225, Thermo Fisher Scientific), and subjected to 10% SDS-PAGE under denaturing conditions. The samples were transferred to nitrocellulose filter membranes (Merck Millipore, Burlington, MA, USA). Membranes were blocked for 1 hour with 5% milk PBS Tween- 20 (PBST) buffer. Afterwards, membranes were incubated overnight at 4°C with primary antibodies against PRL-3 (generated by our laboratory).13 RAP1 (A300-306A-2, Bethyl Laboratories, Montgomery, TX, USA), GAPDH (ab8245, Abcam, Cambridge, UK), PGC-1α (#2178, Cell Signaling Technology, Danvers, MA, USA), and c-Myc (sc-40, Santa Cruz Biotechnology, Dallas, TX, USA). Then, membranes were washed three times with PBST buffer, followed by incubation in goat anti-rabbit or anti-mouse secondary antibody conjugated with horseradish peroxidase for 1 hour at room temperature. After washing with PBST three times, protein bands were visualized using an eECL WB kit (CWBIO, Beijing, China).
Assessment of superoxide anion, mitochondrial membrane potential
In this study, the oxidation-sensitive fluorescent probes dihydroethidium (DHE) (GMS10111.1, Genmed, Plymouth, MN, USA), and MitoSOX Red (M36008, Thermo Fisher Scientific) were used to monitor the production of cytosolic superoxide anions and mitochondrial superoxide anions, respectively. The cationic dye, tetramethylrhodamine ethyl ester perchlorate (TMRE) (#13296, Cell Signaling Technology) was used to detect the mitochondrial membrane potential in cells. HCT116 and SW480 cells were cultured in 24-well plates. Some cells were treated with dimethyl sulfoxide (DMSO) as control (vehicle, 1:1,000), or 20 µM carbonyl cyanide 3-chlorophenylhydrazone (CCCP) (#13296, Cell Signaling Technology) for 30 minutes. The cells were incubated with 5 µM DHE or 5 µM MitoSOX™ reagent working solution, or 200 nM TMRE for 20 minutes at 37°C in the dark. After washing thrice with warm PBS, the fluorescence was imaged with a Leica SP2 confocal system (Leica Microsystems, Wetzlar, Germany).
Flow cytometry analysis of cell cycle
Cells were pretreated with DMSO (control) or 5 mM N-acetyl-l-cysteine (NAC) (616-91-1, Sigma, St. Louis, MO, USA) for 24 hours. Cells were collected after washing with cold PBS twice and then fixed in ice-cold 70% ethanol and stained with propidium iodide. The cell cycle profiles were assayed using the Elite ESP flow cytometry at 488 nm, and the data were analyzed using the CELL Quest software (BD Biosciences, San Jose, CA, USA).
Human RAP1 gene Promoter-Luciferase reporter constructs and luciferase assay
The RAP1 gene promoter fragment (–1,500/+150 bp) or (–500/+150 bp) was cloned into the polylinker site upstream of the luciferase gene (luc) in the pGL-Basic vector. One recombinant clone for each of the constructs was chosen, and the plasmid DNA was extracted and purified using Qiagen Miniprep kit (27106, Qiagen, Hilden, Germany) following the manufacturer’s instructions.
HCT116 and HEK293 cells were seeded in 6-well plates (5×105 cell/well) with 2 mL of DMEM and 10% FBS and grown until 80% confluence (24–36 hour). Cells were cotransfected with the appropriate rap1 promoter-reporter constructs and the pcDNA3-myc-PRL-3 expression plasmid by using 1 µg/well of luciferase reporter construct, 0.25 µg/well of renilla reporter, and 1 µg/well of pcDNA3-myc-PRL-3 (or empty pcDNA3 vector as a negative control). After 48 hours, cells were lysated and assayed for luciferase activity. Luciferase activity was measured using the Dual-Luciferase® Reporter Assay System (Thermo Fisher Scientific). All transfections were performed in duplicate, and experiments were repeated at least three times. The firefly luciferase activity was normalized according to the renilla luciferase activity.
Chromatin immunoprecipitation (ChIP)
Cells were seeded in plates at 5×106 cells/plate. After 24 hours, cells were cross-linked with 2 mM disuccinimidyl glutarate (Thermo Fisher Scientific) for 45 minutes, washed with PBS, and fixed with 1% freshly prepared solution of formaldehyde in PBS/Mg (v/v) for 15 minutes at room temperature. Then fixed cells were washed with PBS, scraped, transferred into Eppendorf tubes, and lysed for 15 minutes on ice. The samples were sonicated on ice for 5 minutes in cycles of 30 seconds on/30 seconds off. Then the sample was incubated with the PRL-3 antibody or negative mouse IgG coupled to magnetic beads overnight at 4°C with rotation. Precipitated complexes were eluted with elution buffer for 30 minutes at room temperature with rotation. Samples were decross-linked in 200 mM NaCl, 50 mM Tris pH 6.8, 10 mM EDTA adding 200 µg/mL proteinase K and incubated at 65°C for 2 hours 30 minutes. DNA was extracted by ChIP DNA purification kit from Qiagen (28104, Qiagen) and used for qPCR analysis. qPCR primers are shown in Table 2.
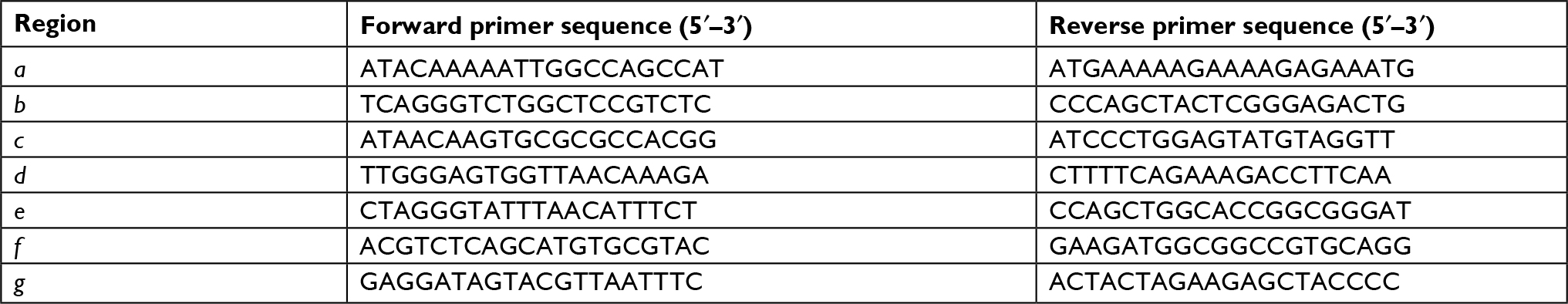 | Table 2 The primers of ChIP-qPCR Abbreviation: ChIP, chromatin immunoprecipitation. |
Gene expression correlation analysis
The gene expression correlation analysis between PRL-3 and RAP1, PRL-3 and superoxide dismutase 2 (SOD2), RAP1 and SOD2 in The Cancer Genome Atlas (TCGA) data and Genotype-Tissue Expression (GTEx) data was performed through Gene Expression Profiling Interactive Analysis (GEPIA), http://gepia.cancer-pku.cn/index.html, a recently developed interactive web server for analyzing the RNA sequencing expression.24
Mitochondrial DNA content analysis
Mitochondrial DNA content was determined in duplicate runs using qRT-PCR targeting the nuclear GAPDH gene and the mitochondrial MT-ND1. The ratio of the mtDNA compared with the nuclear DNA was used as an index for measuring the mtDNA content.25 Primers targeting GAPDH and MT-ND1 are shown in Table 1.
Statistical analysis
Statistical analysis was performed with SPSS11.5 software. Data were presented as mean ± SD from three independent experiments. Univariate comparisons of means were evaluated using the Student’s t-test and/or one-way ANOVA with Tukey’s post hoc adjustment for multiple comparisons when appropriate. Correlation between different gene expressions was tested using Pearson’s correlation test. P<0.05 was considered a statistically significant difference.
Result
Knockdown of PRL-3 increases mitochondrial superoxide anion and induces cell cycle arrest
To examine the function of PRL-3, we generated PRL-3 knockdown cells through lentivirus infection in HCT116 cells and CRISPR-Cas 9 system in SW480 cells as described previously.13 Western blot result showed that PRL-3 was significantly decreased in both HCT116 and SW480 cells after infection (Figure 1A). Knockdown of PRL-3 greatly increased superoxide anion level detected by DHE (Figure 1B), which is a specific probe of superoxide anion. As superoxide anion mainly originates from mitochondria.26 We then detected superoxide anion with MitoSOX, which is a specific detecting probe of superoxide anion generated from mitochondria, and that PRL-3 knockdown cells showed greatly increased MitoSOX fluorescence, suggesting PRL-3 knockdown-induced superoxide anion was mainly generated from mitochondria (Figure 1B). Previous studies have shown that increase of mitochondrial membrane potential results in formation of superoxide anion in mitochondria.27 Here, we stained the mitochondria with TMRE dye, which was widely used to measure the mitochondrial transmembrane potential. We discovered that knockdown or knockout of PRL-3 greatly enhanced the mitochondrial transmembrane potential in both HCT116 and SW480 cells (Figure 1C). We further treated the cells with CCCP, which is a mitochondrial membrane potential disruptor. We found that treatment with CCCP clearly suppressed PRL-3 knockdown strengthened MitoSOX staining (Figure 1D), implying PRL-3 knockdown-promoted superoxide anion generation in mitochondria is dependent on its effect on mitochondrial transmembrane potential.
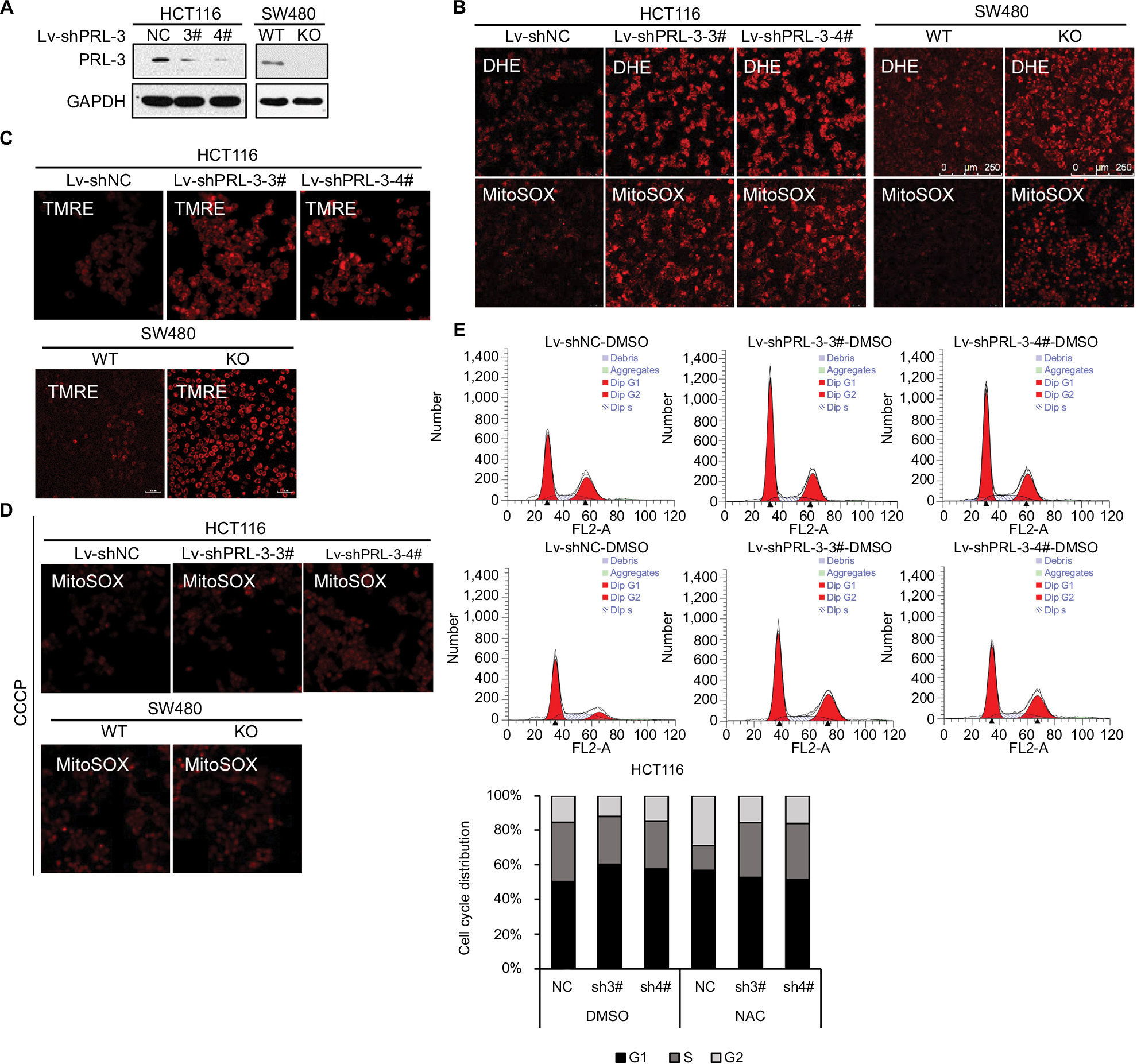 | Figure 1 Knockdown of PRL-3 increases mitochondrial superoxide anion and induces cell cycle arrest (A) Knockdown of PRL-3 in HCT116 cells, and knockout of PRL-3 in SW480 cells. “WT” is wild-type cells, which is the control for PRL-3 knockout cells. “NC” is negative control, which is shRNA control that encodes for a scrambled sequence. (B) Representative DHE and MitoSOX staining in PRL-3 knockdown or knockout cells. (C) Detecting mitochondria membrane potential through TMRE staining in PRL-3 knockdown or knockout cells. (D) MitoSOX staining after PRL-3 knockdown cells were pretreated with CCCP. Treating the PRL-3 knockdown or knockout cells with 20 µM CCCP for 30 minutes at 37°C with 5% CO2, and then staining the cells with MitoSOX. (E) Representative cell cycle analysis and summarized flow cytometry data. Cells were pretreated with DMSO (control) or 5 mM NAC for 24 hours at 37°C with 5% CO2. Abbreviations: CCCP, carbonyl cyanide 3-chlorophenylhydrazone; DHE, dihydroethidium; NAC, N-acetyl-l-cysteine; PRL, phosphatase of regenerating liver; TMRE, tetramethylrhodamine, ethyl ester. |
ROS has been broadly proven to affect cell signaling pathway, immune response, and other physiological responses.28 High levels of ROS could cause damage to DNA, protein or lipids and further restrain cell proliferation and induce cell death. Here, we measured PRL-3’s effect on cell cycle arrest. Knockdown of PRL-3 could induce apparent G1 phase arrest (Figure 1E). However, treatment of the cells with 5 mM NAC for 24 hours significantly diminished PRL-3 knockdown-caused G1 phase arrest (Figure 1E), indicating decreased PRL-3-induced cell cycle arrest is related to increased superoxide anion, which is consistent with our previous study.29
PRL-3 transcriptionally regulates the expression of RAP1
As a transcriptional regulator, RAP1 could affect the expression of PGC-1α, which is crucial for mitochondria function and glucose, fatty acid metabolism, and ROS metabolism.30–32 Our former study showed that PRL-3 could interact with RAP1 and affect its expression,23 but the specific mechanism is not fully studied. Here, we examined whether PRL-3 could transcriptionally modulate the expression of RAP1. Overexpression of PRL-3 in HCT116 and SW480 cells both increased the protein (Figure 2A, left) and mRNA (Figure 2B, up) levels of RAP1. In turn, decreased PRL-3 reduced the protein (Figure 2A, right) and mRNA (Figure 2B, bottom) levels of RAP1. We next investigated the mechanism of PRL-3 regulating RAP1 expression through luciferase reporter assay. We constructed pGLB-RAP1-promoter (–1,500/+150) plasmid and pGLB-RAP1-promoter (–500/+150) plasmid. Luciferase reporter assays showed that overexpression of PRL-3 markedly increased the luciferase activity of pGLB-RAP1-promoter (–1,500/+150) but not pGLB-RAP1-promoter (–500/+150) (Figure 2C), indicating PRL-3 could bind to the promoter (−1,500/–500) of RAP1 gene and regulate RAP1 expression. Consistently, our ChIP assay using PRL-3 antibody and qPCR result showed that PRL-3 could bind to the b and d regions between –1,500 bp and –500 bp of RAP1 gene promoter (Figure 2D). We also conducted expression correlation analysis in TCGA data and GTEx data through GEPIA, and the expression level of PRL-3 was positively correlated with that of RAP1 in both TCGA data and GTEx data (Figure 2E). These results revealed that PRL-3 could positively regulate RAP1 expression through binding to the promoter of RAP1 gene.
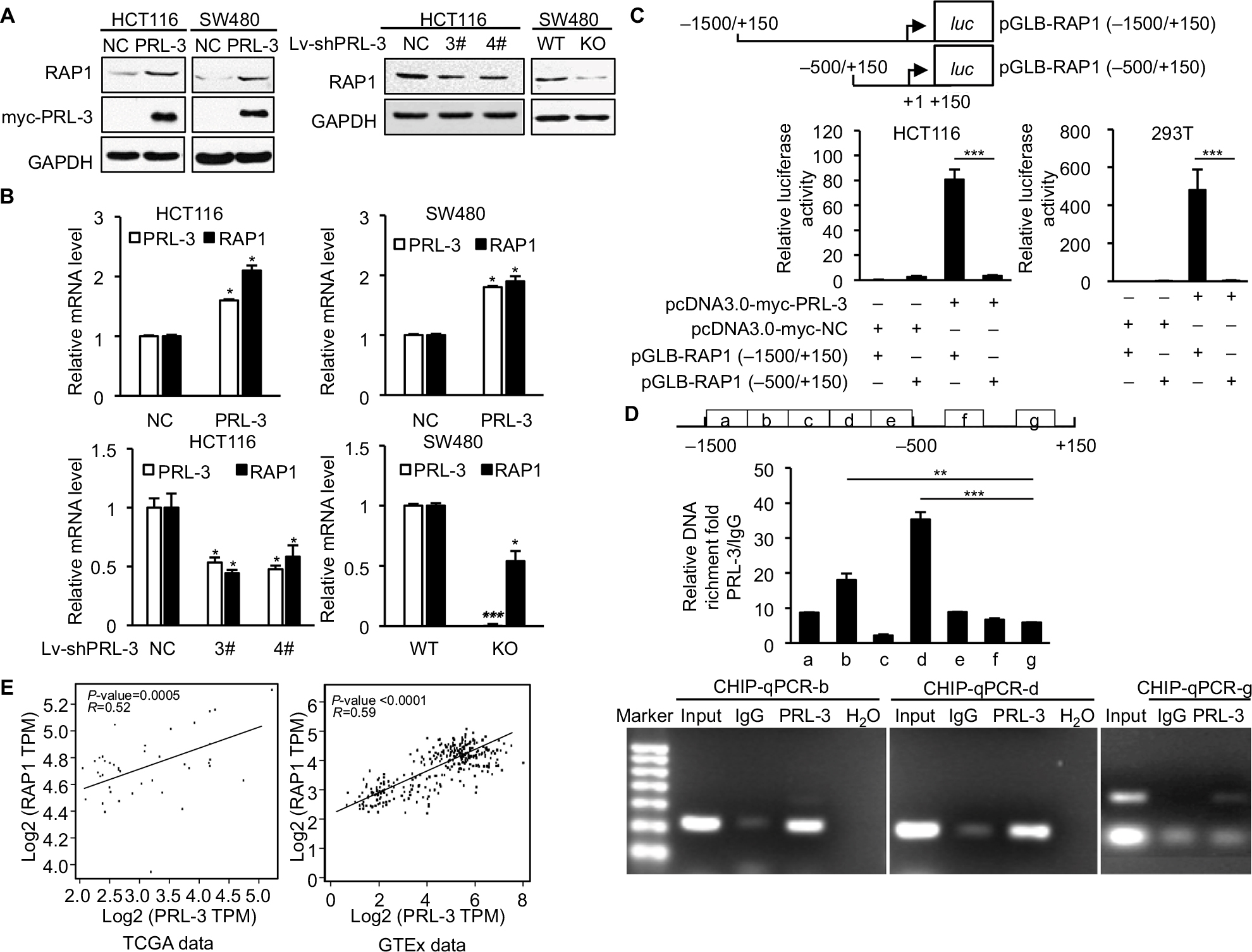 | Figure 2 PRL-3 transcriptionally regulates the expression of RAP1. Notes: (A) Western blot results showed that overexpression of PRL-3 increased the RAP1 level (left), and silencing of PRL-3 decreased RAP1 level (right). (B) RT-PCR results showed that overexpression of PRL-3 increased RAP1 level (upper) and silencing of PRL-3 decreased RAP1 level (lower). (C) Dual luciferase reporter assays of rap1 gene promoter in HCT116 and 293 T cells expressing pcDNA3.0-myc-NC/PRL-3 and pGLB-RAP1(–1,500/+150)/ (–500/+150). Results were normalized to the activity obtained in cells transfected with pcDNA3.0-myc-NC and pGLB-RAP1 (–1,500/+150). (D) ChIP assay using PRL-3 or IgG in HCT116 cells, and qPCR of different promoter regions of rap1 gene. Values correspond to the ratio between the PRL-3 antibody immunoprecipitated DNA with respect to the IgG immunoprecipitated DNA. (E) Gene expression correlation analysis between PRL-3 and RAP1 in TCGA data (P=0.0005, R=0.52) and GTEx data (P<0.0001, R=0.59). Mean ± SD of three independent experiments. *P<0.05, **P<0.01, and ***P<0.001. Abbreviations: IgG, immunoglobulin G; PRL, phosphatase of regenerating liver; RT-PCR, real-time PCR; RAP1, repressor activator protein 1; TMRE, tetramethylrhodamine, ethyl ester. |
Overexpression of RAP1 attenuates PRL-3 knockdown induced mitochondrial superoxide anion
Next, we tried to find whether PRL-3 knockdown induced mitochondrial superoxide anion is associated with RAP1. We first explored RAP1’s effect on superoxide anion level. The expression of RAP1 was suppressed through shRNAs in both HCT116 and SW480 cells (Figure 3A). We detected superoxide anion level using DHE and MitoSOX. Knockdown of RAP1 resulted in increased superoxide anion level in both HCT116 and SW480 cells (Figure 3B).
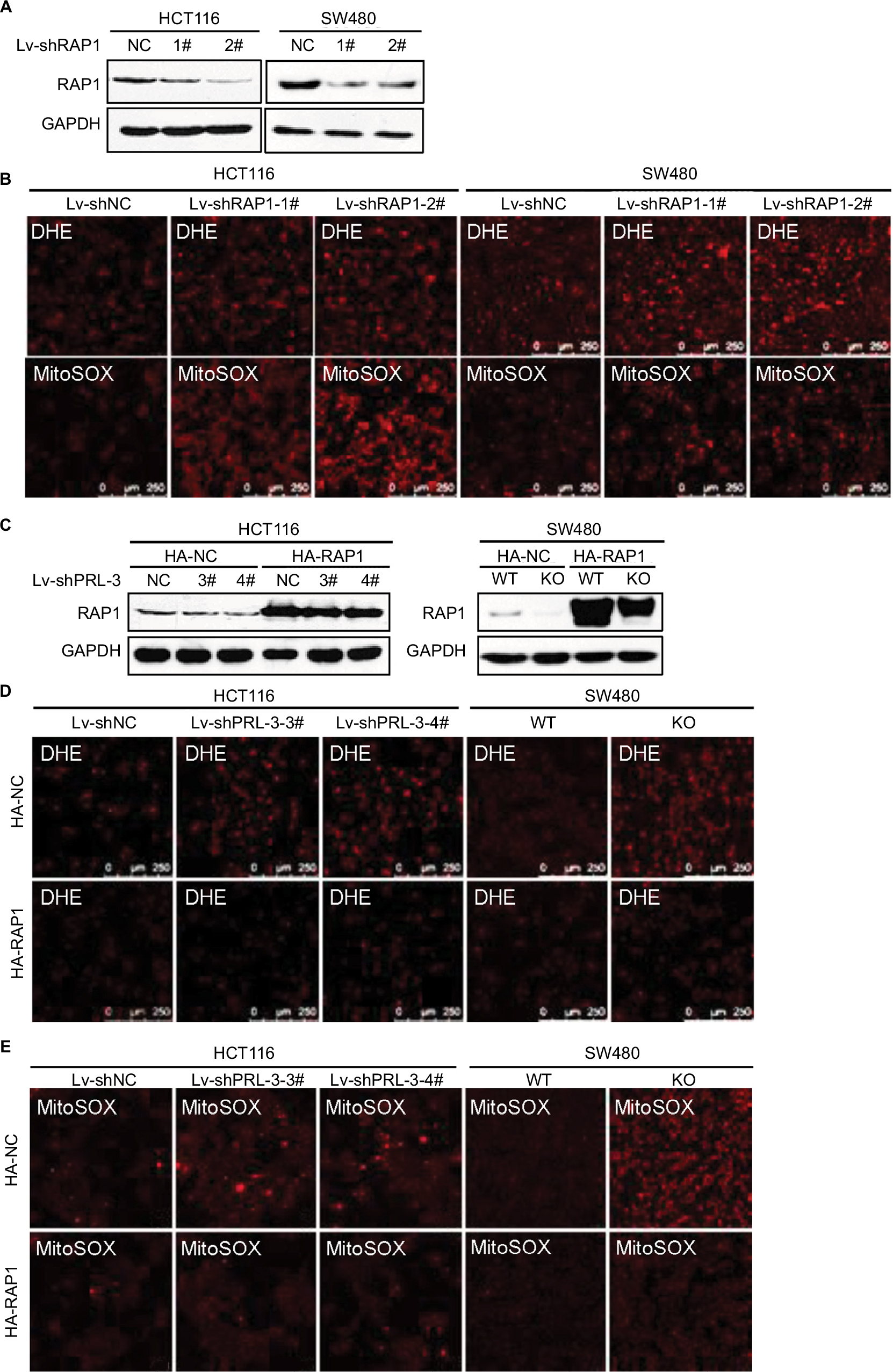 | Figure 3 Overexpression of RAP1 attenuates PRL-3 knockdown induced mitochondrial superoxide anion. Notes: (A) Knockdown of RAP1 in HCT116 and SW480 cells. (B) Representative DHE and MitoSOX staining in RAP1 knockdown cells. (C) Overexpression of RAP1 in PRL-3 knockdown or KO cells. pcDNA3-RAP1-HA and pcDNA3-vector-HA were transfected in PRL-3 knockdown cells. (D) Representative DHE staining after overexpression of RAP1 in PRL-3 knockdown or KO cells. (E) Representative MitoSOX staining after overexpression of RAP1 in PRL-3 knockdown or KO cells. Abbreviations: DHE, dihydroethidium; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; KO, knockout; PRL, phosphatase of regenerating liver; RAP1, repressor activator protein 1; WT, wild-type. |
Then, we detected the superoxide anion after overexpressing RAP1 in PRL-3 knockdown cells. The result showed that overexpressing RAP1 could attenuate PRL-3 knockdown-strengthened DHE and MitoSOX staining (Figure 3C–E), suggesting that PRL-3 knockdown-induced mitochondrial superoxide anion is associated with down-regulated RAP1.
Knockdown of PRL-3 decreases the expression of PGC-1α and its downstream genes
Through Western blot and qRT-PCR, we confirmed that in HCT116 and SW480 cells, knockdown of RAP1 significantly decreased the protein and mRNA levels of PGC-1α (Figure 4A), which is consistent with previous reports.18,19 It is interesting that knockdown of PRL-3 also decreased the protein and mRNA levels of PGC-1α (Figure 4B). Due to PRL-3’s effect on RAP1 expression, we hypothesize that RAP1 is involved in PRL-3’s regulation of PGC-1α. We first overexpressed PRL-3 in HCT116 and SW480 cells, and PRL-3 overexpression increased the expression of PGC-1α (Figure 4C). Then we knockdown RAP1 in the PRL-3 overexpressing and control cells. Knockdown of RAP1 in PRL-3 overexpressing cells suppressed PRL-3-induced upregulation of PGC-1α (Figure 4C), suggesting PRL-3 regulates the expression of PGC-1α through RAP1. PGC-1α is a transcriptional co-activator, which binds to various transcription factors and regulates oxidative metabolism and mitochondrial biogenesis. Knockdown of PGC-1α results in reduced mitochondrial DNA.33 Then we tested the effect of decreased PRL-3 and RAP1 on mitochondria biogenesis through measuring mitochondria DNA content. The result showed that knockdown of either PRL-3 or RAP1 reduced mitochondria DNA content (Figure 4D), which is probably related with decreased PGC-1α.
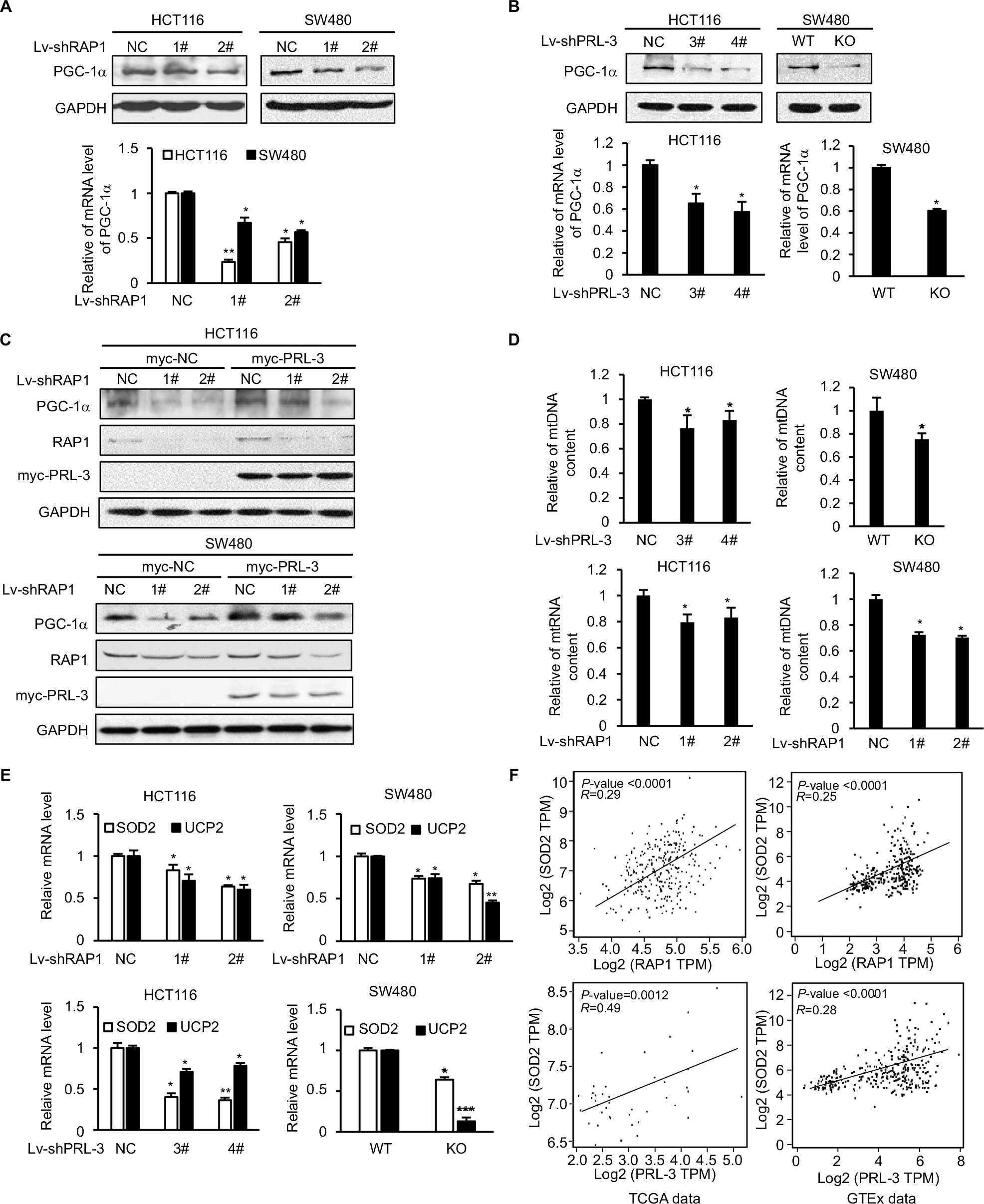 | Figure 4 Knockdown of PRL-3 decreases the expression of PGC-1α targeted SOD2 and UCP2. Notes: (A) Western blot (up) and RT-PCR (bottom) results showed that knockdown of RAP1 decreased the expression of PGC-1α. (B) Western blot (up) and RT-PCR (bottom) results showed that knockdown of PRL-3 decreased the expression of PGC-1α. (C) Knockdown of RAP1 in PRL-3 overexpressing cells suppressed the upregulation of PGC-1α. PRL-3 was overexpressed through transfection of pcDNA3-PRL-3-myc, and pcDNA3-vector-myc as control. Then RAP1 was knockdown through shRNA lentivirus infection. (D) Measurement of relative mtDNA by RT-PCR after knockdown of either PRL-3 or RAP1. (E) Knockdown of either RAP1 (up) or PRL-3 (bottom) decreased the mRNA level of SOD2 and UCP2. (F) Gene expression correlation analysis between PRL-3 and SOD2, RAP1 and SOD2 in TCGA data (PRL-3-SOD2: P=0.0012, R=0.49; RAP1-SOD2: P<0.0001, R=0.29) and GTEx data (PRL-3-SOD2: P<0.0001, R=0.28; RAP1-SOD2: P<0.0001, R=0.25). Mean ± SD of three independent experiments. *P<0.05, **P<0.01, and ***P<0.001. Abbreviations: RAP1, repressor activator protein 1; RT-PCR, real-time PCR; SOD2, superoxide dismutase 2; UCP2, uncoupling protein 2. |
We further studied PRL-3 and RAP1’s effect on PGC-1α’s downstream genes, SOD2 and uncoupling protein 2 (UCP2), which are two important ROS-detoxifying proteins identified in mitochondria to reduce the damaging effect resulted from excessive oxidative stress.32,34,35 Knockdown of either PRL-3 or RAP1 decreased the expression of SOD2 and UCP2 (Figure 4E). We also analyzed the expression correlation between PRL-3 and SOD2, RAP1 and SOD2. The result showed that the expression levels of both PRL-3 and RAP1 are positively correlated with that of SOD2 in TCGA data and GTEx data (Figure 4F). These data suggest that decreased PGC-1α and its downstream genes, SOD2 and UCP2, may contribute to increased superoxide anion induced by knockdown of either PRL-3 or RAP1.
Discussion
Our study showed that knockdown of PRL-3 could highly increase mitochondrial superoxide anion and mitochondrial membrane potential, and cause cell cycle arrest. We discovered that PRL-3 could transcriptionally regulate RAP1. We further concluded that PRL-3 knockdown-induced mitochondrial superoxide anion is associated with the regulation of RAP1 and PGC-1α.
PRL-3 was mainly reported to promote cancer cell metastasis and invasion through activating diverse oncogenic signaling pathways, and as a biomarker of poor prognosis.2–4 Previous studies about PRL-3’s effect on cell proliferation are conflicting.7,12,14,15,17 Both overexpression and knockdown of PRL-3 have been shown to inhibit mouse embryonic fibroblasts cell proliferation, suggesting the complex role of PRL-3’s expression in cell cycle regulation.12 In our recent study, we discovered that knockdown of PRL-3 significantly decreased cell proliferation through enhanced production of superoxide anion without obvious effect on the telomere integrity.13 Our result is consistent with previous reports showing decreased PRL-3 expression could reduce cell proliferation.5,12,15,17 In this study, we further explored the mechanism of superoxide anion generation after PRL-3 knockdown. Mitochondrial respiration is inhibited at high mitochondrial membrane potential, accompanied by increased ROS formation.27 We found that knockdown of PRL-3 could largely increase the mitochondrial membrane potential, which promotes the production of superoxide anion in mitochondria. However, the mechanism of decreased PRL-3 increasing mitochondrial membrane potential is not clear. In our study, we found that knockdown of PRL-3 could inhibit the expression of UCP2, which was reported to reduce mitochondrial membrane potential hyperpolarization and prevent ROS production under stress,36,37 suggesting that UCP2 might participate in PRL-3’s role in regulating mitochondria membrane potential. Superoxide anion is a main form of ROS, which could react to form the other two ROS, such as hydroxyl radical (OH∙) and nonradical oxygen derivatives. Mitochondria are the major organelles that produce ROS within cells.38 Various reports showed that increased ROS from mitochondria could cause cell senescence and apoptosis.39–41 In our previous study, we treated PRL-3 knockdown cells with ROS scavenger, NAC or glutathione (GSH), and then PRL-3 knockdown-induced cell senescence was compromised.13 Here, in this study, we further revealed that knockdown of PRL-3-induced mitochondrial superoxide anion is responsible for PRL-3 knockdown-caused G1 phase arrest.
RAP1 has been reported to be involved in lipid metabolism through regulation of PPARα and PGC-1α.18,42 PGC-1α plays a pivotal role in the regulation of ROS metabolism and mitochondrial biogenesis.32 It is reasonable that RAP1 may have a role in mitochondrial superoxide anion production. Our previous study showed that PRL-3 affects the expression of RAP1, so we supposed that knockdown of PRL-3-induced superoxide anion is very possibly associated with RAP1. Then we investigated how PRL-3 mechanically regulates RAP1 expression. Through luciferase reporter and ChIP assays, we confirmed that PRL-3 could bind to the promoter of RAP1 gene and enhance the transcription of RAP1 gene, revealing that PRL-3 could act as a transcriptional enhancer to upregulate the expression of RAP1. Previous studies about PRL-3’s role in cellular signaling were mainly focused on the proteins indirectly activated by PRL-3, such as ATK,10 SRC,7 and EGFR.43 Our study provided a new possible mechanism that PRL-3 may regulate cell process through acting as a transcriptional factor. However, it is not clear whether PRL-3 binds to the RAP1 promoter DNA directly or through the interaction with other DNA binding proteins. PRL-3 does not contain a DNA binding domain, and our previous study showed that PRL-3’s binding to telomere DNA is dependent on its interaction with RAP1,13 so it is very likely that PRL-3 binds to rap1 promoter DNA through the interaction with other DNA binding proteins.
We next detected the effect of knockdown of RAP1 on the production of mitochondrial superoxide anion and found that knockdown of RAP1 highly increased mitochondrial superoxide anion as PRL-3 did. Overexpression of RAP1 in PRL-3 knockdown cells inhibited the increase of mitochondrial superoxide anion, demonstrating that RAP1 truly participates in the regulation of mitochondrial superoxide anion production, and knockdown of PRL-3-induced superoxide anion is associated with RAP1. We also discovered that knockdown of either RAP1 or PRL-3 significantly decreased the protein and mRNA levels of PGC-1α. And, knockdown of RAP1 could inhibit PRL-3’s upregulation of PGC-1α, implying that PRL-3’s effect on the expression of PGC-1α is mediated by RAP1. Suppression of PGC-1α prevents the induction of SOD2 and UCP2 in response to oxidative stress.32 We further found both SOD2 and UCP2 were decreased after knockdown of either PRL-3 or RAP1. As SOD2 is a critical superoxide dismutase, and UCP2 could stabilize mitochondria membrane potential and then reduce the production of ROS,36,37 decreased SOD2 and UCP2 are probably the main cause of increased mitochondrial superoxide anion in both PRL-3 and RAP1 knockdown cells.
In summary, our study revealed a heretofore undescribed role for PRL-3 in mitochondrial superoxide anion generation as a transcriptional factor. We discovered that knockdown of PRL-3 could increase mitochondrial superoxide anion and mitochondria membrane potential, which induces G1 phase arrest in colon cancer cell lines. We also showed that knockdown of RAP1 could increase mitochondrial superoxide anion, which is in accord with its role in metabolism. We proved that PRL-3 could act as a transcriptional factor as it regulates the expression of RAP1 through binding to the promoter of rap1 gene. Furthermore, PRL-3 affects the expression of PGC-1α and its target genes via RAP1 to regulate the accumulation of mitochondrial superoxide anion. Our study will benefit the investigation of PRL-3’s role in cancer progression in the future.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (grant no. 81230046 and 81872309) and the Ministry of Science and Technology of China (grant no. 2015CB553906).
Disclosure
The authors report no conflicts of interest in this work.
References
Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7(11):833–846. | ||
Stephens BJ, Han H, Gokhale V, von Hoff DD. PRL phosphatases as potential molecular targets in cancer. Mol Cancer Ther. 2005;4(11):1653–1661. | ||
Kato H, Semba S, Miskad UA, Seo Y, Kasuga M, Yokozaki H. High expression of PRL-3 promotes cancer cell motility and liver metastasis in human colorectal cancer: a predictive molecular marker of metachronous liver and lung metastases. Clin Cancer Res. 2004;10(21):7318–7328. | ||
Peng L, Xing X, Li W, et al. PRL-3 promotes the motility, invasion, and metastasis of LoVo colon cancer cells through PRL-3-integrin beta1-ERK1/2 and-MMP2 signaling. Mol Cancer. 2009;8:110. | ||
Gari HH, Degala GD, Ray R, Lucia MS, Lambert JR. PRL-3 engages the focal adhesion pathway in triple-negative breast cancer cells to alter actin structure and substrate adhesion properties critical for cell migration and invasion. Cancer Lett. 2016;380(2):505–512. | ||
Wang H, Quah SY, Dong JM, Manser E, Tang JP, Zeng Q. PRL-3 down-regulates PTEN expression and signals through PI3K to promote epithelial-mesenchymal transition. Cancer Res. 2007;67(7):2922–2926. | ||
Liang F, Liang J, Wang WQ, Sun JP, Udho E, Zhang ZY. PRL3 promotes cell invasion and proliferation by down-regulation of Csk leading to Src activation. J Biol Chem. 2007;282(8):5413–5419. | ||
Ming J, Liu N, Gu Y, Qiu X, Wang EH. PRL-3 facilitates angiogenesis and metastasis by increasing ERK phosphorylation and up-regulating the levels and activities of Rho-A/C in lung cancer. Pathology. 2009;41(2):118–126. | ||
Lian S, Meng L, Xing X, Yang Y, Qu L, Shou C. PRL-3 promotes cell adhesion by interacting with JAM2 in colon cancer. Oncol Lett. 2016;12(3):1661–1666. | ||
Walls CD, Iliuk A, Bai Y, Wang M, Tao WA, Zhang ZY. Phosphatase of regenerating liver 3 (PRL3) provokes a tyrosine phosphoproteome to drive prometastatic signal transduction. Mol Cell Proteomics. 2013;12(12):3759–3777. | ||
Yang Y, Lian S, Meng L, Qu L, Shou C. Antibody array revealed PRL-3 affects protein phosphorylation and cytokine secretion. PLoS One. 2017;12(1):e0169665. | ||
Basak S, Jacobs SB, Krieg AJ, et al. The metastasis-associated gene Prl-3 is a p53 target involved in cell-cycle regulation. Mol Cell. 2008;30(3):303–314. | ||
Lian S, Meng L, Yang Y, et al. PRL-3 promotes telomere deprotection and chromosomal instability. Nucleic Acids Res. 2017;45(11):6546–6571. | ||
Cai SR, Wang Z, Chen CQ, et al. Role of silencing PRL-3 expression by miRNA interference in the growth of gastric cancer. Zhonghua Wai Ke Za Zhi. 2008;46(8):618–621. | ||
Gari HH, Degala GD, Lucia MS, Lambert JR. Loss of the oncogenic phosphatase PRL-3 promotes a TNF-R1 feedback loop that mediates triple-negative breast cancer growth. Oncogenesis. 2016;5(8):e255. | ||
Slørdahl TS, Abdollahi P, Vandsemb EN, et al. The phosphatase of regenerating liver-3 (PRL-3) is important for IL-6-mediated survival of myeloma cells. Oncotarget. 2016;7(19):27295–27306. | ||
Matsukawa Y, Semba S, Kato H, Koma Y, Yanagihara K, Yokozaki H. Constitutive suppression of PRL-3 inhibits invasion and proliferation of gastric cancer cell in vitro and in vivo. Pathobiology. 2010;77(3):155–162. | ||
Yeung F, Ramírez CM, Mateos-Gomez PA, et al. Nontelomeric role for Rap1 in regulating metabolism and protecting against obesity. Cell Rep. 2013;3(6):1847–1856. | ||
Martínez P, Gómez-López G, García F, et al. RAP1 protects from obesity through its extratelomeric role regulating gene expression. Cell Rep. 2013;3(6):2059–2074. | ||
Martinez P, Thanasoula M, Carlos AR, et al. Mammalian Rap1 controls telomere function and gene expression through binding to telomeric and extratelomeric sites. Nat Cell Biol. 2010;12(8):768–780. | ||
Teo H, Ghosh S, Luesch H, et al. Telomere-independent Rap1 is an IKK adaptor and regulates NF-kappaB-dependent gene expression. Nat Cell Biol. 2010;12(8):758–767. | ||
Yang Y, Ye C, Wang L, et al. Repressor activator protein 1-promoted colorectal cell migration is associated with the regulation of Vimentin. Tumour Biol. 2017;39(4):1010428317695034. | ||
Lian S, Meng L, Liu C, et al. PRL-3 activates NF-κB signaling pathway by interacting with RAP1. Biochem Biophys Res Commun. 2013;430(1):196–201. | ||
Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102. | ||
Mambo E, Chatterjee A, Xing M, et al. Tumor-specific changes in mtDNA content in human cancer. Int J Cancer. 2005;116(6):920–924. | ||
Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191(2):421–427. | ||
Lee I, Bender E, Kadenbach B. Control of mitochondrial membrane potential and ROS formation by reversible phosphorylation of cytochrome c oxidase. Mol Cell Biochem. 2002;234-235(1–2):63–70. | ||
Zuo L, Zhou T, Pannell BK, Ziegler AC, Best TM. Biological and physiological role of reactive oxygen species--the good, the bad and the ugly. Acta Physiol. 2015;214(3):329–348. | ||
Lian S, Meng L, Yang Y, et al. PRL-3 promotes telomere deprotection and chromosomal instability. Nucleic Acids Res. 2017;45(11):6546–6571. | ||
Knutti D, Kralli A. PGC-1, a versatile coactivator. Trends Endocrinol Metab. 2001;12(8):360–365. | ||
Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24(1):78–90. | ||
St-Pierre J, Drori S, Uldry M, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127(2):397–408. | ||
Lebleu VS, O’Connell JT, Gonzalez Herrera KN, et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promotemetastasis. Nat Cell Biol. 2014;16(10):992–1003. | ||
Nègre-Salvayre A, Hirtz C, Carrera G, et al. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. Faseb J. 1997;11(10):809–815. | ||
Raha S, Mceachern GE, Myint AT, Robinson BH. Superoxides from mitochondrial complex III: the role of manganese superoxide dismutase. Free Radic Biol Med. 2000;29(2):170–180. | ||
Sanderson TH, Reynolds CA, Kumar R, Przyklenk K, Hüttemann M. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol. 2013;47(1):9–23. | ||
Brand MD, Esteves TC. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2005;2(2):85–93. | ||
Patel M. Mitochondrial dysfunction and oxidative stress: cause and consequence of epileptic seizures. Free Radic Biol Med. 2004;37(12):1951–1962. | ||
Bayir H, Kagan VE. Bench-to-bedside review: mitochondrial injury, oxidative stress and apoptosis--there is nothing more practical than a good theory. Crit Care. 2008;12(1):206. | ||
Papaconstantinou J, Hsieh CC. Activation of senescence and aging characteristics by mitochondrially generated ROS: how are they linked? Cell Cycle. 2010;9(19):3831–3833. | ||
Fleury C, Mignotte B, Vayssière JL. Mitochondrial reactive oxygen species in cell death signaling. Biochimie. 2002;84(2-3):131–141. | ||
Martínez P, Gómez-López G, García F, et al. RAP1 protects from obesity through its extratelomeric role regulating gene expression. Cell Rep. 2013;3(6):2059–2074. | ||
Al-Aidaroos AQ, Yuen HF, Guo K, et al. Metastasis-associated PRL-3 induces EGFR activation and addiction in cancer cells. J Clin Invest. 2013;123(8):3459–3471. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.