Back to Journals » International Journal of Nanomedicine » Volume 12
Killing malignant melanoma cells with protoporphyrin IX-loaded polymersome-mediated photodynamic therapy and cold atmospheric plasma
Authors Wang M ![]() , Geilich BM, Keidar M, Webster TJ
, Geilich BM, Keidar M, Webster TJ ![]()
Received 2 December 2016
Accepted for publication 2 March 2017
Published 31 May 2017 Volume 2017:12 Pages 4117—4127
DOI https://doi.org/10.2147/IJN.S129266
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Israel Rubinstein
Mian Wang,1 Benjamin M Geilich,2 Michael Keidar,3 Thomas J Webster1,4
1Department of Chemical Engineering, 2Department of Bioengineering, Northeastern University, Boston, MA, 3Department of Mechanical and Aerospace Engineering, George Washington University, Washington, DC, USA; 4Wenzhou Institute of Biomaterials and Engineering, Wenzhou Medical University, Wenzhou, People’s Republic of China
Abstract: Traditional cancer treatments contain several limitations such as incomplete ablation and multidrug resistance. It is known that photodynamic therapy (PDT) is an effective treatment for several tumor types especially melanoma cells. During the PDT process, protoporphyrin IX (PpIX), an effective photosensitizer, can selectively kill cancer cells by activating a special light source. When tumor cells encapsulate a photosensitizer, they can be easily excited into an excited state by a light source. In this study, cold atmospheric plasma (CAP) was used as a novel light source. Results of some studies have showed that cancer cells can be effectively killed by using either a light source or an individual treatment due to the generation of reactive oxygen species and electrons from a wide range of wavelengths, which suggest that CAP can act as a potential light source for anticancer applications compared with UV light sources. Results of the present in vitro study indicated for the first time that PpIX can be successfully loaded into polymersomes. Most importantly, cell viability studies revealed that PpIX-loaded polymersomes had a low toxicity to healthy fibroblasts (20% were killed) at a concentration of 400 µg/mL, but they showed a great potential to selectively kill melanoma cells (almost 50% were killed). With the application of CAP posttreatment, melanoma cell viability significantly decreased (80% were killed) compared to not using a light source (45% were killed) or using a UV light source (65% were killed). In summary, these results indicated for the first time that PpIX-loaded polymersomes together with CAP posttreatment could be a promising tool for skin cancer drug delivery with selective toxicity toward melanoma cells sparing healthy fibroblasts.
Keywords: melanoma, polymersomes, protoporphyrin IX, cold atmospheric plasma, photodynamic therapy
Introduction
Cancer is the most distressing and life-threatening disease of modern times that continues to cause severe death worldwide.1 The most common treatment for cancer is chemotherapy, which is usually accompanied by a number of significant drawbacks, such as undesirable toxic effects to normal tissue, nonspecified distribution of anticancer drugs, and multidrug resistance.2 For example, chemotherapy kills drug-sensitive cells, but leaves behind a higher proportion of drug-resistant cells. Among the many kinds of cancers, malignant melanoma is known to be one of the most difficult metastatic cancers to cure.2 This is because melanoma is generally less sensitive to cytotoxic chemotherapeutic agents and radiation than other types of tumors.3 In other words, melanoma is strongly resistant to traditional chemotherapy and radiation therapy.
One of the most promising skin cancer treatments is photodynamic therapy (PDT), which is a relatively new therapeutic method that combines the administration of a photosensitizer, and subsequent targeted irradiation with visible light to generate an oxygen-dependent destruction of the diseased tissue.4 However, skin cancer also shows resistance to this new therapeutic tool. One of the melanoma-resistant mechanisms to PDT is low encapsulation of nanoparticles (used as photosensitizers) and an accompanied low uptake ratio in cells, which leads to a reduction in accumulation of the intracellular photosensitizers. This reduced uptake of photosensitizers clearly reduces the effect of PDT on killing melanoma cells.
To improve on the ability of PDT to kill melanoma cells, in this study, protoporphyrin IX (PpIX) was encapsulated into polymersome nanoparticles. PpIX is a naturally occurring porphyrin constituent of hemoglobin, cytochrome C, and other biologically relevant molecules.5 PpIX can absorb wavelengths around 400 nm, and then the photo-excited photosensitizer molecules trigger the formation of singlet oxygen molecules which subsequently lead to cancer cell death. Furthermore, the degradation rate of PpIX in cancer cells is different from normal cells. PpIX metabolism in all types of healthy cells is based on the bypass of negative feedback controls in the heme biosynthetic pathway.6 In this way, PpIX in normal healthy cells is usually maintained at a constant level and quickly degrades if it exceeds the equilibrium level. However, this feedback mechanism does not work well in cancer cells where PpIX degrades at a slow rate resulting in accumulation to very high concentrations leading to cancer cell toxicity due to a lack of enough ferrochelatase in cancer cells. Ferrochelatase is one of the enzymes involved in converting PpIX into heme, and thus, plays an important role after PpIX is endocytosed by cells. Ferrochelatase remains at a low activity level in cancer cells and results in a limited availability of iron that partially contributes to the high concentration of PpIX in these abnormal cells.7,8 According to previous research, PpIX can be totally degraded within 2–4 hours in normal cells, while it will last 12–24 hours in cancer cells.9 However, while promising, it is difficult to deliver hydrophobic PpIX to cancer cells, since aggregation in water is a large barrier for its efficacy.
Polymersomes can be used as a novel carrier for PpIX since they are synthesized from amphiphilic-block copolymers. They have advantages over other drug delivery vesicles, such as liposomes, due to enhanced stability and longer circulation times, and most importantly, they have an ability to carry both hydrophilic and hydrophobic drug molecules.10 Because they have higher molecular weights than single polymers, the bilayer membranes of polymersomes are generally thicker and stronger, resulting in greater stability than conventional drug delivery vesicles, such as liposomes and micelles.
Another barrier to the use of PDT to fight cancer is the light source. Traditional light sources such as lasers and X-rays damage both tumors and their surrounding healthy cells. In this study, cold atmospheric plasma (CAP) was used in combination with the above polymersomes as a light source. CAP is an ionized gas where the ion temperature is close to room temperature. It contains electrons, charged particles, radicals, various excited molecules, UV photons, and transient electrical fields.11 These various compositional elements have the potential to either enhance and promote cellular activity or disrupt and destroy cells based on different cellular structures and different cell resistance to this treatment. For example, a combined treatment may easily improve cancer cell permeability since cancer cell membranes are more vulnerable than healthy cells resulting in a greater uptake of nanoparticles. Based on this unique property, CAP could offer a minimally invasive surgical approach allowing for specific cancer cell or tumor tissue removal without influencing healthy cells.12 In addition, CAP spectra cover the range of 250–800 nm, where especially at 400 nm, CAP has a very strong peak and can induce PpIX release inside cells.13
Therefore, the objective of this in vitro study was for the first time to design a new nano-carrier (PpIX embedded in polymersomes) and use it in combination with a new light source (CAP) to selectively kill melanoma cells. In this study, two different light sources (CAP and UV light) were compared to highlight the efficiency of CAP posttreatment.
Materials and methods
Materials
PpIX and dialysis bags (molecular weight: 10,000 Da) were purchased from Sigma-Aldrich (St Louis, MO, USA). Monomethoxy poly (ethylene glycol)-b-poly(D,L-lactide) (mPEG-PDLLA) copolymers were purchased from Polyscitech (West Lafayette, IN, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and all other reagents were obtained from Sigma at reagent grade or higher and were used without further purification in the studies.
Polymersome synthesis and physical characterization
Nanoparticle synthesis
PpIX was encapsulated inside polymersomes by self-assembly.14 First, 10 mg of the mPEG-PDLLA copolymer (Polyscitech) was added to 1 mL of tetrahydrofuran (THF; Sigma-Aldrich), which was ultrasonicated until the copolymer was completely dissolved. Then, PpIX was added to the mixture and again ultrasonicated until the drugs/polymers were dissolved in THF. This organic drug/polymer solution was then injected through a syringe atomizer (MAD300; LMA, San Diego, CA, USA) into a phosphate-buffered saline (PBS) solution (Sigma-Aldrich) in a 15 mL glass round-bottom tube with stirring. Finally, the entire polymersome solution was transferred to a 50 kDa dialysis tube (Spectra/Por Float-A-Lyzer G2; Spectrum Labs, Rancho Dominguez, CA, USA) and was allowed to dialyze against pure PBS for 48 hours, with two buffer changes, to remove all traces of the organic solvent and unencapsulated drug.
Physical characterization of polymersomes
Particle size was determined using dynamic light scattering (DLS) (Wyatt, Santa Barbara, CA, USA). Size measurements were performed in triplicate followed by diluting the polymersome suspension 10 times in distilled water at 25°C. Zeta potential was measured in the same instrument at 25°C using the above protocol. All the measurements were performed in triplicate.
Polymersomes were fixed to sample stubs with double-sided carbon tape and sputter coated with platinum for viewing by a scanning electron microscope (XL30 ESEM; FEI Company, Hillsboro, OR, USA). The internal structure of the polymersomes was determined by transmission electron microscopy (TEM) measurements, for which a drop of a diluted solution of the polymersomes loaded with and without PpIX was placed in carbon-coated copper transmission electron microscope grids (150 mesh; Ted Pella Inc., Redding, CA, USA), negatively stained with 1% uranyl acetate (w/v) for 10 minutes, and was allowed to air-dry. The samples were imaged using a Philips 201 transmission electron microscope (FEI Company) at 120 kV.
Fourier transform infrared (FTIR) spectra (PerkinElmer, Waltham, MA, USA) were taken in order to investigate the possible chemical interactions between the PpIX and the polymers. Blank polymersomes, free PpIX, and PpIX-loaded polymersomes were measured by applying a pressure of 300 kg/cm2. FTIR spectra of the above samples were obtained by averaging 32 interferograms with a resolution of 4 cm−1 and 100 scans per sample in the range of 1,000–4,000 cm−1. X-ray diffraction (XRD) (Bruker 9XS; G8ADVANCE) analysis was completed to determine the crystallographic structure of the PpIX-loaded polymersome formulations. The patterns of free PpIX, blank polymersomes, and PpIX-loaded polymersomes were obtained using an X-ray diffractometer. The measurements were performed at a voltage of 40 kV and 25 mA. The scanned angle was set from 3°≤2θ≤40°, and the scan rate was 2 minute−1.
Analysis of photophysical properties of PpIX loaded in polymersomes and free PpIX
Encapsulation and binding of PpIX between the hydrophilic layers during polymersome formulation was further examined by spectroscopic analysis. The absorbance and fluorescence spectra were measured for both free PpIX (dissolved in PBS) and PpIX loaded in polymersomes (PpIX was dissolved in a THF solution) for different concentrations (Particle Size Analyzer 90 Plus; Brookhaven Instruments Company, Holtsville, NY, USA). Since the ratio of the drug to the polymer was 1–5 when polymersomes were made, the same PpIX concentration should hold for the free PpIX and PpIX-loaded polymersomes. Therefore, the concentrations of free PpIX measured were 50, 25, 12.5, 6.25, and 3.125 μg/mL, while the corresponding concentrations of PpIX-loaded polymersomes were 250, 125, 62.5, 31.25, and 15.625 μg/mL. Aqueous free PpIX solutions and PpIX-loaded polymersome suspensions were vortexed followed by sonication for 1 minute to obtain a well-dispersed solution for spectroscopic studies. Fluorescence emission spectra of different concentrations of free PpIX and PpIX-loaded polymersomes were recorded from 500 to 700 nm with an excitation wavelength of 405 nm (PerkinElmer).
Cell studies
Cell culture
The cells used in the present study were purchased through a commercial vendor (American Type Culture Collection, ATCC, Manassas, VA, USA). Malignant melanoma cells A375 (ATCC CRL-1619) were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Human healthy dermal fibroblast cells (ATCC PCS-201-012) were maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. All cells were incubated in a standard incubator at 5% CO2 at 37°C. Both cells were used at population numbers less than 3.
Cytotoxicity
For cell viability studies, melanoma A375 cells and human dermal fibroblasts were separately seeded on a 96-well plate at a density of 30,000 cells/cm2. MTT assays were used to evaluate the cytotoxicity of the prepared polymersomes, as this is a colorimetric test based on the selective ability of viable cells to reduce the tetrazolium component of MTT into purple-colored formazan crystals. Five different concentrations of PpIX-loaded polymersomes (0, 50, 100, 300, and 500 μg/mL) were prepared by dilution with cell media. Since the ratio of drug to the polymer was 1–5, the corresponding concentrations of free PpIX and blank polymersomes were also prepared. For free PpIX, the concentrations were 10, 20, 60, and 100 μg/mL, while blank polymersome concentrations were 40, 80, 240, and 400 μg/mL. The media for the melanoma cells and fibroblasts were used as controls. Since PpIX fluoresces, polymersomes were suspended in cell culture medium alone and were used as a negative control. After being cultured for 24 hours, 3 days, and 5 days, the cells were washed with a PBS buffer, and different concentrations of PpIX-loaded polymersomes, free PpIX, and blank polymersomes (100 μL) were added and incubated for 4 hours. Fifteen microliters of the MTT solution was added and further incubated for 4 hours. About 100 μL of the solubilization solution (10% Triton X-100, 0.1 N HCl, and isopropanol) was added to each well and incubated at room temperature overnight to dissolve the formazan crystals. The optical density of the solution was measured at a wavelength of 570 nm using a UV spectra reader (UV-1700 Pharma Spec). All samples were analyzed in triplicate for each experiment.
Photodynamic therapies
Melanoma A375 cells were seeded into a 96-well plate at a density of 5×104 cell/mL and were incubated overnight at 37°C in a humidified 5% CO2 atmosphere. After being rinsed with PBS, the cells were incubated with 100 μL of varying concentrations of PpIX-loaded polymersomes for 24 hours under the same conditions. Then, cells with medium were placed under the CAP effluent. CAP was used in this study at a 3.7 kV output voltage. The distance between the CAP nozzle and plate bottom was 20 mm during all treatment conditions. All treatments were conducted in a laminar flow hood at room temperature. Immediately after incubation for 24 hours, the medium was discarded and washed with PBS. After removing the PBS, 100 μL of fresh medium with 15 μL of the MTT reagents was added to each well. MTT results were measured by a UV spectrophotometer. Samples were analyzed in triplicate for each experiment.
Confocal laser microscopy
Melanoma cells were grown in sterile coverglasses until 60% confluent, and then were exposed to the polymersomes as described above. After culture and PDT, cells were fixed with a 4% formaldehyde solution. Fixed cells were visualized using confocal laser microscopy. Confocal imaging was performed with a near-infrared reflectance confocal laser scanning microscope (CompuCyte Corporation, Westwood, MA, USA) with laser excitation at 488 nm using a 60× oil immersion objective.
Statistical analysis
The cellular experiments were run in triplicate and repeated three times for each treatment group. Data are presented as the mean value ± standard error of the mean and were analyzed with a Student’s t-test for pair-wise comparison. Statistical significance was considered at P<0.05.
Results
Physical properties of polymersomes loaded with PpIX
After free PpIX was encapsulated by the polymersomes, the polymersomes had an average size of 100±8.6 nm as measured by DLS with a narrow monodispersed unimodal size distribution pattern (Table 1). Empty polymersomes, that is, polymersomes without PpIX, were 70±6.7 nm in diameter (Table 1). The zeta potentials of the empty polymersomes and PpIX-loaded polymersomes were −12±2 and −18±1 mV, respectively (Table 1), which indicated a relatively stable state in solution.
 | Table 1 Properties of PpIX-loaded polymersomes |
TEM confirmed that the resultant polymersomes were spherical (Figure 1). The mean size distribution observed using DLS measurements correlated to that observed with TEM. Most of the polymersomes loaded with PpIX were around 100 nm. SEM images showed that the morphology and shape of the polymersomes were consistent with TEM results (Figure 2). However, the diameter of the polymersomes under SEM was much larger than what was shown using TEM and DLS results. This was expected due to the change of morphology during SEM sample preparation.
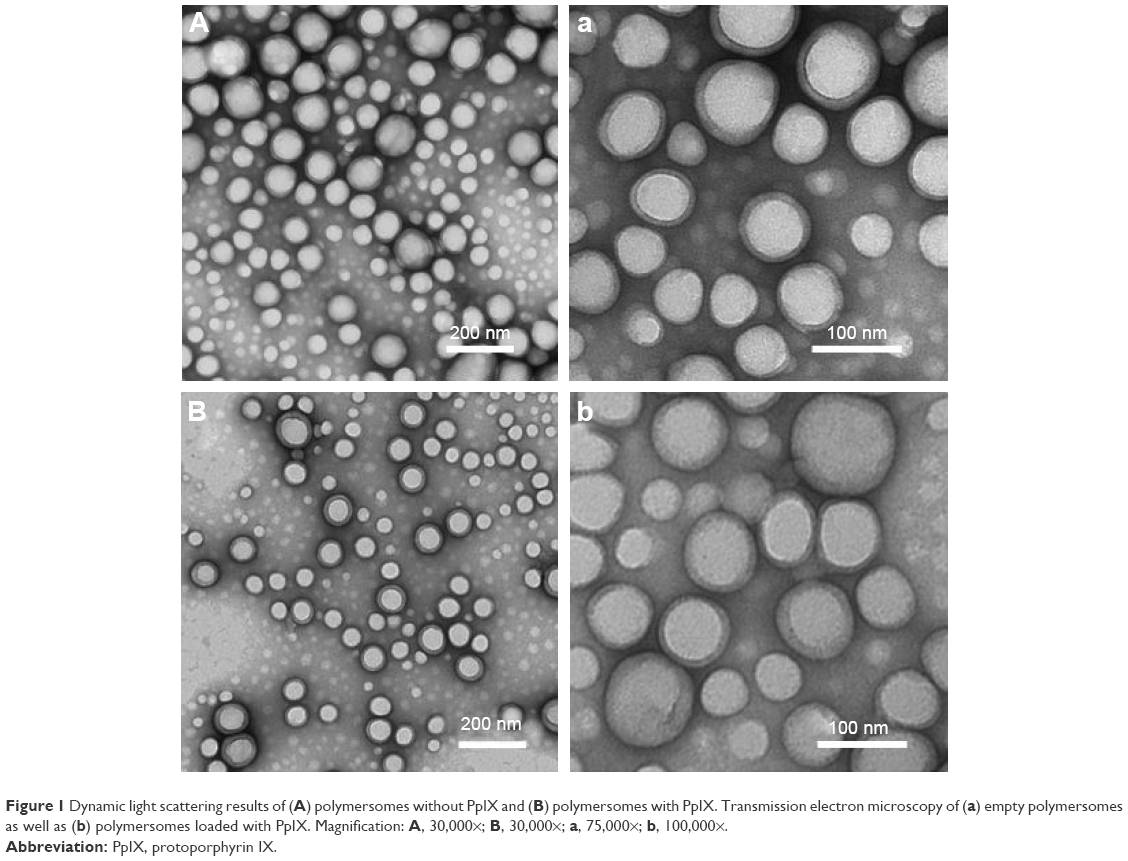 | Figure 1 Dynamic light scattering results of (A) polymersomes without PpIX and (B) polymersomes with PpIX. Transmission electron microscopy of (a) empty polymersomes as well as (b) polymersomes loaded with PpIX. Magnification: A, 30,000×; B, 30,000×; a, 75,000×; b, 100,000×. |
 | Figure 2 Scanning electron microscopy of (A) empty polymersomes and (B) polymersomes loaded with PpIX. |
Besides demonstrating a small size, results also showed a higher zeta potential for the PpIX-loaded polymersomes at around −20 mV. Such higher zeta potential values can help repel polymersomes to guarantee long-term stability and avoid aggregation. Further, to confirm the presence of PpIX in polymersomes, FTIR was used and showed strong peaks at 2,360 and 668 cm−1, characteristic peaks of PpIX, which were not found for the blank polymersomes (Figure 3).13
 | Figure 3 Fourier transform infrared spectroscopy of blank polymersomes, polymersomes loaded with PpIX, and free PpIX. |
Similarly, XRD demonstrated a highly crystalline structure of PpIX and the polymer. The same crystalline structure was found for the blank polymersomes (Figure 4). However, there were no characteristic peaks observed for PpIX when entrapped in polymersomes. This absence of detectable crystallinity of PpIX in polymersomes provides evidence that PpIX was encapsulated by the polymersomes. This is because if there was no PpIX, the curve would show peaks of the polymer, and only PpIX and the polymer mixture would show no peak on XRD, similar to the results shown. The XRD results were, thus, consistent with the FTIR results.
 | Figure 4 X-ray diffraction patterns of blank polymersomes, polymersomes loaded with PpIX, and free PpIX. |
Analysis of photophysical properties of polymersomes loaded with PpIX and without PpIX
To investigate the encapsulation of PpIX as well as binding of PpIX between the hydrophilic layers of the polymersomes, the photophysical properties of PpIX were taken into consideration. The results (Figure 5A and B) showed that both free PpIX in PBS and polymersomes loaded with PpIX dissolved in THF showed the same absorbance peaks and similar tendency. Free PpIX in PBS showed absorbance peaks at around 615 and 663 nm. The absorbance of free PpIX in PBS was relatively low when compared with free PpIX in the organic solvent. The same PpIX concentration with free PpIX and polymersome loaded with PpIX was investigated. For example, 50 μg/mL of the free PpIX had the same PpIX concentration as 250 μg/mL polymersomes loaded with PpIX.
 | Figure 5 Results of photophysical property analysis of different concentrations of (A) free PpIX and (B) PpIX encapsulated in polymersomes. |
The results from the photophysical studies showed that after PpIX was encapsulated by polymersomes, the intensity distinctly increased compared to free PpIX in PBS. For example, the intensity of 50 μg/mL free PpIX in PBS was around 1.2×106 absorbance, while the intensity of the corresponding concentration of polymersomes loaded with PpIX increased to around 4.3×106 absorbance. While observing the fluorescence spectra of PpIX (from 500 to 750 nm) at an excitation wavelength of 435 nm, it was found that the intensity of the free PpIX was not as obvious as that of the polymersomes loaded with PpIX. In addition, polymersomes loaded with PpIX showed a sharp fluorescence peak at 663 nm, a slight shift from 680 nm as was demonstrated for the free PpIX in PBS. This slight shift could be due to binding of PpIX between polymersome hydrophilic layers during the nanoparticulate PpIX formulation.
In vitro cell viability study
The biological activity of PpIX-loaded polymersomes against malignant melanoma A375 and human dermal fibroblast cells was quantified using MTT cytotoxicity assays (Figures 6 and 7). The cytotoxicity of PpIX-loaded polymersomes was compared with free PpIX and empty polymersomes. The PpIX-loaded polymersomes were found to be more toxic to melanoma A375 cells with polymersome concentrations increasing from 50 to 500 μg/mL after 24 hours of culture. The dose of 500 μg/mL showed the highest toxicity to melanoma cells – almost 55% of melanoma cells were killed – whereas the dose of 300 μg/mL of polymersomes loaded with PpIX killed nearly 45% of the population. Consistent with previous reports, PpIX did show some toxicity to normal tissue cells.12 The results showed that PpIX-loaded polymersomes had no significant toxicity when using a low dose of polymersomes. Specifically, polymersome concentrations below 100 μg/mL were less toxic than at a concentration of 300 μg/mL. Seventy-five percent of the population of fibroblasts lived even at the dose of 300 μg/mL. However, a dose of 500 μg/mL of polymersomes loaded with PpIX killed nearly 40% of fibroblasts, which indicated a pretty high cytotoxicity to fibroblasts.
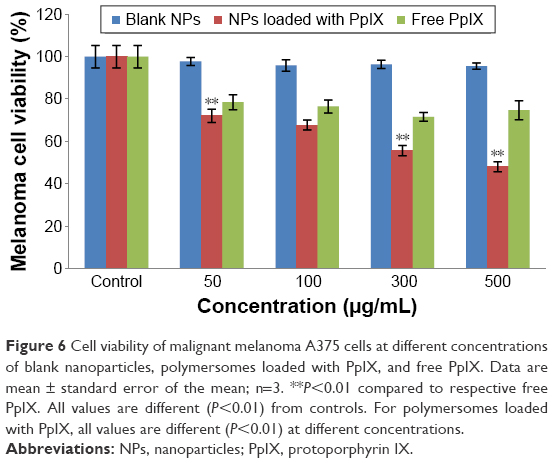 | Figure 6 Cell viability of malignant melanoma A375 cells at different concentrations of blank nanoparticles, polymersomes loaded with PpIX, and free PpIX. Data are mean ± standard error of the mean; n=3. **P<0.01 compared to respective free PpIX. All values are different (P<0.01) from controls. For polymersomes loaded with PpIX, all values are different (P<0.01) at different concentrations. |
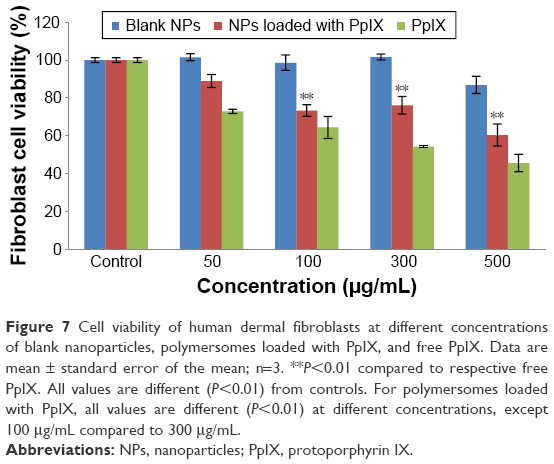 | Figure 7 Cell viability of human dermal fibroblasts at different concentrations of blank nanoparticles, polymersomes loaded with PpIX, and free PpIX. Data are mean ± standard error of the mean; n=3. **P<0.01 compared to respective free PpIX. All values are different (P<0.01) from controls. For polymersomes loaded with PpIX, all values are different (P<0.01) at different concentrations, except 100 μg/mL compared to 300 μg/mL. |
In addition, empty polymersomes did not have a significant influence on the growth or viability of cells in culture for either melanoma cells or human dermal fibroblasts.
As expected, the treatment of melanoma cells with free PpIX was less cytotoxic compared with loading free PpIX into polymersomes. The results showed a similar tendency with all concentrations. However, results were different when measuring the response of fibroblasts to free PpIX. The results implied that free PpIX was more toxic to fibroblasts than PpIX-loaded polymersomes, which means that the polymersomes were helpful in lowering the toxicity of free PpIX. In addition, free PpIX had pretty high cytotoxicity to fibroblasts when compared with that to melanoma cells. Especially at a 500 μg/mL concentration of free PpIX, the viable population of fibroblasts decreased to 45%. The treatment with free PpIX was not found to be more potent when administered simultaneously with polymersomes. The results of cell viability at 1, 3, and 5 days showed that PpIX-loaded polymersomes had a sustained anti-melanoma cell effect (Figure 8A). After 3 and 5 days, PpIX-loaded polymersomes could significantly decrease tumor cell populations. A concentration of 100 μg/mL of PpIX-loaded polymersomes killed more than 65% of tumor cells. By contrast, more than 63% of fibroblasts were alive at the same concentration and conditions (Figure 8B). In addition, concentrations more than 100 μg/mL could not kill more tumor cells than the 100 μg/mL concentration of PpIX-loaded polymersomes. However, this study did show high cytotoxicity to fibroblasts with a sustained treatment with high concentrations (300 and 500 μg/mL). For example, only 15% of fibroblasts were alive after treatment with 500 μg/mL PpIX-loaded polymersomes after 5 days, while 30% of melanoma cells were alive at the same condition, which was unexpected.
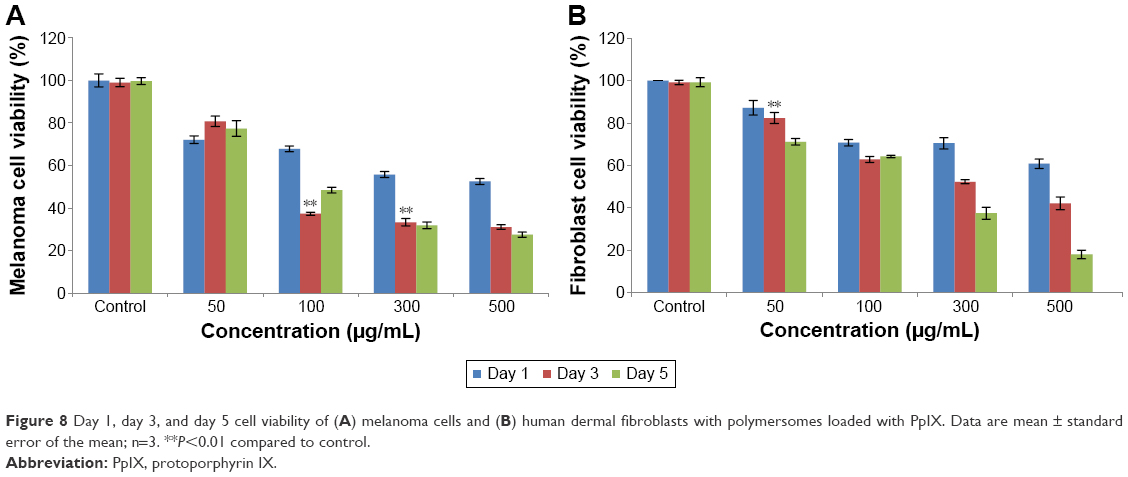 | Figure 8 Day 1, day 3, and day 5 cell viability of (A) melanoma cells and (B) human dermal fibroblasts with polymersomes loaded with PpIX. Data are mean ± standard error of the mean; n=3. **P<0.01 compared to control. |
The low toxicity of blank polymersomes seen in this study implies that this carrier has a promising future for skin cancer applications (Figure 9A and B). With time, it was also observed that melanoma cells adapted more easily to the drug carriers than fibroblasts which needs to be further studied. The results observed for free PpIX-treated melanoma cells and fibroblasts after 1, 3, and 5 days showed that the free PpIX had high toxicity to both cells (Figure 10). For melanoma cells, cell viability decreased quickly after the first day and then started to remain stable after 5 days when concentrations were below 500 μg/mL. However, fibroblasts were more sensitive to free PpIX than melanoma cells. Furthermore, without nanoparticle protection, PpIX was exposed directly to fibroblasts. This direct interaction of fibroblasts with PpIX may have presented serious damage to fibroblasts as PpIX could not be easily degraded by the cells. For instance, only 13% of fibroblasts were alive after 5 days of treatment.
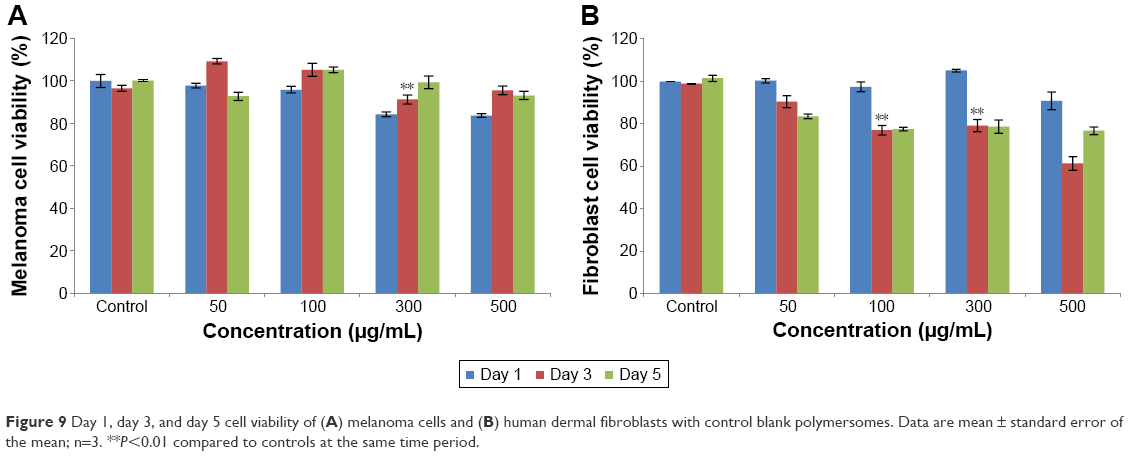 | Figure 9 Day 1, day 3, and day 5 cell viability of (A) melanoma cells and (B) human dermal fibroblasts with control blank polymersomes. Data are mean ± standard error of the mean; n=3. **P<0.01 compared to controls at the same time period. |
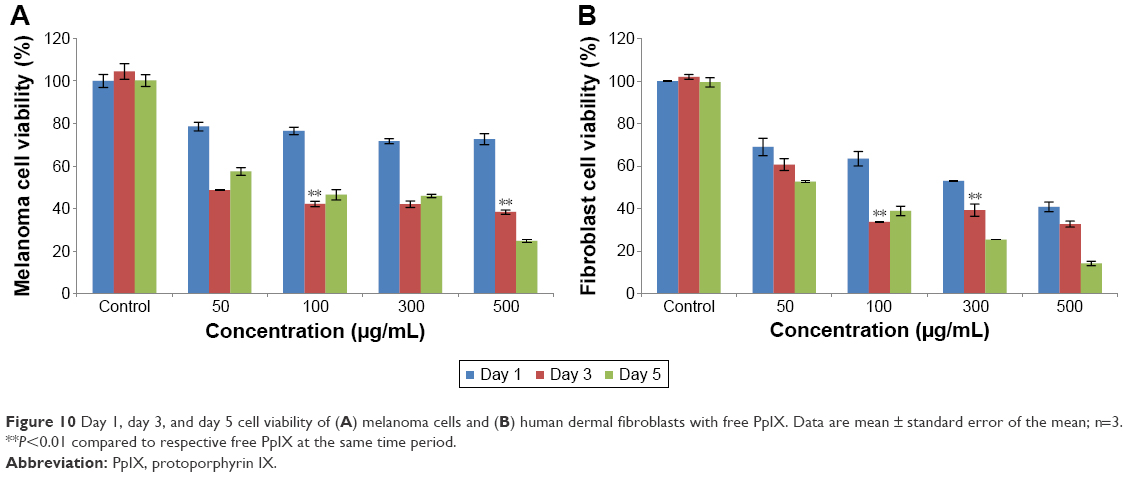 | Figure 10 Day 1, day 3, and day 5 cell viability of (A) melanoma cells and (B) human dermal fibroblasts with free PpIX. Data are mean ± standard error of the mean; n=3. **P<0.01 compared to respective free PpIX at the same time period. |
For combined therapy, a synergistic effect can be found after posttreatment with CAP and UV light. Without nanoparticles, CAP posttreatment can significantly reduce tumor cells viability when compared with UV posttreatment. CAP killed about 35% of tumor cells, while less than 10% of tumor cells were killed under UV light treatment (Figure 11). After the combined therapy with polymersomes, CAP treatment did not have much of an antitumor effect at low concentrations (below 100 μg/mL), but decreased tumor cell viability significantly at higher concentrations (higher than 300 μg/mL). Almost 30% of tumor cells treated with CAP at a concentration of 50 μg/mL were killed, which is the same result observed with UV treatment. However, when the concentration increased to 500 μg/mL, more than 70% of melanoma cells were killed by CAP, while less than 60% of cells were eliminated under UV light exposure. Confocal laser microscopy images showed that many more polymersomes were attached to cell membranes or encapsulated by melanoma cells after CAP treatment, which is consistent with cell viability results (Figure 12). A previous study demonstrated that CAP treatment could create small pores on the cell membrane,14 and in this way, encapsulation of nanoparticles by cells would increase with CAP treatment. This is a possible reason that CAP posttreatment is much better than UV exposure.
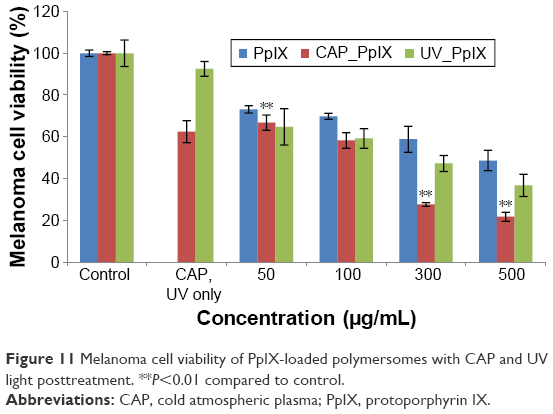 | Figure 11 Melanoma cell viability of PpIX-loaded polymersomes with CAP and UV light posttreatment. **P<0.01 compared to control. |
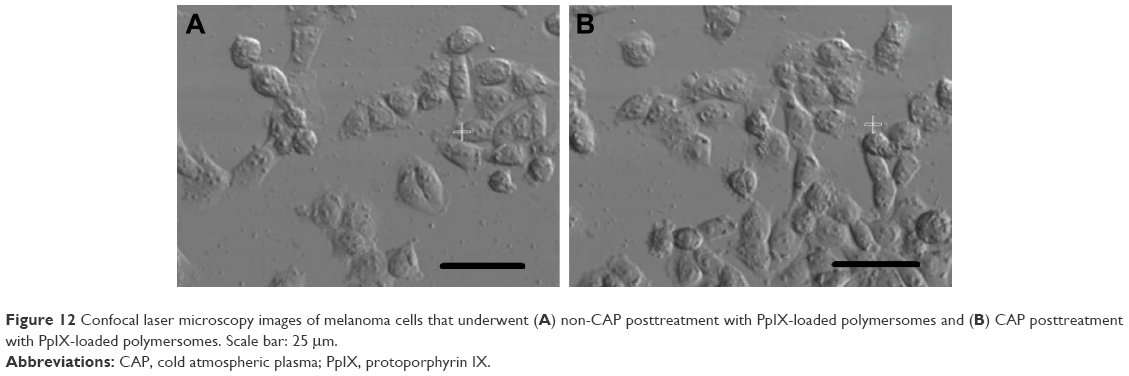 | Figure 12 Confocal laser microscopy images of melanoma cells that underwent (A) non-CAP posttreatment with PpIX-loaded polymersomes and (B) CAP posttreatment with PpIX-loaded polymersomes. Scale bar: 25 mm. |
Discussion
In this study, PpIX was successfully encapsulated by polymersomes. After successful formulation, the physicochemical characterization of the drug delivery system was taken into consideration as it influences physical stability, cellular viability, and biodistribution.15–17 In this regard, the size distribution and surface charge of polymersomes were investigated. It is well known that small sizes of nanoparticles have advantages for passive targeting to tumor cells via the enhanced permeability and retention effect, as well as zeta potential, to have an influence on polymersome suspension stability and cellular uptake.18,19 Therefore, PpIX-loaded polymersomes were anticipated to be of smaller diameter and higher surface charge, which could enhance circulation time as well as evade the reticuloendothelial system.16 As mentioned by previous researchers, the zeta potential of polymersomes is an indicator of particle attachment to surfaces.20 Polymersome assembly was ideal at a less negative zeta potential, since at a more negative zeta potential, the polymersome-to-polymersome repellent forces are higher, thus preventing a tight packing of polymersomes.21 In our case, the zeta potential increased after loading with PpIX, thus achieving excellent negatively charged stable polymersomes. Furthermore, the FTIR and XRD analysis clearly demonstrated the chemical integrity of PpIX and its interaction with the polymersomes. The photophysical properties of PpIX-loaded polymersomes overcame PpIX aggregation in PBS. The results of photophysical property analysis further confirmed that PpIX can be encapsulated by polymersomes and locate between bilayers.
Most importantly, there was no toxicity induced by the polymersomes alone as observed in the cell viability study. MTT results demonstrated a comparatively better uptake of polymersomes loaded with PpIX over free PpIX in melanoma cells. In fact, a 500 μg/mL concentration of polymersomes loaded with PpIX killed 56% of melanoma cells, while the corresponding concentration of PpIX killed 25% of melanoma cells. This is due to the aforementioned fact that polymersomes can protect PpIX from decomposition by melanoma cells. However, free PpIX showed more toxicity to human dermal fibroblasts when compared to PpIX-loaded polymersomes. As MTT results showed, 500 μg/mL of the free PpIX killed 55% of the fibroblasts, while PpIX-loaded polymersomes only killed 40%; the opposite trend was observed with melanoma cells. Reasons for this may be due to the fact that polymersomes can protect PpIX from decomposition by fibroblasts.
It is well known that PDT is a noninvasive therapeutic method that is effective against tumors using light and a photosensitizer. When photosensitizers in cells are exposed to specific wavelengths, they are transformed from a singlet state to an excited singlet state. The energy transferred from the low state to the high state generates reactive oxygen species (ROS). During this process, ROS causes cellular damage that will lead to tumor cell death through either necrosis or apoptosis.22 Apparently, when CAP or UV is applied to posttreated tumor cells, ROS generated by an intracellular photosensitizer state transforms. This is the reason why the antitumor efficiency improved after applying CAP or UV light posttreatment. However, different compositions of CAP and UV light result in different cell responses. UV has a simple composition, while CAP contains ROS, reactive nitrogen species, UV, electrons, and other kinds of charged particles.13 Those unique components enable CAP to kill tumor cells in different ways, one of which is ROS generated by CAP which can create oxidized stress leading to DNA damage to cells. As a PDT light source, CAP induces photosensitizer activation, which also generates ROS inside the cells. Meanwhile, CAP treatment promotes cell encapsulation of nanoparticles by creating small pores in cell membranes, which is another reason for this synergistic effect. What is more, this combined therapy actually has a synergistic effect to kill tumor cells without showing fatal damage to healthy cells.
Conclusion
This study investigated polymersomes loaded with PpIX as a novel drug delivery system in PDT using CAP as a light source for potentially treating skin cancer. PpIX was successfully encapsulated in polymersomes maintaining a 100 nm diameter and a high zeta potential and was incorporated into the bilayer of the polymersomes. An in vitro study of melanoma cells and fibroblasts provided the first evidence that CAP can be used as an effective light source for antitumor PDT applications. PDT with CAP treatment showed a significant decrease in tumor viability, especially at high nanoparticle concentrations (300–500 μg/mL), when compared with UV posttreatment. This combined treatment could be a promising tool for skin cancer drug delivery without bringing fatal damage to healthy cells. All of these results indicated that PpIX-loaded polymersomes with CAP posttreatment can be effectively used in PDT for antitumor applications.
Acknowledgments
The authors would like to acknowledge Northeastern University for funding and Dr William H Fowle of Northeastern University for helping with obtaining TEM images. They would also like to thank Drs Homa Homayoni and Eno Ebong for thoughtful discussions on some aspects of the research. Prior conferences are as follows: 2014 AIChE at Atlanta, GA; 2014 BMES at San Antonio, TX; 2014 ITNANO at Boston, MA; and 2014 MRS at Boston, MA.
Disclosure
The authors report no conflicts of interest in this work.
References
Sahoo SK, Labhasetwar V. Nanotech approaches to drug delivery and imaging. Drug Discov Today. 2003;8(24):1112–1120. | ||
Na R, Stender IM, Wulf HC. Can autofluorescence demarcate basal cell carcinoma from normal skin? A comparison with protoporphyrin IX fluorescence. Acta Derm Venerol. 2001;81(4):246–249. | ||
Bastiaens MT, Hoefnagel JJ, Bruijn JA, Westendorp RG, Vermeer BJ, Bouwes Bavinck JN. Differences in age, site distribution, and sex between nodular and superficial basal cell carcinoma indicate different types of tumors. J Invest Dermatol. 1998;110(6):880–884. | ||
Bonnett R. Photosensitizers of the porphyrin and phthalocyanine series for photodynamic therapy. Chem Soc Rev. 1995;24(1):19–33. | ||
Rossi LM, Silva PR, Vono LL, Fernandes AU, Tada DB, Baptista MS. Protoporphyrin IX nanoparticle carrier: preparation, optical properties, and singlet oxygen generation. Langmuir. 2008;24(21):12534–12538. | ||
Moan J, Peng Q. An outline of the hundred-year history of PDT. Anticancer Res. 2003;23(5A):3591–3600. | ||
Dolmans D, Fukumura D, Jain RK. Photodynamic therapy for cancer. Nat Rev Cancer. 2003;3(5):380–387. | ||
Kurwa HA, Barlow RJ. The role of photodynamic therapy in dermatology. Clin Exp Dermatol. 1999;24(3):143–148. | ||
Boiy A, Roelandts R, de Witte PA. Photodynamic therapy using topically applied hypericin: comparative effect with methyl-aminolevulinic acid on UV induced skin tumours. J Photochem Photobiol B. 2011;102(2):123–131. | ||
Morton CA, Brown SB, Collins S, et al. Guidelines for topical photodynamic therapy: report of a workshop of the British Photodermatology Group. Br J Dermatol. 2002;146(4):552–567. | ||
Bermudez H, Brannan AK, Hammer DA, Bates FS, Discher DE. Molecular weight dependence of polymersome membrane structure, elasticity, and stability. Macromolecules. 2002;35(21):8203–8208. | ||
Fridman G, Brooks A, Balasubramanian M, et al. Comparison of direct and indirect effects of non-thermal atmospheric-pressure plasma on bacteria. Plasma Process Polym. 2007;4(4):370–375. | ||
Wang M, Holmes B, Cheng X, Zhu W, Keidar M, Zhang L. Cold atmospheric plasma for selectively ablating metastatic breast cancer cells. PLoS One. 2013;8(9):e73741. | ||
Keidar M, Walk R, Shashurin A, et al. Cold plasma selectivity and the possibility of a paradigm shift in cancer therapy. Br J Cancer. 2011;105(9):1295–1301. | ||
Jeon SI, Lee JH, Andrade JD, De Gennes P. Protein-surface interactions in the presence of polyethylene oxide: I. Simplified theory. J Colloid Interface Sci. 1991;142(1):149–158. | ||
Acharya S, Dilnawaz F, Sahoo SK. Targeted epidermal growth factor receptor nanoparticle bioconjugates for breast cancer therapy. Biomaterials. 2009;30(29):5737–5750. | ||
Sahu A, Kasoju N, Bora U. Fluorescence study of the curcumine-casein micelle complexation and its application as a drug nanocarrier to cancer cells. Biomacromolecules. 2008;9(10):2905–2912. | ||
Saho SK, Panyam J, Prabha S, Labhasetwar V. Residual polyvinyl alcohol associated with poly(D,L-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake. J Control Release. 2002;82(1):105–114. | ||
Pan CJ, Tang JJ, Shao ZY, Wang J, Huang N. Improved blood compatibility of rapamycin-eluting stent by incorporating curcumin. Colloids Surf B Biointerfaces. 2007;59(1):105–111. | ||
Lo CT, Van Tassel PR, Saltzman WM. Poly(lactide-co-glycolide) nanoparticle assembly for highly efficient delivery of potent therapeutic agents from medical devices. Biomaterials. 2010;31(13):3631–3642. | ||
Vakarelski IU, McNamee CE, Higashitani K. Deposition of silica nanoparticles on a gold surface via a self-assembled monolayer of (3-mercaptopropyl)trimethoxysilane. Colloids Surf A Physicochem Eng Asp. 2007;295(1–3):16–20. | ||
Volotskova O, Hawley TS, Stepp MA, Keidar M. Targeting the cancer cell cycle by cold atmospheric plasma. Sci Rep. 2012;2:636. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.