Back to Journals » International Medical Case Reports Journal » Volume 9
Kikuchi-Fujimoto disease and systemic lupus erythematosus
Authors Baenas D, Diehl F, Haye Salinas M, Riva V, Diller A, Lemos P
Received 14 February 2016
Accepted for publication 16 March 2016
Published 29 June 2016 Volume 2016:9 Pages 163—167
DOI https://doi.org/10.2147/IMCRJ.S106396
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ronald Prineas
Diego F Baenas,1 Fernando A Diehl,1 María J Haye Salinas,2 Verónica Riva,3 Ana Diller,3 Pablo A Lemos1,4
1Clinical Medicine Department, 2Rheumatology Department, 3Pathology Department, Hospital Privado Universitario de Córdoba Medical Center, 4Instituto Universitario de Ciencias Biomédicas, Universitary Institute, Córdoba, Argentina
Abstract: Kikuchi–Fujimoto disease, or histiocytic necrotizing lymphadenitis, is an infrequent idiopathic disorder. It has been associated with autoimmune disorders, of which systemic lupus erythematosus is the most outstanding. The basis of its diagnosis relies on the histological examination of lymph nodes, which typically reveals necrosis surrounded by histiocytes with crescentic nucleus, immunoblasts and plasma cells, and absence of neutrophils. We report the case of a 27-year-old Argentinian female patient without any relevant past medical history to demonstrate the correlation between Kikuchi–Fujimoto disease and systemic lupus erythematosus.
Keywords: histiocytic necrotizing lymphadenitis, systemic lupus erythematosus, autoimmune disorders, febrile syndrome
Introduction
Kikuchi–Fujimoto disease (KFD), or histiocytic necrotizing lymphadenitis, is an infrequent idiopathic disorder.1 It is usually a self-limited condition with a good prognosis because it has a low complication rate.2 It mainly affects young women.3 The most common clinical symptoms are fever and laboratory finding of cervical lymphadenopathy associated with leukopenia.3 Cases of KFD associated with autoimmune disorders, such as systemic lupus erythematosus (SLE), have been reported.4 Herein, we describe a female patient who presented with cervical necrotizing lymphadenitis and febrile syndrome and subsequently met the diagnostic criteria for SLE.5
Case report
A 27-year-old female patient, without any relevant past medical history, presented to the clinic with a 30-day history of voluminous and tender enlargement of cervical and nuchal lymph nodes. Five days after the onset of the symptoms, the patient developed asthenia and fever of 38–39°C that responded poorly to antipyretics. A week before visiting us, the patient had symmetric arthralgias, mainly of proximal and distal interphalangeal joints of hands and elbows.
On physical examination, vital signs were otherwise unremarkable. Multiple indurated, fixed, and tender lymphadenopathies were evident in the cervical and nuchal areas. Arthralgia of the proximal and distal interphalangeal joints of the hands was evident. Cardiovascular and respiratory examinations were within normal parameters. Abdominal examination was also normal. A recent occurrence of an erythematous, indolent node of diameter 5 mm was detected in the right cheek. Mucous membranes did not show any lesion.
The laboratory findings are summarized in Table 1.
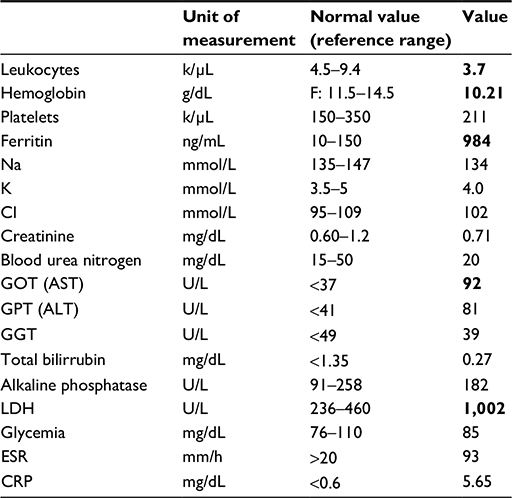 | Table 1 Laboratory findings Note: Bold values represent abnormal values. Abbreviations: F, female; Na, sodium; K, potassium; Cl, chloride; GOT, glutamic oxaloacetic transaminase; AST, aspartate aminotransferase; GPT, glutamic pyruvic transaminase; ALT, alanine aminotransferase; GGT, gamma-glutamyl transferase; LDH, lactate dehydrogenase; ESR, erythrocyte sedimentation rate, CRP, C-reactive protein. |
Laboratory test results showed hypochromic normocytic anemia and elevated lactate dehydrogenase, erythrocyte sedimentation rate, glutamic oxaloacetic transaminase, and glutamic pyruvic transaminase. The urine sediment analysis was normal. Institutional approval by the Hospital Privado Ethics Committee and written informed consent from the patient was obtained to report the findings of this case.
Serology test results for human immunodeficiency virus (HIV), hepatitis B and C, Epstein–Barr virus, toxoplasmosis, cytomegalovirus, and parvovirus B19 were all negative. Blood culture to detect common germs, mycobacteria, and fungi were also negative. An autoimmune screen test was requested.
An ultrasonography of the neck revealed bilaterally enlarged submandibular and jugular chain lymph nodes (the largest was 2 cm in diameter), with some showing signs of necrosis.
Fiberoptic rhinolaryngoscopy and chest X-ray were normal. Cervical and chest computed tomography scans only showed lymph node enlargements as previously described. An abdominal computed tomography scan evidenced infracentimetric inguinal, intercavoaortic, and left lateroaortic lymph nodes of measuring up to 9 mm in diameter. It did not show hepatomegaly or splenomegaly.
A cervical lymph node biopsy was performed. The histopathological report disclosed more than ninety percent of the lymph node sample showed necrosis with distortion of the normal architecture; periodic acid–Schiff, methenamine and Ziehl-Neelsen stainings were negative in the viable tissue. The polymerase chain reaction analysis of the biopsy material was negative for histoplasma, aspergillus, mycobacteria, and cytomegalovirus.
Twelve days after the onset of the symptoms, the patient developed arthritis of the proximal and distal interphalangeal joints of the hands, malar rash, photosensitive erythema in the chest, and painful oral ulcers. The patient then developed generalized erytroderma in the chest, abdomen, and upper and lower limbs, as well as diffuse alopecia.
Pending autoimmune screen test results were received, which revealed positive antinuclear antibodies (Hep2) with a titer of 1:320 and a speckled pattern; negative anti-double-stranded-DNA antibodies; positive anti-Ro antibodies; and negative anti-La, anti-Smith, anti-ribonucleic protein, lupus anticoagulant, and anticardiolipin antibodies. Complement proteins C3 and C4 were within normal limits.
Skin biopsy showed predominantly lymphocytic perivascular dermatitis with few eosinophils and negative immunofluorescence.
Owing to the nonspecific findings of the first lymph node sampling, it was decided to perform a second biopsy guided by ultrasound to select tissue with less necrosis (Figure 1). The histopathological examination of the specimen revealed scarce lymphoid tissue due to extensive geographic necrosis and numerous histiocytes diffusely laid out surrounding the necrotic foci. No polymorphonuclear neutrophils or hematoxylin bodies were observed in the sinusoidal capillaries. Immunohistochemical panels evidenced residual lymphoid tissue that expressed positive CD20, CD3, and bcl-2. AE1/AE3 immunostaining, as well as CD1 immunostaining, was negative. CD68 immunostaining was widely positive, which is consistent with KFD.
 | Figure 1 Cervical lymph node biopsy. Notes: (A) Lymph node with geographic necrosis surrounded by histiocytes (hematoxylin–eosin stain, ×10 magnification). (B) Geographic necrosis and cellular detritus without the presence of polymorphonuclear neutrophils or histiocytes (hematoxylin–eosin stain, ×40 magnification). (C and D) Positive immunostaining in histiocytes with CD68 (hematoxylin–eosin stain, ×10 and ×40 magnifications). (E) Immunostaining with CD20 antibody (hematoxylin–eosin stain, ×10 magnification). (F) Negative immunostaining with CD1a antibody (hematoxylin–eosin stain, ×40 magnification). |
A diagnosis of associated SLE was made because the patient presented malar rash, photosensitivity, oral ulcers, alopecia, arthritis, and positive antinuclear antibody (2012 Systemic Lupus International Collaborating Clinics classification criteria).5
The patient has begun treatment with hydroxychloroquine (200 mg/d) and prednisone (20 mg/d), with a marked improvement in the symptoms 1 week after the treatment was started and a progressive reduction in the lymphadenopathies until complete resolution after 1 month of treatment initiation.
Discussion
Histiocytic necrotizing lymphadenitis was described by Kikuchi and Fujimoto in 1972.3 It was mainly described in young adults <40 years of age.6
This disease is more prevalent in Asia, but some cases have been reported in other continents also.
Its etiology is unknown, although the most accepted hypothesis is its viral–autoimmune origin.7
Three patterns of presentation of this association were found by Medline/PubMed search until 2015: KFD before the onset of SLE (30%), simultaneous occurrence of both disorders (47%), and KFD after SLE (23%). The reported cases are listed in Table 2. Our case is the first report of the coexistence of these disorders in Argentina.
 | Table 2 Reported cases of KFD and SLE Abbreviations: KFD, Kikuchi–Fujimoto disease; SLE, systemic lupus erythematosus. |
Some cases of familial occurrence of KFD were observed in Japan within a short period of time and in a similar setting.8 Only one human leukocyte antigen (HLA)-typing study was found.9 Also, few reports of KFD proposed the possibility of familial clustering. One of them described two nontwin sisters with HLA-identical phenotype who lived contemporarily in the same environment.10 Other report showed two nontwin sisters with HLA-identical phenotype who developed the disease in different locations and 10 years apart from each other.8 A current publication described the development of KFD in four members of the same family (three confirmed cases and one possible case) at different periods of time. Its onset may be acute or subacute and may last 2 or 3 weeks.11 No studies have shown if familial cluster of KFD is present when it links with SLE.
Cervical lymphadenopathy, which is usually tender to palpation, is the main clinical feature. Other lymph node regions can also be involved. 12
The common presenting symptoms are fever, sweating, chills, tender lymph nodes, malaise, weight loss, and cough.12–14 The laboratory and radiologic tests available for the diagnosis are nonspecific. The common laboratory abnormalities are leukopenia, usually neutropenia; anemia; thrombocytopenia; elevated C-reactive protein and erythrocyte sedimentation rate; impaired liver function; and atypical lymphocytes on peripheral blood smear.7,12
The diagnosis is mainly made by histopathological assessment of the lymph node.
Clinical and histological differential diagnoses of KFD should take into account the following diseases: non-Hodgkin lymphomas and other lymphoid malignancies; lymphadenopathies associated with connective disorders such as SLE, rheumatoid arthritis, and Still’s disease; and bacterial or viral infections such as cat scratch disease, infectious mononucleosis, herpes simplex, HIV, toxoplasmosis, tuberculosis, and atypical mycobacterial lymphadenitis.6
Because of the clinical and pathological correlation between KFD and SLE, some authors have postulated that KFD may be a clinical feature or an incomplete phase of lupus lymphadenitis.15–18 However, there are several case reports of KFD without SLE,1 which may support the fact that they are two independent entities that may commonly coexist, as it happens with most of the autoimmune diseases in susceptible subjects.
In KFD, the most common histologic finding is lymph node showing geographic necrosis with foci of apoptotic cells with abundant karyorrhectic fragments surrounded by histiocytes.6 Characteristically, neutrophils and eosinophils are absent.6
Only in those cases in which the pathologist notes hematoxyphilic bodies (clusters of basophilic material within lymph node sinuses), DNA deposits in the wall of the vessels, or areas of vasculitis surrounding the necrotic foci, he or she would suggest the diagnosis of lupus lymphadenitis and not KFD.2 Immunohistochemical analysis has great value and is generally used to exclude hematologic malignancies. CD8+ T cells prevail in KFD. The histiocytes typically express myeloperoxidase, along with lysozyme. Also, positive immunostaining appeared for CD68 and CD4 in histiocytes.
From a clinical approach, it is important to consider the possibility of SLE and KFD comorbidity.
Usually, KFD takes a monophasic and benign course that limits itself in 1–6 months.15–18 However, if it coexists with SLE, it can have a more aggressive course and should be treated to prevent sequelae. A long-term follow-up is necessary to monitor for recurrence.19
In addition, there have been reports of death in patients with KFD and SLE due to severe infections and development of hemophagocytic syndrome.19 Our patient did not have this complication.
In our case, a new ultrasonographically guided biopsy was repeated to select the lymph nodes with less necrotic tissue. We propose this strategy to reach a histological diagnosis. In order to do that, the abovementioned ancillary studies were requested and the lymph node biopsy was repeated to ultrasonographically select the lymph nodes with less necrotic tissue to reach a histological diagnosis.
KFD is one of the differential diagnoses of cervical necrotizing lymphadenitis, and when it is present, it would be wise to suspect, during its clinical course, a possible association with other autoimmune conditions, such as SLE.
Acknowledgments
The authors thank Enrique Caeiro and Santiago Orozco for their valuable contributions to reach the diagnosis.
Disclosure
The authors report no conflicts of interest in this work.
References
Kucukardali Y, Solmazgul E, Kunter E, Oncul O, Yildirim S, Kaplan M. Kikuchi-Fujimoto disease: analysis of 244 cases. Clin Rheumatol. 2007;26(1):50–54. | ||
Diez-Morrondo C, Pantoja-Zarza L, Manjón-Haces JA. Kikuchi Fujimoto’s disease appearing as systemic lupus erythematosus. Inicio de lupus eritematoso sistémico como enfermedad de Kikuchi-Fujimoto. Reumatol Clin. 2012;8:153–155. | ||
Ruaro B, Sulli A, Alessandri E, Fraternali-Orcioni G, Cutolo M. Kikuchi-Fujimoto’s disease associated with systemic lupus erythematous: difficult case report and literature review. Lupus. 2014;23(9):939–944. | ||
Cheng CY, Sheng WH, Lo YC, Chung CS, Chen YC, Chang SC. Clinical presentations, laboratory results and outcomes of patients with Kikuchi’s disease: emphasis on the association between recurrent Kikuchi’s disease and autoimmune diseases. J Microbiol Immunol Infect. 2010;43(5):366–371. | ||
Petri M, Orbai AM, Alarcón GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677–2686. | ||
Blake C, Wang E. Kikuchi-Fujimoto disease. Arch Pathol Lab Med. 2010;134(2):289–293. | ||
Yu HL, Lee SS, Tsai HC, et al. Clinical manifestations of Kikuchi’s disease in southern Taiwan. J Microbiol Inmunol Infect. 2005;38(1):35–40. | ||
Amir AR, Amr SS, Sheikh SS. Kikuchi-Fujimoto’s disease: report of familial occurrence in two human leucocyte antigen-identical non-twin sisters. J Intern Med. 2002;252(1):79–83. | ||
Kikuchi M, Takeshita M, Eimoto T, Iwasaki H, Minamishima Y, Maeda Y. Histiocytic necrotizing lymphadenitis: clinicopathologic, immunologic, and HLA typing study. In: Hanaoka M, Kadin M, Mikata A, editors. Lymphoid Malignancy. New York: Field and Wood; 1990:251–257. | ||
Stasiuk A, Teschke S, Williams GJ, Seftel MD. Kikuchi-Fujimoto disease: lymphadenopathy in siblings. CMAJ. 2011;183(1):E58–E60. | ||
Krishnappa J, Bharath Reddy D, Harsha PJ, Prasad CSB. Familial Kikuchi-Fujimoto disease. J Clin Sci Res. 2015;4:40–44. | ||
Santana A, Lessa B, Glarao L, Lima I, Santiago M. Kikuchi-Fujimoto’s disease associated with systemic lupus erythematosus: case report and review of the literature. Clin Rheumatol. 2005;24(1):60–63. | ||
Zuo Y, Foshat M, Qian Y-W, Kelly B, Harper B, Karnath B. A rare case of Kikuchi Fujimoto’s disease with subsequent development of systemic lupus erythematosus. Case Rep Rheumatol. 2012;2012:325062. | ||
Di Lernia V, Bajocchi G, Piana S. Subacute cutaneous lupus erythematosus onset preceded by Kikuchi-Fujimoto disease. Dermatol Pract Concept. 2014;4(1):47–49. | ||
Cramer J, Schmiedel S, Alegre NG, Schafer H, Burchard GD, Merz H. Necrotizing lymphadenitis: Kikuchi-Fujimoto disease alias lupus lymphadenitis? Lupus. 2010;19(1):89–92. | ||
Murthy SC, Dandin SM, Dandin AS, Patwardan MY. Kikuchi’s disease associated with systemic lupus erythematosus. Indian J Dermatol Venereol Leprol. 2005;71(5):338–341. | ||
Rao GS, Vohra D, Kuruvilla M. Is Kikuchi-Fujimoto disease a manifestation of systemic lupus erythematosus? Int J Dermatol. 2006;45(4):454–456. | ||
Gordon JK, Magro C, Lu T, et al. Overlap between systemic lupus erythematosus and Kikuchi Fujimoto disease: a clinical pathology conference held by the Department of Rheumatology at Hospital for Special Surgery.HSS J. 2009;5(2):169–177. | ||
Kampitak T. Fatal Kikuchi-Fujimoto disease associated with SLE and hemophagocytic syndrome: a case report. Clin Rheumatol. 2008;27(8):1073–1075. | ||
Hoffmann A, Kirn E, Kuerten A, Sander C, Krueger GR, Ablashi DV. Active human herpesvirus-6 (HHV-6) infection associated with Kikuchi-Fujimoto disease and systemic lupus erythematosus (SLE). IHYPERLINK “http://www.ncbi.nlm.nih.gov/pubmed/1654149”\o “In vivo(Athens, Greece).” n Vivo. 1991;5(3):265–9. | ||
Chua SH, Giam YC, Sim CS. Systemic lupus erythematosus with erythema multiforme-like lesions and histiocytic necrotizing lymphadenitis–a case report. Ann Acad Med Singapore. 1996;25(4):599–601. | ||
Eisner MD, Amory J, Mullaney B, Tierney L Jr, Browner WS. Necrotizing lymphadenitis associated with systemic lupus erythematosus. Semin Arthritis Rheum. 1996;26(1):477–82. | ||
Jimenez Saenz JM, Llorente Arenas EM, Fuentes Solsona F, de Miguel GF, Alvarez Alegret R. Kikuchi-Fujimoto’s disease and the association with systemic lupus erythematosus. An Med Interna. 2004;18(8):429–31. | ||
Quintas-Cardama A, Fraga M, Cozzi SN, Caparrini A, Maceiras F, Forteza J.Fatal Kikuchi-Fujimoto disease: the lupus connection. Ann Hematol. 2003;82(3):186–8. | ||
Tanasescu C, Nitescu D, Staniceanu F, Ardeleanu C, Atanasiu C. Kikuchi’sdisease associated with systemic lupus erythematosus and autoimmune-like hepatitis. Rom J Intern Med. 2003;41(3):299–305. | ||
Leyral C, Camou F, Perlemoine C, Caubet O, Pellegrin JL, Viallard JF. Pathogenic links between Kikuchi’s disease and lupus: a report of three new cases. Rev Med Interne. 2005;26(8):651–5. | ||
Yilmaz M, Camci C, Sari I, et al. Histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto’s disease) mimicking systemic lupus erythematosus: a review of two cases. Lupus. 2006;15(6):384–7. | ||
Mahajan T, Merriman RC, Stone MJ. Kikuchi-Fujimoto disease (histiocytic necrotizing lymphadenitis): report of a case with other autoimmune manifestations. Proc (Bayl Univ Med Cent). 2007;20(2):149–51. | ||
Frikha F, Marzouk S, Frigui M, et al. Kikuchi-Fujimoto’s disease and connective tissue disease: a report of three cases. Rev Med Internet. 2008;29(2):129–34. | ||
Gallien S, Lagrange-Xelot M, Crabol Y, Briere J, Galicier L, Molina JM. Systemic lupus erythematosus and Kikuchi-Fujimoto disease mimicking tuberculosis. Med Mal Infect. 2008;38(7):392–5. | ||
Shusang V, Marelli L, Beynon H, et al. Autoimmune hepatitis associated with Kikuchi-Fujimoto’s disease. Eur J Gastroenterol Hepatol. 2008;20(1):79–82. | ||
Aota N, Hirahara K, Kano Y, Fukuoka T, Yamada A, Shiohara T. Systemic lupus erythematosus presenting with Kikuchi-Fujimoto’s disease as a long-term sequela of drug-induced hypersensitivity syndrome. A possible role of Epstein-Barr virus reactivation. Dermatology. 2009;218(3):275–7. | ||
Gionanlis L, Katsounaros M, Bamihas G, Fragidis S, Veneti P, Sombolos K. Kikuchi-Fujimoto disease and systemic lupus erythematosus: the EBV connection? Ren Fail. 2009;31(2):144–8. | ||
Sopena B, Rivera A, Vazquez-Trinanes C, et al. Autoimmune manifestations of Kikuchi disease. Semin Arthritis Rheum. 2012;41(6):900–6. | ||
Smith LW, Petri M. Diffuse lymphadenopathy as the presenting manifestation of systemic lupus erythematosus. J Clin Rheumatol. 2013;19(7):397–9. | ||
Chen YH, Lan JL. Kikuchi disease in systemic lupus erythematosus: clinical features and literature review. J Microbiol Immunol Infect. 1998;31(3):187–92. | ||
Wano Y, Ebata K, Masaki Y, et al. Histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto’s disease) accompanied by hemophagocytosis and salivary gland swelling in a patient with systemic lupus erythematosus. Rinsho Ketsueki. 2000; 41(1):54–60. | ||
Bachi R. Kikuchi disease in a connective tissue disorder. Med J Malaysia.2002;57(3):357–60. | ||
Pace-Asciak P, Black MA, Michel RP, Kost K. Case Series: raising awareness about Kikuchi-Fujimoto disease among otolaryngologists: is it linked to systemic lupus erythematosus? J Otolaryngol Head Neck Surg.2008;37(6):782–7. | ||
Londhey VA, Buche AS, Kini SH, Rajadhyaksha GC. Kikuchi Fujimoto disease and systemic lupus erythematosus–a rare association. J Assoc Physicians India. 2010;58:642–3. | ||
el-Ramahi KM, Karrar A, Ali MA. Kikuchi disease and its association with systemic lupus erythematosus. Lupus.1994;3(5):409–11. | ||
Sanpavat A, Wannakrairot P, Assanasen T. Necrotizing non-granulomatous lymphadenitis: a clinicopathologic study of 40 Thai patients. Southeast Asian J Trop Med Public Health. 2006;37(3):563–70. | ||
Alijotas-Reig J, Casellas-Caro M, Ferrer-Oliveras R, Cabero-Roura L,Vilardell-Tarres M. Recurrent Kikuchi-Fujimoto disease during pregnancy:report of case evolving into systemic lupus erythematosus and review of published work. J Obstet Gynaecol Res. 2008;34(4 Pt 2):595–8. | ||
Goldblatt F, Andrews J, Russell A, Isenberg D. Association of Kikuchi-Fujimoto’s disease with SLE. Rheumatology (Oxford). 2008;47(4):553–4. | ||
Paradela S, Lorenzo J, Martinez-Gomez W, Yebra-Pimentel T, Valbuena L,Fonseca E. Interface dermatitis in skin lesions of Kikuchi-Fujimoto’s disease: a histopathological marker of evolution into systemic lupus erythematosus? Lupus. 2008;17(12):1127–35 | ||
Ogata S, Bando Y, Saito N, Katsuoka K, Ishii M. Kikuchi-Fujimoto disease developed into autoimmune disease: a report of two cases. Mod Rheumatol.2010;20(3):301–5. | ||
Patra A, Bhattacharya SK (2013) SLE developing in a follow-Up patient of Kikuchi’s disease: a rare disorder. J Clin Diagn Res. 2013;7(4):752–3. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.