Back to Journals » International Medical Case Reports Journal » Volume 7
Keratitis–ichthyosis–deafness syndrome: first affected family reported in the Middle East
Authors Al-Fahaad HA ![]()
Received 28 November 2013
Accepted for publication 22 January 2014
Published 25 March 2014 Volume 2014:7 Pages 63—66
DOI https://doi.org/10.2147/IMCRJ.S58432
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Hamad Al Fahaad
Department of Dermatology, College of Medicine, Najran University, Najran, Saudi Arabia
Introduction: Keratitis–ichthyosis–deafness (KID) syndrome is a rare congenital multisystem disorder affecting certain tissues of ectodermal origin such as epidermis, cochlea, and cornea, leading mainly to palmoplantar hyperkeratosis, ichthyosiform scaling, deafness, and blindness. The author reports for the first time in the Middle East three family members suffering from KID syndrome in the southwestern part of Saudi Arabia.
Case presentation: Three patients from one family (ages 26, 16, and 14 years) of apparently normal parents, with the two eldest being females and the youngest being male. All three patients were referred from a peripheral hospital to our dermatology clinic due to recurrent cutaneous fungal infections on their trunk, forearms, legs, and nails. On full assessment, they also found to have nearly similar cutaneous problems manifested by palmoplantar hyperkeratosis, generalized ichthyosiform scaling, subungual hyperkeratosis, and nail dystrophies. All patients suffered from total hearing loss in both ears since childhood as confirmed by pure tune audiometry. However, there was no blindness in any case; blepharitis with marked photophobia was the only ocular complaint. All these features are classically suggestive of KID syndrome.
Keywords: connexin 26, GJB2, ichthyosis, KID syndrome, palmoplantar hyperkeratosis
Introduction
Keratitis–ichthyosis–deafness (KID) syndrome is a rare congenital multisystem disorder due to mutations in the gene encoding connexin 26 protein affecting certain tissues of ectodermal origin such as epidermis, cochlea, and cornea. The cutaneous manifestations include palmoplantar hyperkeratosis with leather grain-like keratoderma, nail dystrophy, alopecia, and ichthyosiform scaling. Bilateral sensorineural hearing loss and slowly progressing keratitis culminates in deafness and blindness, respectively, at or before puberty.1 Most cases have been sporadic but autosomal recessive and dominant cases are reported; it is genetically heterogeneous and is caused by missense mutations in the GJB2 (gap junction β-2) gene encoding for connexin 26 protein, which clusters at chromosome 13q12.2 Germinal mosaicism – a mechanism by which some of the germ cells of the gonad are of a form not present in either parent, because of mutation in an intermediate progenitor of these cells – may also be present in cases with KID syndrome.2 The treatment consists of a keratolytic, topical emollients and antiseptic, antibiotic, antimycotic and systemic retinoids. Here, the author reports for the first time in the Middle East three family members suffering from KID syndrome in the southwestern part of Saudi Arabia.
Case report
Three patients from one family (ages 26, 16, and 14 years) with the two eldest being females and the youngest being male of apparently normal parents with close consanguinity (first cousins) were diagnosed as having KID syndrome in a remote area of Saudi Arabia. The patients have a very poor economic background and had not been able to seek medical advice before. Despite immense counseling and reassurance, the patients and father were not willing to have skin biopsies. Cases were diagnosed solely on clinical criteria. All of the patients showed good improvement on usual treatment and were started on short courses of a small dose of isotretinoin. Systemic alitretinoin was not used because of the fact that no complications such as follicular occlusion and dissecting cellulitis were noticed in any case.
Case one
The eldest was a female patient of 26 years of age. She presented with history of recurrent cutaneous fungal infections on the trunk, forearms, and legs. Detailed history revealed that she suffered from total hearing loss in both ears since her childhood. Pulse, blood pressure, and respiratory rate were normal. Skin examination showed palmoplantar hyperkeratosis, generalized ichthyosiform scaling, and subungual hyperkeratosis with nail dystrophies as shown in Figures 1–3. Audiogram revealed no response bilaterally at 90 dB. Examination of eyes showed blepharitis with marked photophobia but with normal vision.
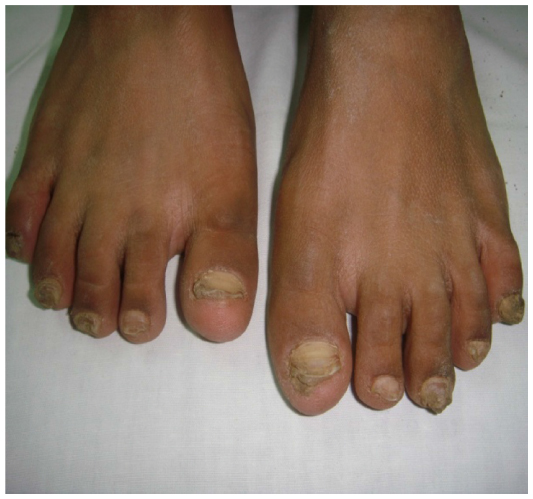 | Figure 1 Subungual hyperkeratosis with nail dystrophy in patient with KID syndrome. |
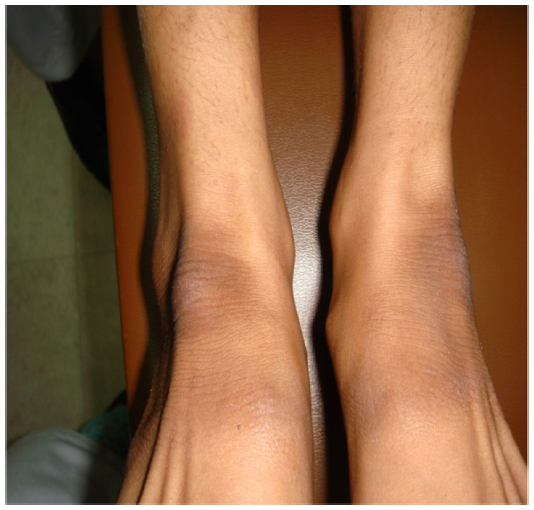 | Figure 2 Ichthyosiform scaling in patient with KID syndrome. |
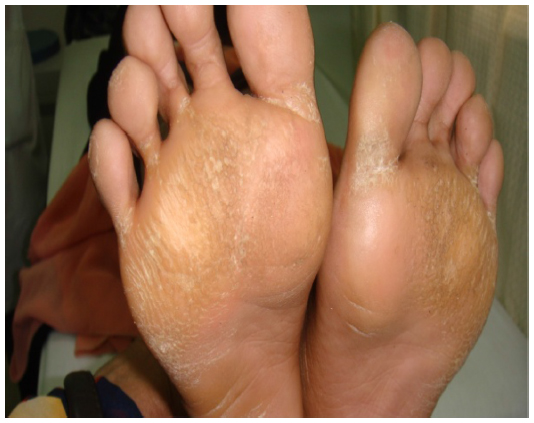 | Figure 3 Plantar hyperkeratosis involving plantar aspect of feet. |
Case two
The next eldest patient was also female and of 16 years of age. She was found to be vitally stable. Systemic examination was unremarkable. Her skin examination showed palmoplantar hyperkeratosis and generalized ichthyosiform scaling. Vision was also normal in this case and there was no hearing at all in both ears.
Case three
The youngest patient was a male of 13 years of age. This patient had a similar spectrum of symptomatology to both cases above, suggestive of KID syndrome. Recurrent cutaneous candida infections of the trunk and forearms were also noticed. The audiogram showed no response in both ears. The key classical triad suggestive of diagnosis was ichthyosis, deafness, and blepharitis in all these cases.
Discussion
KID syndrome is a very uncommon congenital syndrome with reported prevalence of less than one per 1,000,000. Only eleven families affected by this disorder were described in the literature.3 Autosomal dominant patterns of inheritance have been noticed in familial cases with GJB2 gene found to be the common culprit. This gene encodes connexin 26 protein, which is a component of gap junction channels in epidermis and stria vascularis of the cochlea. These channels play a role in the coordinated exchange of molecules and ions occurring in a wide spectrum of cellular activities.4
Involvement of the epidermis is found in almost all cases manifested by the presence of hyperkeratotic skin lesions. These lesions, when coupled with infections, are the sole presenting features in the dermatology clinic. Various case reports mentioned a wide variety of skin lesions for KID syndrome. The most commonly described features are marked by discrete erythematous scaly thickened plaques affecting skin on the trunk and acral parts of the body. The palms of the hands and soles of the feet typically have thickened, hardened, and dry skin.5 Less common reported features are nail involvement, alopecia, abnormal teeth, reduced sweating, and recurrent infections.6 In our cases we saw nail abnormalities in all three cases. Recurrent fungal infections affecting the skin of exposed parts on the face, hands, and legs were also seen in our cases. Patients with this syndrome are considered to be at lifelong risk of development of squamous cell carcinoma of the skin or of the tongue at an unusually younger age.7
Severe sensorineural hearing loss in KID syndrome is usually present since birth. This problem leads to social isolation and decreased attention on the part of relatives, culminating in developmental and language delays. In contrast to the skin lesions that are given medical attention for treatment, deafness is accepted as an inborn error with no appreciable positive approach to improve it. Cochlear implant may be an option in these cases if feasible in all respects.8
Most affected individuals develop different eye problems – usually keratitis and blepharitis – that can result in increased sensitivity to light and progressive vision loss. Other ocular signs include loss of eyebrows and lashes, thickened and keratinized lids, trichiasis, recurrent corneal epithelial defects, superficial and deep corneal stromal vascularization with scarring, and keratoconjunctivitis sicca with variable severity independent of the age of the patient.9 Blepharitis seen in our cases was mild and responded well to topical treatment.
Recently, some researchers showed success in antenatal diagnosis of KID syndrome in mothers who already had KID syndrome in their family.10 Genetic testing is also available now to establish the responsible gene involvement in KID syndrome. In our cases, we could not perform genetic testing because of multiple issues such as unwillingness, poor socioeconomic status, lack of education, and ethical concerns on the part of the patient and attendants.
Hyperkeratotic skin lesions in KID syndrome show improvement to systemic retinoids (acitretin), but this treatment can worsen vision due its ability to stimulate corneal neovascularization. We administered short courses of isotretinoin (shorter half-life than acitretin) as prophylaxis for possible cutaneous complications and it also helped in management of their main skin lesions. Hearing impairment is usually permanent, except by the benefit provided by cochlear implant.
KID syndrome, although rare, predisposes patients to recurrent cutaneous infections and malignant degeneration with additional profound loss of hearing and vision. Early diagnosis, proper treatment, and regular clinical follow up can greatly improve the quality of life in these patients, making them a normal or a near normal member of society.
Disclosure
The author reports no conflicts of interest in this work.
References
Enei ML, Cassettari A, Córdova S, Torres O, Paschoal F. [Do you know this syndrome?] An Bras Dermatol. 2011;86:819–821. Portuguese. | |
Iossa S, Marciano E, Franzé A. GJB2 gene mutations in syndromic skin diseases with sensorineural hearing loss. Curr Genomics. 2011;12:475–785. | |
Markova TG, Brazhkina NB, Bliznets EV, Poliakov AV, Tavartkiladze GA. [Diagnostics of keratitis-ichthyosis-deafness syndrome (KID-syndrome)]. Vestn Otorinolaringol. 2012;3:58–61. Russian. | |
Terrinoni A, Codispoti A, Serra V, et al. Connexin 26 (GJB2) mutations as a cause of the KID syndrome with hearing loss. Biochem Biophys Res Commun. 2010;395:25–30. | |
Gonzalez ME, Tlougan BE, Price HN, Patel R, Kamino H, Schaffer JV. Keratitis-ichthyosis-deafness (KID) syndrome. Dermatol Online J. 2009;15:11. | |
De Raeve L, Bonduelle M, Deconinck H, Roseeuw D, Stene JJ. Trichothiodystrophy-like hair abnormalities in a child with keratitis ichthyosis deafness syndrome. Pediatr Dermatol. 2008;25:466–469. | |
Natsuga K, Akiyama M, Shimizu H. Malignant skin tumours in patients with inherited ichthyosis. Br J Dermatol. 2011;165:263–268. | |
Smyth CM, Sinnathuray AR, Hughes AE, Toner JG. Cochlear implantation in keratitis-ichthyosis-deafness syndrome: 10-year follow-up of two patients. Cochlear Implants Int. 2012;13:54–59. | |
Messmer EM, Kenyon KR, Rittinger O, Janecke AR, Kampik A. Ocular manifestations of keratitis-ichthyosis-deafness (KID) syndrome. Ophthalmology. 2005;112:e1–e6. | |
Sbidian E, Feldmann D, Bengoa J, et al. Germline mosaicism in keratitis-ichthyosis-deafness syndrome: pre-natal diagnosis in a familial lethal form. Clin Genet. 2010;77:587–592. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.