Back to Journals » Hepatic Medicine: Evidence and Research » Volume 17
Ischemia Reperfusion Injury Induced Systemic Inflammatory Response Following Interspecies Liver Transplantation
Authors Rao JS, Rangarajan P, Shetty A ![]() , Steinhoff M, Tran A, Crane AT
, Steinhoff M, Tran A, Crane AT ![]() , Low WC, Steer CJ
, Low WC, Steer CJ ![]() , Ramachandran S
, Ramachandran S ![]()
Received 11 April 2025
Accepted for publication 21 November 2025
Published 1 December 2025 Volume 2025:17 Pages 161—176
DOI https://doi.org/10.2147/HMER.S533805
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Gerry Lake-Bakaar
Joseph Sushil Rao,1,2 Parthasarathy Rangarajan,2 Anala Shetty,3 Magie Steinhoff,2 Anna Tran,2 Andrew T Crane,4 Walter C Low,5,6 Clifford J Steer,3,4,6 Sabarinathan Ramachandran2
1Division of Solid Organ Transplantation, Department of Surgery, University of Minnesota, Minneapolis, MN, USA; 2Schulze Diabetes Institute, Department of Surgery, University of Minnesota, Minneapolis, MN, USA; 3Molecular, Cellular, Developmental Biology, and Genetics Graduate Program, University of Minnesota, Minneapolis, MN, USA; 4Division of Gastroenterology, Hepatology and Nutrition, Department of Medicine, University of Minnesota, Minneapolis, Minnesota, USA; 5Department of Neurosurgery, University of Minnesota, Minneapolis, MN, USA; 6Stem Cell Institute, University of Minnesota, Minneapolis, MN, USA
Correspondence: Joseph Sushil Rao, Division of Solid Organ Transplant & Schulze Diabetes Institute, Department of Surgery, University of Minnesota, 420 Delaware St SE, Minneapolis, MN, 55455, USA, Email [email protected] Sabarinathan Ramachandran, Schulze Diabetes Institute, Department of Surgery, University of Minnesota, 420 Delaware St. SE, Minneapolis, MN, 55455, USA, Email [email protected]
Introduction: Xenotransplantation has advanced through porcine genetic modifications to enhance graft survival and immunological compatibility. More recently, generating of exogenic organs from recipient-derived stem cells via blastocyst complementation has emerged as a promising strategy to achieve graft-specific tolerance and reduce dependence on long-term immunosuppression. However, ischemia-reperfusion-injury (IRI) during organ donation and transplantation activates inflammatory and innate immune pathways that compromise graft function and survival. To better understand the immunological response following interspecies transplantation, we compared xeno-adaptive and acute inflammatory responses between mouse-to-rat heterotopic liver transplants and rat allotransplants. This model will serve as baseline parameters for future studies of anti-inflammatory mechanisms to mitigate IRI, especially while testing exogenic livers.
Methods: Livers from retired breeder male C57BL/6 mice were heterotopically transplanted into retired breeder male Lewis rats. After 90 minutes of reperfusion, transplanted liver, recipient blood and spleen were recovered for immune and inflammatory analyses. As a control to xenografts, Sprague Dawley donor livers were transplanted into Lewis recipients to represent a clinically relevant allotransplant model. Plasma cytokine and chemokine levels were quantified, and mixed leukocyte reactions (MLR) were performed to assess CD4+ and CD8+ T cell proliferation.
Results: Xenografts exhibited elevated plasma levels of IL-2 (p = 0.04), IL-10 (p < 0.01), IL-13 (p = 0.01), IL-17A (p < 0.01) TNF-a (p < 0.05), and RANTES (p < 0.05) compared with naïve controls. Although xenografts demonstrated higher levels after transplant, they were not statistically distinct to allotransplant results. MLR assays revealed significantly increased CD8+ T cell proliferation (p = 0.01) when rat splenocytes were stimulated with mouse non-parenchymal liver cells, whereas CD4+ T cell proliferation was not significant in either model.
Discussion: Although xenograft induced heightened inflammatory and adaptive responses, these were comparable to allotransplants. For successful exogenic organ transplantation without maintenance immunosuppression, future strategies must focus on mitigating reperfusion-induced systemic inflammation to improve graft tolerance and survival.
Keywords: ischemia reperfusion injury, systemic inflammatory response, interspecies transplant, blastocyst complementation
Introduction
The standard of care for end-stage liver disease and malignancies is liver transplantation.1,2 However, a common etiology for primary graft dysfunction, non-function and chronic rejection is ischemic-reperfusion injury (IRI).3 Although IRI following liver transplantation cannot be completely prevented, reducing the injury associated with IRI could result in effective utilization of marginal liver grafts and thereby expand the donor pool. Similarly, regenerative medicine and genetic modifications have enabled organ transplantation without the use of long-term immunosuppression by exploiting the inhibitory pathways to reduce T cell-mediated responses.4 Although the fields of interspecies chimerism and blastocyst complementation can be used to generate donor-derived exogenic organs in distinct host species,5–7 to mitigate allo/xenogenic adaptive immune responses, the presence of IRI following liver transplantation is most likely inevitable. In the initial phase of IRI following liver transplantation, the ischemic insult exposes the parenchymal and non-parenchymal cells to oxygen deprivation, altered pH and ATP depletion.8 These events initiate the production of reactive oxygen species (ROS), augment intracellular calcium concentrations, and enhance organelle damage, ultimately leading to hepatic cell death.9 The reperfusion at transplant following a period of ischemia alters the liver metabolism and initiates a complex interconnected inflammatory cascade that further aggravates hepatocellular damage. Host innate immune activation is central in the cytotoxic cycle that progresses towards an immunologically inflamed organ.10,11 The inflammatory cascade releases cytokines and chemokines that result in activation of neutrophils, macrophages and other cell types that accelerate tissue destruction.12,13
In the hope of creating an interspecies exogenic liver through blastocyst complementation, we preemptively describe the ischemia-reperfusion injury with non-specific inflammatory markers following liver transplantation in a non-survival heterotopic/auxiliary C57Bl/6 mouse to Lewis rat heterotopic liver transplant model and compare the results to heterotopic Sprague Dawley to Lewis rat allo-transplantation. Figure 1 demonstrates the schematic of the study, detailing the liver transplant model and the use of tissue samples to evaluate the allo- and xeno-immune response and non-specific inflammatory response. The reperfusion injury markers identified will lay the groundwork for conditioning the graft as well as institute an induction/receiver conditioning protocol that can mitigate the IRI with the goal to improve graft survival in long-term exogenic liver transplant models that spare maintenance immunosuppression.
 |
Figure 1 Schematic of Study. (A) Mouse to rat heterotopic liver transplantation and sampling of blood and tissue for post-transplant analysis of immune response and non-specific inflammatory response. (B) Naïve splenocytes, liver non-parenchymal cells and plasma as controls for the study. (C) Mixed leukocyte reaction following transplantation to study the immune response to transplanted liver graft. Created in BioRender. Rao, J. (2025) https://BioRender.com/9zebguh. |
Materials and Methods
Surgical Steps in Liver Donation
Animal experiments and surgical techniques were approved by the Institutional Animal Care and Use Committee (IACUC 2304–40989A) at the University of Minnesota as required by the Health Research Extension Act of 1985 (Public Law 99–158). All experiments in this study were performed according to the provisions of the Animal Welfare Act of 1966 (Public law 89–554), and its subsequent amendments, as well as the standards set forth by NIH in “The Guide for the Care and Use of Laboratory Animals” (NIH Publication No. 85–23, revised 2011). Retired breeder C57Bl/6 female mice (Charles Rivers, Wilmington, MA) weighing between 25–35 g served as liver donors for xenotransplant and Sprague Dawley rats (Charles River, Wilmington, MA) weighting 300–350 g served as liver allografts for control transplants. Following the administration of 2% isoflurane through a nose cone, the abdomen of the mouse was prepared for incision. A cruciate incision was performed to expose the liver and porta hepatis. The coronary, triangular, falciform, round, hepatogastric, hepatoduodenal ligaments were incised to mobilize the liver from all its attachments. Both renal veins and arteries were ligated at the inferior vena cava and the vena cava was mobilized from lumbar branches posteriorly. Care was also taken to ligate the right adrenal vein. The portal vein was identified and mobilized beyond the splenic and superior mesenteric vein. Care was taken to prevent injury to the pancreas and prevent significant blood loss. The bile duct was identified and mobilized at the duodenum. Supra-hepatic vena cava was mobilized, and the hepatic veins identified, and the left phrenic vein was ligated using 9–0 prolene (ETHICON; D9567). The abdominal aorta below the renal artery was mobilized and the donor was systemically heparinized. Splenic vein and superior mesenteric artery were ligated; and the supra-hepatic and infra-hepatic vena cava were vented, while aortic and portal vein cannulas were inserted, to flush the donor liver with University of Wisconsin (UW) solution at 4°C. The hepatic artery was ligated, and the liver explanted (Figure 2A and B). A similar technique and protocol were used for Sprague Dawley donor rats.
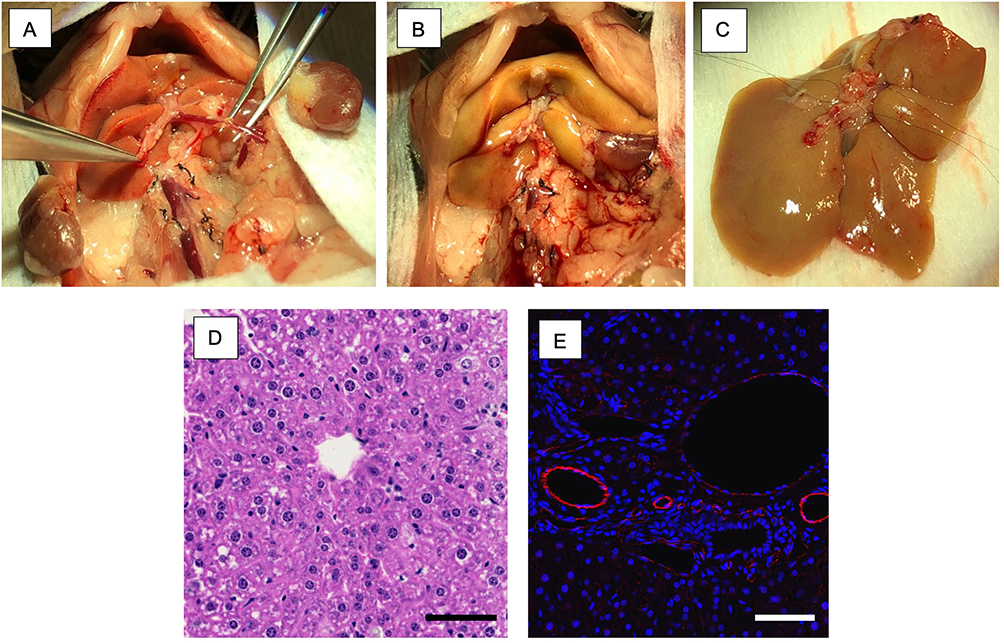 |
Figure 2 Surgical model of mouse donor liver recovery and microscopy. (A) Retroperitoneal dissection and preparation of mouse liver for recovery; (B) Donor liver flushed with University of Wisconsin solution at 4°C; (C) Back bench preparation of donor liver for transplantation; (D) Hematoxylin and Eosin staining of mouse donor liver. (E) Anti-CD31 rabbit primary antibody (Abcam; Catalogue number ab281583) and secondary goat anti-rabbit IgG H&L Alexa Flour 488 (Abcam; Catalogue number ab150077) expression of endothelium by immunofluorescence confocal imaging. Scale bar at 100µm. |
The donor liver graft was examined on the back table for any leaks in the supra-hepatic vena cava, infra hepatic vena cava, and portal vein. The infra-hepatic vena cava was ligated, and the portal vein flushed to confirm no leaks. The liver was preserved in UW at 4°C for 20–30 minutes until transplanted (Figure 2C) and hematoxylin and eosin (H&E) (Figure 2D) and CD31 labeling of the endothelium (Figure 2E) confirm intact microscopic and microvascular architecture.
Recipient Heterotopic Liver Transplant Model
Two-month-old male Lewis rats (Charles River, Wilmington, MA) weighting 150–200 g were used as recipients. No immunosuppression or pain medications were administered to any of the recipients as they were all non-survival surgeries euthanized by exsanguination after 90 minutes of reperfusion. Under isoflurane anesthesia, the abdomen of the rat was prepared for incision. A midline incision was performed, and hemostasis is achieved with electrocoagulation and 6–0 silk suture ties (Fischer Scientific; Catalogue No NC9201232). The abdominal aorta and infra-hepatic inferior vena cava were mobilized from lumbar branches and collaterals. Left renal vein was mobilized by dividing the left adrenal vein and mobilizing from the adjacent renal artery. Microvascular clamps were applied on the inferior vena cava below the level of the renal veins and another proximal to the bifurcation of iliac veins. A venotomy was made and the lumen flushed immediately with heparinized saline. The donor mouse liver was brought into the field for anastomosis. Using 10–0 prolene (ETHICON, 2790G) the portal vein was anastomosed to the inferior vena cava in an end-to-side fashion (Figure 3A–C). Another set of microvascular clamps were applied onto the left renal vein and a venotomy was made and again flushed immediately with heparinized saline. The donor liver’s supra-hepatic vena cava was anastomosed to the left renal vein in an end-to-side fashion using 10–0 prolene. The recipient’s anterior wall of infra hepatic vena cava was constricted using 10–0 prolene to narrow the luminal diameter thereby diverting more blood into the donor portal vein (Figure 3A). Bile production at the cut end of the donor bile duct was assessed to confirm liver function and a wedge biopsy to confirm adequate perfusion (Figure 3D). Hemostasis was achieved through absorbable hemostat (Sugicel® SNOW™). The graft was monitored for 90 min after which blood, spleen and the transplanted liver were recovered for analysis and the recipient euthanized by exsanguination under anesthesia. The same surgical technique was used for the allo-transplant model. H&E staining and CD31 labeling (Figure 3E and F) of the transplanted liver were used to assess microscopic and microvascular architecture.
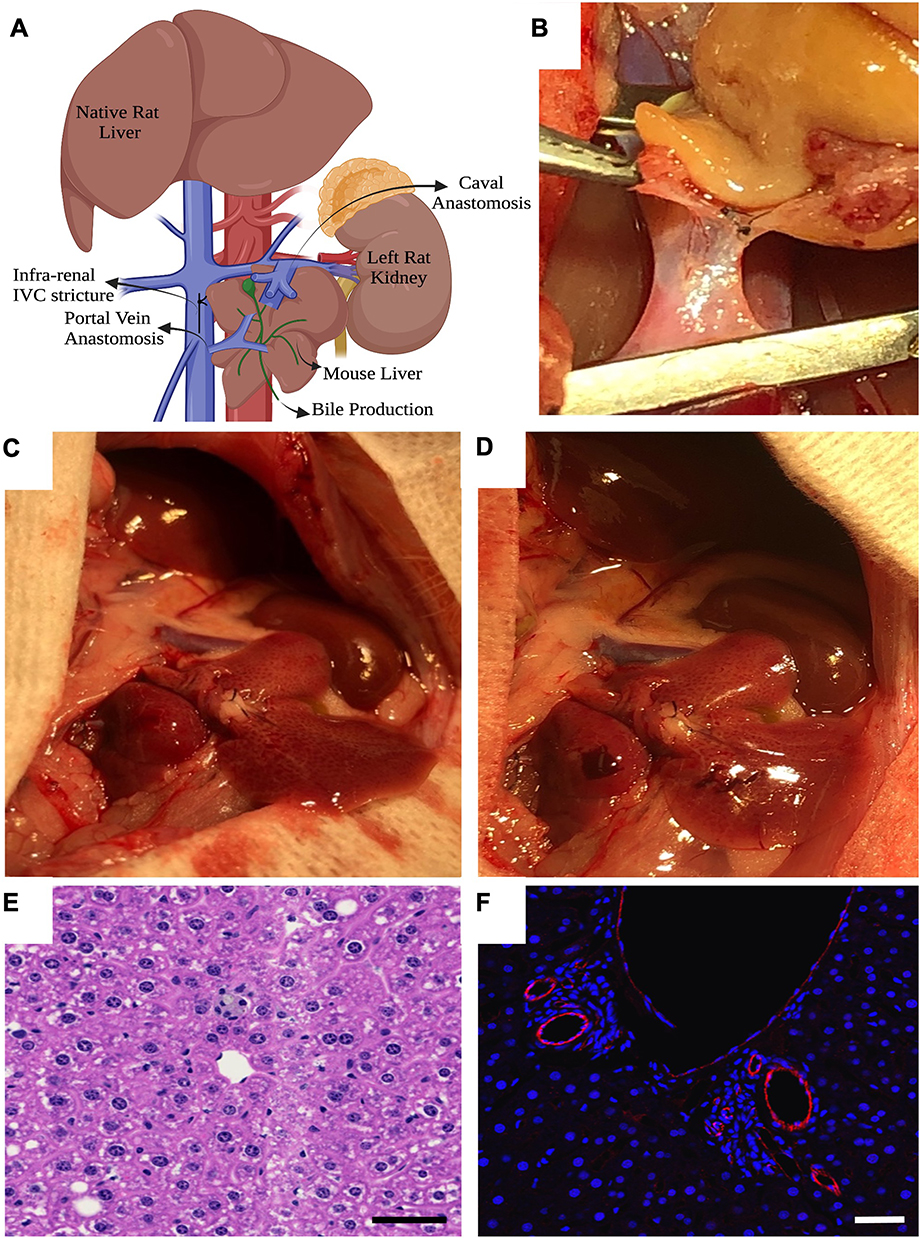 |
Figure 3 Surgical model of recipient liver transplant and microscopy. (A) Schematic of heterotopic/auxiliary liver transplant; (B) Donor mouse liver portal vein anastomosis to recipient rat infra-hepatic vena cava; (C) Reperfusion of donor graft at reperfusion; (D) Bleeding from liver biopsy confirms adequate perfusion at 60 minutes; (E) H&E of graft after 90 minutes of reperfusion; (F) Anti-CD31 rabbit primary antibody (Abcam; Catalogue number ab281583) and secondary goat anti-rabbit IgG H&L Alexa Flour 488 (Abcam; Catalogue number ab150077) expression of endothelium by immunofluorescence confocal imaging at explant. Scale bar at 100µm. Panel A Created in BioRender. Rao, J. (2025) https://BioRender.com/hflj147. |
Rat and Mouse Plasma Preparation
Peripheral blood samples from both control and transplanted animals were collected into sodium heparin tubes. The samples were centrifuged at 2000 × g for 10 min, separating the blood components and leaving plasma at the top. The plasma was carefully aspirated, transferred to labeled vials, and stored at −80°C to maintain stability until analysis. Upon thawing on ice, the frozen plasma samples were mixed by vortexing to ensure uniformity and then centrifuged again at 10,000 × g for 10 min to remove any particulates. Finally, the prepared plasma samples were subjected to cytokine assays.14
Splenocyte Isolation
Rat spleens were processed to obtain a single-cell suspension through manual mincing with a 10-mL rubber syringe plunger and filtration using a 20-μm sterile nylon mesh. Following filtration, cells were collected via centrifugation at 1500 rpm for 5 min, and the supernatant was discarded. The cells were then resuspended in PBS and centrifuged a second time. After discarding the supernatant, the cells were treated with ACK red blood cell (RBC) lysing buffer as per the manufacturer’s instructions (Thermo Scientific, Catalog No A1049201) to remove RBCs. Once the RBCs were removed, the splenocytes underwent two washes with PBS with 2% BSA, followed by filtration through a 20μM vacuum top tube filter (Millipore, Catalog No SCNY00020), centrifugation and resuspension in an appropriate volume of complete RPMI cell culture medium.15
Liver Non-Parenchymal Cell (LNPC) Isolation
Naïve and post-transplant rat livers were isolated, minced initially to small pieces and treated with HEPES-buffered Hanks balanced salt solution at pH 7.5. The livers were treated with 0.05% collagenase and incubated in RPMI-1640 medium containing 5% fetal bovine serum under constant agitation. The resulting suspension underwent filtration through multiple layers of nylon mesh to eliminate undigested clumps, followed by two rounds of centrifugation at 50 × g for 1 minute to remove parenchymal cells. The remaining cell suspension was then subjected to discontinuous Ficoll-paque density gradient centrifugation at 500 × g for 30 min. LNPCs, which accumulated at the interface layer, were carefully removed, and washed twice with complete RPMI-1640 medium and used for MLR cultures.16
Mixed Lymphocyte Reaction
Mixed Lymphocyte Reactions (MLR) were performed on splenocytes isolated from naïve control rats (Male Lewis rats not exposed) and liver recipients 90 min following reperfusion. Responder splenocyte (500,000 cells) samples from naïve (controls) and Tx rats were labeled with 2.5 μM Carboxylfluorescein succinimidyl ester (CFSE) (Invitrogen, Catalogue No C34554) and co-cultured with irradiated (3000 cGy) VPD-450-labeled (BD, Catalogue No 562158) stimulator splenocytes (500,000 cells) from naïve mouse splenocytes, mouse non-parenchymal liver cells from control and post-transplant. After five days of incubation at 37°C with 5% CO2 the proliferated cells were stained for CD4+, CD8+ T Cells and CD20+ B Cells. The percentage proliferation of T and B cells was analyzed using multiparametric flowcytometric analysis where the proliferation percentage was determined by the level of fluorescence intensity of CFSE in the responders. On flowcytometric analysis, cells were gated on FSC-H vs FSC-A and then on SSC-H vs SSC-A to discriminate doublets. Lymphocytes were further gated based on well-characterized SSC-A and FSC-A characteristics. Dead cells were excluded based on viability dye. The following phenotypic characteristics were used to define the immune cell populations representing CD4+ T cells: CD4+/CD8−; CD8+ T cells: CD8+/CD4− (Figure 4A and B). The same protocols were used in analyzing the MLR in allo-transplantation (No mouse samples were used; Donor – Sprague Dawley rat spleen and liver; Recipient – Lewis rat spleen and transplanted allograft of donor liver served as LNPCs).
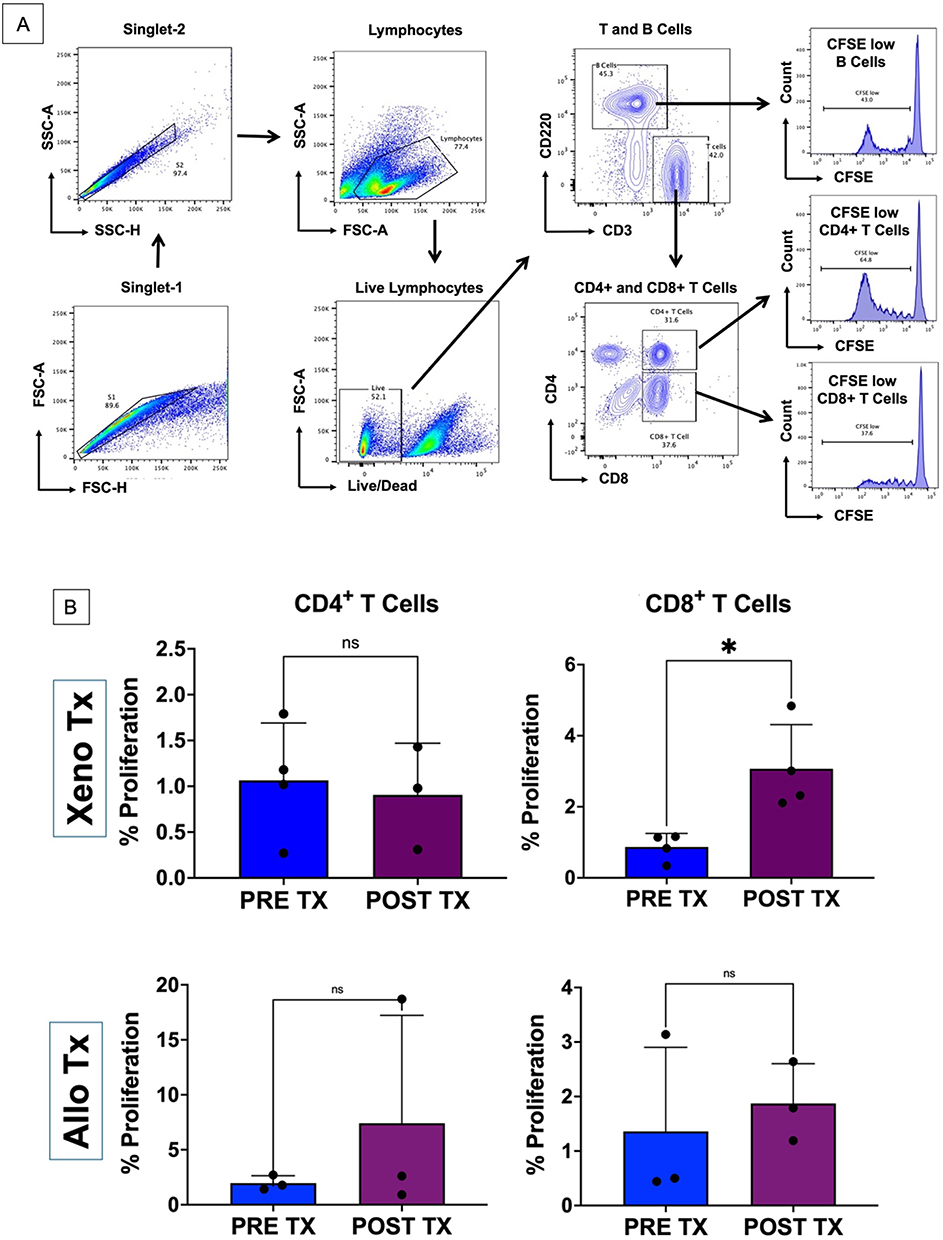 |
Figure 4 T cell proliferation following allo- and xenotransplantation. (A) Mixed Leukocyte Reaction gating strategy. (B) CD4+ and CD8+ T cell proliferation following 7-day culture to non-parenchymal liver donor cells comparing post-reperfusion (Post Tx) and naïve (Pre Tx) immunological state in xeno and allo transplant model. (*) symbolizes statistical significance (p < 0.05) and (ns) symbolized not significant. |
Cytokine Analysis by Luminex Immunoassay
We measured the levels of cytokines and chemokines in the plasma of mouse to rat liver transplant recipients after 90 min of reperfusion. Samples from non-transplanted control rats were used to analyze the change in expression levels of IRI markers. MILLIPLEX MAP Rat Cytokine/Chemokine Magnetic Bead Panel - Immunology Multiplex Assay (RECYTMAG-65K) kits were purchased from Millipore Sigma (Burlington, MA, USA). The magnetic bead panel detects autoantibodies to G-CSF, Eotaxin, GM-CSF, IL-1α, MIp-1α, IL-4, IL-1β, IL-2, IL-6, EGF, IL-13, IL-10, IL-12p70, IFN-γ, IL-5, IL-17α, IL-18, MCP-1, IP-10, GRO, VEGF, Fractalkine, LIX, MIP-2, TNF-α, and RANTES via signal specific magnetic beads (Figure 5A). Plasma samples were diluted at 1:2, 1:4 and 1:8 in assay specific buffer and incubated together with antibody coated beads in 96-well plates for 2 hours at 37°C in a rocker platform after they were normalized by volume. The plates were washed three times with wash buffer. PE-IgG conjugate was added to each well and incubated for 90 min at room temperature. After washing three times with wash buffer, Sheath Fluid PLUS was added to each well and the plates were read on the Luminex® platform (Instrument - IFLEXS22334001; Instrument software version – 2.0.1017 and Analysis software version Belysa 1.0.19). Luminescence was measured in picograms per mL and median fluorescent intensity (MFI) data were obtained for statistical analysis. Comparisons between xeno and allotransplants were represented as a heatmap (Figure 5B).
 |
Figure 5 Chemoattractants and cytokines following graft reperfusion. (A) Luminex analysis of cytokine and chemokines following xenotransplantation comparing levels in pre-transplant (naïve) to post transplant levels (Tx). (B) Heat map compares the fold change expression between pre- and post-transplant in the allo- and xeno- transplant model. Color scale on the far right corresponds to the fold change. There was no statistical significance in the fold change (pre- vs post-transplant) between allo- and xeno- transplantation. |
Statistical analysis was performed using Graph Pad Prism v10.0. Two-tailed unpaired t-test was used to define statistical significance and p < 0.05 was considered statistically significant.
Development of Rat-Mouse Interspecies Chimeras
Mouse zygotes were extracted from the oviduct of super ovulated time mated C57/Bl6 females, and the embryos were cultured in vitro until E3.5 stage. To generate rat-mouse chimeras, eight to ten eGFP-labelled rat donor embryonic stem cells (ESCs) (cell line, F344eGFP) were injected into the inner cell mass of the mouse E3.5 embryos. The injected chimeric embryos were then transferred into E2.5 pseudo pregnant CD1 surrogate dams (15–20 embryos/dam) and were allowed to develop in utero until E12.5. At E12.5 (10 days after transfer), the embryos were extracted. Whole embryos were imaged for GFP expression, which indicates the contribution of rat GFP+ donor cells in the rat-mouse chimeric embryos. The embryos were then fixed, embedded in OCT blocks, and stored at −80°C for further analysis (Figure 6A–C).
 |
Figure 6 Albumin Staining in HHEX KO + rESC Embryo. (A) Generation of rat-mouse chimeras by injection of GFP+ rat ESCs into the E3.5 mouse blastocysts; (B) Microinjection of rat ESCs into E3.5 mouse embryo cavity. Imaging of chimeric embryo at E6.5 to confirm contribution of the GFP+ rat cells to the chimeric embryo; (C) Whole embryo imaging of E12.5 embryos to detect presence of GFP+ rat cells in the chimeric embryos; (D) Liver sections of E12.5 chimeric embryos (n = 3) validating integration of GFP+ rat donor cells into the chimeric livers and expression of the key hepatocyte-specific gene, albumin, in the rat cells. Panel A Created in BioRender. Rao, J. (2025) https://BioRender.com/ehwa5vr. |
Immunofluorescence Staining of the Rat-Mouse Chimeras
The fixed rat-mouse embryos were sectioned and stained with primary antibody Albumin (Proteintech; Catalog No 16475-1-AP), secondary antibody Donkey anti-Rabbit (Abcam, Catalog No 150074), and DAPI. The embryo sections were imaged under a fluorescence microscope (Leica DM16000 B) and processed using QuPath and ImageJ software (Figure 6D).
Results
Heterotopic Liver Transplantation
A total of four retired breeder C57Bl/6 female mice were used as liver donors for xenotransplantation and a total of three Sprague Dawley male rats were used as liver donors for allotransplantation. Cold ischemia time was kept constant at under one hour for all donor grafts. Warm ischemia time (Vascular anastomosis time) was restricted to 30 minutes for both xeno as well as allotransplants. Upon reperfusion all livers reperfused immediately upon releasing clamps (Figure 3C). There was minimal bleeding, and hemostasis was achieved using Surgicel SNoW™ hemostatic agents. A biopsy performed at 60 min confirmed satisfactory perfusion (Figure 3D). All livers were adequately perfused for the entire duration of 90 min before explant. Bile production was present following reperfusion in all seven transplants.
Microscopy with hematoxylin and eosin (H&E) of donor livers before (Figure 2D) and after transplant (Figure 3E) did not show changes associated with necrosis or congestion. Architecture of the liver lobule was maintained with well-defined borders and cell morphology. The portal triad and central vein demonstrated intact endothelium with anti-CD31 as seen in Figures 2E and 3F.
T Cell Mediated Immune Response Following Transplantation
NPLC were isolated from the transplanted mouse (xeno) and rat (allo) livers after 90 min of reperfusion. Naïve rat and mouse livers served as controls for the assay. We performed a one-way CFSE MLR by stimulating recipient rat splenocytes with irradiated naïve donor rat NPLCs. Cell tracking by multiparametric flow cytometry demonstrated that the CD8+ T cell proliferation was significantly higher (p = 0.015) following stimulation with recipient NPLC (3.07 ± 1.24%) compared to stimulation with naïve mouse NPLC (0.87 ± 0.38%). In contrast, no significant difference (p = 0.744) in the proliferation of CD4+ T cells was observed (Naïve was 1.07 ± 0.63% and post-transplant was 0.91 ± 0.56%). Following allotransplant, the CD4+ T cell proliferation was 7.41 ± 9.82 and CD8+ T cell proliferation was 1.87 ± 0.73 and neither met statistical significance (p = 0.392 and 0.629 respectively) (Figure 4).
Chemokines and Cytokines Mediated Ischemia Reperfusion Injury
Systemic inflammatory response following mouse to rat liver transplantation was quantified from the plasma following 90 min of reperfusion using the Luminex® platform. Interferon-γ (INF-γ), a true marker of reperfusion injury was significantly elevated (p = 0.022) post reperfusion (3831.19 ± 700.6 pg/mL) compared to controls (2240.82 ± 532.15 pg/mL). Interleukin-2 (IL-2), a critical cytokine playing an important role in recruiting CD8+ T cells, was significantly elevated (p = 0.049) post reperfusion compared to controls (pre-transplant was 1032.38 ± 224.80 pg/mL and post-transplant was 1745.83 ± 534.64 pg/mL). In contrast, the levels of anti-inflammatory IL-4 with tissue protecting roles were observed to be significantly lower (p = 0.046) in the transplanted recipients (305.97 ± 67.84 pg/mL) when compared to controls (438.07 ± 43.3 pg/mL). Macrophage Inflammatory Protein-2 (MIP-2) is a CXC chemokine ligand that affects recruitment of neutrophils and is a mediator of inflammation in acute liver injury. Although elevated following reperfusion, it was not statistically significant (p = 0.076) post-transplant (524 ± 133.94 pg/mL) compared to fresh controls (294 ± 26.05 pg/dl) but trended towards statistical significance. IL-6 plays a complex role in IRI through pro-inflammation and repair but was not statistically significant (p = 0.333) between reperfusion (11197.43 ± 1919.08 pg/mL) and control (9807.83 ± 1301.86 pg/mL). IL-13, similar to MIP-2 facilitates neutrophil and macrophage activation and was observed to be statistically significant (p = 0.015) following reperfusion injury (807.88 ± 68.25 pg/mL) compared to controls (613.53 ± 44.44 pg/mL). Monocyte Chemoattractant Protein-1 (MCP-1) facilitates migration of monocytes across the endothelium following inflammation and was significantly higher (p = 0.021) following reperfusion (4683.97 ± 299.20 pg/mL) in comparison to controls (4047.33 ± 191 pg/mL). IL-17A is key mediator of inflammation and results in amplification of response to IRI and was significantly increased (p = 0.0003) following reperfusion (704.63 ± 29.46 pg/mL) compared to controls (341.03 ± 43.28 pg/mL). IL-18 is an early mediator of inflammatory response and is responsible for stimulating other pro-inflammatory cytokines and was significantly elevated (p = 0.047) following reperfusion (1883.55 ± 777.27 pg/mL) as compared to controls (673.90 ± 58.73 pg/mL). Tumor Necrosis Factor-α (TNF-α), one of the earliest markers of IRI especially in transplantation is a critical cytokine released from neutrophils, macrophages and endothelial cells. TNF-α was significantly elevated (p = 0.007) following transplantation (513.2 ± 131.26 pg/mL) compared to controls 232.68 ± 55.01 pg/mL). Regulated upon Activation, Normal T Cell Expressed and Secreted (RANTES) also known as CCL5 is a chemoattractant for T cells, monocytes, basophils and eosinophils was significantly elevated (p = 0.005) in the transplant recipients (3250.77 ± 188.07 pg/mL) when compared to controls (2309.33 ± 311.26 pg/mL). Lipopolysaccharide-induced CXC chemokine (LIX) is a potent chemoattractant for neutrophils. The recruitment of neutrophils by CXCL5 further amplifies the inflammatory response and was observed to be significantly elevated (p = 0.003) post-transplant (5888.30 ± 716.91 pg/mL) compared to controls (3233.35 ± 134.52 pg/mL). Interferon inducible-protein 10 (IP-10) produced during IRI is upregulated in response to tissue damage and facilitates recruitment of cells to the site of injury, was significantly elevated (p = 0.0002) in transplant recipients (597.43 ± 4.8 pg/mL) compared to controls (320.23 ± 49.23 pg/mL). (Figure 5). Other chemokines such as Fractalkine, a soluble chemokine and membrane bound adhesion molecule and a potent chemoattractant for natural killer and T cells were significantly elevated (p = 0.015) following reperfusion (282.7 ± 63.54 pg/mL) in comparison to controls (118.83 ± 9.06 pg/mL). IL-1β (Naïve 171.70 ± 10.14 pg/mL; Post-transplant 211.2 ± 37.44 pg/mL), IL-6 (Naïve 9807.83 ± 1301.86 pg/mL; Post-transplant 11197.43 ± 1919.08 pg/mL), growth regulated oncogenes (GRO) such as CXCL1, 2 and 3 levels following reperfusion (746.88 ± 108.77 pg/mL) were not observed to not be significantly different (p = 0.638) from that of controls (6989.33 ± 150.42 pg/mL). In addition, the circulating levels of endothelial growth factor (EGF) and vascular endothelial growth factor (VEGF) were significantly higher (p = 0.0014 and 0.0016 respectively) in the liver transplant recipients (14.47 ± 2.55 pg/mL and 243.13 ± 33.02 pg/mL respectively) compared to controls (5.8 ± 0.96 pg/mL and 135.98 ± 11.19 pg/mL respectively).
In the allotransplant arm of the study, post-transplant chemoattractant levels had a 0.54 ± 0.02-fold change from naïve controls with RANTES, 0.08 ± 0.004 with MCP-1 and 0.3 ± 0.26 with LIX and did not achieve statistical significance (p = 0.535, 0.692, 0.864 respectively). Other cytokine markers were altered without achieving statistical significance. Similarly, there was a 0.98 ± 0.04 fold change in INF-γ between the naïve and post-transplant (p = 0.351), 1.57 ± 0.36 times in IL-13 (p = 0.161), 0.04 ± 0.02 times in IL-18 (p = 0.405), 0.02 ± 0.01 times in TNFα (p = 0.752), 1.55 ± 0.23 times in IL1β (p = 0.076), 1.51 ± 1.12 times in IL-6 (p = 0.278), 0.98 ± 0.25 times in IL-10 (p = 0.422), 0.9 ± 0.03 times in IL-4 (p = 0.745). However, a fold change of 0.64 ± 0.05 between naïve and post-transplant in IL-2 was deemed statistically significant (p = 0.0009).
Chimeric Liver Generation Through Blastocyst Complementation
Rat-mouse interspecies chimeras at E12.5 were generated as described in the methods. Six out of nine embryos (66%) recovered at E12.5 were GFP+ (Figure 6C). The E12.5 chimeric embryos demonstrated normal growth, comparable to WT E12.5 embryos. Liver sections of the chimeric embryos showed a high degree of chimerism in the liver (>50%) as observed by the GFP+ rat cells in the liver. These GFP+ rat cells also expressed albumin, a signatory biomarker for functional, and well-differentiated hepatocytes (Figure 6D). These results demonstrated successful generation of rat-mouse chimeras where rat ESCs significantly contributed to and differentiated into functional hepatocytes in the developing E12.5 rat-mouse chimeric liver.
Discussion
The mismatch between available donor livers and recipients on the waitlist has been of concern and a major motivation to develop alternative sources for organ transplantation.17–22 Recent breakthroughs in the development of humanized porcine donors have demonstrated successful short-term survival rates but continue to be threatened by chronic inflammation and immunological incompatibility that impose a need for potent immunosuppression associated with inherent complications.23–29 Genetic modifications with CRISPR technology30 have profoundly diminished the incidence of hyperacute rejection following xenotransplant by eliminating a few xeno-antigens and humanizing porcine donors. However, the systemic inflammatory response to the xenograft and long-standing chronic inflammation cannot be mitigated by currently available immunosuppression31 and can impact long-term graft survival. Our goal is to develop livers from the recipient’s iPSCs in a porcine donor by knocking out the donor’s haematopoietically expressed homeobox (HHEX) gene. Then, through blastocyst complementation of human iPSCs containing wild-type HHEX, develop a human liver in the pig that is syngeneic to the recipient and requires no immunosuppression medications following transplantation. In this study, we quantified the systemic inflammatory response following interspecies liver transplantation using a mouse to rat auxiliary heterotopic liver transplant model to develop a baseline for exogenic organ transplant and identify potential targets for intervention.
Mouse to rat orthotopic liver transplantation is challenging and results in high mortality following reperfusion.32 To overcome the challenges and improve the outcomes of inter-species transplant, we developed a heterotopic auxiliary model with no native hepatectomy. The heterotopic auxiliary liver transplant model has no impact on venous return or bowel congestion associated with blood flow during transplant. The cuff techniques of mouse to rat orthotopic liver transplantation between the critical anhepatic phase and reperfusion has demonstrated significantly higher mortality.32 Our technique resulted in no mortality during or after reperfusion. The transplanted mouse livers demonstrated good histological architecture with no necrosis or injured vascular endothelium assuring adequate reperfusion and blood supply.
Immune response following transplantation involves the innate and adaptive system, however, in light of interspecies transplantation, in addition to the innate immune response and antibody mediated rejection, the xeno-immune response plays a significant role.12,17 At reperfusion, neutrophils infiltrate grafts and release neutrophil extracellular traps (NETs) that induce injury through generation of reactive oxygen species (ROS) and release of digestive enzymes.33,34 Macrophages recognize the NETs as damage associated molecular patterns (DMAPs) and release cytokines and other inflammatory markers34 and continue the vicious cycle of xenograft destruction through T-cell mediated cytotoxicity systemic inflammatory response.35 Antigen presenting cells, especially macrophages play a central role in the activation of T-cells (CD4+ and CD8+) to reject the transplanted allo- or xeno-grafts. The effect of organ recovery and reperfusion initiates tissue inflammation and release of chemokines36,37 that trigger the migration of donor-specific antigen presenting cells (APC), dendritic cells through lymphatic channels to encounter T cells.38,39 In addition, the donor-antigenic peptides presented by the major histocompatibility (MHC) molecule are recognized by the T cell receptors (TCR) expressed on the recipient T cells to initiate the rejection process.40
On reperfusion, chemoattractants such as RANTES, MCP-1 and LPS-induced CXC chemokine (LIX) attract T cells, monocytes, basophils, and eosinophils. RANTES and TNF-α synergistically induce the expression of adhesion molecules on endothelial cells, such as ICAM-1 and VCAM-1, promoting the adhesion and transmigration of leukocytes into the tissue. This process is critical in the pathogenesis of IRI. The oxidative stress causes direct cellular damage and contributes to the overall tissue injury during reperfusion.41 In parallel, the CD4+ T cells play a central role in cellular rejection. Activated naïve T cells subsequently differentiate into specific subsets under the influence of cytokines generated by innate and stromal cells during the immune process.42 The development of effector response often requires the presence of chemokines and cytokines that route the host T cells into the graft43 at the time of IRI.36,44 CD8+ T cells affect the graft through cytotoxicity leading to apoptosis45,46 and have been proven to be a significant barrier to inducing transplantation tolerance.47 In our study, we observed a significant increase in proliferation of CD8+ T cells post reperfusion following stimulation with NPLCs isolated from the transplanted liver. Brain death of the donor, warm/cold ischemia during the organ recovery for transplantation and reperfusion in the recipients have been shown to induce a significant increase in the stress and injury in the transplanted organs in clinical setting and animal models of transplantation. This results in the alterations of the tissue proteome and immunopeptidomes resulting in the presentation of cryptic or neoantigens by the MHC molecules resulting in the activation of weak T cell responses.48–50
RANTES, LIX and MCP-1 initiate the release of cytokines that are either pro-inflammatory or anti-inflammatory. MCP-1 can induce the activation of endothelial cells, leading to increased vascular permeability and leukocyte adhesion facilitating the migration of immune cells from the bloodstream into the tissue. Prolonged MCP-1 expression contributes to the development of fibrosis and tissue remodeling. Excessive deposition of extracellular matrix proteins can impair tissue function and lead to long-term organ dysfunction. Due to its central role in mediating inflammation and tissue damage during IR injury, MCP-1 has emerged as a potential therapeutic target. Strategies aimed at inhibiting MCP-1 signaling or neutralizing its effects have shown promise in preclinical studies for attenuating tissue damage and improving outcomes following IR injury. MCP-1 plays a crucial role in orchestrating the inflammatory response and tissue damage associated with IRI. Targeting MCP-1 or its downstream signaling pathways may offer therapeutic benefits in preventing or attenuating IRI as we continue to develop exogenic organs.51
IFN-γ is known to play a significant role in IRI by exacerbating the inflammatory response, promoting immune cell infiltration, inducing cell death and generating reactive nitrogen and oxygen species. With the development of exogenic organs, the role of T cell mediated immune response can significantly be diminished, however therapeutic approaches to mitigate the IFN-γ signaling pathways are critical. Similarly, IL-2 is a crucial cytokine that in synergy with IFN-γ plays its role in promoting the activation and proliferation of T cells, particularly CD4+ helper T cells and CD8+ cytotoxic T cells.52 IL-2 also plays a key role in regulating the balance between different subsets of T cells. While it promotes the differentiation of effector T cells, it also stimulates the development and maintenance of regulatory T cells (Tregs).53 In our study, IFN-γ and IL-2 levels were significantly elevated corresponding to increases following IRI. This could lead to increased tissue injury as well as activation of the T cell immune responses to the cryptic and neo-epitopes of the donor antigen impacting the survival and function of the transplanted organ.
The combined pro-inflammatory effects of IL-13 and IL-2 induce oxidative stress and apoptosis, further contributing to tissue damage. Excessive production of IL-13 can amplify the inflammatory cascade further increasing cellular injury as seen in our study. IL-13 can also modulate the balance between pro-inflammatory and anti-inflammatory pathways, either exacerbating inflammation or promoting the resolution of inflammation and tissue repair.54–57 Overall, IL-13’s effects following liver transplantation and IRI are context-dependent and can vary based on factors such as the timing and magnitude of its expression, the specific cell types involved, and the microenvironment of the affected tissue. While IL-13’s immunomodulatory and tissue repair functions may be beneficial in certain settings, its pro-inflammatory and pro-fibrotic effects highlight the need for careful consideration of its therapeutic targeting in transplantation and IR injury contexts, especially in genetically manipulated livers.58 In addition, IL-18 is rapidly released in response to tissue ischemia and reperfusion and acts as an early mediator of the inflammatory response by stimulating the production of such cytokines as TNF-α, IL-1β, and IL-6.
TNF-α is a critical cytokine and one of the earliest cytokines released following IRI. It induces the expression of adhesion molecules (eg, ICAM-1, VCAM-1) on endothelial cells, enhancing the adhesion and transmigration of leukocytes into the tissue contributing to increased vascular permeability and edema. TNF-α can directly damage endothelial cells, exacerbating the inflammatory response and promoting further tissue injury. TNF-α can induce apoptosis and necrosis and specifically in hepatocytes. Persistent TNF-α signaling contributes to chronic graft rejection by promoting inflammation, fibrosis, and vascular changes within the graft, leading to long-term graft dysfunction. TNF-α also enhances the expression of MHC molecules and costimulatory signals on APCs, facilitating the activation of T cells and the immune response against the graft. Targeted therapeutic intervention such as anti-TNF monoclonal antibodies (infliximab) and soluble TNF receptors (etanercept) have been explored to reduce inflammation and improve graft survival in transplant recipients.59 Similarly, IL-18 can modulate cell death pathways, including apoptosis and pyroptosis (a highly inflammatory form of cell death). Due to its central role in mediating inflammation and tissue injury during IRI, IL-18 has also emerged as a potential therapeutic target. Strategies aimed at inhibiting IL-18 signaling or neutralizing its effects have shown promise in preclinical studies for mitigating tissue damage and improving outcomes following IR injury.60 The impact of preventing pyroptosis in exogenic livers can significantly inhibit chronic inflammation and improve long-term graft survival rates.
IL-6 plays a complex role in IRI. IL-6 is involved in promoting inflammation as well as initiating tissue repair processes. The balance between its pro-inflammatory and pro-repair functions may influence the severity and outcome of IR injury in different organs and tissues. Therapeutic strategies targeting IL-6 or its downstream signaling pathways may hold promise for mitigating IR injury in various clinical settings.61 IL-4 plays an anti-inflammatory and tissue-protective role in IRI and transplantation. IL-4 promotes the polarization of macrophages towards the M2 phenotype, which is associated with tissue repair and anti-inflammatory effects. This helps in reducing the inflammatory response, minimizing tissue damage, and enhancing healing during IR injury.62,63 Along with IL-4 and IL-2, IL-10 helps modulating the immune response during IRI. During IRI, IL-10 helps to suppress the production of pro-inflammatory cytokines and reduces the infiltration of neutrophils and macrophages in the affected tissue. IL-10 is pivotal in promoting the transition of macrophages from a pro-inflammatory M1 phenotype to an anti-inflammatory M2 phenotype. M2 macrophages secrete IL-10 and other factors that aid in tissue repair and remodeling by inhibiting inflammatory responses and fostering an environment conducive to healing.64
A limitation of this study is the small size limiting statistical power. A future study with a larger sample size comparing xeno and allotransplants will increase the statistical power. In addition, the use of anti-inflammatory interventions such as Toll-Like Receptor (TLR) inhibitors,65 IL-10 therapy,66 complement inhibitors67 and TNF-a.68 In addition, long-term survival studies with exogenic livers can determine the correlation of liver enzymes (AST/ALT) to cytokine markers and histology to better understand the impact of IRI for long-term graft survival and function.
Conclusion
In conclusion, our model developed for heterotopic auxiliary mouse to rat liver transplant is reliable and offers a platform to study the systemic inflammatory response to interspecies liver transplantation. As we continue to develop the HHEX-KO and develop a chimeric exogenic liver to avoid long standing maintenance immunosuppression, we quantify the systemic inflammatory response that currently cannot be mitigated by available immunosuppression and continue to assess avenues to prevent systemic inflammation and thereby study the impact of diminished chronic inflammation on long-term graft survival. This model will lay the groundwork to study the impact of systemic inflammatory response in an exogenic rodent model wherein the insult of IRI will determine graft survival in the absence of a xenogenic immune response. The use of anti-inflammatory protocols could potentially play a critical role in maximizing graft survival of exogenic livers in the future.
Funding
This work was supported, in part, by a grant from the National Institutes of Health, National Institute of Allergy and Infectious Diseases (NIH/NIAID 1R01AI173804-01) to CJS, WCL.
Disclosure
The authors report no conflicts of interest in this work. Biorender was used to develop scientific illustrations.
References
1. Dutkowski P, Linecker M, DeOliveira ML, et al. Challenges to liver transplantation and strategies to improve outcomes. Gastroenterology. 2015;148(2):307–323. doi:10.1053/j.gastro.2014.08.045
2. Wertheim JA, Petrowsky H, Saab S, et al. Major challenges limiting liver transplantation in the United States. Am J Transplant. 2011;11(9):1773–1784. doi:10.1111/j.1600-6143.2011.03587.x
3. Zhai Y, Petrowsky H, Hong JC, et al. Ischaemia-reperfusion injury in liver transplantation--from bench to bedside. Nat Rev Gastroenterol Hepatol. 2013;10(2):79–89. doi:10.1038/nrgastro.2012.225
4. Jain A, Bansal R. Applications of regenerative medicine in organ transplantation. J Pharm Bioallied Sci. 2015;7(3):188–194. doi:10.4103/0975-7406.160013
5. Wang J, Xie W, Li N, et al. Generation of a humanized mesonephros in pigs from induced pluripotent stem cells via embryo complementation. Cell Stem Cell. 2023;30(9):1235–1245.e6. doi:10.1016/j.stem.2023.08.003
6. Usui J, Kobayashi T, Yamaguchi T, et al. Generation of kidney from pluripotent stem cells via blastocyst complementation. Am J Pathol. 2012;180(6):2417–2426. doi:10.1016/j.ajpath.2012.03.007
7. Kobayashi T, Yamaguchi T, Hamanaka S, et al. Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell. 2010;142(5):787–799. doi:10.1016/j.cell.2010.07.039
8. Selzner M, Selzner N, Jochum W, et al. Increased ischemic injury in old mouse liver: an ATP-dependent mechanism. Liver Transpl. 2007;13(3):382–390. doi:10.1002/lt.21100
9. Guan LY, Fu PY, Li PD, et al. Mechanisms of hepatic ischemia-reperfusion injury and protective effects of nitric oxide. World J Gastrointest Surg. 2014;6(7):122–128. doi:10.4240/wjgs.v6.i7.122
10. Eltzschig HK, Eckle T. Ischemia and reperfusion--from mechanism to translation. Nat Med. 2011;17(11):1391–1401. doi:10.1038/nm.2507
11. Nakamura K, Kageyama S, Ke B, et al. Sirtuin 1 attenuates inflammation and hepatocellular damage in liver transplant ischemia/Reperfusion: from mouse to human. Liver Transpl. 2017;23(10):1282–1293. doi:10.1002/lt.24821
12. Lu L, Zhou H, Ni M, et al. Innate immune regulations and liver ischemia-reperfusion injury. Transplantation. 2016;100(12):2601–2610. doi:10.1097/TP.0000000000001411
13. Reiniers MJ, van Golen RF, van Gulik TM, Heger M, van Golen RF. Reactive oxygen and nitrogen species in steatotic hepatocytes: a molecular perspective on the pathophysiology of ischemia-reperfusion injury in the fatty liver. Antioxid Redox Signal. 2014;21(7):1119–1142. doi:10.1089/ars.2013.5486
14. Xiong W, Dong H, Wang J, et al. Analysis of plasma cytokine and chemokine profiles in patients with and without tuberculosis by liquid array-based multiplexed immunoassays. PLoS One. 2016;11(2):e0148885. doi:10.1371/journal.pone.0148885
15. Qaisar N, Arowosegbe A, Derr AG, et al. Type I IFN-driven immune cell dysregulation in rat autoimmune diabetes. Immunohorizons. 2021;5(10):855–869. doi:10.4049/immunohorizons.2100088
16. Hozumi K, Masuko T, Hashimoto Y. Pre-Kupffer like CD4/CD8 double positive mononuclear cells present in rat liver. J Biochem. 1994;115(5):904–908. doi:10.1093/oxfordjournals.jbchem.a124438
17. Rao JS, Hosny N, Kumbha R, et al. HLA-G1(+) expression in GGTA1KO pigs suppresses human and monkey anti-Pig T, B and NK cell responses. Front Immunol. 2021;12:730545. doi:10.3389/fimmu.2021.730545
18. Zhan L, Rao JS, Sethia N, et al. Pancreatic islet cryopreservation by vitrification achieves high viability, function, recovery and clinical scalability for transplantation. Nat Med. 2022;28(4):798–808. doi:10.1038/s41591-022-01718-1
19. Han Z, Rao JS, Gangwar L, et al. Vitrification and nanowarming enable long-term organ cryopreservation and life-sustaining kidney transplantation in a rat model. Nat Commun. 2023;14(1):3407. doi:10.1038/s41467-023-38824-8
20. Sharma A, Rao JS, Han Z, et al. Vitrification and nanowarming of kidneys. Adv Sci. 2021;8(19):e2101691. doi:10.1002/advs.202101691
21. Sharma A, Lee CY, Namsrai BE, et al. Cryopreservation of whole rat livers by vitrification and nanowarming. Ann Biomed Eng. 2023;51(3):566–577. doi:10.1007/s10439-022-03064-2
22. Gao Z, Namsrai B, Han Z, et al. Vitrification and rewarming of magnetic nanoparticle-loaded rat hearts. Adv Mater Technol. 2022;7(3)
23. Russo MW, Wheless W, Vrochides D. Management of long-term complications from immunosuppression. Liver Transpl. 2024;30(6):647–658. doi:10.1097/LVT.0000000000000341
24. Hosny N, Matson AW, Kumbha R, et al. 3’UTR enhances hCD47 cell surface expression, self-signal function, and reduces ER stress in porcine fibroblasts. Xenotransplantation. 2021;28(1):e12641. doi:10.1111/xen.12641
25. Hosny N, Rao JS, Burlak C. Endoplasmic reticulum stress and cellular homeostasis in genetically engineered porcine donors for xenotransplantation. Exp Clin Transplant. 2023;21(5):387–396. doi:10.6002/ect.2022.0357
26. Moazami N, Stern JM, Khalil K, et al. Pig-to-human heart xenotransplantation in two recently deceased human recipients. Nat Med. 2023;29(8):1989–1997. doi:10.1038/s41591-023-02471-9
27. Montgomery RA, Stern JM, Lonze BE, et al. Results of two cases of pig-to-human kidney xenotransplantation. N Engl J Med. 2022;386(20):1889–1898. doi:10.1056/NEJMoa2120238
28. Rao JS, Burlak C. Xenotransplantation literature update for September - October 2020. Xenotransplantation. 2021;28(1):e12665. doi:10.1111/xen.12665
29. Rao JS, Matson AW, Taylor RT, Burlak C. Xenotransplantation literature update January/February 2021. Xenotransplantation. 2021;28(3):e12685. doi:10.1111/xen.12685
30. Watanabe M, Nagashima H. Genome editing of pig. Methods Mol Biol. 2023;2637:269–292.
31. Graham ML, Ramachandran S, Singh A, et al. Clinically available immunosuppression averts rejection but not systemic inflammation after porcine islet xenotransplant in cynomolgus macaques. Am J Transplant. 2022;22(3):745–760. doi:10.1111/ajt.16876
32. Oldani G, Lacotte S, Orci LA, et al. Efficient nonarterialized mouse liver transplantation using 3-dimensional-printed instruments. Liver Transpl. 2016;22(12):1688–1696. doi:10.1002/lt.24637
33. Uchida Y, Freitas MC, Zhao D, et al. The protective function of neutrophil elastase inhibitor in liver ischemia/reperfusion injury. Transplantation. 2010;89(9):1050–1056. doi:10.1097/TP.0b013e3181d45a98
34. Kashir J, Ambia AR, Shafqat A, et al. Scientific premise for the involvement of neutrophil extracellular traps (NETs) in vaccine-induced thrombotic thrombocytopenia (VITT). J Leukoc Biol. 2022;111(3):725–734. doi:10.1002/JLB.5COVR0621-320RR
35. Cadili A, Kneteman N. The role of macrophages in xenograft rejection. Transplant Proc. 2008;40(10):3289–3293. doi:10.1016/j.transproceed.2008.08.125
36. Hancock WW, Gao W, Faia KL, Csizmadia V. Chemokines and their receptors in allograft rejection. Curr Opin Immunol. 2000;12(5):511–516. doi:10.1016/S0952-7915(00)00130-8
37. el-Sawy T, Fahmy NM, Fairchild RL. Chemokines: directing leukocyte infiltration into allografts. Curr Opin Immunol. 2002;14(5):562–568. doi:10.1016/S0952-7915(02)00382-5
38. Lakkis FG, Arakelov A, Konieczny BT, Inoue Y. Immunologic ‘ignorance’ of vascularized organ transplants in the absence of secondary lymphoid tissue. Nat Med. 2000;6(6):686–688. doi:10.1038/76267
39. Larsen CP, Morris PJ, Austyn JM. Migration of dendritic leukocytes from cardiac allografts into host spleens. A novel pathway for initiation of rejection. J Exp Med. 1990;171(1):307–314. doi:10.1084/jem.171.1.307
40. Talmage DW, Dart G, Radovich J, Lafferty KJ. Activation of transplant immunity: effect of donor leukocytes on thyroid allograft rejection. Science. 1976;191(4225):385–388. doi:10.1126/science.1082167
41. Schenk M, Zipfel A, Schulz C, et al. RANTES in the postoperative course after liver transplantation. Transpl Int. 2000;13(Suppl 1):S147–S149.
42. El-Ouaghlidi A, Jahr H, Pfeiffer G, et al. Cytokine mRNA expression in peripheral blood cells of immunosuppressed human islet transplant recipients. J Mol Med. 1999;77(1):115–117. doi:10.1007/s001090050315
43. Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008;29(6):848–862. doi:10.1016/j.immuni.2008.11.002
44. Nelson PJ, Krensky AM. Chemokines, chemokine receptors, and allograft rejection. Immunity. 2001;14(4):377–386. doi:10.1016/S1074-7613(01)00118-2
45. Bergese SD, Klenotic SM, Wakely ME, et al. Apoptosis in murine cardiac grafts. Transplantation. 1997;63(2):320–325. doi:10.1097/00007890-199701270-00025
46. Ito H, Kasagi N, Shomori K, et al. Apoptosis in the human allografted kidney. Analysis by terminal deoxynucleotidyl transferase-mediated DUTP-botin nick end labeling. Transplantation. 1995;60(8):794–798. doi:10.1097/00007890-199510270-00006
47. Koyama I, Nadazdin O, Boskovic S, et al. Depletion of CD8 memory T cells for induction of tolerance of a previously transplanted kidney allograft. Am J Transplant. 2007;7(5):1055–1061. doi:10.1111/j.1600-6143.2006.01703.x
48. Li Q, Lan P. Activation of immune signals during organ transplantation. Signal Transduct Target Ther. 2023;8(1):110. doi:10.1038/s41392-023-01377-9
49. Sharland AF, Hill AE, Son ET, et al. Are induced/altered self-peptide antigens responsible for de novo autoreactivity in transplantation? Transplantation. 2023;107(6):1232–1236. doi:10.1097/TP.0000000000004499
50. Ramachandran S, Subramanian V, Mohanakumar T. Immune responses to self-antigens (autoimmunity) in allograft rejection. Clin Transpl. 2012;2012:261–272.
51. Shah RJ, Diamond JM, Lederer DJ, et al. Plasma monocyte chemotactic protein-1 levels at 24 hours are a biomarker of primary graft dysfunction after lung transplantation. Transl Res. 2012;160(6):435–442. doi:10.1016/j.trsl.2012.08.003
52. Peralta C, Jiménez-Castro MB, Gracia-Sancho J. Hepatic ischemia and reperfusion injury: effects on the liver sinusoidal milieu. J Hepatol. 2013;59(5):1094–1106. doi:10.1016/j.jhep.2013.06.017
53. Sosa RA, Zarrinpar A, Rossetti M, et al. Early cytokine signatures of ischemia/reperfusion injury in human orthotopic liver transplantation. JCI Insight. 2016;1(20):e89679. doi:10.1172/jci.insight.89679
54. Wang Y, Yang Y, Wang M, et al. Eosinophils attenuate hepatic ischemia-reperfusion injury in mice through ST2-dependent IL-13 production. Sci Transl Med. 2021;13(579). doi:10.1126/scitranslmed.abb6576
55. Alvarez-Argote S, Paddock SJ, Flinn MA, et al. IL-13 promotes functional recovery after myocardial infarction via direct signaling to macrophages. JCI Insight. 2024;9(2). doi:10.1172/jci.insight.172702
56. Mannon P, Reinisch W. Interleukin 13 and its role in gut defence and inflammation. Gut. 2012;61(12):1765–1773. doi:10.1136/gutjnl-2012-303461
57. Liu Y, Munker S, Müllenbach R, Weng HL. IL-13 signaling in liver fibrogenesis. Front Immunol. 2012;3:116. doi:10.3389/fimmu.2012.00116
58. Slegtenhorst BR, Dor FJ, Rodriguez H, et al. Ischemia/reperfusion injury and its consequences on immunity and inflammation. Curr Transplant Rep. 2014;1(3):147–154. doi:10.1007/s40472-014-0017-6
59. Grenz A, Schenk M, Zipfel A, Viebahn R. TNF-alpha and its receptors mediate graft rejection and loss after liver transplantation. Clin Chem Lab Med. 2000;38(11):1183–1185. doi:10.1515/CCLM.2000.184
60. Wu C, Xu J, Zhang Z, et al. The effects of IL-23/IL-18-polarized neutrophils on renal ischemia-reperfusion injury and allogeneic-skin-graft rejection in mice. Biomedicines. 2023;11(12):3148. doi:10.3390/biomedicines11123148
61. Chandran S, Tang Q. Impact of interleukin-6 on T cells in kidney transplant recipients. Am J Transplant. 2022;22(4):18–27. doi:10.1111/ajt.17209
62. Daseke MJ, Tenkorang-Impraim MAA, Ma Y, et al. Exogenous IL-4 shuts off pro-inflammation in neutrophils while stimulating anti-inflammation in macrophages to induce neutrophil phagocytosis following myocardial infarction. J Mol Cell Cardiol. 2020;145:112–121. doi:10.1016/j.yjmcc.2020.06.006
63. Deng M, Wang J, Wu H, et al. IL-4 alleviates ischaemia-reperfusion injury by inducing Kupffer cells M2 polarization via STAT6-JMJD3 pathway after rat liver transplantation. Biomed Res Int. 2020;2020(1):2953068. doi:10.1155/2020/2953068
64. Soares ROS, Losada DM, Jordani MC, et al. Ischemia/Reperfusion injury revisited: an overview of the latest pharmacological strategies. Int J Mol Sci. 2019;20(20). doi:10.3390/ijms20205034
65. Hsieh YC, Lee KC, Wu PS, et al. Eritoran attenuates hepatic inflammation and fibrosis in mice with chronic liver injury. Cells. 2021;10(6):1562. doi:10.3390/cells10061562
66. Dinant S, Veteläinen RL, Florquin S, et al. IL-10 attenuates hepatic I/R injury and promotes hepatocyte proliferation. J Surg Res. 2007;141(2):176–182. doi:10.1016/j.jss.2006.09.018
67. Troise D, Allegra C, Cirolla LA, et al. Exploring potential complement modulation strategies for ischemia-reperfusion injury in kidney transplantation. Antioxidants. 2025;14(1):66. doi:10.3390/antiox14010066
68. Matsumoto S, Takita M, Chaussabel D, et al. Improving efficacy of clinical islet transplantation with iodixanol-based islet purification, thymoglobulin induction, and blockage of IL-1β and TNF-α. Cell Transplant. 2011;20(10):1641–1647. doi:10.3727/096368910X564058
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.