Back to Journals » OncoTargets and Therapy » Volume 12
IRF1 Negatively Regulates Oncogenic KPNA2 Expression Under Growth Stimulation and Hypoxia in Lung Cancer Cells
Authors Huang JX, Wu YC, Cheng YY, Wang CL, Yu CJ ![]()
Received 5 July 2019
Accepted for publication 11 December 2019
Published 27 December 2019 Volume 2019:12 Pages 11475—11486
DOI https://doi.org/10.2147/OTT.S221832
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjeev K. Srivastava
Jie-Xin Huang,1 Yi-Cheng Wu,2 Ya-Yun Cheng,1 Chih-Liang Wang,3,4,* Chia-Jung Yu1,4–6,*
1Graduate Institute of Biomedical Sciences, College of Medicine, Chang Gung University, Taoyuan, Taiwan; 2Department of Thoracic Surgery, Chang Gung Memorial Hospital, Taoyuan, Taiwan; 3School of Medicine, College of Medicine, Chang Gung University, Taoyuan, Taiwan; 4Division of Pulmonary Oncology and Interventional Bronchoscopy, Department of Thoracic Medicine, Chang Gung Memorial Hospital, Taoyuan, Taiwan; 5Department of Cell and Molecular Biology, College of Medicine, Chang Gung University, Taoyuan, Taiwan; 6Molecular Medicine Research Center, Chang Gung University, Taoyuan, Taiwan
*These authors contributed equally to this work
Correspondence: Chia-Jung Yu
Department of Cell and Molecular Biology, College of Medicine, Chang Gung University, 259 Wen-Hwa 1st Road, Guishan District, Taoyuan City, Taiwan
Tel +886-3-2118800 ext. 3424
Fax +886-3-2118042
Email [email protected]
Chih-Liang Wang
Division of Pulmonary Oncology and Interventional Bronchoscopy, Department of Thoracic Medicine, Chang Gung Memorial Hospital, Linkou, 5 Fuxing Street, Guishan District, Taoyuan City, Taiwan
Email [email protected]
Purpose: Karyopherin alpha 2 (KPNA2) has been reported as an oncogenic protein in numerous human cancers and is currently considered a potential therapeutic target. However, the transcriptional regulation and physiological conditions underlying KPNA2 expression remain unclear. The aim of the present study was to investigate the role and regulation of interferon regulatory factor-1 (IRF1) in modulating KPNA2 expression in lung adenocarcinoma (ADC).
Materials and methods: Bioinformatics tools and chromatin immunoprecipitation were used to analyze the transcription factor (TF) binding sites in the KPNA2 promoter region. We searched for a potential role of IRF1 in non-small-cell lung cancer (NSCLC) using Oncomine and Kaplan-Meier Plotter datasets. qRT-PCR was applied to examine the role of IRF1 and signaling involved in regulating KPNA2 transcription. Western blotting was used to determine the effects of extracellular stimulation and intracellular signaling on the modulation of KPNA2-related TF expression.
Results: IRF1 was identified as a novel TF that suppresses KPNA2 gene expression. We observed that IRF1 expression was lower in cancerous tissues than in normal lung tissues and that its low expression was correlated with poor prognosis in NSCLC. Notably, both ataxia telangiectasia mutated (ATM) and mechanistic target of rapamycin (mTOR) inhibitors reduced KPNA2 expression, which was accompanied by increased expression of IRF1 but decreased expression of E2F1, a TF that promotes KPNA2 expression in lung ADC cells. IRF1 knockdown restored the reduced levels of KPNA2 in ATM inhibitor-treated cells. We further demonstrated that epidermal growth factor (EGF)-activated mTOR and hypoxia-induced ATM suppressed IRF1 expression but promoted E2F1 expression, which in turn upregulated KPNA2 expression in lung ADC cells.
Conclusion: IRF1 acts as a potential tumor suppressor in NSCLC. EGF and hypoxia promote KPNA2 expression by simultaneously suppressing IRF1 expression and enhancing E2F1 expression in lung ADC cells. Our study provides new insights into targeted therapy for lung cancer.
Keywords: lung adenocarcinoma, KPNA2, IRF1, E2F1, EGF, hypoxia
Introduction
Karyopherin alpha 2 (KPNA2, also known as importin α1) is a member of the importin α family and transports cargo containing a canonical nuclear localization signal by forming an importin α/β/cargo heterotrimer.1,2 Due to its function in nucleocytoplasmic transport, KPNA2 is involved in many cellular processes, including differentiation, development, viral infection, the immune response, transcriptional regulation and cellular maintenance.3 Recently, several studies have linked KPNA2 to cancer. During the past decade, KPNA2 overexpression has been reported in at least 18 human cancer types, such as lung, breast, colon and bladder cancer. A high level of KPNA2 is positively associated with cancer invasiveness and poor prognosis in patients, thus establishing KPNA2 as a potentially relevant therapeutic target.3,4 We previously identified KPNA2 as a potential biomarker for lung ADC, and we observed that KPNA2 overexpression promotes the proliferation and migration of lung ADC cells.5 We applied proteomic approaches to search for differentially expressed protein profiles and invasiveness-associated KPNA2−vimentin−pErk complexes in lung ADC cells with siRNA-mediated knockdown of KPNA2.6,7 Notably, KPNA2 transports the oncogenes c-Myc and E2F1 and the tumor suppressor genes p53, NBS1 and BRCA1 into the nucleus, suggesting that spatiotemporal regulation of KPNA2 is crucial for its role in tumorigenesis.6,8–10 Our recent study showed that the mTOR pathway is involved in the regulation of KPNA2 protein turnover and correlates with Dp1/E2F1-mediated KPNA2 transcription.11 However, the upstream signaling pathway and the transcription factor (TF) responsible for regulating KPNA2 expression are still unclear.
Interferon regulatory factor-1 (IRF1), a TF belonging to the IRF family, regulates IFN-β and IFN-related gene expression.12 Accumulating evidence supports the notion that IRF1 has multiple functions in gene expression regulation during inflammation, immune responses, cell proliferation, cell cycle progression, T cell differentiation, and DNA damage.13–15 Notably, IRF1 is also involved in cancer biology, but its role in cancer progression is controversial. Gene alteration and/or low expression of IRF1 are correlated with poorer clinical outcomes, high cancer susceptibility and low immunotherapy response, suggesting that IRF1 is a tumor suppressor in multiple cancer types, such as leukemia, breast cancer, cervical cancer and colorectal cancer.16–19 However, the oncogenic ability of IRF1 in hepatocellular carcinoma and esophageal cancer was recently reported.20–22 These studies suggest that the role of IRF1 in cancer is cancer-type specific.
In the present study, we identified IRF1 as a novel transcriptional suppressor of KPNA2 in lung ADC cells. We further investigated the signaling pathways and physiological conditions involved in IRF1-mediated KPNA2 expression in lung ADC cells.
Materials and Methods
Reagents and Antibodies
Epidermal growth factor (EGF), rapamycin, ATM inhibitor and β-actin antibody (MAB1501) were purchased from Millipore (Bedford, MA, USA). KPNA2 (sc-55538), E2F1 (sc-251), IRF1 (sc-497) and ATM (sc-23921) antibodies were obtained from Santa Cruz (California, USA). Phospho-ATM (Ser1981), p70S6K, phospho-p70S6K (Thr389), mTOR, phospho-mTOR (Ser2448), IRF1 and Slug antibodies were obtained from Cell Signaling (Beverly, MA, USA). Hypoxia inducible factor 1α (HIF-1α) and lactate dehydrogenase A (LDHA) antibodies were purchased from GeneTex (Irvine, California, USA) and Abcam (Cambridge, Massachusetts, USA), respectively.
Cell Culture
A549 ADC, NCI-H520 squamous cell carcinoma (SCC) and NCI-H460 large-cell carcinoma (LCC) cell lines were purchased from Food Industry Research and Development Institute (Hsinchu, Taiwan). CL1-5 ADC cell line was derived from one man with poorly differentiated lung ADC23 and kindly provided by Professor P.C. Yang (Department of Internal Medicine, National Taiwan University Hospital, Taipei, Taiwan). A549 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Invitrogen, Carlsbad, CA, USA), and CL1-5, NCI-H520 and NCI-H460 cells were cultured in RPMI 1640 (Gibco). All media were supplemented with 10% fetal bovine serum (Gibco), 100 units/mL penicillin-streptomycin (Gibco), and 2 mM L-glutamine (Gibco). The cells were incubated at 37°C in a humidified atmosphere comprising 95% air/5% CO2.
Small Interfering RNA Transfection
Cells were transfected for 24 h with 10 nM negative control (NC) small interfering RNA (siRNA), siRNA against IRF1 (GGGCUCAUCUGGAUUAAUA, GCUCAGCUGUGCGAGUGUA, Dharmacon, Lafayette, USA) or siRNA against KPNA2 (GAAAUGAGGCGUCGCAGAA, GAAGCUACGUGGACAAUGU, AAUCUUACCUGGACACUUU, GUAAAUUGGUCUGUUGAUG, Dharmacon, Lafayette, USA) with Lipofectamine RNAiMAX Transfection Reagent (Invitrogen, Carlsbad, CA, USA).
Quantitative Reverse Transcription Polymerase Chain Reaction
Total RNA was extracted from cells using TRIzol reagent (Invitrogen, Grand Island, NY, USA), and cDNA was synthesized using a ToolsQuant II Fast RT kit (TOOLS Biotechnology Co., Ltd. Taiwan). Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed with Power SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA). The SYBR Green fluorescence intensity was determined using a QuantStudio® 3 System (Applied Biosystems), and the expression levels of each mRNA were normalized to those of β-actin. The sequences of the primers used are available upon request.
Chromatin Immunoprecipitation Assay
Chromatin immunoprecipitation (ChIP) assays were performed as described previously.11 Briefly, the crosslinked, sonicated chromatin obtained from lung cancer cells was precleared with Dynabeads Protein G (Invitrogen) before the chromatin samples were incubated with E2F1 or IRF1 antibodies on a rotator at 4°C overnight. Dynabeads Protein G were added for an additional 15 min. Mouse IgG antibody (Santa Cruz Biotechnology) was used as the immunoprecipitation control. After extensive washes, the immunecomplexes were treated with proteinase K and decrosslinked at 65°C for 6 h. Bound DNA was extracted with a PCR purification kit (Qiagen, Chatsworth, CA, USA) and subjected to PCR analysis using KPNA2 primers designed to span the E2F1 and IRF1 binding sites. The sequences of primers are listed as follows: IRF1 #1, sense 5ʹ-AGATAGCAAATTGTAAGGAGGG-3ʹ and antisense 5ʹ-GTGCCAGGGCAATAAATTC-3ʹ; IRF1 #2, sense 5ʹ-GAGCCTCCTGAGGATCT-3ʹ and antisense 5ʹ-CCAAATAGTATTTCAACGTGTTATCA-3ʹ; IRF1 #3, sense 5ʹ-CTCCAGGAAGTCTCAGC-3ʹ and antisense 5ʹ-TATGAGACAAAGGGAGAAAGCTAA-3ʹ; and E2F1, sense 5ʹ-ATGGGCACACAGCTTAG-3ʹ and antisense 5ʹ-CTGAGTCTGTACCTGCGAA-3ʹ.
Cell Viability Assay
Cells (5×103 cells/well) were plated in 24-well plates and cultured for the indicated time intervals. After culture, cell viability was evaluated with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenytetrazolium bromide (MTT) colorimetric growth assay as described previously.5
Western Blotting Assay
Protein extracts were resolved by SDS-PAGE and transferred to PVDF membranes using a Tris-glycine buffer system. The membranes were incubated with primary antibodies followed by horseradish peroxidase-conjugated secondary antibodies and developed using a chemiluminescent HRP substrate (Merck Millipore, Darmstadt, Germany).
Hypoxia Treatment
Cells were seeded in 6-well plates, incubated under normoxic conditions overnight and then transferred to hypoxia chambers (SCI-tive; Baker Ruskinn, Sanford MA) containing 1% O2 for the indicated times. Oxygen dissolved in the media was removed for 4 h before it was replaced under hypoxic conditions. Cells were collected and lysed for Western blotting.
Statistics
All quantitative data are shown as the mean ± standard deviation (SD). Significant differences in mRNA expression between the two groups were assessed by the Mann–Whitney test. Significant differences in protein expression in response to different inhibitor treatments were assessed by unpaired t-test analysis. For cell viability assay, two-way ANOVA was used. All data were processed using GraphPad Prism 5.01 software. A p value of less than 0.05 was considered statistically significant.
Results
IRF1 Is a Potentially Suppressive TF Regulating KPNA2 Expression in Lung Adenocarcinoma Cells
To the best of our knowledge, E2F1/Dp1 and E2F7 are the best-known TFs involved in KPNA2 gene expression.11,24,25 To identify the TF responsible for KPNA2 transcription, we applied the available online software programs PROMO26 and TFBIND27 to analyze putative TF binding sites in the KPNA2 promoter region (NC_000017.11., range from 68,035,602 to 68,046,859). Accordingly, 64 and 138 TF candidates were predicted via PROMO and TFBIND, respectively, to bind to the KPNA2 promoter sequences. Among these candidates, 20 TFs overlapped between these two programs: AP-1, ATF, C/EBPalpha, CREB, c-Jun, Elk-1, GATA-1/2/3, GR, HNF-1A, IRF-1, IRF-2, NF-1, NF-Y, p53, Pax-5, SRY, XBP-1 and YY1. Specifically, eleven canonical IRF1-binding elements on the KPNA2 promoter region were predicted; the region −854~-4 was predicted as the IRF1 binding region by both PROMO and TFBIND (Figure 1A). We then performed a ChIP assay with an IRF1 antibody and primers that spanned three putative IRF1 binding sites located in −854~-4 of the KPNA2 promoter region. Figure 1B and C demonstrate the positive amplification of two pairs of IRF1 primers in the A549 and CL1-5 cell lines, confirming the associations between IRF1 binding sites in the KPNA2 promoter. The positive binding of E2F1 to the KPNA2 promoter was included as a positive control. Next, RT-qPCR analysis of cells with IRF1 knockdown revealed that the transcriptional activity of KPNA2 increased by 50% and 13% in IRF1-knockdown A549 and CL1-5 cells, respectively (Figure 1D and E). These results support a potential role of IRF1 in negatively regulating KPNA2 transcription in lung ADC cells. Our previous study showed that KPNA2 knockdown significantly reduced the viability of CL1-5 cells. Therefore, we performed a cell viability assay to examine the potentially suppressive role of IRF1 in cell proliferation and observed that IRF1 knockdown enhanced cell viability, whereas KPNA2 knockdown significantly diminished the increased viability induced by IRF1 knockdown (Figure 1F). This result suggests that IRF1 may inhibit lung cancer cell proliferation through suppression of KPNA2 expression.
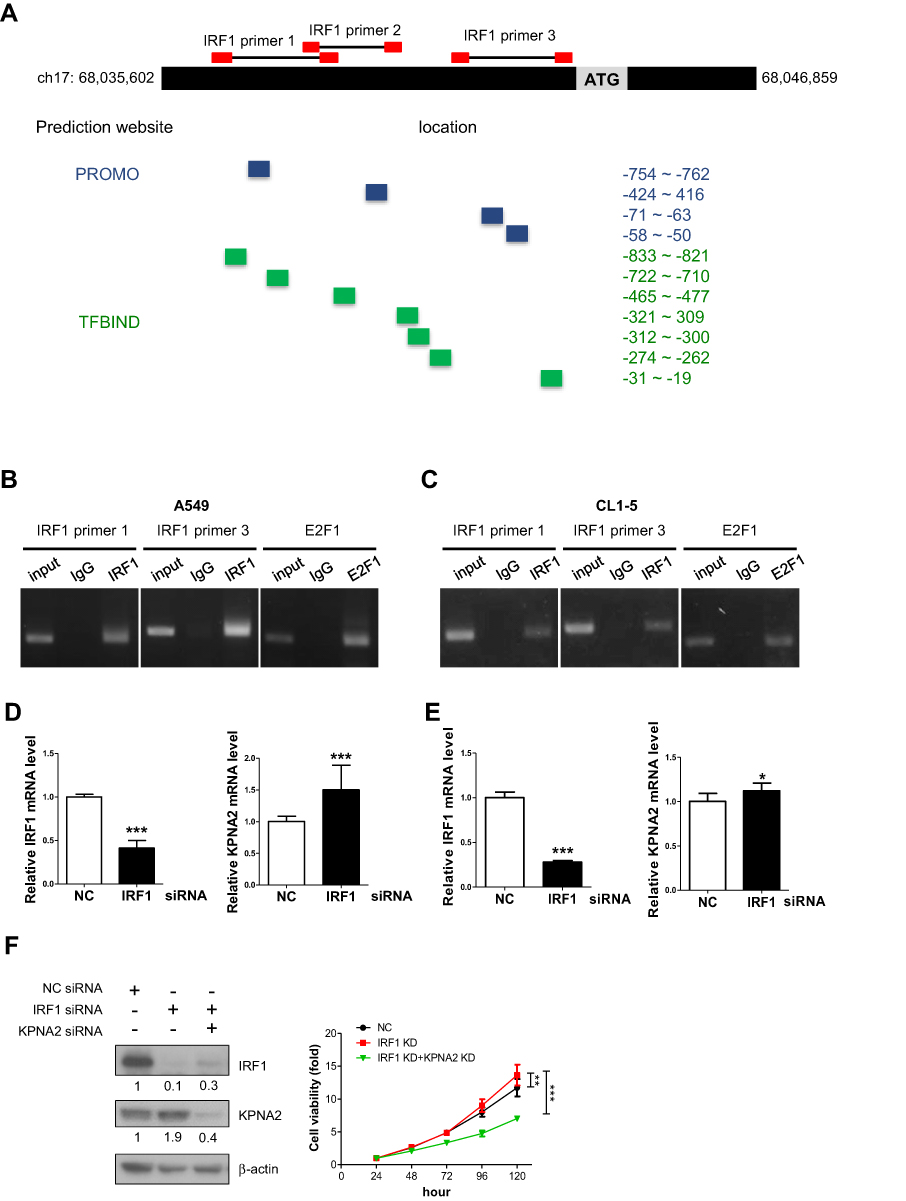 |
Figure 1 IRF1 binds to the promoter region of KPNA2 and negatively modulates KPNA2 transcription. (A) Diagram depicting the regions of predicted IRF1 binding sites (highlighted with blue and green boxes) within the KPNA2 promoter region (NC_000017.11, ranging from 68,035,602 to 68,046,859) based on two software packages, PROMO and TFBIND. The red boxes indicate the location of primer pairs used in the ChIP assay. (B–C) The ChIP assay detected positive binding of IRF1 to the KPNA2 promoter sequence. Fragmented chromatin prepared from A549 (B) and CL1-5 cells (C) was immunoprecipitated with IgG, IRF1 or E2F1 antibodies, as indicated. An anti-E2F1 antibody was used as a positive control. DNA isolated from the immunoprecipitated material was amplified by PCR, and the resulting fragments were analyzed by agarose gel electrophoresis. (D–E) IRF1 knockdown increased the mRNA level of KPNA2 in lung ADC cells. A549 or CL1-5 cells were transfected with negative control (NC) or IRF1 siRNA for 24 h, and total RNA was purified and subjected to qRT-PCR using IRF1 and KPNA2 primers. mRNA levels were calculated as a ratio relative to the control treatment. The data are presented as the mean ± SD from three independent experiments. *p<0.05, ***p<0.0001. (F) KPNA2 knockdown suppressed the enhanced cell viability induced by IRF1 knockdown. CL1-5 cells were transfected with NC, IRF1 siRNA or KPNA2 siRNA as indicated. The cell viability was determined via the MTT colorimetric growth assay at the indicated time intervals. **p<0.01 and ***p<0.001. |
IRF1 Is Downregulated in Cancerous Tissues, and Its Low Expression Correlates with Poor Survival in Lung Cancer Patients
Both the diagnostic and prognostic values of IRF1 in lung cancer are unclear. We therefore determined the clinical significance of IRF1 in lung cancer using Oncomine (www.oncomine.org), a cancer microarray database with web-based data mining characteristics, and Kaplan-Meier Plotter (http://kmplot.com), a database that integrates mRNA expression and clinical data from five types of human cancers. NSCLC, which includes ADC, SCC, LCC, and some rare subtypes, is the most common type of lung cancer, comprising approximately 85% of all cases.28,29 ADC is the most common subtype of lung cancer worldwide and is the predominant histological type of NSCLC. SCC accounts for approximately 20–30% of all lung cancers, representing the second most common type of NSCLC. LCC accounts for approximately 3–10% of NSCLC.29,30 Based on the Oncomine 4.5 database, we found that the IRF1 mRNA expression level was significantly lower (1.5-fold decreased, p<0.05) in NSCLC tissues than in normal lung tissues in six ADC, four SCC, and one LCC microarray datasets. These lung datasets were initially published by Bhattacharjee,31 Su,32 Stearman,33 Beer,34 Selamat,35 Hou,36 Wachi37 and Garber38 (Figure 2A). Furthermore, the Kaplan-Meier Plotter showed that lower IRF1 mRNA expression was correlated with poor overall survival (OS) in 673 ADC patients and first progression survival (FPS) in 443 ADC patients (Figure 2B). This low expression of IRF1 in cancer tissues was also significantly correlated with worse FPS in 141 SCC patients but was not associated with OS in 271 SCC patients (Figure 2C). We also confirmed the IRF1 gene expression and outcome association based on TCGA RNA-seq data, which contains 979 NSCLC cancer tissues (483 ADC and 486 SCC) and 109 normal lung samples. We found that IRF1 gene expression levels in these NSCLC cancer tissues were significantly lower than those detected in normal tissues. The Kaplan-Meier Plotter analysis also demonstrated that low IRF1 expression was associated with a worse prognosis, including shorter OS (log-rank p=0.025) and disease-free survival (log-rank p=0.092), in 482 ADC and SCC patients. These results collectively support the suppressive role of IRF1 in lung cancer progression.
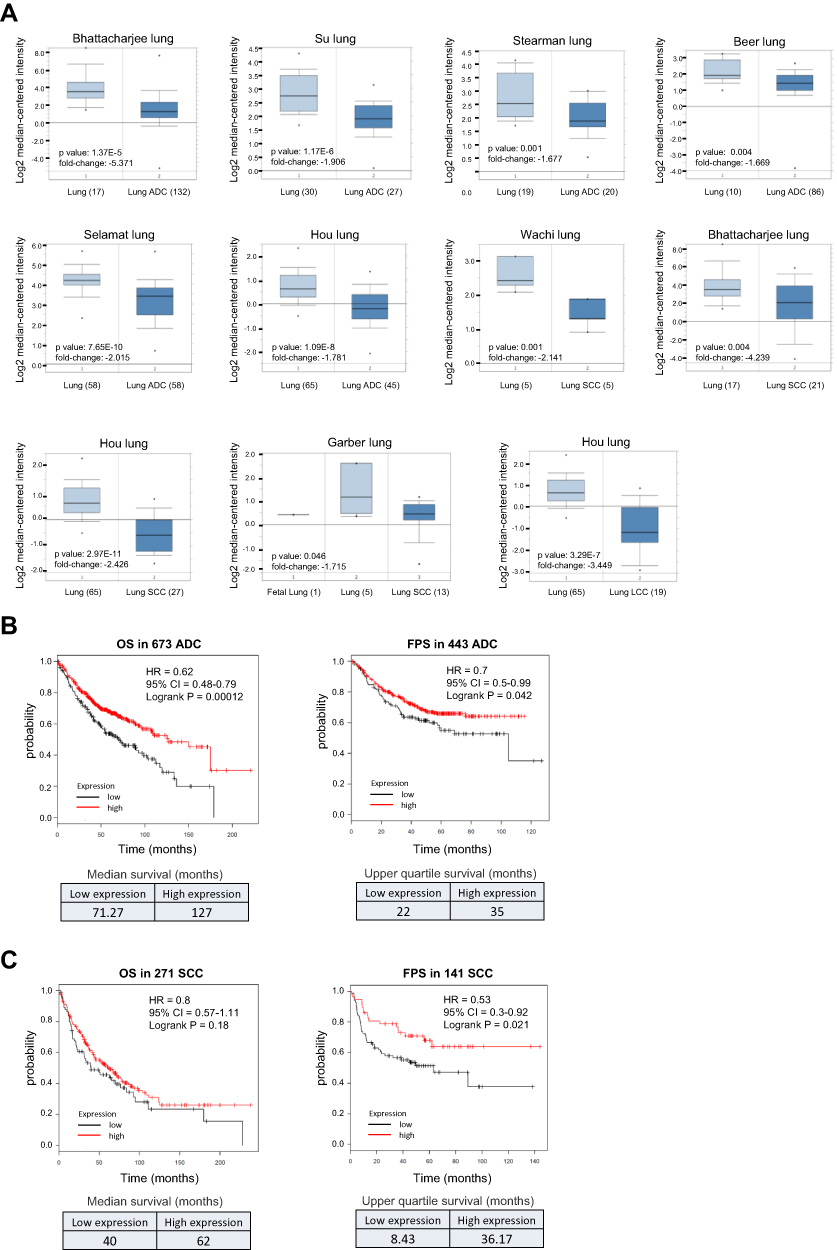 |
Figure 2 Low expression of IRF1 correlates with poor prognosis in NSCLC patients. (A) IRF1 gene expression levels in NSCLC tissues (ADC, SCC and LCC) were significantly lower than those detected in normal tissues. Box plots were derived from the mRNA expression datasets from Oncomine that compared IRF1 mRNA expression in NSCLC tissues and normal lung tissues. The numbers shown in the parentheses indicate the case number used in the microarray analysis. (B–C) Low expression of IRF1 was positively associated with a worse outcome in NSCLC patients. Survival curves from Kaplan-Meier plot profiles for cancer patients stratified by high and low mRNA expression of IRF1 in lung ADC patients (B) and SCC patients (C). Patients were split by autoselecting the best cutoff, and the best JetSet probe set was used for the IRF1 probe. The median and upper quartile survival values are shown in the lower panel to indicate a positive correlation between IRF1 expression and survival in NSCLC patients. |
Both the mTOR and ATM Pathways are Involved in Regulating KPNA2 Transcription by Modulating E2F1 and IRF1 Expression in Lung Cancer Cells
Our quantitative proteomics analysis revealed that KPNA2 is involved in the DNA damage stimulation response, DNA metabolic processes, DNA repair, cell cycle and cell migration.6 The serine/threonine kinase ATM is an important cell cycle checkpoint kinase and a sensor to DNA damage and oxidative stress.39–41 Our previous study also indicated that the mTOR pathway contributes to E2F1/Dp1-mediated KPNA2 transcription in lung cancer cells.11 In this study, we treated cells with the mTOR inhibitor rapamycin (Rap) and an ATM inhibitor (ATMi) to test whether the mTOR and ATM pathways play a role in regulating E2F1/Dp1- and/or IRF1-mediated KPNA2 transcription. Figure 3A shows that these two inhibitors significantly reduced KPNA2 mRNA levels in two lung ADC cell lines. However, NCI-H520 SCC and NCI-H460 LLC cells showed only minor changes in the modulation of KPNA2 gene expression in response to both mTOR and ATM inhibitors. Importantly, the protein level of IRF1 was dramatically increased, but E2F1 expression was decreased in two lung ADC cell lines after Rap and ATMi treatment (p<0.05, unpaired t-test) (Figure 3B and C). Similar results were observed in NCI-H520 cells treated with mTOR inhibitor. Unexpectedly, Rap increased E2F1 expression in NCI-H460 cells (Figure 3B and C). IRF1 knockdown restored the reduced level of KPNA2 in ATMi-treated A549 cells, further supporting the suppressive role of IRF1 in regulating KPNA2 expression in ADC cells (Figure 3D). These results collectively suggest that the mTOR and ATM pathways modulate E2F1 and IRF1 expression in NSCLC cells. However, IRF1 and E2F1 may play a specific and potent role in the regulation of KPNA2 gene transcription in lung ADC cells. We therefore propose that mTOR and ATM activation contribute to KPNA2 overexpression by suppressing IRF1 expression and enhancing E2F1 expression in lung ADC cells (Figure 4E).
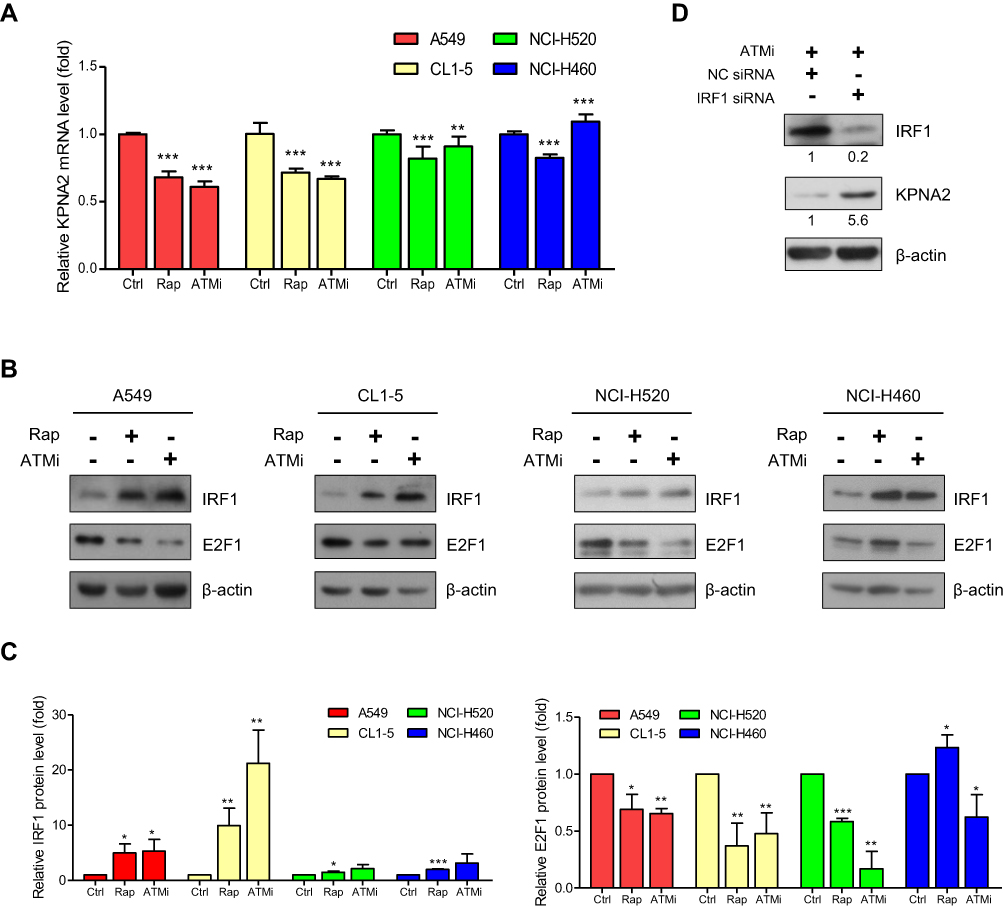 |
Figure 3 Both the mTOR and ATM pathways are involved in regulating KPNA2 transcription by modulating E2F1 and IRF1 expression in lung ADC cancer cells. (A) mTOR and ATM inhibitors significantly reduced KPNA2 mRNA levels in lung ADC and SCC cells. NSCLC cells were treated with rapamycin (Rap) or an ATM inhibitor (ATMi) for 24 h. Total RNA was purified from the cells and processed for qRT-PCR with KPNA2 primers. The KPNA2 mRNA level was calculated as a ratio relative to the level in the control treatment. (B–C) mTOR and ATM inhibitors significantly increased IRF1 protein levels but reduced E2F1 protein levels in lung ADC cells. (B) NSCLC cells were treated with Rap or ATMi. After 24 h, the cell lysates were prepared for Western blotting using anti-E2F1 and anti-IRF1 antibodies, as indicated. (C) Quantification of the IRF1 and E2F1 expression levels derived from panel B. (D) IRF1 knockdown restored the reduced levels of KPNA2 in ATM inhibitor-treated cells. A549 cells were transfected with NC or IRF1 siRNA. After 24 h of transfection, cells were treated with ATMi (10 μM) for an additional 24 h, followed by Western blotting with anti-IRF1 and anti-KPNA2 antibodies. β-actin was used as an internal control. The data are presented as the mean ± SD from three independent experiments. *p<0.05, **p<0.01 and ***p<0.0001. |
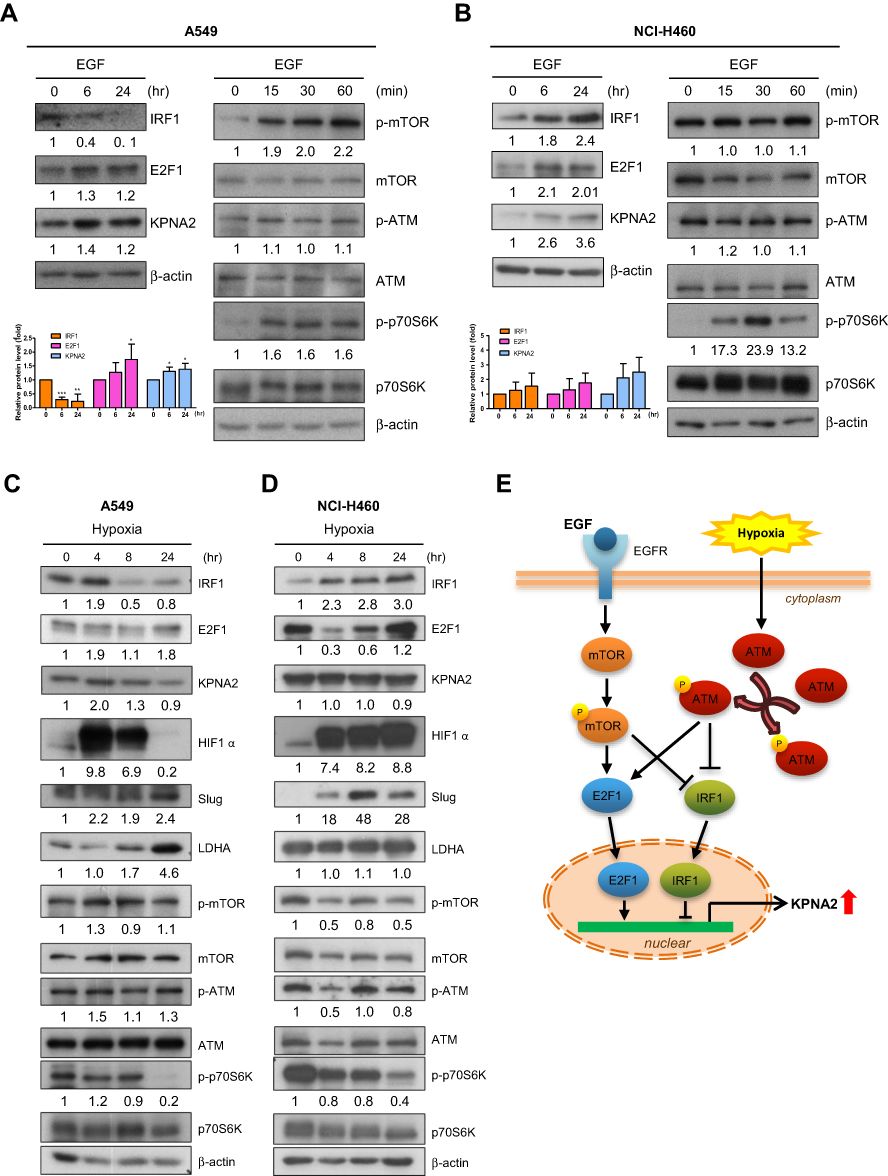 |
Figure 4 EGF stimulation and hypoxia contribute to the transcriptional regulation of KPNA2 by suppressing IRF1 expression and promoting E2F1 expression. (A–B) EGF induced KPNA2 and E2F1 expression but suppressed IRF1 protein expression in lung ADC cells. A549 (A) and NCI-H460 (B) cells were cultured in the presence or absence of EGF (25 µg/mL) for the indicated times. Cell lysates were prepared and subjected to Western blotting using antibodies for the indicated proteins. β-actin was used as an internal control, and phospho-p70S6K was used as a positive control for mTOR activation. The data are presented as the mean ± SD from three independent experiments. *p<0.05, **p<0.01 and ***p<0.0001. (C–D) Hypoxia induced KPNA2 and E2F1 protein expression but suppressed IRF1 protein expression in lung ADC cells. A549 (C) and NCI-H460 (D) cells were cultured in normoxic or hypoxic conditions for the indicated times. Cell lysates were prepared and subjected to Western blotting as described above. The reduction in phospho-p70S6K levels and the increases in HIF-1α, Slug and LDHA levels represent molecular changes in response to hypoxia. (E) Proposed working model of the mTOR and ATM signaling pathways in regulating KPNA2 transcription in lung ADC cells. Both EGF-activated mTOR and hypoxia-induced ATM signaling promote KPNA2 transcription by simultaneously enhancing E2F1 expression and suppressing IRF1 expression in lung ADC cells. |
EGF Stimulation and Hypoxia Contribute to the Transcriptional Regulation of KPNA2 by Suppressing IRF1 Expression and Promoting E2F1 Expression
EGF and hypoxia are common in microenvironments that contribute to lung cancer progression. We tested whether these two factors promote KPNA2 expression by modulating IRF1 and E2F1 expression. We observed that EGF treatment enhanced KPNA2 and E2F1 but suppressed IRF1 protein expression in A549 ADC cells (Figure 4A). In contrast, EGF treatment significantly increased IRF1 levels in NCI-H460 LCC cells (Figure 4B). Western blotting revealed that the mTOR but not the ATM pathway was upregulated upon EGF treatment. We also found that hypoxia induced KPNA2 and E2F1 expression but reduced IRF1 expression in A549 ADC cells but not in NCI-H460 LCC cells (Figure 4C and D). The reduction in phospho-p70S6K levels and elevation of HIF-1α, Slug and LDHA levels represent the molecular changes induced by hypoxic stress.42–45 Accordingly, we propose that both EGF-activated mTOR and hypoxia-induced ATM signaling promote KPNA2 transcription by enhancing E2F1 expression and suppressing IRF1 expression in lung ADC cells (Figure 4E).
Discussion
IRF1 is one of the best-known IRF genes involved in cancer biology. Both oncogenic and antioncogenic roles of IRF1 have been documented.16,18 Specifically, IRF1 activates several target genes associated with cell cycle regulation, growth suppression, apoptosis induction and immune responses, and these genes are responsible for the antitumor activity of IRF1. KPNA2 is a common oncogenic protein in numerous cancers, and KPNA2 overexpression is involved in the malignant transformation of cancer cells.3,4,46 Herein, we identified IRF1 as a novel and negative regulator of KPNA2 transcription, which supports the theory that IRF1 plays an antioncogenic role by modulating the susceptibility of cells to transformation via oncogenes such as KPNA2.
As a tumor suppressor, IRF-1 can be inactivated by a loss of heterozygosity at the DNA level, exon skipping at the mRNA level, and SUMOylation or oncoprotein interaction at the protein level.18 We show that activation of both mTOR and ATM was involved in negatively regulating IRF1 expression. The knowledge regarding the regulation between ATM and IRF1 is limited. In human fibroblasts and malignant melanoma cells, IRF1 is upregulated and stabilized through an ATM-dependent pathway in response to irradiation or genotoxic stress.18,47 In addition, mTOR signaling has been reported to be suppressed by ATM-induced LBK1/AMPK/TSC2 under oxidative stress independent of DNA damage.48 These studies and ours suggest that the regulation of IRF1 is dependent on the cell type. The molecular mechanisms of mTOR- and/or ATM-mediated suppression of IRF1 in cancer cells require further exploration in the future.
Hypoxic stress triggers alterations of the microenvironment surrounding the tumor tissue via different adaptive mechanisms that promote cancer progression, including cell proliferation, glucose metabolism, inflammation, angiogenesis, and epithelial-mesenchymal transition.49,50 Specifically, HIF is the master TF in response to hypoxia, and hundreds of genes have been reported as HIF targets in different cell types. HIF-1 overexpression enhances the expression of Slug, LDHA, GLUT, TWIST, c-Myc, and VEGF in tumorigenesis, which has been well documented.42–45 Hypoxia also induces ATM activation in the absence of DNA damage, but severe hypoxia activates ATM signaling by inducing DNA damage.51 A recent study showed that ATM can phosphorylate S696 on the inhibitory domain of HIF-1α, which enhances HIF-1α activity under hypoxic conditions.52 Furthermore, this mediation is mTORC1-dependent, in which phosphorylation and activation of HIF-1α by ATM require mTORC1 suppression.53 In addition, Guerra et al reported that Myc is required for the activation of the ATM-dependent checkpoints in response to DNA damage.43 The interplay between oncogenic c-Myc and HIF-1α enhances the metabolic needs of the tumor, which in turn contribute to cancer cell adaptation and survival under hypoxic stress.54,55 In the current study, we found that hypoxia might induce KPNA2 expression by regulating the mTOR and ATM pathways, which is consistent with our previous finding that KPNA2 is involved in cell cycle progression, DNA metabolism, DNA repair, transport of cellular components and cell migration.6
The genetic background of cancer cells is heterogeneous, and defective tumor suppressors or mutated oncogenes promote lung cancer formation and progression. Constitutive activation of the epidermal growth factor receptor (EGFR) caused by EGFR gene mutations was observed in over 60% of cases of lung ADC in Eastern Asia.56,57 Several lines of evidence support the hypothesis that ATM kinase is a tumor suppressor gene.40,58,59 Recently, Petersen et al reported that loss of tumor-specific ATM protein expression is an independent prognostic factor in early resected lung cancer.59 ATM polymorphisms associated with increased lung cancer risk and ATM gene mutations with a concomitant increase in tumor tendency were also described.58 However, we observed that hypoxia induced ATM activation, which in turn suppressed IRF1, suggesting that the microenvironment plays a critical role in modulating KPNA2 transcription. Although the detailed regulation between EGFR and ATM signaling requires further investigation, the present study reveals that growth stimulation and hypoxia can modulate oncogenic KPNA2 transcription by simultaneously enhancing the expression of its positive regulator E2F1 and suppressing the expression of its negative regulator IRF1 in lung ADC cells.
Abbreviations
ADC, adenocarcinoma; ATM, ataxia telangiectasia mutated; KPNA2, karyopherin alpha 2; IRF1, interferon regulatory factor-1; TF, transcription factor; DMEM, Dulbecco’s Modified Eagle Medium; mTOR, mechanistic target of rapamycin; NC, negative control; ChIP, chromatin immunoprecipitation; SD, standard deviation; Rap, rapamycin; ATMi, ATM inhibitor; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; SCC, squamous cell carcinoma; LCC, large-cell carcinoma; OS, overall survival; FPS, first progression survival; NSCLC, non-small-cell lung cancer; HIF-1α, hypoxia inducible factor 1α; LDHA, lactate dehydrogenase A.
Acknowledgments
We thank Hsiang-Pu Feng for performing the Western blotting and quantitative analysis of image. Hsiang-Pu Feng also contributes to the critical review and final approval of this study. This work was financially supported by grants from Chang Gung Memorial Hospital, Taoyuan, Taiwan (CMRPG3A0661, CMRPD1H0641, CMRPD1H0081-2, CLRPD190019 and BMRP894) and the Ministry of Science and Technology, Taiwan (105-2320-B-182-035-MY3).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Goldfarb DS, Corbett AH, Mason DA, Harreman MT, Adam SA. Importin alpha: a multipurpose nuclear-transport receptor. Trends Cell Biol. 2004;14(9):505–514. doi:10.1016/j.tcb.2004.07.016
2. Lange A, Mills RE, Lange CJ, Stewart M, Devine SE, Corbett AH. Classical nuclear localization signals: definition, function, and interaction with importin alpha. J Biol Chem. 2007;282(8):5101–5105. doi:10.1074/jbc.R600026200
3. Christiansen A, Dyrskjot L. The functional role of the novel biomarker karyopherin α 2 (KPNA2) in cancer. Cancer Lett. 2013;331(1):18–23. doi:10.1016/j.canlet.2012.12.013
4. Zhou LN, Tan Y, Li P, et al. Prognostic value of increased KPNA2 expression in some solid tumors: a systematic review and meta-analysis. Oncotarget. 2017;8(1):303–314. doi:10.18632/oncotarget.13863
5. Wang CI, Wang CL, Wang CW, et al. Importin subunit alpha-2 is identified as a potential biomarker for non-small cell lung cancer by integration of the cancer cell secretome and tissue transcriptome. Int J Cancer. 2011;128(10):2364–2372. doi:10.1002/ijc.25568
6. Wang CI, Chien KY, Wang CL, et al. Quantitative proteomics reveals regulation of karyopherin subunit alpha-2 (KPNA2) and its potential novel cargo proteins in nonsmall cell lung cancer. Mol Cell Proteomics. 2012;11(11):1105–1122. doi:10.1074/mcp.M111.016592
7. Wang CI, Wang CL, Wu YC, et al. Quantitative proteomics reveals a novel role of karyopherin alpha 2 in cell migration through the regulation of vimentin-pERK protein complex levels in lung cancer. J Proteome Res. 2015;14(4):1739–1751. doi:10.1021/pr501097a
8. Chen CF, Li S, Fau - Chen Y, et al. The nuclear localization sequences of the BRCA1 protein interact with the importin-alpha subunit of the nuclear transport signal receptor. J Biol Chem. 1996;271(51). doi:10.1074/jbc.271.51.32863
9. Huang L, Wang HY, Li JD, et al. KPNA2 promotes cell proliferation and tumorigenicity in epithelial ovarian carcinoma through upregulation of c-Myc and downregulation of FOXO3a. Cell Death Dis. 2013;4:e745. doi:10.1038/cddis.2013.256
10. Kim IS, Kim DH, Han SM, et al. Truncated form of importin alpha identified in breast cancer cell inhibits nuclear import of p53. J Biol Chem. 2000;275(30):23139–23145. doi:10.1074/jbc.M909256199
11. Wang CI, Chen YY, Wang CL, Yu JS, Chang YS, Yu CJ. mTOR regulates proteasomal degradation and Dp1/E2F1- mediated transcription of KPNA2 in lung cancer cells. Oncotarget. 2016;7(18):25432–25442. doi:10.18632/oncotarget.8170
12. Fujita T, Kimura Y, Miyamoto M, Barsoumian EL, Taniguchi T. Induction of endogenous IFN-alpha and IFN-beta genes by a regulatory transcription factor, IRF-1. Nature. 1989;337(6204):270–272. doi:10.1038/337270a0
13. Frontini M, Vijayakumar M, Garvin A, Clarke N. A ChIP-chip approach reveals a novel role for transcription factor IRF1 in the DNA damage response. Nucleic Acids Res. 2009;37(4):1073–1085. doi:10.1093/nar/gkn1051
14. Sato M, Taniguchi T, Tanaka N. The interferon system and interferon regulatory factor transcription factors – studies from gene knockout mice. Cytokine Growth Factor Rev. 2001;12(2–3):133–142. doi:10.1016/S1359-6101(00)00032-0
15. Romeo G, Fiorucci G, Chiantore MV, Percario ZA, Vannucchi S, Affabris E. IRF-1 as a negative regulator of cell proliferation. J Interferon Cytokine Res. 2002;22(1):39–47. doi:10.1089/107999002753452647
16. Chen FF, Jiang G, Xu K, Zheng JN. Function and mechanism by which interferon regulatory factor-1 inhibits oncogenesis. Oncol Lett. 2013;5(2):417–423. doi:10.3892/ol.2012.1051
17. Walch-Ruckheim B, Pahne-Zeppenfeld J, Fischbach J, et al. STAT3/IRF1 pathway activation sensitizes cervical cancer cells to chemotherapeutic drugs. Cancer Res. 2016;76(13):3872–3883. doi:10.1158/0008-5472.can-14-1306
18. Alsamman K, El-Masry OS. Interferon regulatory factor 1 inactivation in human cancer. Bioscience Reports. 2018;38(3):BSR20171672. doi:10.1042/BSR20171672
19. Hong M, Zhang Z, Chen Q, et al. IRF1 inhibits the proliferation and metastasis of colorectal cancer by suppressing the RAS-RAC1 pathway. Cancer Manag Res. 2019;11:369–378. doi:10.2147/CMAR.S186236
20. Lin YH, Wu MH, Liao CJ, et al. Repression of microRNA-130b by thyroid hormone enhances cell motility. J Hepatol. 2015;62(6):1328–1340. doi:10.1016/j.jhep.2014.12.035
21. Yu M, Xue H, Wang Y, et al. miR-345 inhibits tumor metastasis and EMT by targeting IRF1-mediated mTOR/STAT3/AKT pathway in hepatocellular carcinoma. Int J Oncol. 2017;50(3):975–983. doi:10.3892/ijo.2017.3852
22. Zhou Y, Wang Q, Chu L, et al. FOXM1c promotes oesophageal cancer metastasis by transcriptionally regulating IRF1 expression. Cell Prolif. 2019;52(2):e12553. doi:10.1111/cpr.12553
23. Chu YW, Yang PC, Yang SC, et al. Selection of invasive and metastatic subpopulations from a human lung adenocarcinoma cell line. Am J Respir Cell Mol Biol. 1997;17(3):353–360. doi:10.1165/ajrcmb.17.3.2837
24. Xiang S, Wang Z, Ye Y, et al. E2F1 and E2F7 differentially regulate KPNA2 to promote the development of gallbladder cancer. Oncogene. 2019;38(8):1269–1281. doi:10.1038/s41388-018-0494-7
25. van der Watt PJ, Ngarande E, Leaner VD. Overexpression of kpnβ1 and kpnα2 importin proteins in cancer derives from deregulated E2F activity. PLoS One. 2011;6(11):e27723. doi:10.1371/journal.pone.0027723
26. Messeguer X, Escudero R, Farre D, Nunez O, Martinez J, Alba MM. PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics. 2002;18(2):333–334. doi:10.1093/bioinformatics/18.2.333
27. Tsunoda T, Takagi T. Estimating transcription factor bindability on DNA. Bioinformatics. 1999;15(7–8):622–630. doi:10.1093/bioinformatics/15.7.622
28. Chansky K, Sculier JP, Crowley JJ, et al. The international association for the study of lung cancer staging project: prognostic factors and pathologic TNM stage in surgically managed non-small cell lung cancer. J Thorac Oncol. 2009;4(7):792–801. doi:10.1097/JTO.0b013e3181a7716e
29. Barta JA, Powell CA, Wisnivesky JP. Global epidemiology of lung cancer. Ann Glob Health. 2019;85(1). doi:10.5334/aogh.2419
30. Meza R, Meernik C, Jeon J, Cote ML. Lung cancer incidence trends by gender, race and histology in the united states, 1973–2010. PLoS One. 2015;10(3):e0121323. doi:10.1371/journal.pone.0121323
31. Bhattacharjee A, Richards WG, Staunton J, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A. 2001;98(24):13790–13795. doi:10.1073/pnas.191502998
32. Su LJ, Chang CW, Wu YC, et al. Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC Genomics. 2007;8(1):140. doi:10.1186/1471-2164-8-140
33. Stearman RS, Dwyer-Nield L, Zerbe L, et al. Analysis of orthologous gene expression between human pulmonary adenocarcinoma and a carcinogen-induced murine model. American Journal of Pathology. 2005;167(6):1763–1775. doi:10.1016/S0002-9440(10)61257-6
34. Beer DG, Kardia SL, Huang CC, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8(8):816–824. doi:10.1038/nm733
35. Selamat SA, Chung BS, Girard L, et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res. 2012;22(7):1197–1211. doi:10.1101/gr.132662.111
36. Hou J, Aerts J, den Hamer B, et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS One. 2010;5(4):e10312. doi:10.1371/journal.pone.0010312
37. Wachi S, Yoneda K, Wu R. Interactome-transcriptome analysis reveals the high centrality of genes differentially expressed in lung cancer tissues. Bioinformatics. 2005;21(23):4205–4208. doi:10.1093/bioinformatics/bti688
38. Garber ME, Troyanskaya OG, Schluens K, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A. 2001;98(24):13784–13789. doi:10.1073/pnas.241500798
39. Bensimon A, Aebersold R, Shiloh Y. Beyond ATM: the protein kinase landscape of the DNA damage response. FEBS Lett. 2011;585(11):1625–1639. doi:10.1016/j.febslet.2011.05.013
40. Ditch S, Paull TT. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem Sci. 2012;37(1):15–22. doi:10.1016/j.tibs.2011.10.002
41. Stagni V, Cirotti C, Barilà D. Ataxia-telangiectasia mutated kinase in the control of oxidative stress, mitochondria, and autophagy in cancer: a maestro with a large orchestra. Frontiers in Oncology. 2018;8:73. doi:10.3389/fonc.2018.00073
42. Bahrami A, Atkin SL, Majeed M, Sahebkar A. Effects of curcumin on hypoxia-inducible factor as a new therapeutic target. Pharmacol Res. 2018;137:159–169. doi:10.1016/j.phrs.2018.10.009
43. Guerra L, Albihn A, Tronnersjo S, et al. Myc is required for activation of the ATM-dependent checkpoints in response to DNA damage. PLoS One. 2010;5(1):e8924. doi:10.1371/journal.pone.0008924
44. Li L, Kang L, Zhao W, et al. miR-30a-5p suppresses breast tumor growth and metastasis through inhibition of LDHA-mediated Warburg effect. Cancer Lett. 2017;400:89–98. doi:10.1016/j.canlet.2017.04.034
45. Park J, Kim DH, Shah SR, et al. Switch-like enhancement of epithelial-mesenchymal transition by YAP through feedback regulation of WT1 and Rho-family GTPases. Nat Commun. 2019;10(1):2797. doi:10.1038/s41467-019-10729-5
46. Noetzel E, Rose M, Bornemann J, Gajewski M, Knuchel R, Dahl E. Nuclear transport receptor karyopherin-α2 promotes malignant breast cancer phenotypes in vitro. Oncogene. 2012;31(16):2101–2114. doi:10.1038/onc.2011.403
47. Pamment J, Ramsay E, Kelleher M, Dornan D, Ball KL. Regulation of the IRF-1 tumour modifier during the response to genotoxic stress involves an ATM-dependent signalling pathway. Oncogene. 2002;21:7776. doi:10.1038/sj.onc.1205981
48. Alexander A, Cai SL, Kim J, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A. 2010;107(9):4153–4158. doi:10.1073/pnas.0913860107
49. Samanta D, Semenza GL. Metabolic adaptation of cancer and immune cells mediated by hypoxia-inducible factors. Biochim Biophys Acta Rev Cancer. 2018;1870(1):15–22. doi:10.1016/j.bbcan.2018.07.002
50. Li L, Liang Y, Kang L, et al. Transcriptional regulation of the Warburg effect in cancer by SIX1. Cancer Cell. 2018;33(3):368e367–385e367. doi:10.1016/j.ccell.2018.01.010
51. Olcina MM, Grand RJ, Hammond EM. ATM activation in hypoxia - causes and consequences. Mol Cell Oncol. 2014;1(1):e29903. doi:10.4161/mco.29903
52. Kietzmann T, Mennerich D, Dimova EY. Hypoxia-inducible factors (HIFs) and phosphorylation: impact on stability, localization, and transactivity. Front Cell Dev Biol. 2016;4:11. doi:10.3389/fcell.2016.00011
53. Cam H, Easton JB, High A, Houghton PJ. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1α. Mol Cell. 2010;40(4):509–520. doi:10.1016/j.molcel.2010.10.030
54. Huang LE. Carrot and stick: HIF-α engages c-Myc in hypoxic adaptation. Cell Death Differ. 2008;15(4):672–677. doi:10.1038/sj.cdd.4402302
55. Podar K, Anderson KC. A therapeutic role for targeting c-Myc/HIF-1-dependent signaling pathways. Cell Cycle. 2010;9(9):1722–1728. doi:10.4161/cc.9.9.11358
56. Wu JY, Yu CJ, Chang YC, Yang CH, Shih JY, Yang PC. Effectiveness of tyrosine kinase inhibitors on “uncommon” epidermal growth factor receptor mutations of unknown clinical significance in non-small cell lung cancer. Clin Cancer Res. 2011;17(11):3812–3821. doi:10.1158/1078-0432.CCR-10-3408
57. Seo JS, Ju YS, Lee WC, et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012;22(11):2109–2119. doi:10.1101/gr.145144.112
58. Xu Y, Gao P, Lv X, Zhang L, Zhang J. The role of the ataxia telangiectasia mutated gene in lung cancer: recent advances in research. Ther Adv Respir Dis. 2017;11(9):375–380. doi:10.1177/1753465817725716
59. Petersen LF, Klimowicz AC, Otsuka S, et al. Loss of tumour-specific ATM protein expression is an independent prognostic factor in early resected NSCLC. Oncotarget. 2017;8(24):38326–38336. doi:10.18632/oncotarget.16215
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.