Back to Journals » Journal of Experimental Pharmacology » Volume 12
Investigational Treatment Agents for Recurrent Clostridioides difficile Infection (rCDI)
Authors Kullar R, Tran MCN, Goldstein EJC
Received 26 June 2020
Accepted for publication 13 August 2020
Published 9 October 2020 Volume 2020:12 Pages 371—384
DOI https://doi.org/10.2147/JEP.S242959
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bal Lokeshwar
Ravina Kullar,1 Mai-Chi N Tran,2,3 Ellie JC Goldstein4,5
1Expert Stewardship, Inc., Newport Beach, CA, USA; 2Pharmacy Department, Keck Medical Center of USC, Los Angeles, CA, USA; 3Clinica Juan Pablo Medical Group, Los Angeles, CA, USA; 4R.M. Alden Research Laboratory, Santa Monica, CA, USA; 5David Geffen School of Medicine, Los Angeles, CA, USA
Correspondence: Ellie JC Goldstein Tel +1-310-315-1511
Email [email protected]
Abstract: Clostridioides difficile infection (CDI) is a major cause of nosocomial diarrhea that is deemed a global health threat. C. difficile strain BI/NAP1/027 has contributed to the increase in the mortality, severity of CDI outbreaks and recurrence rates (rCDI). Updated CDI treatment guidelines suggest vancomycin and fidaxomicin as initial first-line therapies that have initial clinical cure rates of over 80%. Unacceptably high recurrence rates of 15– 30% in patients for the first episode and 40% for the second recurrent episode are reported. Alternative treatments for rCDI include fecal microbiota transplant and a human monoclonal antibody, bezlotoxumab, that can be used in patients with high risk of rCDI. Various emerging potential therapies with narrow spectrum of activity and little systemic absorption that are in development include 1) Ibezapolstat (formerly ACX-362E), MGB-BP-3, and DS-2969b-targeting bacterial DNA replication, 2) CRS3213 (REP3123)-inhibiting toxin production and spore formation, 3) ramizol and ramoplanin-affecting bacterial cell wall, 4) LFF-571-blocking protein synthesis, 5) Alanyl-L-Glutamine (alanylglutamine)-inhibiting damage caused by C. difficile by protecting intestinal mucosa, and 6) DNV3837 (MCB3681)-prodrug consisting of an oxazolidinone–quinolone combination that converts to the active form DNV3681 that has activity in vitro against C. difficile. This review article provides an overview of these developing drugs that can have potential role in the treatment of rCDI and in lowering recurrence rates.
Keywords: Clostridioides difficile, CDI, C. difficile, C. difficile infection, investigational drugs
Plain Language Summary
Clostridioides difficile infection (CDI) is the leading cause of healthcare-associated infections. The Centers for Disease Control and Prevention (CDC) and the World Health Organization (WHO) deem CDI as an urgent public health threat due to the increasing incidence and high recurrence rates. With high recurrence rates and limited treatment options, there is a need for development of novel therapies in CDI that have a narrow spectrum of activity, little systemic absorption and minimal disruption to the gut microbiota. The developing therapeutic agents have unique mechanisms of action such as preserving host colonization, targeting toxin activity/sporulation, inhibiting DNA replication, eliminating C. difficile, inhibition of bacterial growth, and causing the bacterial cell wall lysis. This article provides an overview of developing drugs that may have a potential role in the treatment of CDI.
Introduction
Clostridioides difficile, the leading cause of nosocomial infections, is considered an urgent global and national public health threat, by the Centers for Disease Control and Prevention (CDC) and the World Health Organization (WHO).1 This infection is a diarrhea caused by C. difficile toxins that usually respond well to antibiotic treatments; however, the main challenge is recurrences, partly due to persistent dysbiosis caused by antibiotics. There are greater than 200,000 C. difficile infections (CDI) reported annually in hospitalized patients in the US in 2017 and ~12,800 deaths.1 The emergence of fluoroquinolone-resistant ribotype 027 (BI/NAP1/027) isolates has been correlated with and in increase in complicated CDI cases, such as toxic megacolon and increased mortality.2 The use of antibiotics is the primary risk factor for the development of CDI, as well as for the prolongation of or perpetuation of symptoms.3 Current first-line treatment for primary CDI includes the use of vancomycin or fidaxomicin4 with initial clinical cure rates >80%.5,6 However, following antimicrobial treatment, up to 30% of patients experience a first recurrent CDI (rCDI)5–8 and ~40% will have a second rCDI.7 Several host factors have been associated with an increased risk of rCDI or CDI-related adverse outcomes, including age ≥65 years, immunosuppression, severe CDI, prior CDI episode(s), and infection with the BI/NAP1/027 strain.9 These recurrences are associated with an increased readmission in 28.3% of patients (75.2% vs 46.9% CDI-infected compared to non-infected, respectively).10 rCDI is one of the most challenging infections to treat; however, treatment choices in this patient population are limited.
The high incidence and severity of disease, increased recurrence rates, and the lack of optimal treatment options for CDI, especially rCDI, have created a critical need for new therapeutic agents. We previously published a review article on investigational agents in CDI,11 but several new compounds have been added since the prior review and for those few compounds that appear in both, we have provided updated data in this review. We, therefore, provide an updated review of investigational agents under development for CDI (Table 1).
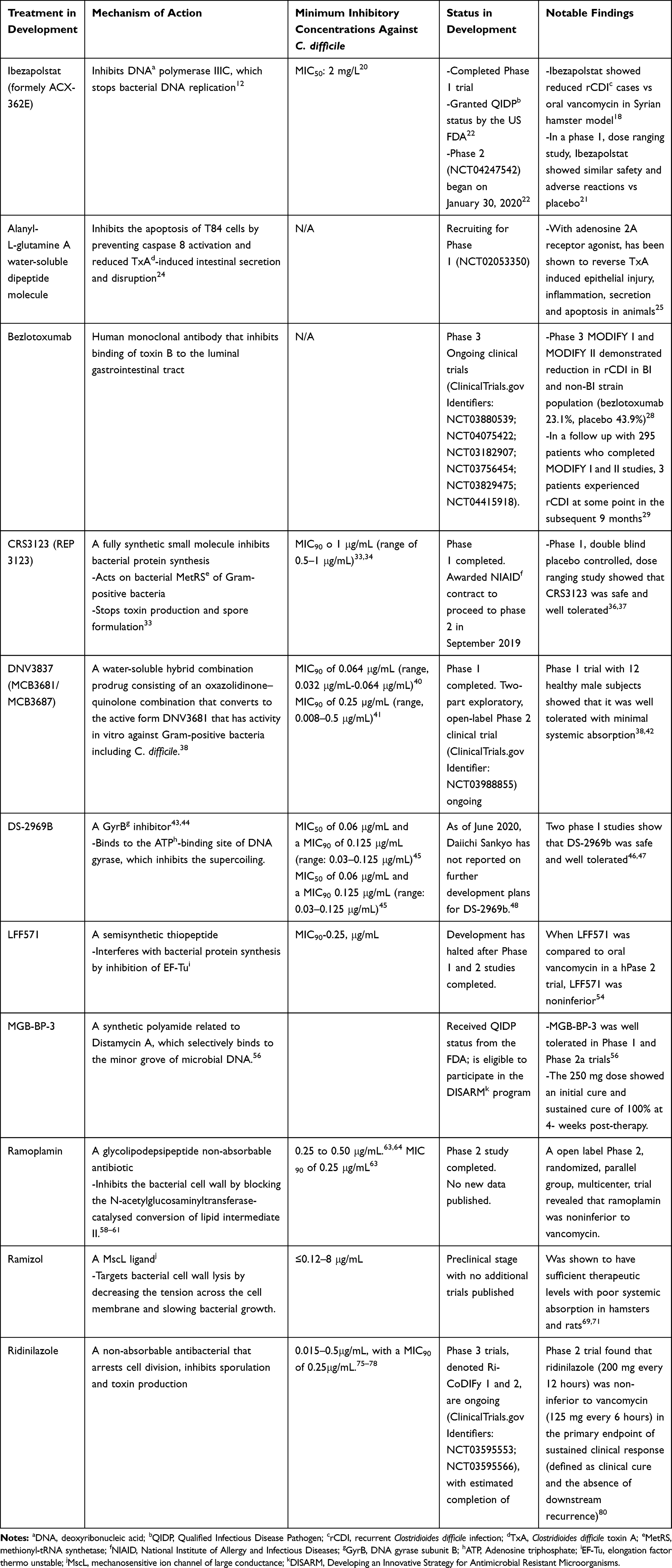 |
Table 1 Therapeutics in Development for Clostridioides difficile Infection |
Methods
We searched MEDLINE, PubMed, Embase, Web of Science, Cochrane Central Register of Controlled Trials, and ClinicalTrials.gov for C. difficile and for agents in early stages of clinical development until June 1, 2020. Keywords used included search terms “Clostridioides difficile”, “Clostridium difficile”, “Clostridioides difficile infection” or “Clostridium difficile infection” with “investigational drugs”, “treatment”, “therapy” or “drugs”. We also reviewed pertinent references from recently published manuscripts.
Investigational Agents
Ibezapolstat (ACX-362E)
Synthetic small molecules that inhibit bacterial DNA polymerase IIIC have led to the development of a variety of novel agents that diminish the propensity of cross drug resistance with the existing drug classes.12 DNA polymerase IIIC (pol IIIC) is essential for replicative DNA synthesis in aerobic, low guanine-cytosine (G-C) ratio Gram-positive bacteria, but has no effect on human cells. Pol IIIC-specific genes of several Gram-positive bacteria have been cloned and expressed13–15 and these enzymes share a unique capacity to be inhibited by 6-anilinouracils, 2-phenylguanines (PG), and related compounds which are analogs of 2ʹ-deoxyguanosine 5ʹ-triphosphate (dGTP).16,17 ACX-362E is a new agent with an N7-substituted guanine inhibitor of DNA polymerase IIIC (N7-morpholino-ethyl-N2-DCGB). ACX-362E (GLS362E), a closely related dichloro-benzyl guanine inhibitor of pol IIIC that is under development for CDI therapy.18
ACX-362E inhibits purified C. difficile pol IIIC with a Ki of 0.325 µM.16 In addition, a whole cell study involving the measurement of chromosomal DNA replication demonstrated gene dosage results that suggest inhibition of DNA replication by ACX-362E has an active-site domain that incorporates a unique pocket to achieve anti-C. difficile activity.19 ACX-362E has shown in vitro activity against strains of C. difficile, with an MIC50 = 2 µg/mL,20 and in vivo activity in the hamster model.18 In a Syrian hamster model study, ACX-362E (10 mg/kg twice daily for 10 days) was compared to vancomycin (10 mg/kg twice daily for 10 days) and showed a reduced number of rCDI cases.18
ACX-362E is poorly absorbed from the gastrointestinal (GI) tract and has limited solubility.12 Garey et al21 reported a randomized, double-blind placebo-controlled, single (150, 300, 600, and 900 mg) and multiple-ascending dose (300 and 450 mg twice daily for 10 days) Phase 1 study in healthy subjects and found safety signals that were similar to the placebo group and with similar transitory adverse reactions. ACX-362E had low systemic concentrations (<1 ng/mL with 300 mg dose) and delayed and lower plasma concentrations in the fed group compared to the fasting group. Achievable fecal levels of 4000 and 6000 ug/gm of stool for the 300 mg and 450 mg dose respectively, were achieved.21 Additionally, when compared to vancomycin (125 mg four times a day) there was less disruption of the fecal microbiota, including Bacteroides and Firmicutes, with distinct differences in abundance and beta diversity. Alpha diversity changes showed increased Actinobacteria with ACX-362-E compared to increased Proteobacteria in the vancomycin group.
ACX-362E was granted Qualified Infectious Disease Product (QIDP) status by the US FDA and has received an Investigational New Drug (IND).22 Phase 1 trials began in December 2018 and Phase 2 trials began January 30, 2020 (NCT04247542).22
Alanyl-Glutamine
Alanyl-L-Glutamine (alanylglutamine) (C8H15N3O4) is a water-soluble dipeptide molecule, composed of 2 amino acids of L-glutamine and L-alanine with a molecular weight of 217.22 g/mol that has been used as a dietary supplement, before prolonged physical exercise to enhance electrolyte absorption, and improve endurance.23 Glutamine and alanyl-glutamine were shown to inhibit the apoptosis of T84 cells by preventing caspase 8 activation and reduced C. difficile toxin A(TxA)-induced intestinal secretion and disruption.24 In combination therapy with an adenosine 2A receptor agonist it was effective in reversing TxA-induced epithelial injury, inflammation, secretion and apoptosis in animals and, therefore, has therapeutic potential for the management of CDI.25 Working locally in the GI tract it could protect the integrity of the intestinal mucosa as well as maintain intestinal barrier functions, thus reducing bacterial translocation, the risk of infection, infection-induced inflammatory damage and infection-associated symptoms, such as diarrhea, dehydration, malabsorption and electrolyte imbalances.23
It may also increase the absorption of other chemicals. Posted on March 12, 2020, at ClinicalTrials.gov (ClinicalTrials.gov Identifier: NCT02053350), the study is using oral Alanyl-glutamine as a supplement given along with standard therapy to treat CDI to potentially decrease C. difficile diarrhea, mortality and disease recurrence. This double-blind, placebo-controlled trial to determine optimal dose (4, 24 and 44 g for 10 days) and safety has not yet begun recruiting patients. It seeks to study hospitalized patients aged 65 or older with the first episode of CDI.
Bezlotoxumab
Bezlotoxumab is a fully human monoclonal antibody that is already marketed for use in patients with a high risk of rCDI.26 In general, it reduced rCDI rates over 12 weeks post treatment and showed greater efficacy in patients with multiple (>1-3) risk factors. It binds toxin B with an equilibrium dissociation constant (Kd) of <1×10-9M and inhibits the binding of toxin B, but not toxin A, thereby preventing attachment to luminal GI tract cells. Bezlotoxumab had a mean volume of distribution of 7.33 L (16%), and elimination half-life (t½) of approximately 19 days (28%).26
We include a brief review of active study protocols and updates on interesting and evolving data gathered from Phase 3 MODIFY I and MODIFY II trials.27 In both trials, bezlotoxumab was given to patients with CDI receiving antibiotic treatment (metronidazole, vancomycin, or fidaxomicin). The control group received a matching placebo infusion along with antibiotic treatment. Johnson et al28 reported on the efficacy of bezlotoxumab in 2559 patients with CDI, including 328 with the REA type BI strain that is associated with poor outcomes (predominantly ribotype 027, but also includes ribotypes 176 and 198). Bezlotoxumab (given alone or with actoxumab, a monoclonal antibody that binds toxin A) was associated with reduced rCDI in BI and non-BI strain subpopulations (bezlotoxumab 23.1%, placebo 43.9%). Goldstein et al29 reported on 295 patients who completed the 12-week study (MODIFY I and II) study and were monitored for new episodes of CDI for an additional 9 months by monthly telephone calls. Additionally, C. difficile colonization was assessed at months 6, 9, and 12. In total, 3 of 168 patients who had achieved sustained clinical at month 3 experienced rCDI at some point in the subsequent 9 months (0 bezlotoxumab, 2 actoxumab + bezlotoxumab, 1 placebo). C. difficile colonization rates ranged from 16% to 32% and the ribotype isolated from surveillance stool samples collected at months 6, 9, and 12 were the same as a previous isolate in 68 of 122 (55.7%) paired positive samples. This suggested that the efficacy of bezlotoxumab appears to be due to prevention rather than just the delay in onset of recurrence.
Various studies have recently been conducted in special populations.30,31 A post hoc analysis of CDI-related outcomes was conducted in subgroups of MODIFY I/II participants of 382 patients and compared those with vs. without cancer. Bezlotoxumab treatment had no effect on initial clinical cure rate compared with placebo (76.8% vs 71.9%), but resulted in a statistically significant reduction in rCDI vs placebo (17.8% vs 30.4%; absolute difference, −12.6%; 95% CI, –22.5% to –2.7%).32 In addition, there are several other clinical trials underway (ClinicalTrials.gov Identifiers: NCT03880539; NCT04075422; NCT03182907; NCT03756454; NCT03829475; NCT04415918).
CRS3123 (REP3123)
CRS3123 (REP3123) is a novel, fully synthetic small molecule that inhibits C. difficile toxin production and spore formation by acting on bacterial methionyl-tRNA synthetase (MetRS) of Gram-positive bacteria. It has shown activity against C. difficile B1/NAP1/027 strains33 with a MIC90 of 1 μg/mL (range of 0.5–1 μg/mL).33,34 CRS3123 does not have activity against most Gram-negative bacteria or intestinal organisms such as Bacteroides, Prevotella, bifidobacteria, actinobacteria or lactobacilli.34 A hamster GI infection model showed CRS3123 caused >10-fold reduction of the sporulation of C. difficile and was superior to vancomycin in protection against C. difficile recurrence.35 A Phase I, double-blind, placebo-controlled, single dose-escalation study (100 to 1200 mg) evaluated the safety and systemic exposure of CRS3123 in 40 healthy adults showed it was safe and well tolerated.36 Its bioavailability declined with increasing dose because absorption is not proportional to the dose. Common adverse effects were decreased hemoglobin, headache, and abnormal urinalysis. A multiple-ascending dose (200, 400, or 600 mg twice daily for 10 days) phase 1 study37 in 30 healthy volunteers aged 18 to 45 years were divided into three groups of 8 plus 2 placebo controls. The study noted the drug was generally safe and well tolerated. Thirty-three percent of the placebo group reported an adverse event compared to 12.5% of the study group all of which were mild and included transient diarrhea, dysgeusia, and mild transaminase elevation. There were no EKG changes. CRS3123 was poorly absorbed with limited but some traceable plasma uptake which increased with increasing dosage (range, 352–654 ng/mL) and by trial day 10 (range, 470 ng/mL–731 ng/mL); the Tmax after the first day ranged from 2 to 3 hours and the geometric mean half-life was 3–4 hours. Fecal levels on day 10 of the trial were 2115 µg/gm feces for the 200 mg dosage, 5390 µg/gm feces for 400 mg dosage 8250 µg/gm feces for the 600 mg dosage all dramatically higher than MIC levels. A small fraction (<2%) of CRS3123 and its glucuronide metabolite were excreted into the urine. After 10 days of treatment, 248 stool samples of CRS3123 treated patients exhibited minimal disruption of the normal fecal microbiome by 16S ribosomal RNA (16S rRNA) gene sequencing and did not impact commensal anaerobes. The microbiota of the treatment group and the placebo controls were similar at the 200 mg dose, in all groups no important phyla were lost and the microbiota returned to normal 7 days after treatment.37 These results support further development and CRS3123 was awarded an National Institute of Allergy and Infectious Diseases (NIAID) contract to proceed to phase 2 in September 2019.
DNV3837 (MCB3681/MCB3687)
DNV3837 (MCB3681) (C31-H32-F2-N4-O8) is a novel water-soluble hybrid combination prodrug consisting of an oxazolidinone–quinolone combination that converts to the active form DNV3681 that has activity in vitro against Gram-positive bacteria including C. difficile.38 It is designed for intravenous administration but actively crosses the GI barrier and accumulates in the intestinal lumen.38,39 Consequently, it might prove useful for patients with reduced GI motility or those unable to take oral antimicrobials.
Using a supplemented Brucella blood agar dilution method, Rashid et al40 reported on the comparative in vitro activity of MCB3681 against 114 toxin B positive C. difficile strains collected between 2008 and 2011. MCB361 demonstrated a MIC 90 of 0.064 μg/mL (range, 0.032 μg/mL-0.064 μg/mL) including 107 ciprofloxacin-resistant, 12 moxifloxacin-resistant and 3 linezolid resistant isolates. No ribotype (RT) 027 strains were studied.40 Comparatively, fidaxomicin had a MIC 90 of 0.125 μg/mL (range, 0.008 μg/mL-0.125 μg/mL) and cadazolid had a MIC 90 of 0.125 μg/mL (range, 0.064 μg/mL-0.064 μg/mL).
Freeman et al41 studied the in vitro activity of MCB3681 and 8 comparator agents against 199 prevalent or emerging European C. difficile RTs isolated between 2011 and 2013 using a Wilkens-Chalgren agar dilution method. MCB3681 was active against all isolates, including RTs 027, 001, 017,018, and 356; there was an MIC90 of 0.25 µg/mL (range, 0.008–0.5 μg/mL) and a geometric mean MIC of 0.12 μg/mL. Fidaxomicin was more active than MCB3681 (P = 0.0001) with a geometric mean MIC of 0.125 μg/mL (range, 0.004–0.25 μg/mL).
In a Phase I trial of 12 healthy male subjects,38,42 MCB3837 was given intravenously (6 mg/kg body weight) and high concentrations were found in the feces with, 16.6 to 275.1 mg/kg feces on day 2 and 98.9 to 226.3 mg/kg feces on day 5. Rashid et al found that MCB3837 did not affect Gram-negative aerobes (E. coli and other Enterobacteriaceae) or anaerobes (Bacteroides spp.), which contributes to maintaining a healthy gut microbiota. The number of bifidobacteria, clostridia, enterococci and lactobacilli were decreased.40,42
DNV3837 has posted a two-part exploratory, open-label phase 2 clinical trial (ClinicalTrials.gov Identifier: NCT03988855) to evaluate its efficacy, safety and pharmacokinetics. In both parts, patients will be infused with the study drug at a constant rate of 0.5 mg/kg BW/hour during a 12-hour infusion once daily for 10 consecutive days. Part 1 was to enroll 10 volunteers with non-severe CDI and part 2 is proposed to enroll 30 subjects with severe CDI in a 2:1 randomization. The study is also to assess fecal colonization with ESBL organisms, VRE and CRE and other microbiome changes using 16S RNA analysis. The sponsor is expecting final results by the end of 2020.
DS-2969b
DS-2969b, (4-chloro-5-ethyl-N(3S,4R))-1-[5(2-hydroxypropan-2-yl)-1,3,4-thiadiazol-2-yl]-3-[methyloxypiperidin-4-yl]-1H-imidazole-2-carboxamide 2/3 hydrate, is a novel GyrB inhibitor that has shown in vitro and in vivo activity against C. difficile including the NAP1/027 strains with minimal effects on the intestinal microbiota.43,44 Unlike fluoroquinolones which bind at cleavage ligation active site at the enzyme DNA interface, DS-2969b binds to the ATP-binding site of DNA gyrase, which inhibits the supercoiling.43 DS-2969b has a 50% inhibitory concentration (IC50) of 20 ng/mL against C. difficile DNA gyrase.
There have been several studies evaluating DS-2969b’s in vitro activity against C difficile. One study found that DS-2969b had a low propensity in developing in vitro resistance to five C. difficile isolates including 3 hypervirulent NAP1 strains.43 DS2969b had a MIC90 of 0.06 µg/mL against 55 isolates of C. difficile and had good activity against the NAP1/027 strain in a hamster model.43 Another study by Tyrrell et al reported a MIC50 of 0.06 μg/mL and a MIC90 of 0.125 μg/mL (range: 0.03–0.125 μg/mL) against 101 North American ribotyped C difficile isolates.45 A fecal level of 10μg/g DS-2969a, which is the free form of DS-2969b, after administration of DS-2969b in rats and monkeys, was adequate for the eradication of C. difficile from the intestine.44
There have been two studies that evaluated the safety, tolerability, pharmacokinetics and effects of DS-2969b in healthy volunteers.46,47 One study examined the effects of three sequential ascending dose (60 mg, 200 mg, and 400 mg) cohort with six subjects and placebo groups with 2 subjects every day for 14 days. One study (n=47) evaluated a 14-day regimen of five sequential ascending-dose (6 mg, 20 mg, 60 mg, 200 mg, and 400 mg) cohort with six subjects and placebo groups with 2 subjects.46 In both studies, some subjects reported mild events, primarily GI-related (ie, lower abdominal pain, diarrhea, and hematochezia).46,47 The mean plasma concentration-time profiles at day 1 and day 14 found that DS2969a (free form of DS-2969b) plasma concentrations increased with increasing doses; however, both the maximum concentration of drug in serum (Cmax) and the area under the concentration–time curve (AUC) increased less than the dose proportionally.46 Both studies found that the target fecal level 10 μg of DS-2969a per gram of feces of DS-2969b (Kumar M et al, unpublished data, 2018) that was sufficient in clearing C. difficile with doses of 60 mg or higher.46,47 DS-2969b reduced the Clostridium coccoides and Bifidobacterium groups with a minimal effect on Bacteroides fragilis, Clostridium leptum, and Prevotella spp, which demonstrates a mild effect on intestinal microbiota. As of June 2020, Daiichi Sankyo has not reported on further development plans for DS-2969b.48
LFF571
LFF571 is a semisynthetic thiopeptide that blocks protein synthesis in Gram-positive bacteria.49 LFF571 inhibits exogenous protein synthesis elongation factor EF-Tu and interferes with the ability for EF-Tu to deliver aminoacylated tRNA to the ribosome. It has been shown to have potent in vitro activity against 50 C.difficile strains (MIC90-0.25, μg/mL) that was one-dilution lower than fidaxomicin (MIC90, 0.5μg/mL).49 LFF571 demonstrated activity against most other Gram-positive rods and cocci (MIC50,90 −0.125/0.25μg/mL) except for bifidobacteria and some species of lactobacilli. LFF571 had reduced active activity against Gram-negative anaerobes with MICs for Bacteroides fragilis of 4 and 8 μg/mL. However, the other species in the B. fragilis group, including Bacteroides thetaiotaomicron, Bacteroides ovatus, and Parabacteroides (Bacteroides) distasonis, Prevotella bivia, Prevotella melaninogenica/denticola, and Veillonella spp were even less susceptible, with an overall MIC90 of >32 μg/mL. It was speculated that LFF571 might have less impact on the normal gut microbiota, which helps maintain colonization resistance.50 It has been shown that spontaneous mutants with reduced susceptibility to LFF571 were selected in vitro in a single step, but not via serial passage.51
A randomized, double-blind, placebo-controlled dose-ranging study evaluated single and multiple ascending oral doses of LFF571 (25 mg, 100 mg, or 200 mg every 6 hours for 10 days) in fifty-six healthy subjects.52 The most common adverse events were diarrhea and gastrointestinal pain in all cohorts. There were high concentrations of LFF571 in feces with minimal systemic absorption with the highest serum drug concentration of 3.2 ng/mL in a subject receiving the maximum dose of 200 mg. Bhansali et al found that the calculated pharmacokinetic parameters from drug concentrations measured in serum and fecal samples showed limited systemic exposure with the highest observed LFF571 serum concentration of 41.7 ng/mL, but fecal levels at the end of treatment between 107 and 12,900 μg/g.53 A phase 2, multicenter, randomized, evaluator-blind, active-controlled study (NCT01232595) evaluated the safety, efficacy, and pharmacokinetics of LFF571 in adults with primary episodes or first relapses of moderate C. difficile infections.54 Subjects were randomized to receive a 10-day course of oral LFF571 200 mg four times a day (n=46) or oral vancomycin 125 mg four times a day (n=26). Results showed that the clinical cure rate of LFF571 was 90.6% (29 out of 32 patients) compared to 78.3% (18 out of 23 patients) in the vancomycin-treated group with 30-day cure rates of 58.7% and 60.0%, respectively. The recurrence rates were analyzed using toxin-confirmed cases and were 19% versus 25%, respectively. LFF571-treated patients tended to have more potential risk factors for poor outcomes such as older age, more first relapses, more severe infections, less effective prior therapy and more concomitant antibiotics when compared to the vancomycin-treated patients. In comparison, the vancomycin-treated patients had a higher likelihood to harbor the NAP1/BI/027 strain of C. difficile and higher usage of proton pump inhibitors. The LFF571-treatment group had slightly more adverse events when compared to vancomycin (76.1% versus 69.2%), but less adverse events suspected to be related to the treatment (32.6% versus 38.5%). Abdominal pain and closely related GI-events had a slightly higher incidence in the LFF571-treated group (15.2%) versus vancomycin-treated group (7.7%). CDI recurrence rates were slightly higher in the LFF571 group, but the interpretation of recurrence rates is limited because a small number of patients relapsed and the study was not designed to compare recurrence rates between treatment arms. As of June 2020, Novartis has halted development of LFF571.55
MGB-BP-3
MGB-BP-3 is a unique, synthetic polyamide related to Distamycin A, which selectively binds to the minor grove of microbial DNA.56 It is highly active against Gram-positive pathogens including C. difficile and is rapidly bactericidal. It kills the vegetative C. difficile cell within the 10 hours, before it is able to sporulate.57 MGB-BP-3 displays strong bactericidal activity against the BI/NAP1/027 strains that are associated with a greater frequency.57 This is in contrast to vancomycin, which is bacteriostatic, and fidaxomicin, which both require more than 24 hours to achieve their maximum effect. This rapid activity could potentially achieve initial cure and, therefore, prevent disease recurrence by reducing the total C. difficile burden.
In the hamster model of CDI, MGB-BP-3 has been shown to protect against death and prolonged post-treatment survival.56 In a phase I study, MGB-BP-3 showed an excellent safety and tolerability profile with no serious adverse events (SAEs) with one subject in the 125 mg dose cohort experiencing transient dizziness. A Phase 2a study56 of sequential ascending dose of 125 mg, 250 mg, and 500 mg twice daily for 10 days with 10 to 12 subjects/dose level with primary or first recurrent Enzyme immunoassay (EIA) toxin positive CDI assessed initial cure at day 12 and followed them for recurrence at 4 and 8-weeks post-treatment. It showed better-than-expected efficacy at the lowest dose level (125mg given twice daily). In this group, quantitative cultures showed suppression to lower limit of detection (LLOD) log 2 at day 10 in 7/8 subjects. Results further improved at a dosage of 250 mg given twice daily. When a 250 mg dose of MGB-BP-3 was given twice daily for 10 days, it achieved an initial cure and sustained cure of 100% at four weeks post therapy. This met the proposed endpoints and the 250 mg dosage was selected for further clinical trials. Changes in the fecal microbiome over time were measured by quantitative polymerase chain reaction (qPCR) and 16S rRNA gene sequencing. In a subset of half of the subjects, quantitative counts of C. difficile burden before, during and after treatment to day 38 assessed MGB-BP-3’s effect on C. difficile in vivo. Preservation of Bacteroidetes and moderate reduction in Cluster IV and XIVa microbes was shown using qPCR.
As recurrence rates are unacceptably high with current bacteriostatic treatments, this compound potentially offers a new therapeutic advantage. Consequently, MGB-BP-3 received QIDP status from the FDA, enabling Fast Track submission. It will also be eligible to participate in the Developing an Innovative Strategy for Antimicrobial Resistant Microorganisms (DISARM) program of prescribing incentives being considered in the US that will increase patient access to new and innovative treatments.
Ramoplanin
Ramoplanin (C106H170ClN21O30; molecular weight 2254.1 g/mol) is a bactericidal glycolipodepsipeptide, nonabsorbable antibiotic derived from Actinoplanes sp. (ATCC 33,076) that inhibits the bacterial cell wall by blocking the N-acetylglucosaminyltransferase-catalysed conversion of lipid intermediate II.58–61 It achieves high fecal concentrations.61,62 After oral doses of 200 mg, 400 mg and 800 mg of ramoplanin given twice daily for 10 days, Montecalvo et al reported fecal concentrations of ramoplanin on day 3 and day 10 of 827 mg/kg and 949 mg/kg, respectively, for the 200 mg dose, 1742 mg/kg and 1417 mg/kg, respectively, for the 400 mg dose, and 1901 mg/kg and 2647 mg/kg, respectively, for the 800 mg dose and was detectable in feces for up to 4 days after the last dose.62
Ramoplanin has bacterial activity against Gram-positive aerobic and anaerobic organisms such as C. difficile with MICs ranging from 0.25 to 0.50 μg/mL.63,64 Citron et al63 reported a MIC 90 of 0.25 μg/mL against 18 C. difficile strains but noted MICs of >256 μg/mL for Bacteroides fragilis gp. spp., Fusobacterium spp. and Veillonella spp. Cross-resistance has not been documented with vancomycin and other glycopeptides. Pelaez et al64 reported that all 105 toxigenic C. difficile isolates, including 8 vancomycin-resistant strains, were susceptible to ramoplanin with a MIC90 of 0.25 μg/mL and a range of 0.03 to 0.5 μg/mL and a geometric mean MIC of 0.22 μg/mL.64 An in vitro gut model showed ramoplanin was effective at reducing cytotoxin production and an in vivo hamster model showed concordance in that it resolved CDI symptoms. Additionally, ramoplanin adheres to the exosporium for a prolonged period so that it is available to attack newly germinating cells potentially augmenting its bactericidal activity against vegetative C. difficile cells.65
An open-label Phase 2, randomized, parallel-group, multicenter, trial was conducted in 86 patients that had CDI and received ramoplanin 200 mg orally twice daily for 10 days (n = 28) or 400 mg orally twice daily (n = 29) or vancomycin 125 mg orally four times daily (n = 29).66 The drug was well tolerated with a response rate at a 1–2 weeks post-therapy visit of 83% in the ramoplanin 200 mg group, 85% in the 400 mg group compared to 86% in the vancomycin arm. The relapse rate was 26.3% for the 200 mg ramoplanin group, 21.7% in the 400 mg group and 20.8% in the vancomycin-treated group. However, the study was poorly powered to show non-inferiority compared to vancomycin. While this trial demonstrated that ramoplanin is efficacious and with limited toxicity, no new studies have been published since then, raising the question of continued future development.
Ramizol
Ramizol, 1,3,5-tris[(1E)-2′-(4″-benzoic acid)vinyl]benzene is a mechanosensitive ion channel of large conductance (MscL) ligands.67 MscL, which only exists in bacteria, decreases the osmotic environment, which protects the bacterial cell wall from lysis. As a potential target for drugs, MscL releases solutes and small protein when it opens, which decreases the tension across cell membrane. Ramizol slows bacterial growth by lowering the threshold of MscL. Ramizol had an MIC range of ≤0.12–8 μg/mL against 100 C. difficile isolates.68 The oral administrations of vancomycin 20 mg/kg, ramizol 50 mg/kg and ramizol 100 mg/kg twice a day for 5 days were evaluated in a C. difficile colitis hamster model and using C. difficile ATCC BAA-1805, a ribotype 027 NAP-1 epidemic strain.69 During a 28-day observation, the survival rates were 43% for ramizol 50 mg/kg, 57% for ramizol 100 mg/kg and 86% for orally administered vancomycin 20 mg/kg. The survival rate increased to 71% for the ramizol 100 mg/kg group when the frequency was changed from twice a day to four times a day. The hamsters treated with ramizol did not have any diarrhea, which suggest minimal effects on the gut flora. Oral ramizol is a potential for treatment of C. difficile due to achieving sufficient therapeutic levels with poor systemic absorption. In order to increase the half life and systemic absorption, ramizol had been developed to be administered as an intramuscular and subcutaneous injection for the treatment of systemic infection.70 A 14-day study was conducted in rats to determine possible toxicity from ramizol administered via oral gavage at repeat dosing that ranged from 50 mg/kg, 500 mg/kg and 1500 mg/kg.71 It was observed that high doses of ramizol at 1500 mg/kg/day were well tolerated. Phase I studies will be required to assess the safety of ramizol in healthy volunteers. As of June 2020, ramizol continues to be in the pre-clinical stage with no clinical trials listed on ClinicalTrials.gov.72
Ridinilazole
Ridinilazole [2,2-bis(4-pyridyl)3H,3ʹH 5.5-bibenzimidazole], (SMT19969), is a new non-absorbable antibacterial that arrests cell division, inhibits sporulation and toxin production, and displays a unique and relatively specific mechanism compared to current antimicrobials for the treatment of C. difficile.73,74 Ridinilazole MICs against C. difficile have ranged from 0.015 to 0.5 μg/mL, with a MIC90 of 0.25 μg/mL.75–78 No differences in MICs across a broad range of C. difficile ribotypes including hyper-virulent strains and no increases in MICs in isolates with reduced susceptibilities to other agents tested have been noted. Ridinilazole exhibits a prolonged post-antibiotic effect (4–20 hours) against C. difficile ribotypes 012, 027 and 078 at 10X MIC concentrations with no growth recovery following 1 hour of treatment 20X MIC75 and significantly reduced toxin A and B concentrations even at 0.5X MIC.74 It has been shown to be inactive in vitro compared to vancomycin and metronidazole against several intestinal Gram-positive and -negative aerobes and anaerobes, suggesting it is sparing of the normal intestinal flora.76,79
In the Phase II trial, Vickers et al80 found that ridinilazole (200 mg every 12 hours) was non-inferior to vancomycin (125 mg every 6 hours) in the primary endpoint of sustained clinical response (defined as clinical cure and the absence of downstream recurrence) [24/36 (66.7%) vs 14/33 (42.4%); P=0.0004] when studied using a non-inferiority margin of 15%. This was driven primarily by a lower recurrence rate of 14.3% vs 34.8% in the ridinilazole arm compared to the vancomycin arm. Ridinilazole was also well tolerated with an adverse event rate similar to the vancomycin arm. There were no documented study drug-related adverse events that led to ridinilazole discontinuation. Thorpe et al recently compared the effects of ridinilazole and vancomycin on fecal microbiota during and after treatment among those in the Phase 2 study. Changes in the microbiota were assessed using 16s rRNA gene profiling on patients’ stools, with the primary comparisons made at baseline and at the end-of-therapy (EOT).81 Given that ridinilazole better preserved the microbiome than vancomycin and secondary bile acid production and the metabolome may represent a critical factor in preventing recurrence of CDI, both these factors may contribute to the lower recurrence rate observed in the clinical trial.82 Additional work from the Phase 2 trial by Qian et al83 showed that in contrast to vancomycin, ridinilazole treatment preserves bile acid composition over the course of therapy, which provides the functional rationale for the observed reduction in recurrences.
Paul et al evaluated ridinilazole’s impact on Health-Related Quality of Life (HRQoL) at baseline, days 5, 10, 12, and 40 using the EQ-5D-3L index, which is a descriptive system comprised five elements–mobility, self care, usual activities, pain/discomfort and anxiety/depression.84 Ridinilazole improved early and 40-day HRQoL compared to vancomycin, with early significant improvements occurring by day 5 on ridinilazole but not vancomycin (P=0.008). By day 40, ridinilazole improved anxiety/depression significantly more than vancomycin (P=0.039). This pioneering study documents improvements in HRQoL after antimicrobial treatment for CDI.
Phase III trials, denoted Ri-CoDIFy 1 and 2, are ongoing (ClinicalTrials.gov Identifiers: NCT03595553; NCT03595566) and have estimated completion dates of September 2021. The primary endpoint being evaluated is sustained clinical response 30 days after end of therapy (EOT). Of note, for this primary endpoint, there is more than 95% power of concluding superiority using a 2-sided test at the 5% significance level, which is a novel statistical level from other clinical trials. Secondary endpoints include evaluation of the relative effects on both the microbiome and bile salt composition and health economic outcome endpoints, including readmission rates and length of hospital stay.
Discussion
C. difficile is one of the most common hospital-acquired infections, leading to inpatient costs of nearly $5 billion.85 A key clinical challenge to management of CDI is rCDI, which typically occurs within 4–6 weeks after completing therapy. The risk of recurrence increases with each episode, with a rate of > 60–65% after ≥3 CDI episodes.86 CDI occurs primarily due to disruption of colonic microbiota. Therefore, the main goal for treating rCDI is to let the normal colonic microbiota to reestablish itself.86 Adding to the difficulty in treating rCDI is the capability of C difficile to change from a vegetative form, which is susceptible to killing by anti-C difficile therapy, to a spore form that is resistant to treatment.87
According to the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA) guidelines, treatment of rCDI is based on the number of episodes of CDI.4 Fidaxomicin, vancomycin are first line and fecal microbiota transplant (FMT) is a recurrence treatment option. Fidaxomicin was compared to vancomycin in 2 randomized double-blind clinical trials, with the clinical cure rates, defined as resolution of diarrhea 2 days after completing therapy, being similar. However, significantly fewer patients treated with fidaxomicin compared to vancomycin developed rCDI within 4 weeks of stopping treatment (15.4% vs 25.3%; P=0.005 and 12.7% vs 26.9%; P=0.002).6,88
There are various promising investigational agents that show some potential, with their unique mechanism of action and narrow-spectrum of activity against C. difficile that keeps the gut microbiota composition more intact. Bezlotoxumab, a monoclonal antibody directed against toxin B, with standard antibiotic treatment may be a good agent to reduce the rate of recurrence. Ridinilazole prevents sporulation and preserves bile acid composition over the course of therapy. Likewise, CRS3123 and ramoplanin prevent C. difficile sporulation. These three agents are promising as they may not only reduce rCDI but may also decrease transmission in the hospital setting. Further, the improved 5-day and 40-day HRQoL of ridinilazole compared to vancomycin shows that patients are recognizing the benefit of ridinilazole. This unique HRQoL metric used may encourage other companies to include in their clinical trials. Albeit several agents are in clinical development, it is discouraging to see four of the eleven agents potentially not moving through. These are potentially costly decisions for the four pharmaceutical companies. A similar decision was observed with cadazolid, which met the primary endpoint in the phase 2 study, but failed to meet the endpoint in the phase 3 trials (IMPACT I and II, NCT01987895 and NCT01983683).89
Conclusions
New agents that are similar to already available agents will be limited in addressing the high rCDI rates, which is a considerable obstacle. One hopes that these newer agents with unique mechanisms of action and protective of the microbiome will enhance patient outcomes and decrease rCDI. Future studies evaluating agents in groups at increased risk of CDI and rCDI are warranted.
Disclosure
RK serves on the Advisory Board for BioK+. EJCG has served on Advisory boards for Acuryx Inc, Merck & Co, Bayer Pharmaceuticals, BioK+, Sanofi-Adventis, Summit Corp. plc, Cutis Pharmaceuticals, Kindred Healthcare Corp., Novartis, Sankyo-Daichi, Paratek Pharma, and Shionogi Inc. EJCG has also been on the Speakers’ bureau for Bayer Inc., Merck & Co, Medicines Co., Allergan Inc and has received research grants from Bayer Inc., Cutis Pharmaceuticals, Entasis Therapeutics, Merck & Co., Micromyx LLC, Parateck Pharmaceuticals, Spero Therapeutics, Tetraphase Inc. The authors report no other conflicts of interest in this work.
References
1. Centers for Disease Control and Prevention. Biggest threats and data; 2020. Atlanta, GA. Available from: https://www.cdc.gov/drugresistance/biggest-threats.html.
2. Pepin J, Valiquette L, Cossette B. Mortality attributable to nosocomial Clostridium difficile-associated disease during an epidemic caused by a hypervirulent strain in Quebec. CMAJ. 2005;173(9):1037–1042. doi:10.1503/cmaj.050978
3. Mullish BH, Williams HR. Clostridium difficile infection and antibiotic-associated diarrhoea. Clin Med (Lond). 2018;18(3):237–241. doi:10.7861/clinmedicine.18-3-237
4. McDonald LC, Gerding DN, Johnson S, et al. Clinical practice guidelines for Clostridium difficile infection in adults and children: 2017 update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin Infect Dis. 2018;66(7):987–994. doi:10.1093/cid/ciy149
5. Johnson S, Louie TJ, Gerding DN, et al. Vancomycin, metronidazole, or tolevamer for Clostridium difficile infection: results from two multinational, randomized, controlled trials. Clin Infect Dis. 2014;59(3):345–354. doi:10.1093/cid/ciu313
6. Louie TJ, Miller MA, Mullane KM, et al. Fidaxomicin versus vancomycin for Clostridium difficile infection. N Engl J Med. 2011;364(5):422–431. doi:10.1056/NEJMoa0910812
7. Sheitoyan-Pesant C, Abou Chakra CN, Pepin J, Marcil-Heguy A, Nault V, Valiquette L. Healthcare burden of multiple recurrences of Clostridium difficile infection. Clin Infect Dis. 2016;62(5):574–580. doi:10.1093/cid/civ958
8. Cornely OA, Miller MA, Louie TJ, Crook DW, Gorbach SL. Treatment of first recurrence of Clostridium difficile infection: fidaxomicin versus vancomycin. Clin Infect Dis. 2012;55(Suppl 2):S154–161. doi:10.1093/cid/cis462
9. Gerding DN, Kelly CP, Rahav G, et al. Bezlotoxumab for prevention of recurrent Clostridium difficile infection in patients at increased risk for recurrence. Clin Infect Dis. 2018;67(5):649–656. doi:10.1093/cid/ciy171
10. Olsen MA, Yan Y, Reske KA, Zilberberg M, Dubberke ER. Impact of Clostridium difficile recurrence on hospital readmissions. Am J Infect Control. 2015;43(4):318–322. doi:10.1016/j.ajic.2014.12.020
11. Tran MN, Kullar R, Goldstein EJC. Investigational drug therapies currently in early-stage clinical development for the treatment of clostridioides (clostridium) difficile infection. Expert Opin Investig Drugs. 2019;28(4):323–335. doi:10.1080/13543784.2019.1581763
12. Xu WC, Silverman MH, Yu XY, Wright G, Brown N. Discovery and development of DNA polymerase IIIC inhibitors to treat Gram-positive infections. Bioorg Med Chem. 2019;27(15):3209–3217. doi:10.1016/j.bmc.2019.06.017
13. Hammond RA, Barnes MH, Mack SL, Mitchener JA, Brown NC. Bacillus subtilis DNA polymerase III: complete sequence, overexpression, and characterization of the polC gene. Gene. 1991;98(1):29–36. doi:10.1016/0378-1119(91)90100-P
14. Pacitti DF, Barnes MH, Li DH, Brown NC. Characterization and overexpression of the gene encoding Staphylococcus aureus DNA polymerase III. Gene. 1995;165(1):51–56. doi:10.1016/0378-1119(95)00377-I
15. Foster KA, Barnes MH, Stephenson RO, et al. DNA polymerase III of Enterococcus faecalis: expression and characterization of recombinant enzymes encoded by the polC and dnaE genes. Protein Expr Purif. 2003;27(1):90–97. doi:10.1016/S1046-5928(02)00577-6
16. Torti A, Lossani A, Savi L, et al. Clostridium difficile DNA polymerase IIIC: basis for activity of antibacterial compounds. Curr Enzym Inhib. 2011;7(3):147–153. doi:10.2174/157340811798807597
17. Xu WC, Wright GE, Brown NC, et al. 7-Alkyl-N(2)-substituted-3-deazaguanines. Synthesis, DNA polymerase III inhibition and antibacterial activity. Bioorg Med Chem Lett. 2011;21(14):4197–4202. doi:10.1016/j.bmcl.2011.05.093
18. Dvoskin S, Xu WC, Brown NC, Yanachkov IB, Yanachkova M, Wright GE. A novel agent effective against Clostridium difficile infection. Antimicrob Agents Chemother. 2012;56(3):1624–1626. doi:10.1128/AAC.06097-11
19. van Eijk E, Boekhoud IM, Kuijper EJ, Bos-Sanders IMJG, Wright G, Smits WK. Genome location dictates the transcriptional response to sub-inhibitory concentrations of PolC-inhibitors in Clostridium difficile. bioRxiv. 2018.
20. van Eijk E, Boekhoud IM, Kuijper EJ, Bos-Sanders I, Wright G, Smits WK. Genome location dictates the transcriptional response to PolC inhibition in Clostridium difficile. Antimicrob Agents Chemother. 2019;63:2. doi:10.1128/AAC.00779-19
21. Garey K. A Randomized, Double Blind, Placebo Controlled, Single and Multiple Ascending Dose Phase 1 Study to Determine the Safety, Pharmacokinetics, Food, and Fecal Microbiome Effects of ACX 362E Administered Orally to Healthy Subjects. Washington DC: IDWeek; 2019.
22. Acurx Pharmaceuticals. ACX-375C. Available from: https://www.acurxpharma.com/pipeline/acx-375c.
23. Wikipedia. Glutamine. Available from: https://en.wikipedia.org/w/index.php?title=glutamine&oldid=956552063.
24. Carneiro BA, Fujii J, Brito GA, et al. Caspase and bid involvement in Clostridium difficile toxin A-induced apoptosis and modulation of toxin A effects by glutamine and alanyl-glutamine in vivo and in vitro. Infect Immun. 2006;74(1):81–87. doi:10.1128/IAI.74.1.81-87.2006
25. Warren CA, Calabrese GM, Li Y, et al. Effects of adenosine A(2)A receptor activation and alanyl-glutamine in Clostridium difficile toxin-induced ileitis in rabbits and cecitis in mice. BMC Infect Dis. 2012;12:13. doi:10.1186/1471-2334-12-13
26. Alonso CD, Mahoney MV. Bezlotoxumab for the prevention of Clostridium difficile infection: a review of current evidence and safety profile. Infect Drug Resist. 2019;12:1–9. doi:10.2147/IDR.S159957
27. Wilcox MH, Gerding DN, Poxton IR, et al. Bezlotoxumab for prevention of recurrent Clostridium difficile infection. N Engl J Med. 2017;376(4):305–317. doi:10.1056/NEJMoa1602615
28. Johnson S, Sambol S, Best E, et al. Efficacy of Bezlotoxumab in Patients Infected with Strains of Clostridium Difficile Associated with Poor Outcomes. IDWeek; 2016.
29. Goldstein EJC, Citron DM, Gerding DN, et al. Bezlotoxumab for the prevention of recurrent Clostridioides difficile infection: 12-month observational data from the randomized phase III trial, MODIFY II. Clin Infect Dis. 2019. doi:10.1093/cid/ciz1151
30. Golan Y, Dupont HL, Aldomiro F, et al. Bezlotoxumab (BEZ) for prevention of Clostridium difficile infection (CDI) recurrence (rCDI): outcomes in patients with substantial renal impairment (SRI). Open Forum Infect Dis. 2017;4(Suppl 1):S387. doi:10.1093/ofid/ofx163.962
31. Kelly CP, Wilcox M, Glerup H, et al. Characteristics and outcomes in patients with C. difficile infection (CDI) and inflammatory bowel disease: bezlotoxumab versus placebo. Gastroenterology. 2017;152(5):S340. doi:10.1016/S0016-5085(17)31400-2
32. Cornely OA, Mullane KM, Birch T, et al. Exploratory evaluation of bezlotoxumab on outcomes associated with Clostridioides difficile infection in MODIFY I/II participants with cancer. Open Forum Infect Dis. 2020;7(2):ofaa038. doi:10.1093/ofid/ofaa038
33. Critchley IA, Green LS, Young CL, et al. Spectrum of activity and mode of action of REP3123, a new antibiotic to treat Clostridium difficile infections. J Antimicrob Chemother. 2009;63(5):954–963. doi:10.1093/jac/dkp041
34. Citron DM, Warren YA, Tyrrell KL, Merriam V, Goldstein EJ. Comparative in vitro activity of REP3123 against Clostridium difficile and other anaerobic intestinal bacteria. J Antimicrob Chemother. 2009;63(5):972–976. doi:10.1093/jac/dkp037
35. Ochsner UA, Bell SJ, O’Leary AL, et al. Inhibitory effect of REP3123 on toxin and spore formation in Clostridium difficile, and in vivo efficacy in a hamster gastrointestinal infection model. J Antimicrob Chemother. 2009;63(5):964–971. doi:10.1093/jac/dkp042
36. Nayak SU, Griffiss JM, Blumer J, et al. Safety, tolerability, systemic exposure, and metabolism of CRS3123, a methionyl-tRNA synthetase inhibitor developed for treatment of Clostridium difficile, in a phase 1 study. Antimicrob Agents Chemother. 2017;61:8. doi:10.1128/AAC.02760-16
37. Lomeli BK, Galbraith H, Schettler J, et al. Multiple-ascending-dose phase 1 clinical study of the safety, tolerability, and pharmacokinetics of CRS3123, a narrow-spectrum agent with minimal disruption of normal gut microbiota. Antimicrob Agents Chemother. 2019;64:1. doi:10.1128/AAC.01395-19
38. Rashid MU, Dalhoff A, Backstrom T, et al. Ecological impact of MCB3837 on the normal human microbiota. Int J Antimicrob Agents. 2014;44(2):125–130. doi:10.1016/j.ijantimicag.2014.03.016
39. Deinove. Available from: http://www.deinove.com.
40. Rashid MU, Dalhoff A, Weintraub A, Nord CE. In vitro activity of MCB3681 against Clostridium difficile strains. Anaerobe. 2014;28:216–219. doi:10.1016/j.anaerobe.2014.07.001
41. Freeman J, Pilling S, Vernon J, Wilcox MH. In vitro activities of MCB3681 and eight comparators against Clostridium difficile isolates with known ribotypes and diverse geographical spread. Antimicrob Agents Chemother. 2017;61:3. doi:10.1128/AAC.02077-16
42. Dalhoff A, Rashid MU, Kapsner T, Panagiotidis G, Weintraub A, Nord CE. Analysis of effects of MCB3681, the antibacterially active substance of prodrug MCB3837, on human resident microflora as proof of principle. Clin Microbiol Infect. 2015;21(8):767e761–764. doi:10.1016/j.cmi.2015.05.025
43. Mathur T, Barman TK, Kumar M, et al. In vitro and in vivo activities of DS-2969b, a novel GyrB inhibitor, against Clostridium difficile. Antimicrob Agents Chemother. 2018;62:4. doi:10.1128/AAC.02157-17
44. Kumar M, Mathur T, Joshi V, Upadhyay DJ, Inoue SI, Masuda N. Effect of DS-2969b, a novel GyrB inhibitor, on rat and monkey intestinal microbiota. Anaerobe. 2018;51:120–123. doi:10.1016/j.anaerobe.2018.04.017
45. Tyrrell KL, Citron DM, Merriam CV, Leoncio E, Goldstein EJC. In vitro activity of DS-2969b and comparator antimicrobial agents against Clostridioides (Clostridium) difficile, methicillin-resistant Staphylococcus aureus, and other anaerobic bacteria. Anaerobe. 2018;54:39–41. doi:10.1016/j.anaerobe.2018.04.010
46. Dennie J, Vandell AG, Inoue S, et al. A phase I, single-ascending-dose study in healthy subjects to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of DS-2969b, a novel GyrB inhibitor. J Clin Pharmacol. 2018;58(12):1557–1565. doi:10.1002/jcph.1151
47. Vandell AG, Inoue S, Dennie J, et al. Phase 1 study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple oral doses of DS-2969b, a novel GyrB inhibitor, in healthy subjects. Antimicrob Agents Chemother. 2018;62:5. doi:10.1128/AAC.02537-17
48. Daiichi-Sankyo. Available from: https://www.daiichisankyo.com/rd/pipeline/development_pipeline/.
49. Leeds JA, Sachdeva M, Mullin S, Dzink-Fox J, Lamarche MJ. Mechanism of action of and mechanism of reduced susceptibility to the novel anti-Clostridium difficile compound LFF571. Antimicrob Agents Chemother. 2012;56(8):4463–4465. doi:10.1128/AAC.06354-11
50. Citron DM, Tyrrell KL, Merriam CV, Goldstein EJ. Comparative in vitro activities of LFF571 against Clostridium difficile and 630 other intestinal strains of aerobic and anaerobic bacteria. Antimicrob Agents Chemother. 2012;56(5):2493–2503. doi:10.1128/AAC.06305-11
51. Leeds JA. Antibacterials developed to target a single organism: mechanisms and frequencies of reduced susceptibility to the novel anti-Clostridium difficile compounds fidaxomicin and LFF571. Cold Spring Harb Perspect Med. 2016;6(2):a025445. doi:10.1101/cshperspect.a025445
52. Ting LS, Praestgaard J, Grunenberg N, Yang JC, Leeds JA, Pertel P. A first-in-human, randomized, double-blind, placebo-controlled, single- and multiple-ascending oral dose study to assess the safety and tolerability of LFF571 in healthy volunteers. Antimicrob Agents Chemother. 2012;56(11):5946–5951. doi:10.1128/AAC.00867-12
53. Bhansali SG, Mullane K, Ting LS, et al. Pharmacokinetics of LFF571 and vancomycin in patients with moderate Clostridium difficile infections. Antimicrob Agents Chemother. 2015;59(3):1441–1445. doi:10.1128/AAC.04252-14
54. Mullane K, Lee C, Bressler A, et al. Multicenter, randomized clinical trial to compare the safety and efficacy of LFF571 and vancomycin for Clostridium difficile infections. Antimicrob Agents Chemother. 2015;59(3):1435–1440. doi:10.1128/AAC.04251-14
55. Novartis. Available from: https://www.novartis.com/our-science/novartis-global-pipeline.
56. Louie T, Morgan A, Larios O, Ravic M. Phase IIa Dose Escalation Treatment of Clostridioides Difficile-Associated Diarrhea with MGB-BP-3, a Novel First in Class DNA Minor Grove Binding Antibiotic. ECCMID; 2020.
57. MGB biopharma. Available from: http://www.mgb-biopharma.com.
58. Cavalleri B, Pagani H, Volpe G, Selva E, Parenti F. A-16686, a new antibiotic from Actinoplanes. I. Fermentation, isolation and preliminary physico-chemical characteristics. J Antibiot (Tokyo). 1984;37(4):309–317. doi:10.7164/antibiotics.37.309
59. McCafferty DG, Cudic P, Frankel BA, Barkallah S, Kruger RG, Li W. Chemistry and biology of the ramoplanin family of peptide antibiotics. Biopolymers. 2002;66(4):261–284. doi:10.1002/bip.10296
60. Boger DL. Vancomycin, teicoplanin, and ramoplanin: synthetic and mechanistic studies. Med Res Rev. 2001;21(5):356–381. doi:10.1002/med.1014
61. Farver DK, Hedge DD, Lee SC. Ramoplanin: a lipoglycodepsipeptide antibiotic. Ann Pharmacother. 2005;39(5):863–868. doi:10.1345/aph.1E397
62. Montecalvo MA. Ramoplanin: a novel antimicrobial agent with the potential to prevent vancomycin-resistant enterococcal infection in high-risk patients. J Antimicrob Chemother. 2003;51(Suppl 3):iii31–35. doi:10.1093/jac/dkg274
63. Citron DM, Merriam CV, Tyrrell KL, Warren YA, Fernandez H, Goldstein EJ. In vitro activities of ramoplanin, teicoplanin, vancomycin, linezolid, bacitracin, and four other antimicrobials against intestinal anaerobic bacteria. Antimicrob Agents Chemother. 2003;47(7):2334–2338. doi:10.1128/AAC.47.7.2334-2338.2003
64. Pelaez T, Alcala L, Alonso R, et al. In vitro activity of ramoplanin against Clostridium difficile, including strains with reduced susceptibility to vancomycin or with resistance to metronidazole. Antimicrob Agents Chemother. 2005;49(3):1157–1159. doi:10.1128/AAC.49.3.1157-1159.2005
65. Kraus CN, Lyerly MW, Carman RJ. Ambush of Clostridium difficile spores by ramoplanin: activity in an in vitro model. Antimicrob Agents Chemother. 2015;59(5):2525–2530. doi:10.1128/AAC.04853-14
66. Pullman JPJ, Leach TS Ramoplanin versus vancomycin in the treatment of Clostridium difficile diarrhea: a phase 2 study.
67. Iscla I, Wray R, Blount P, et al. A new antibiotic with potent activity targets MscL. J Antibiot (Tokyo). 2015;68(7):453–462. doi:10.1038/ja.2015.4
68. Wolfe C, Pagano P, Pillar CM, Shinabarger DL, Boulos RA. Comparison of the in vitro antibacterial activity of Ramizol, fidaxomicin, vancomycin and metronidazole against 100 clinical isolates of Clostridium difficile by broth microdilution. Diagn Microbiol Infect Dis. 2018;92(3):250–252. doi:10.1016/j.diagmicrobio.2018.06.002
69. Rao S, Prestidge CA, Miesel L, Sweeney D, Shinabarger DL, Boulos RA. Preclinical development of Ramizol, an antibiotic belonging to a new class, for the treatment of Clostridium difficile colitis. J Antibiot (Tokyo). 2016;69(12):879–884. doi:10.1038/ja.2016.45
70. Wright L, Rao S, Thomas N, Boulos RA, Prestidge CA. Ramizol((R)) encapsulation into extended release PLGA micro- and nanoparticle systems for subcutaneous and intramuscular administration: in vitro and in vivo evaluation. Drug Dev Ind Pharm. 2018;44(9):1451–1457. doi:10.1080/03639045.2018.1459676
71. Sibley K, Chen J, Koetzner L, et al. A 14-day repeat dose oral gavage range-finding study of a first-in-class CDI investigational antibiotic, in rats. Sci Rep. 2019;9(1):158. doi:10.1038/s41598-018-36690-9
72. Boulos & Cooper Pharmaceuticals. Available from: https://bouloscooper.com/research-and-development/antibiotic-drugs/.
73. Kelly. Comparing the Effects of SMT19969, Vancomycin and Fidaxomicin on Sporulation of Several Clostridium Difficile Ribotypes. Copenhagen, Denmark: ECCMID; 2015.
74. Basseres E, Endres BT, Khaleduzzaman M, et al. Impact on toxin production and cell morphology in Clostridium difficile by ridinilazole (SMT19969), a novel treatment for C. difficile infection. J Antimicrob Chemother. 2016;71(5):1245–1251. doi:10.1093/jac/dkv498
75. Corbett D, Wise A, Birchall S, et al. In vitro susceptibility of Clostridium difficile to SMT19969 and comparators, as well as the killing kinetics and post-antibiotic effects of SMT19969 and comparators against C. difficile. J Antimicrob Chemother. 2015;70(6):1751–1756. doi:10.1093/jac/dkv006
76. Goldstein EJ, Citron DM, Tyrrell KL, Merriam CV. Comparative in vitro activities of SMT19969, a new antimicrobial agent, against Clostridium difficile and 350 gram-positive and gram-negative aerobic and anaerobic intestinal flora isolates. Antimicrob Agents Chemother. 2013;57(10):4872–4876. doi:10.1128/AAC.01136-13
77. Snydman DR, McDermott LA, Thorpe CM, et al. In Vitro Activity of Ridinilazole and Comparators Against Isolates of Clostridium Difficile Obtained from Stools of Patients as Part of US Surveillance in 2014. ASM Microbe; 2017.
78. Snydman DR, McDermott LA, Thorpe CM, et al. Antimicrobial susceptibility and ribotypes of Clostridium difficile isolates from a Phase 2 clinical trial of ridinilazole (SMT19969) and vancomycin. J Antimicrob Chemother. 2018;73(8):2078–2084. doi:10.1093/jac/dky135
79. Baines SD, Crowther GS, Freeman J, Todhunter S, Vickers R, Wilcox MH. SMT19969 as a treatment for Clostridium difficile infection: an assessment of antimicrobial activity using conventional susceptibility testing and an in vitro gut model. J Antimicrob Chemother. 2015;70(1):182–189. doi:10.1093/jac/dku324
80. Vickers RJ, Tillotson GS, Nathan R, et al. Efficacy and safety of ridinilazole compared with vancomycin for the treatment of Clostridium difficile infection: a phase 2, randomised, double-blind, active-controlled, non-inferiority study. Lancet Infect Dis. 2017;17(7):735–744. doi:10.1016/S1473-3099(17)30235-9
81. Thorpe CM, Kane AV, Chang J, Tai A, Vickers RJ, Snydman DR. Enhanced preservation of the human intestinal microbiota by ridinilazole, a novel Clostridium difficile-targeting antibacterial, compared to vancomycin. PLoS One. 2018;13(8):e0199810. doi:10.1371/journal.pone.0199810
82. Theriot CM, Koenigsknecht MJ, Carlson PE, et al. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun. 2014;5:3114. doi:10.1038/ncomms4114
83. Qian X, Yanagi K, Kane AV, et al. Effect of Broad Vs. Narrow Spectrum Clostridioides Difficile Treatment on Human Stool Bile Acid Composition Over Time. IDWeek; 2019.
84. Paul S, Vickers RJ, Garey KW, et al. Quality of Life Changes in Patients with Clostridium Difficile Infection (CDI): A Randomized, Double-Blind Trial of Ridinilazole (RDZ) Compared to Vancomycin (VAN). IDWeek; 2019.
85. Guh AY, Mu Y, Winston LG, et al. Trends in U.S. Burden of Clostridioides difficile infection and outcomes. N Engl J Med. 2020;382(14):1320–1330. doi:10.1056/NEJMoa1910215
86. Kelly CP, LaMont JT. Clostridium difficile–more difficult than ever. N Engl J Med. 2008;359(18):1932–1940. doi:10.1056/NEJMra0707500
87. Kelly CP. Can we identify patients at high risk of recurrent Clostridium difficile infection? Clin Microbiol Infect. 2012;18(Suppl 6):21–27. doi:10.1111/1469-0691.12046
88. Cornely OA, Crook DW, Esposito R, et al. Fidaxomicin versus vancomycin for infection with Clostridium difficile in Europe, Canada, and the USA: a double-blind, non-inferiority, randomised controlled trial. Lancet Infect Dis. 2012;12(4):281–289. doi:10.1016/S1473-3099(11)70374-7
89. Gerding DN, Cornely OA, Grill S, et al. Cadazolid for the treatment of Clostridium difficile infection: results of two double-blind, placebo-controlled, non-inferiority, randomised phase 3 trials. Lancet Infect Dis. 2019;19(3):265–274. doi:10.1016/S1473-3099(18)30614-5
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.