Back to Journals » Drug Design, Development and Therapy » Volume 20
Investigation of Potent Anti-Mycobacterium tuberculosis Agents Derived from Pyridine Derivatives Targeting the Enoyl Acyl Carrier Protein Reductase (InhA): Design, Synthesis, and Computational Analysis
Authors Sabt A ![]() , Korycka-Machala M, Kassem AF, The Son N, Ha NX, Brzostek A, Thabit MG, Kawka M, Dziadek B, Elshamy AI, Atwa AM, Dziadek J
, Korycka-Machala M, Kassem AF, The Son N, Ha NX, Brzostek A, Thabit MG, Kawka M, Dziadek B, Elshamy AI, Atwa AM, Dziadek J ![]()
Received 30 September 2025
Accepted for publication 23 December 2025
Published 8 January 2026 Volume 2026:20 566020
DOI https://doi.org/10.2147/DDDT.S566020
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Ahmed Sabt,1 Małgorzata Korycka-Machala,2 Asmaa F Kassem,3 Ninh The Son,4 Nguyen Xuan Ha,4 Anna Brzostek,2 Mohamed G Thabit,5 Malwina Kawka,6 Bożena Dziadek,6 Abdelsamed I Elshamy,1 Ahmed M Atwa,7,8 Jarosław Dziadek2
1Chemistry of Natural Compounds Department, Pharmaceutical and Drug Industries Research Institute, National Research Centre, Cairo, 12622, Egypt; 2Laboratory of Genetics and Physiology of Mycobacterium, Institute of Medical Biology of the Polish Academy of Sciences, Lodz, Poland; 3Department of Chemistry, College of Science and Humanities in Al-Kharj, Prince Sattam Bin Abdulaziz University, Al-Kharj, Saudi Arabia; 4Institute of Chemistry, Vietnam Academy of Science and Technology, Hanoi, 10000, Vietnam; 5Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Pharos University (PUA), Alexandria, Egypt; 6Department of Molecular Microbiology, Faculty of Biology and Environmental Protection, University of Lodz, Lodz, Poland; 7College of Pharmacy, Al-Ayen Iraqi University, AUIQ, An Nasiriyah, Iraq; 8Department of Pharmacology and Toxicology, Faculty of Pharmacy, Egyptian Russian University, Cairo, 11829, Egypt
Correspondence: Ahmed Sabt, Email [email protected] Jarosław Dziadek, Email [email protected]
Introduction: Tuberculosis is a very complicated disease because of how the TB bacteria behaves in the human body. This makes it hard to diagnose, treat, and control. Because of this, the World Health Organization’s latest reports show that there are still very few good treatment options for drug-resistant TB.
Methods: A novel series of pyridine-derived compounds were rationally designed and synthesized to evaluate their potential as antitubercular agents. These derivatives were specifically developed to target the enoyl acyl carrier protein reductase (InhA), and molecular docking studies were performed to predict binding modes with InhA.
Results: All compounds exhibited notable antitubercular activity, with minimum inhibitory concentrations (MIC) ranging from 0.5 to 2.0 μg mL− 1 against Mycobacterium tuberculosis H37Rv.
Discussion: Derivative 6 was the most potent compound (MIC: 0.5 μg mL− 1), inhibiting intracellular bacteria, disrupting biofilms, and potently targeting InhA (IC50: 0.36 μM). Its pyridine-thiazole scaffold was key for stable binding, as shown by molecular modeling.
Keywords: pyridine, thiazole derivatives, Mycobacterium tuberculosis, InhA enzyme, docking and molecular dynamics
Introduction
Tuberculosis (TB) is a chronic airborne infectious disease caused by the acid-fast bacterium Mycobacterium tuberculosis (Mtb). While the primary site of infection is the lungs, the pathogen can disseminate via the bloodstream or lymphatic system, leading to extrapulmonary manifestations of TB. Consequently, the global burden of tuberculosis remains substantial, with a high incidence of cases and mortality annually. The emergence of drug-resistant strains, such as multidrug-resistant TB (MDR-TB) and extensively drug-resistant TB (XDR-TB), poses an increasingly severe threat, particularly in low- and middle-income countries. In 2021, approximately 241,399 new cases of MDR-TB and 12,861 new cases of XDR-TB were reported worldwide.1–9 Although anti-TB drugs such as isoniazid (INH), rifampicin (RIF), ethambutol (EMB), and pyrazinamide (PZA) are effective in treating TB infections, they do not achieve optimal outcomes due to several drawbacks. These include lengthy treatment periods, the possibility of serious side effects, risks of drug interactions, and difficulties with patient compliance, all of which can contribute to the emergence of drug-resistant strains.10,11 Therefore, it is crucial to develop new anti-TB medications that are well-tolerated, effective against both drug-sensitive and drug-resistant Mycobacterium tuberculosis strains, have low toxicity, and require shorter treatment durations.
Recent studies have concentrated on the enzyme an enoyl acyl carrier protein reductase (InhA) as a promising target for novel anti-TB drug development.12,13 InhA, this enzyme from Mycobacterium tuberculosis, plays a central role in the type II fatty acid synthesis (FAS II) pathway by catalyzing the Nicotinamide adenine dinucleotide (NADH)-dependent reduction of 2-trans-enoyl-ACP (acyl carrier protein) to produce NAD+ and the reduced enoyl thioester-ACP substrate.14,15 This reaction is critical for the biosynthesis of mycolic acids, which are essential components of the mycobacterial cell envelope. Importantly, InhA represents an excellent therapeutic target because the FAS II system is absent in human cells, thereby minimizing potential off-target effects.16–18 Thus, disrupting the synthesis of mycolic acid is vital for eradicating these bacteria and ultimately overcoming TB.19,20 Furthermore, the clinical efficacy of isoniazid (INH) I, (Figure 1) one of the most potent anti-TB drugs, is largely attributed to its inhibition of InhA. However, resistance to isoniazid has emerged, primarily due to mutations in KatG, a catalase-peroxidase enzyme responsible for activating isoniazid within the bacterium,21,22 In response to this challenge, numerous efforts have been undertaken, resulting in the synthesis and evaluation of various analogs based on the structures of isoniazid and pyridine as potential antimycobacterial agents.23,24 Pyridine represents a fundamental heterocyclic framework prevalent in various natural compounds, including alkaloids, e.g trigonelline, vitamins like B3 and B6, and coenzymes exemplified by nicotinamide adenine dinucleotide. Owing to its multifaceted properties-namely, water solubility, notable chemical stability, and the capacity to form hydrogen bonds.25 Consequently, derivatives of pyridine display a range of biological activities26,27 and have been shown to possess significant antitubercular properties.28,29 For instance, lansoprazole II, (Figure 1) a drug containing the pyridine moiety, inhibits gastric acid secretion through proton-pump receptor binding and exhibits intracellular efficacy against Mycobacterium tuberculosis (Mtb).30 Similarly, pyridomycin III, (Figure 1) a naturally derived antibiotic featuring a pyridine structure, shows strong activity against various Mycobacteria, including Mtb through its inhibition of the InhA enzyme.31 In addition, Almasirad et al prepared a new series of 2-(phenylthio)benzoylarylhydrazones and tested their antimycobacterial activity against Mycobacterium tuberculosis H37Rv. Among the compounds, Compound IV (Figure 1) exhibited the highest activity with a minimum inhibitory concentration (MIC) of 2.96 μg/Ml.32 Likewise, Matsa et al described a 2-hydrazinylthiazole derivative built on a pyridine framework, which showed strong antimycobacterial activity against the Mtb H37Rv strain, with MIC of 6.40 µM for compound V33 (Figure 1).
 |
Figure 1 Illustrates previously reported antitubercular agents featuring bioactive cores such as pyridine derivatives (I–V), thiazole derivatives (VI–X), and hydrazone groups (XI and XII), alongside the novel target compounds synthesized in this study, designated as 4–6 and 7a-f. |
Another key scaffold is thiazole and its derivatives, which constitute a class of compounds with considerable potential as antitubercular agents because of their target specificity.34 Beyond their role in tuberculosis treatment, thiazole-containing compounds have demonstrated diverse pharmacological effects, such as anticancer,35 antimalarial,36 anti-inflammatory,37 antimicrobial,38 and antihyperlipidemic activities.39 Structurally, the thiazole moiety shares similarity with thiolactomycin VI, (Figure 1) a naturally occurring antibiotic that poses synthetic challenges. It exerts its antibacterial effect by inhibiting FAS-II β-ketoacyl-ACP synthase (KasA) Throughout the biosynthetic process of the Mycobacterium tuberculosis (Mtb) cell wall, ultimately resulting in bacterial cell death.40 Also, nitazoxanide (NTZ) VII, (Figure 1) an orally administered, FDA-approved drug containing a thiazole ring, is used to treat protozoal infections and has been shown to significantly inhibit intracellular Mtb growth.41,42 Furthermore, tizoxanide VIII, (Figure 1) a metabolite of NTZ has demonstrated inhibitory activity against both replicative and non-replicative Mtb strains.43 Notably, Boraste et al demonstrated that derivatives of thiadiazole-linked thiazole have been synthesized and characterized as effective anti-tubercular agents. Among these, compound IX (Figure 1) exhibited the potent activity, with MIC of 7.81 μg/mL against Mycobacterium tuberculosis H37Ra.44 Additionally, Makam and Kannan synthesized a series of 2-aminothiazole connected to a pyridine framework using the Hantzsch thiazole synthesis method. The inhibitory potential of these compounds was evaluated against Mycobacterium tuberculosis (Mtb), strain H37Rv. Among them, compound X, (Figure 1) demonstrated strong antimycobacterial activity, with MIC of 6.25 μM.45
The hydrazone functional group (R1R2C=NNH2) represents a significant and pharmacologically valuable scaffold in drug design. The amine nitrogen’s lone pair electrons participate in conjugation with the adjacent imine moiety, a key electronic feature of hydrazone derivatives that influences their reactivity and biological activity. Importantly, atom of nitrogen in hydrazones acts as a nucleophilic center, while the carbon atom exhibits both electrophilic and nucleophilic behavior.46 These unique functional characteristics make hydrazones versatile compounds with a diverse array of biological activities has been observed, encompassing anti-HIV, antimicrobial, antifungal, anticancer, analgesic, antioxidant and anti-inflammatory effects.38,47–49 Additionally, several well-known antitubercular agents, including rifampicin XI and compound XII, (Figure 1) incorporate the hydrazone moiety within their molecular structures.11,50 Because of their distinctive hydrogen bond donor and acceptor capabilities, hydrazones have gained significant interest as effective anti-tuberculosis (anti-TB) agents.51,52
Building upon the previously reported data and our ongoing research on the synthesis of novel antituberculosis agents,11,12,51–54 the present study employed a hybrid pharmacophore strategy to design and synthesize thiazolidinone and thiazole derivatives conjugated to a pyridine scaffold via a hydrazone linkage Figure 1. The objective was to develop potent antituberculosis compounds exhibiting inhibitory activity against the InhA enzyme. Selected promising candidates were further assessed for their antimycobacterial efficacy within human macrophages, as well as their impact on mycobacterial biofilm formation. Among these, the most active compound, designated as compound 6, underwent additional evaluation to assess the inhibitory efficacy of the compound against the Mycobacterium tuberculosis InhA enzyme, comprehensive biochemical assays were conducted. Additionally, in silico analyses, including molecular docking and molecular dynamics (MD) simulations, were employed to characterize the binding modes and intermolecular interactions of the bioactive analogues with the InhA active site.
Materials and Methods
Chemistry
Melting points were measured using the Electrothermal IA 9000 apparatus, and no corrections were applied to these values. High-resolution mass spectrometry (HR-TOF-ESI-MS) data for all compounds were acquired utilizing the JEOL JMS-700 instrument, based in Tokyo, Japan. Nuclear magnetic resonance (NMR) analysis, encompassing both 1H and 13C-NMR spectra, was conducted with Bruker 500 NMR spectrometers located at the Faculty of Pharmaceutical Science, Tokushima Bunri University, Japan. Chemical shifts are reported in δ (ppm), while coupling constants are expressed in Hz. Thin-layer chromatography (TLC) was employed to monitor the reactions, utilizing silica gel on aluminum sheets (60 F254, Merck) with a chloroform/methanol (9.8:0.2 v/v) eluent, which was subsequently visualized using iodine-potassium spray. Compounds 3 and 6 was synthesized according to previously established methods.55,56
Methodology for the Synthesis of the Target Compound 4,5,6 and 7a-e
A solution containing equimolar amounts (0.001 mol each) of thiosemicarbazone 3 and selected α-halocarbonyl compounds (chloroacetone, ethyl bromoacetate, ethyl-2-chloroacetoacetate, or phenacyl bromide derivatives) was prepared in absolute ethanol (15 mL). The reaction mixture was then treated with a catalytic amount of anhydrous sodium acetate (0.002 mol) and refluxed for 8–10 hours. After cooling, the precipitated product was isolated via filtration and purified by recrystallization from ethanol, affording the desired derivatives 4, 5, 6, and 7a–f in pure form.
4-Methyl-2-(2-(1-(Pyridin-2-Yl)ethylidene)hydrazinyl)thiazole (4)
Brown powder, m.p. 267–268°C, yield (74%). 1H NMR (500 MHz, CD3Cl): δ = 2.43 (CH3), 2.52 (CH3-thiazole), 6.80 (s, 1H, CH-thiazole), 8.00–8.02 (m, 1H, H-Ar), 8.38 (d, 1H, J = 8.0 Hz, H-Ar), 8.58–8.61 (m, 1H, H-Ar), 8.89 (dd, 1H, J = 0.5 and 5.5 Hz, H-Ar), 9.95 (s, 1H, NH). 13C-NMR (126 MHz, CD3Cl) δ=12.01 (CH3), 12.81 (CH3), 105.62 (C5-thiazole), 125.13, 126.79, 138.99, 143.11, 145.00, 146.51, 147.13, 167.27 (C=N-thiazole). LREIMS: 232.0; HREIMS: 232.0781 (Calcd. for C11H12N4S; 232.0783) (see Supplementary Figures 1–3).
2-(2-(1-(Pyridin-2-Yl)ethylidene)hydrazinyl)thiazol-4(5H)-One (5)
Yellowish brown powder, m.p. 177–179°C, yield (82%). 1H NMR (500 MHz, CD3Cl): δ = 2.42 (s, 3H, CH3), 4.50 (s, 2H, CH2-thiazolidinone), 7.43–7.51 (m, 1H, H-Ar), 7.87–7.91 (m, 1H, H-Ar), 8.12 (d, 1H, J = 8.0 Hz, H-Ar), 8.89 (dd, 1H, J = 0.5 and 5.5 Hz, H-Ar), 10.02 (s, 1H, NH). 13C NMR (126 MHz, CD3Cl) δ = 14.21 (CH3), 32.79 (CH2), 122.78, 125.46, 139.48, 146.64, 147.29, 153.81, 167.24 (C=N), 172.12 (C=O). LREIMS: 234.0; HREIMS: 234.0577 (Calcd. for C10H10N4OS; 234.0575) (see Supplementary Figures 4–6).
Ethyl-4-Methyl-2-(2-(1-(Pyridin-2-Yl)ethylidene)hydrazinyl)thiazole-5-Carboxylate (6)
Off-white powder, m.p. 180–181°C, yield (87%). 1H NMR (500 MHz, DMSO): δ = 1.21 (t, 3H, J = 7.5 Hz, CH3), 2.44 (s, 3H, CH3), 2.68 (s, 3H, CH3), 4.08 (q, 2H, J = 7.5 Hz, CH2), 7.35–7.37 (m, 1H, H-Ar), 7.77–7.80 (m, 1H, H-Ar), 8.13 (d, 1H, J = 8.0 Hz, H-Ar), 8.53 (d, 1H, J = 5.5 Hz, H-Ar), 11.19 (s, 1H, NH). 13C NMR (126 MHz, DMSO) δ 12. 67 (CH3), 13.21 (CH3), 18.29 (CH3), 61.43 (CH2), 121.20, 122.66, 124.17, 136.46, 148.20, 151.49, 155.78, 162.02, 163.23 (C=O), 169.42 (C=N). LREIMS: 305 (M+H) (see Supplementary Figures 7–9).
4-Phenyl-2-(2-(1-(Pyridin-2-Yl)ethylidene)hydrazinyl)thiazole (7a)
Brown powder, m.p. 127–128°C, yield (76%). 1H NMR (500 MHz, CD3Cl): δ = 2.62 (s, 3H, CH3), 6.88 (s, 1H, CH-thiazole), 7.33–7.35 (m, 1H, H-Ar), 7.40–7.43 (m, 1H, H-Ar), 7.46–7.48 (m, 2H, H-Ar), 7.78–7.80 (m, 3H, H-Ar), 8.09 (d, 1H, J = 8.0 Hz, H-Ar), 8.64 (d, 1H, J = 4.5 Hz, H-Ar), 11.85 (s, 1H, NH). 13C-NMR (126 MHz, CDCl3) δ 12.97 (CH3), 102.31 (C5-thiazole), 120.94, 124.29, 125.83, 129.30, 129.61, 129.87, 136.83, 144.65, 148.53, 153.75, 169.56 (C=N). LREIMS: 294.0; HREIMS: 294.0942 (Calcd. for C16H14N4S; 294.0939) (see Supplementary Figures 10–12).
2-(2-(1-(Pyridin-2-Yl)ethylidene)hydrazinyl)-4-(p-Tolyl)thiazole (7b)
Red powder, m.p. 289–290°C, yield (78%). 1H NMR (500 MHz, CD3Cl): δ = 2.39 (s, 3H, CH3), 2.63 (s, 3H, CH3), 6.75 (s, 1H, CH-thiazole), 7.31–7.35 (m, 2H, H-Ar), 7.64 (d, 2H, J = 7.5 Hz, H H-Ar), 7.75–7.82 (m, 1H, H-Ar), 8.08 (d, 1H, J = 8.0 Hz, H-Ar), 8.63 (d, 1H, J = 5.0 Hz, H-Ar), 11.04 (s, 1H, NH). 13C-NMR (126 MHz, CDCl3) δ 13.24 (CH3), 21.38 (CH3), 101.13 (C5-thiazole), 120.91, 122.23, 124.30, 125.34, 125.74, 126.73, 129.91, 130.02, 131.23, 136.43, 140.01, 144.39, 145.32, 149.09, 169.75 (C=N). LREIMS: 308.0; HREIMS: 308.1094 (Calcd. for C17H16N4S; 308.1096) (see Supplementary Figures 13–15).
4-(4-Fluorophenyl)-2-(2-(1-(Pyridin-2-Yl)ethylidene)hydrazinyl)thiazole (7c)
Yellow powder, m.p. 146–147°C, yield (88%). 1H NMR (500 MHz, CD3Cl): δ = 2.44 (s, 3H, CH3), 6.64 (s, 1H, CH-thiazole), 7.06–7.11 (m, 2H, H-Ar), 7.28–7.35 (m, 1H, H-Ar), 7.73–7.77 (m, 3H, H-Ar), 8.13 (d, 1H, J = 8.0 Hz, H-Ar), 8.56 (d, 1H, J = 5.0 Hz, H-Ar), 10.98 (s, 1H, NH). 13C-NMR (126 MHz, CDCl3) δ 11.55 (CH3), 103.76 (C5-thiazole), 115.60, 115.78, 120.90, 123.65, 127.74, 127.80, 130.72, 136.99, 147.05, 148.19, 154.97, 161.71, 163.68, 169.68 (C=N). LREIMS: 312.0; HREIMS: 312.0845 (Calcd. for C16H13N4SF; 312.0845) (see Supplementary Figures 16–18).
4-(4-Chlorophenyl)-2-(2-(1-(Pyridin-2-Yl)ethylidene)hydrazinyl)thiazole (7d)
Red powder, m.p. 237–238°C, yield (85%). 1H NMR (500 MHz, CD3Cl): δ = 2.48 (s, 3H, CH3), 6.89 (s, 1H, CH-thiazole), 7.35–7.38 (m, 3H, H-Ar), 8.68 (d, 2H, J = 8.5 Hz, H-Ar), 7.79–7.88 (m, 1H, H-Ar), 8.11 (d, 1H, J = 8.0 Hz, H-Ar), 8.80 (d, 1H, J = 5.0 Hz, H-Ar), 11.12 (s, 1H, NH). 13C-NMR (126 MHz, CDCl3) δ 12.19 (CH3), 104.57 (C5-thiazole), 121.48, 123.61, 124.01, 127.33, 129.07, 129.14, 131.79, 134.25, 138.36, 147.30, 153.80, 169.28 (C=N). LREIMS: 328.0; HREIMS: 328.0548 (Calcd. for C16H13N4SCl; 328.0549) (see Supplementary Figures 19–21).
4-(4-Bromophenyl)-2-(2-(1-(Pyridin-2-Yl)ethylidene)hydrazinyl)thiazole (7e)
Red powder, m.p. 202–204°C, yield (68%). 1H NMR (500 MHz, CD3Cl): δ = 2.44 (s, 3H, CH3), 6.92 (s, 1H, CH-thiazole), 7.50–7.54 (m, 3H, H-Ar), 7.64–7.74 (m, 3H, H-Ar), 8.12 (d, 1H, J = 8.0 Hz, H-Ar), 8.54 (d, 1H, J = 5.0 Hz, H-Ar), 11.02 (s, 1H, NH). 13C-NMR (126 MHz, CDCl3) δ 11.05 (CH3), 104.47 (C5-thiazole), 121.91, 123.48, 127.45, 129.25, 131.83, 132.26, 136.26, 137.46, 147.56, 153.29, 154.79, 168.71 (C=N). LREIMS: 372.0; HREIMS: 372.0044 (Calcd. for C16H13N4SBr; 372.0044) (see Supplementary Figures 22–24).
4-(4-Nitrophenyl)-2-(2-(1-(Pyridin-2-Yl)ethylidene)hydrazinyl)thiazole (7f)
Yellow powder, m.p. > 300°C, yield (77%). 1H NMR (500 MHz, CD3Cl): δ = 2.45 (s, 3H, CH3), 7.15 (s, 1H, CH-thiazole), 7.71–7.76 (m, 1H, H-Ar), 7.96 (d, 2H, J = 9.0 Hz, H-Ar), 8.13 (d, 1H, J = 8.0 Hz, H-Ar), 8.24–8.27 (m, 3H, H-Ar), 8.59 (d, 1H, J = 4.5 Hz, H-Ar), 10.89 (s, 1H, NH). 13C NMR (126 MHz, CDCl3) δ 10.95 (CH3), 108.05 (C5-thiazole), 120.35, 123.45, 123.56, 124.09, 124.17, 126.37, 136.29, 137.43, 140.61, 148.61, 154.75, 168.89 (C=N). LREIMS: 339.0; HREIMS: 339.0797 (Calcd. for C16H13N5O2S; 339.0790) (see Supplementary Figures 25–27).
Biological Activity
All the biological assessments of the tested compounds were evaluated in vitro approach with all the required ethical tools and according to the techniques below.
Antitubercular Activity
MIC Determination
Minimum inhibitory concentration (MIC) evaluation against M. tuberculosis H37Rv and M. abscessus was performed using liquid 7H9/OADC medium (Middlebrook, Difco, Baltimore, MD, United States) containing different concentrations of the tested compounds. Test substances were dissolved in dimethyl sulfoxide (DMSO) before addition to the culture medium. DMSO concentration in the final medium was kept at maximum 0.1% (vol/vol) to prevent any impact on bacterial growth. MIC determination was carried out using the Micro-plate Alamar Blue Assay (MABA) method described by Franzblau et al.57 Bacterial susceptibility was assessed by monitoring the indicator color transition from blue to pink through visual examination. Each experiment included suitable control wells with bacteria only, medium only, or compound only, and the MABA procedure was independently repeated three times.
In vitro Cytotoxicity Assay
Cytotoxicity evaluation in vitro was performed following international guidelines (ISO 10993-5:2009(E)) using L929 cell lines and MTT assay protocols. Additionally, the toxic effects of the investigated compounds were assessed using human monocyte-derived macrophages through 48-hour exposure studies.
Bactericidal Activity
The bactericidal activity of the investigated compounds was evaluated by monitoring optical density (OD600) changes and enumerating colony-forming units (CFU) in M. tuberculosis H37Rv cultures treated with the test substances. M. tuberculosis cultures were adjusted to an OD600 of 0.1 using liquid 7H9/OADC (Middlebrook, Difco, Baltimore, MD, United States) medium containing 0.05% Tween 80. Test compounds were applied at various concentrations in duplicate, alongside untreated control cultures. The bacterial cultures were incubated at 37°C, and optical density measurements were taken at 7 and 14 days following treatment. For colony-forming unit (CFU) quantification, Middlebrook 7H10/OADC agar (Difco, Baltimore, MD, USA) supplemented with 0.5% glycerol was used. Bacterial suspensions from culture bottles underwent serial dilution in 7H9/OADC broth containing 0.05% Tween 80, followed by plating onto solid agar medium at days 1, 7, and 14 of the study. Colony counts were determined following 3–5 weeks of incubation at 37°C to establish CFU values.
Biofilm Formation
M. tuberculosis biofilm formation was conducted according to established protocols with slight modifications.58 M. tuberculosis cultures were grown to an OD600 of 1.0 in 7H9/OADC medium containing 0.05% tyloxapol. The bacterial suspension was then diluted 1:100 (v/v) in Sauton’s medium and dispensed into 24-well plates at 2.5 mL per well. The plates were sealed using parafilm and incubated at 37°C under humid conditions for a duration of five weeks to facilitate biofilm formation. After biofilm maturation, the culture medium was substituted with fresh medium supplemented with 0.1% casitone and different concentrations of the test compounds, followed by 48-hour incubation at 37°C. Bacterial viability within biofilms was determined using resazurin-based fluorescence assays, with 375 µL of 0.02% resazurin added per well and incubated for 90 minutes. The fluorescence intensity was quantified using a SpectraMax® i3 multi-mode microplate reader (Syngen) with excitation and emission wavelengths set at 550 nm and 590 nm, respectively. Data were normalized and reported as a percentage of viability relative to untreated Mycobacterium tuberculosis biofilm controls.
Quantifying the Interaction Between Rifampicin (RMP) and 7a; 7c; 7d Using a Two-Dimensional Dilution Matrix
Drug interaction studies were conducted using the checkerboard assay to evaluate potential interactions between rifampicin (RMP) and the tested compounds. Serial dilutions of rifampicin were prepared along one axis of 96-well plates, while the investigated compounds were serially diluted along the perpendicular axis, creating a two-dimensional dilution matrix. Plates were inoculated with M. tuberculosis and incubated under standard conditions. Bacterial growth was measured using resazurin detection methods. The Fractional Inhibitory Concentration Index (FICI) was calculated by first determining individual minimum inhibitory concentrations (MICs) for each compound, then identifying the lowest effective combination concentrations from the checkerboard assay. FICI values were calculated as the sum of FIC_A and FIC_B, where FIC_A represents the concentration of rifampicin in the effective combination divided by its MIC when used alone, and FIC_B represents the concentration of the test compound in the effective combination divided by its individual MIC. Interactions were categorized based on the fractional inhibitory concentration index (FICI) as follows: synergistic when FICI was less than or equal to 0.5, additive when FICI ranged between 0.5 and 1, indifferent when FICI was greater than 1 but less than or equal to 4, and antagonistic when FICI exceeded 4.
Isolation and Differentiation of Human Monocyte-Derived Macrophages (MDMs), Followed by Assessment of the Bactericidal Activity of the Tested Compounds Against Intracellular Mycobacterium tuberculosis
Human monocyte-derived macrophage (MDM) were obtained from buffy coats of healthy blood donors (Regional Blood Donation Station, Lodz, Poland) and isolated according to established procedures.59 Following extensive washing to eliminate non-adherent cells, differentiated MDMs were allowed to rest overnight before treatment with varying concentrations of test compounds. Macrophage viability was determined after 48-hour incubation using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma, St. Louis, MO, United States). For intracellular infection studies, MDMs were infected with M. tuberculosis at a multiplicity of infection (MOI) of 1:10 following the methodology of Korycka-Machala et al.60 Extracellular bacteria were eliminated by washing with culture medium two hours post-infection, followed by one-hour treatment with gentamicin (1 g/L) (Sigma, St. Louis, MO, United States) and triple washing with Iscove’s medium containing 2% human AB serum (Sigma, St. Louis, MO, United States). Infected macrophages were then cultured in medium containing test compounds at 1×BC concentrations or without compounds (control) for 48 hours at 37°C in a humidified atmosphere (10% CO2–90% air). Macrophages were subsequently lysed using 0.1% SDS, and colony-forming unit (CFU) enumeration was performed as previously described.61
Assessment of InhA Inhibition
The assay is performed by preparing InhA (10 nM), NADH (200 µM), DD-CoA (100 µM), and inhibitor serial dilutions, then assembling 50 µL reactions in a 96-well plate containing assay buffer, inhibitor/DMSO, NADH (final 40 µM), InhA (2 nM), and DD-CoA (20 µM) added in that order to start the reaction, with positive controls lacking inhibitor and negative controls lacking enzyme. Reactions are incubated at 37°C for 30 min, stopped by adding an equal volume (50 µL) of NAD/NADH-Glo™ reagent, incubated 30 min at room temperature in the dark, and luminescence is recorded (0.5–1 s integration). Data are normalized to positive (100%) and negative (0%) controls, IC50 values obtained from log-dose response curves using a 4-parameter fit, and assay quality checked via Z′ (>0.5), while ensuring ≤1% DMSO, fresh NADH, and including a known inhibitor such as triclosan, following the protocol established in prior studies62 (see Supplementary Figures 28 and 29).
Molecular Docking and Molecular Dynamics Simulation
The three-dimensional crystal structure of the Mycobacterium tuberculosis InhA protein was retrieved from the RCSB Protein Data Bank (https://www.rcsb.org/structure/4TZK) in *.pdb format.63 Before docking, the structure was prepared by eliminating all co-crystallized molecules except NAD, and then hydrogen atoms were added. The prepared structure was then converted into *.pdbqt format using AutoDockTools for molecular docking. The 2D and 3D structures of compound 6 and triclosan were generated according to previously established methodologies.64 Specifically, their geometries were initially sketched in 2D using Marvin JS and then converted into 3D models within the same software. Molecular docking simulations were conducted using AutoDock Vina version 1.2.3,65 with the exhaustiveness parameter set to 400. The docking grid was defined as 25 × 25 × 25 Å3, centered on the position of the co-crystallized ligand inside the active site of the InhA protein (PDB ID: 4TZK), as also reported in previous studies.53,54 The docking poses and interactions of the selected compound were analyzed and visualized using Discovery Studio Visualizer.
To assess the stability and dynamic interactions of the docked complexes under biologically relevant conditions, molecular dynamics (MD) simulations were conducted using GROMACS 2023.66 The AMBER99SB-ILDN force field was utilized to generate the topology files for the InhA protein (PDB ID: 4TZK), while ligand parameters were derived using ACPYPE and AmberTools 22, following established protocols.53,54 The simulation setup involved embedding the complexes (compound 6 and the co-crystallized ligand) in a triclinic box, solvated with TIP3P water molecules. The system was neutralized by adding Na⁺/Cl− ions as required. Energy minimization was performed using the steepest descent algorithm, with a convergence threshold of 1000 kJ•mol−1•nm−1 for maximum force. Following minimization, the system underwent equilibration in NVT and NPT ensembles (2 ns each) before a 200 ns production run. Temperature was maintained at 300 K using the V-rescale thermostat, while pressure was controlled (where applicable) with the C-rescale barostat. The LINCS algorithm was applied for bond constraints, and long-range electrostatics were handled via the Particle Mesh Ewald (PME) method. Simulations employed a 2-fs time step and a 1.2 nm van der Waals cutoff. Post-simulation analyses including RMSD (root-mean-square deviation), RMSF (root-mean-square fluctuation), and Rg (radius of gyration) were processed and visualized using XMGRACE software.
Results and Discussion
Chemistry
The preparation of the target compounds 3, 4, 5, 6, and 7a–f was proceeded through the following steps. First, thiosemicarbazone 3 was prepared through the reaction of 2-acetylpyridine (1) with thiosemicarbazide (2). Then, heterocyclization of the synthesized thiosemicarbazone 3 with various α-halocarbonyl compounds produced the corresponding thiazole and thiazolidinone derivatives through an S-alkylation reaction subsequently accompanied by the removal of a water or alcohol molecule. Specifically, the cyclocondensation of thiosemicarbazone 3 with chloroacetone, ethyl-2-chloroacetoacetate, and/or phenacyl bromide derivatives in ethanol, using a catalytic amount of anhydrous sodium acetate, resulted in the formation of the corresponding thiazole compounds: 4-methylthiazolines 4, 4-methylthiazolyl-5-carboxylates 6, and 4-(R)-phenylthiazoles 7a-f. Furthermore, Thiazolidinones 5 were synthesized via the condensation reaction of the corresponding thiosemicarbazones 3 with ethyl bromoacetate, employing anhydrous sodium acetate and absolute ethanol as the reaction medium (Scheme 1). The chemical structures of the newly designed compounds were determined and verified through comprehensive spectroscopic analysis proton nuclear magnetic resonance (1H NMR), carbon-13 nuclear magnetic resonance (13C NMR), and high-resolution time-of-flight electrospray ionization mass spectrometry (HR-TOF-ESI-MS), as detailed in the experimental section.
 |
Scheme 1 Synthesis of the target compounds 3–6 and 7a-f. |
Biological Activity
Antimycobacterial Screening
The synthesized compounds (3–6 and 7a–f) were evaluated for their antitubercular potential and cytotoxic effects against Mycobacterium tuberculosis H37Rv, along with fast-growing, non-tuberculous mycobacterial species, including Mycobacterium abscessus. Primary screening was performed at a standardized concentration of 125 μg/mL to determine their inhibitory activity, and the tested compounds did not show any activity against M. abscessus.67 However, these derivatives demonstrated inhibitory activity against M. tuberculosis (Table 1).
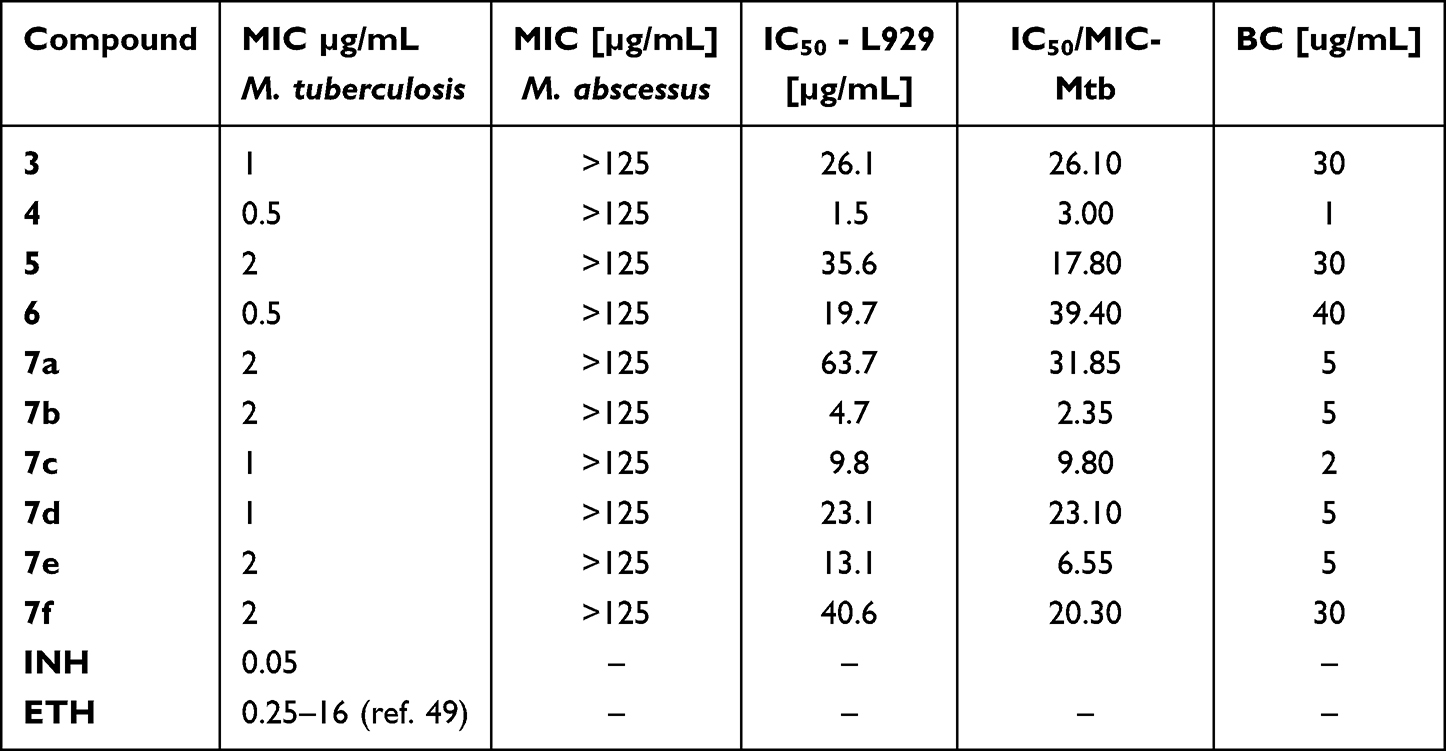 |
Table 1 The Antimycobacterial Effect of Target Compound 3–7a-f |
For the derivatives active against M. tuberculosis, MIC was assessed utilizing the microplate Alamar Blue assays (MABA). The findings demonstrated that the synthesized compounds exhibited remarkable activity by inhibiting the growth of M. tuberculosis at concentrations ranging from 0.5 to 2 µg/mL.
Following the initial screening, the chosen compounds underwent additional cytotoxicity assessment in accordance with international guidelines (ISO 10993–5:2009(E)), employing L929 fibroblast cells and the MTT assay.68 The IC50 values of the compounds were found to be 1.3–39.4 times higher than their respective MIC values (Table 1).
The MIC value represents the lowest concentration of a compound that inhibits the metabolic activity of bacteria. Therefore, to assess the ability of the tested derivatives to eliminate M. tuberculosis, the bactericidal value was determined by evaluating the number of viable bacteria forming colonies on a plate after incubation with the tested compounds. The compounds demonstrating bactericidal effects at the lowest concentrations were the synthesized derivatives, for target compounds Figure 2.
 |
Figure 2 Evaluation of the bactericidal activity of the tested compounds through CFU analysis. The Y-axis represents % survival, the X-axis shows the control strain M. tuberculosis H37Rv cultured without compound (Rv_control), and in the presence of individual compounds indicating compound number_used concentration in μg/mL_incubation time in hours. A one-way ANOVA followed by Dunnett’s post hoc test was used to compare the untreated M. tuberculosis control strain (Rv_control) with bacterial cultures treated with the test compounds at specified concentrations and time points. The analysis revealed statistically significant differences, with adjusted *p*-values of **** <0.0001, * 0.0304, and 0.0128 for compounds 5 and 6, respectively. All statistical evaluations and data visualizations were performed using GraphPad Prism 9 (Version 10.4.1). |
Evaluation of the Antimycobacterial Efficacy of Selected Compounds in Human Macrophage Models
Tuberculosis (TB) is an airborne disease that begins when the pathogen enters the alveolar regions of the host’s lungs. Following infection, M. tuberculosis is engulfed by alveolar macrophages, specialized cells responsible for the intracellular destruction of bacteria. However, the tubercle bacilli have the ability to survive and replicate within human macrophages by interfering with their infection response. Consequently, effective antituberculosis drugs must penetrate human macrophages without damaging them to assess the efficacy of the tested compounds targeting intracellular bacterial pathogens, we examined their cytotoxicity toward human macrophages at concentrations ranging from 2 to 10 times the minimum inhibitory concentration (MIC). Human macrophages (monocyte-derived macrophages, MDMs) were produced by differentiating monocytes isolated from the buffy coats of healthy blood donors. Among the tested compounds, only three (3, 5, and 6) exhibited low cytotoxicity against human macrophages at a level below 25%. Therefore, these three compounds were selected for further investigation of their activity against M. tuberculosis residing inside macrophages. All of these compounds, at a concentration of 10 × MIC, demonstrated the ability to kill intracellular tubercle bacilli Figure 3.
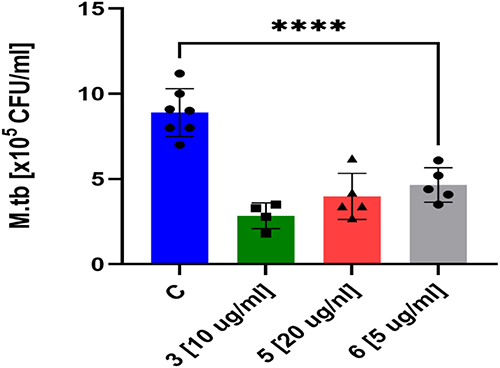 |
Figure 3 Evaluation of Anti-Tubercular Activity in Human Monocyte-Derived Macrophages (MDMs) Human MDMs were infected with Mycobacterium tuberculosis and subsequently treated with compounds 3, 5, and 6 at a concentration equivalent to 10× the minimum inhibitory concentration (MIC). To assess the efficacy of these compounds, intracellular bacterial load was quantified and compared to untreated controls using a standard one-way analysis of variance (ANOVA). Statistical significance was confirmed for all tested compounds (adjusted *p* < 0.0001, Dunnett’s multiple comparisons test). Data analysis and visualization were performed using GraphPad Prism 9 (version 9.3.1). |
Assessing the Effectiveness of Compounds Against Mycobacterial Biofilms
Mycobacterial biofilms represent a defense mechanism that challenges therapeutic interventions, creating robust microbial communities resistant to both pharmaceutical treatments and immune responses.69 We evaluated all selected compounds in concern to their antimicrobial potential against M. tuberculosis biofilms. Using Sauthon’s medium, we cultivated bacterial cultures for five weeks and exposed established biofilms to the compounds at bactericidal concentration. Cell survival assays indicated a slight yet meaningful reduction in bacterial viability. The results indicate that 3 and 6 show modest potential in disrupting bacterial biofilms, but struggles to comprehensively penetrate these complex microbial structures (Figure 4).
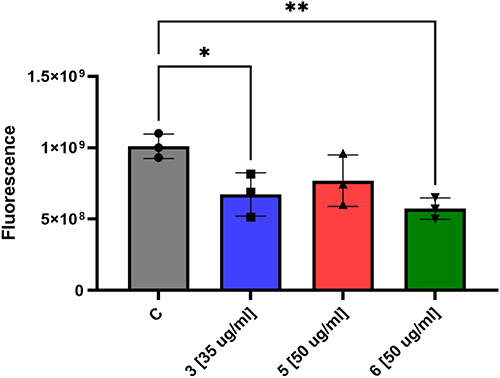 |
Figure 4 The effects of selected compounds on pre-formed biofilms of M. tuberculosis were evaluated. Statistical analyses were performed using ordinary one-way analysis of variance (ANOVA) to compare the quantity of bacilli in untreated M. tuberculosis biofilm with biofilms treated with compounds 3, 5, and 6. * and ** represent 0.0334 and 0.009 P value, respectively. Statistical analyses and graphical visualizations were conducted utilizing GraphPad Prism version 9.3.1. |
Testing of Additive, Synergistic or Indifferent Interaction Between Selected Compounds and Rifampicin
Tuberculosis therapy is a multi-drug process; hence the assessment of interactions between potential new drugs and those already used in therapy is extremely important. The most important anti-tuberculosis drug is rifampicin (RMP), therefore we decided to check whether the simultaneous application of this drug with the tested compounds would have a synergistic, additive, antagonistic effect, or whether no interactions would be observed. To evaluate these relationships, the checkerboard assay was employed in this study. The analysis revealed different interaction patterns between rifampicin and the tested compounds. Compounds 7d and 7c demonstrated additive effects when combined with rifampicin, both showing FICI values of 0.75. In contrast, compounds 7a exhibited no significant interaction with rifampicin, displaying FICI values of 1.25, indicating indifferent relationships. No synergistic or antagonistic interactions were observed for any of the tested compound combinations with rifampicin.
Inhibition of the Mtb InhA Enzyme
To evaluate the efficacy of the most promising candidate, derivative 6 was further investigated for its ability to inhibit the InhA enzyme. The findings revealed that this compound exhibited significant InhA inhibition, demonstrating nearly twofold greater potency than the standard inhibitor, triclosan. Specifically, the thiazole pyridine ester (compound 6) displayed robust inhibitory activity, achieving an IC50 value of 0.36 µM, which is notably lower than triclosan’s IC50 of 0.62 µM, indicating superior inhibitory performance Table 2.
Molecular Docking and Dynamics Simulations
To enhance comprehension of the binding mechanism between compound 6 with Mycobacterium tuberculosis InhA protein, molecular docking simulations were performed. The docking results revealed that the binding affinity of compound 6 to M. tuberculosis InhA protein was −7.77 kcal/mol, while the reference compound triclosan displayed a score of –7.26 kcal/mol. These values are highly comparable, indicating that compound 6 interacts with the InhA active site with a binding strength similar to that of triclosan, which is consistent with the in vitro findings presented in Table 2. Protein–ligand interaction analysis was conducted to provide insights into the affinity potential of the investigated derivative with the target protein. The detailed interactions are illustrated in Figure 5. Compound 6 primarily formed hydrophobic interactions, specifically π-alkyl and alkyl interactions with Met103, Phe149, Met161, Ala198, and Ile215. Additionally, compound 6 established a π-anion interaction with Tyr158 and a π-sulfur interaction with Met199. Meanwhile, the reference compound triclosan also interacted with residues Ile215, Tyr158, Met199, Met161, and Phe149, indicating that compound 6 exhibits a favorable binding capability to the InhA protein.
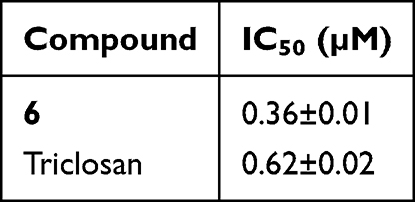 |
Table 2 InhA Inhibition Effect of the Most Active Compound 6 |
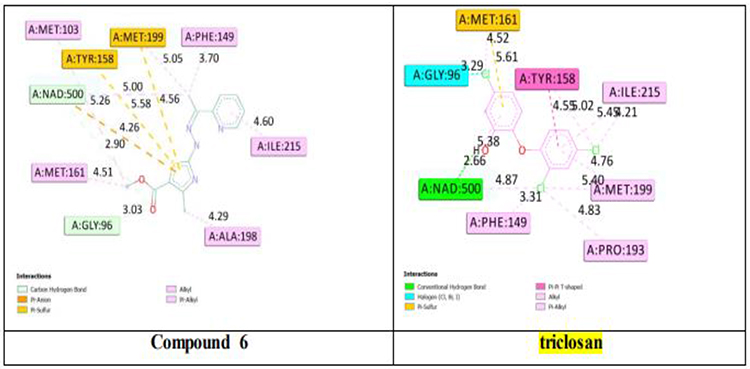 |
Figure 5 2D representation of InhA residues interacting to compound 6 and reference triclosan (carbon hydrogen bonds: light blue; π–anion interactions: dark Orange; π–sulfur: yellow-brown; Alkyl and Pi–alkyl: light purple, halogen interactions (Cl, Br): cyan; π–π T-shaped: pink-purple). |
While molecular docking provides an initial approximation of the binding affinity and interaction profile, it does not account for the dynamic and flexible nature of biomolecular systems. To address this limitation and further confirm the stability of the predicted binding poses under conditions that more closely resemble a biological environment, molecular dynamics (MD) simulations were subsequently performed. This methodology enabled the assessment of the structural stability of the complexes as well as the durability of critical protein–ligand interactions throughout the duration of the study. The proteins within these complexes demonstrated minimal fluctuations during the entire simulation, as illustrated by the RMSD plot in Figure 6a, for both the reference complex and the compound 6 complex. The average RMSD values of InhA-6 and InhA-triclosan were 1.75 Å and 2.17 Å, respectively. As noted, both complexes maintained RMSD values below 3 Å, with the InhA protein complex with compound 6 showing lower fluctuations than the reference. The findings suggest that the interaction of compound 6 with the protein did not induce any notable conformational alterations. Furthermore, the protein within these complexes demonstrated greater stability in RMSD values relative to both the quinoline-isatin hybrid compound, 3-(2-(Quinolin-2-yl)hydrazono)-5-(trifluoromethoxy)indolin-2-one, and the co-crystallized ligand (PDB ID: 4TZK).54 Moreover, the stability of the ligands within the active site throughout the simulation was assessed by analyzing RMSD plots of the ligands, as illustrated in Figure 6b. The ligands in the InhA-6 complexes exhibited lower average RMSD values than that of InhA-triclosan, specifically 1.331 Å and 1.747 Å, respectively. In conclusion, both ligands demonstrated consistently stable RMSD values throughout their binding interactions within the protein’s active site, providing additional evidence for the overall stability of these complexes. Throughout the simulation, particular amino acid residues were examined, and RMSF plots were created to show the average fluctuations of these residues over time, as shown in Figure 6c. The analysis of these plots demonstrated that the relative stability of all two complexes was consistently preserved throughout the duration of the simulation, with the exception of a minor loop segment spanning residues 100 to 120, which demonstrated marginally increased fluctuations in the triclosan-bound complex as well as in the previously characterized apoprotein.54 This observation suggests that the reference compound and compound 6 exert similarly stabilizing effects on this region of the protein. The radius of gyration (Rg) reflects the compactness of the protein structure. A stable Rg value throughout the simulation suggests that the protein conformation tends to remain preserved. Notably, Rg remained stable during the entire simulation for both complexes. The Rg values of InhA-6 and InhA-triclosan were 18.368 Å and 18.364 Å, respectively, Figure 6d. The Rg plots displayed that InhA-6 maintained a similarly stable folded structure as the apoprotein.54 Collectively, these findings demonstrate that compound 6 forms a stable complex with the InhA protein, with favorable binding affinity and persistent interactions throughout the simulation period.
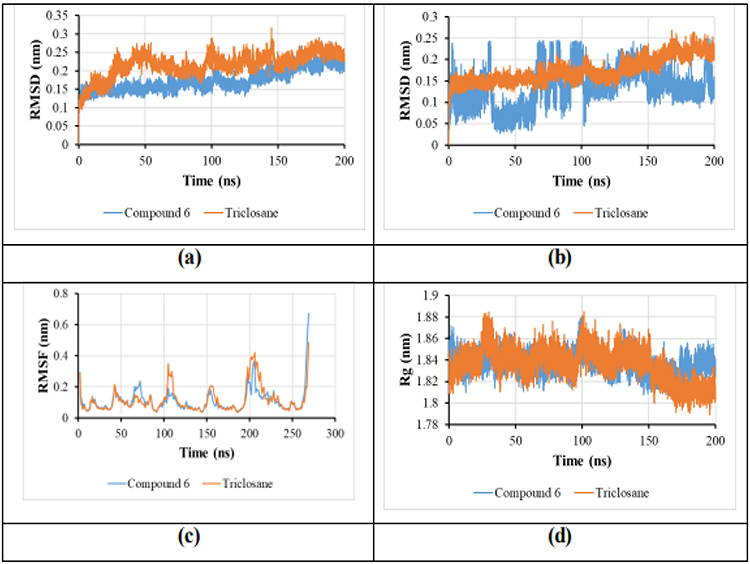 |
Figure 6 (a) The RMSD plot for protein in complexes InhA-6 and InhA-triclosan; (b) The RMSD plot for ligands complexes with InhA; (c) RMSF plot for studied complexes; (d) Radius of gyration of complex 6, and co-crystalized. |
Conclusion
This research outlines the design and synthesis of innovative pyridine-thiazolidinone/thiazole hybrid compounds as potential InhA inhibitors for tuberculosis (TB) treatment. The synthesized derivatives demonstrated strong antimycobacterial activity, with minimum inhibitory concentrations (MICs) between 0.5 and 2 μg/mL. Among these, compound 6 emerged as the most potent candidate, exhibiting an MIC of 0.5 μg/mL against Mycobacterium tuberculosis while maintaining low cytotoxicity in L929 cell lines, indicating a favorable safety profile. Furthermore, compound 6 displayed significant anti-TB efficacy, including antibiofilm activity, effectively penetrating and disrupting bacterial biofilms. The derivatives also exhibited strong InhA inhibition, a critical enzyme for M. tuberculosis survival, with an IC50 value of 0.36 μM superior to the reference drug. To further elucidate the mechanism of action, molecular docking studies were performed, revealing the binding interactions of the most active compounds within InhA’s active site, providing valuable insights into their inhibitory potential. The findings of this study establish a foundation for multiple promising avenues of research and optimization. Subsequent investigations may concentrate on further structural modification of the most potent compound, molecule 6, to improve its efficacy and selectivity against mycobacterial InhA. Furthermore, the exploration of combination therapy strategies could facilitate the development of more effective treatment protocols by assessing potential synergistic interactions between these compounds and currently available antituberculosis agents. Additionally, conducting in vivo studies using animal models represents a critical next step to evaluate the safety profiles, pharmacokinetic characteristics, and therapeutic effectiveness of the most promising molecules within relevant tuberculosis models. Collectively, these research directions hold considerable potential to advance the treatment of mycobacterial infections significantly.
Ethical Approval
The manuscript does not require ethical approval as all biological assessments were conducted in vitro. Generally, ethical approval is not necessary for standard in vitro assays, given that these procedures do not involve living human subjects or sentient animals. Consequently, fundamental research ethics principles—such as the protection of human dignity, autonomy, and animal welfare—are not applicable within these controlled, non-living biological systems.
Acknowledgments
JD, AB & MK-M were supported by the Ministry of Science and Higher Education, POLOPENSCREEN, 2024/WK/06, and National Science Centre, Poland, UMO-2023/49/B/NZ7/01421.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation. Also, they took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding.
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. The authors alone are responsible for the content and writing of the paper.
References
1. Bussi C, Gutierrez MG. Mycobacterium tuberculosis infection of host cells in space and time. FEMS Microbiol Rev. 2019;43(4):341–17. doi:10.1093/femsre/fuz006
2. Falzon D, Zignol M, Bastard M, Floyd K, Kasaeva T. The impact of the COVID-19 pandemic on the global tuberculosis epidemic. Front Immunol. 2023;14:1234785. doi:10.3389/fimmu.2023.1234785
3. Al Khatib A, Hassanein S, Almari M, Koubar M, Fakhreddine S. Tuberculosis morbidity and mortality during the COVID-19 pandemic: a life-threatening complex challenge. Front Cell Infect Microbiol. 2024;14:1423081. doi:10.3389/fcimb.2024.1423081
4. Raad R, Dixon J, Gorsky M, Hoddinott G. Cycles of antibiotic use and emergent antimicrobial resistance in the South African tuberculosis programme (1950-2021): a scoping review and critical reflections on stewardship. Glob Public Health. 2024;19(1):2356623. doi:10.1080/17441692.2024.2356623
5. Garcia-Bereguiain MA, Rodriguez-Pazmiño AS, Franco-Sotomayor G, Orlando SA, González M, Ugarte-Gil C. “The end TB strategy” pathway in South America: out of track for 2025 milestones and 2035 eradication. Lancet Reg Health Am. 2025;44:101045. doi:10.1016/j.lana.2025.101045
6. Sabt A, Khaleel EF, Shaldam MA, et al. Discovery of new quinoline derivatives bearing 1-aryl-1, 2, 3-triazole motif as influenza H1N1 virus neuraminidase inhibitors. Bioorg Chem. 2024;151:107703. doi:10.1016/j.bioorg.2024.107703
7. Kufa M, Finger V, Kovar O, et al. Revolutionizing tuberculosis treatment: breakthroughs, challenges, and hope on the horizon. Acta Pharmaceutica Sinica B. 2025;15(3):1311–1332. doi:10.1016/j.apsb.2025.01.023
8. El-Naggar ME, Radwan EK, Rashdan HR, El-Wakeel ST, Koryam AA, Sabt A. Simultaneous removal of Pb 2+ and direct red 31 dye from contaminated water using N-(2-hydroxyethyl)-2-oxo-2 H-chromene-3-carboxamide loaded chitosan nanoparticles. RSC Adv. 2022;12(29):18923–18935. doi:10.1039/D2RA02526D
9. Shleider Carnero Canales C, Marquez Cazorla J, Furtado Torres AH, et al. Advances in diagnostics and drug discovery against resistant and latent tuberculosis infection. Pharmaceutics. 2023;15(10):2409. doi:10.3390/pharmaceutics15102409
10. Majrashi TA, Sabt A, Abd el salam HA, Al-Ansary GH, Hamissa MF, Eldehna WM. An updated review of fatty acid residue-tethered heterocyclic compounds: synthetic strategies and biological significance. RSC Adv. 2023;13(20):13655–13682. doi:10.1039/D3RA01368E
11. Sharma A, Sharma V, Sharma S, Sharma S, Sharma M, Sivanesan I. Advanced nanosystems and emerging therapies: innovations in tuberculosis treatment and drug resistance. Pharmaceutics. 2025;17(11):1459. doi:10.3390/pharmaceutics17111459
12. Joshi SD, Dixit SR, More UA, Aminabhavi T, Kulkarni VH, Gadad AK. Enoyl ACP reductase as effective target for the synthesized novel antitubercular drugs: a-state-of-the-art. Mini Rev Med Chem. 2014;14(8):678–693. doi:10.2174/1389557514666140820112524
13. Ebaid MS, Korycka-Machala M, Shaldam MA, et al. Exploring antitubercular activity of new coumarin derivatives targeting enoyl acyl carrier protein reductase (InhA): synthesis, biological evaluation and computational studies. J Mol Struct. 2025;1336:142074. doi:10.1016/j.molstruc.2025.142074
14. Al-Warhi T, Sabt A, Korycka-Machala M, et al. Benzenesulfonohydrazide-tethered non-fused and fused heterocycles as potential anti-mycobacterial agents targeting enoyl acyl carrier protein reductase (InhA) with antibiofilm activity. RSC Adv. 2024;14(41):30165–30179. doi:10.1039/D4RA05616G
15. Teneva Y, Simeonova R, Valcheva V, Angelova VT. Recent advances in anti-tuberculosis drug discovery based on hydrazide–hydrazone and thiadiazole derivatives targeting InhA. Pharmaceuticals. 2023;16:484. doi:10.3390/ph16040484
16. North EJ, Jackson M, Lee RE. New approaches to target the mycolic acid biosynthesis pathway for the development of tuberculosis therapeutics. Curr Pharm Des. 2014;20(27):4357–4378. doi:10.2174/1381612819666131118203641
17. Chebaiki M, Delfourne E, Tamhaev R, et al. Discovery of new diaryl ether inhibitors against Mycobacterium tuberculosis targeting the minor portal of InhA. Eur J Med Chem. 2023;259:115646. doi:10.1016/j.ejmech.2023.115646
18. Diab A, Dickerson H, Al Musaimi O. Targeting the heart of Mycobacterium: advances in anti-tubercular agents disrupting cell wall biosynthesis. Pharmaceuticals. 2025;18(1):70. doi:10.3390/ph18010070
19. Kuang W, Zhang H, Wang X, Yang P. Overcoming Mycobacterium tuberculosis through small molecule inhibitors to break down cell wall synthesis. Acta Pharmaceutica Sinica B. 2022;12(8):3201–3214. doi:10.1016/j.apsb.2022.04.014
20. Sachan RS, Mistry V, Dholaria M, et al. Overcoming Mycobacterium tuberculosis drug resistance: novel medications and repositioning strategies. ACS Omega. 2023;8(36):32244–32257. doi:10.1021/acsomega.3c02563
21. Vilchèze C, Jacobs WR. The isoniazid paradigm of killing, resistance, and persistence in Mycobacterium tuberculosis. J Mol Biol. 2019;431(18):3450–3461. doi:10.1016/j.jmb.2019.02.016
22. Shaaban MM, Mahran MA, Teleb M, Ragab HM. The key players in the arsenal of combating TB; reviewing the lead InhA inhibitors. J Adv Pharm Sci. 2024;1(2):18–33. doi:10.21608/japs.2024.270897.1016
23. Lourenco MC, de Souza MV, Pinheiro AC, et al. Evaluation of anti-tubercular activity of nicotinic and isoniazid analogues. Arkivoc. 2007;15:181–191. doi:10.3998/ark.5550190.0008.f18
24. Mahajan NS, Dhawale SC. Linked pyridinyl-thiadiazoles: design and synthesis as potential candidate for treatment of XDR and MDR tuberculosis. Eur J Med Chem. 2015;102:243–248. doi:10.1016/j.ejmech.2015.07.039
25. Ling Y, Hao ZY, Liang D, Zhang CL, Liu YF, Wang Y. The expanding role of pyridine and dihydropyridine scaffolds in drug design. Drug Des Devel Ther. 2021;15:4289–4338. doi:10.2147/DDDT.S329547
26. Alizadeh SR, Ebrahimzadeh MA. Antiviral activities of pyridine fused and pyridine containing heterocycles, a review (from 2000 to 2020). Mini Rev Med Chem. 2021;21(17):2584–2611. doi:10.2174/1389557521666210126143558
27. Mohammad Abu-Taweel G, Ibrahim MM, Khan S, et al. Medicinal importance and chemosensing applications of pyridine derivatives: a review. Crit Rev Anal Chem. 2024;54(3):599–616. doi:10.1080/10408347.2022.2089839
28. Patel H, Jadhav H, Ansari I, Pawara R, Surana S. Pyridine and nitro-phenyl linked 1, 3, 4-thiadiazoles as MDR-TB inhibitors. Eur J Med Chem. 2019;167:1–9. doi:10.1016/j.ejmech.2019.01.073
29. Samanta S, Kumar S, Aratikatla EK, Ghorpade SR, Singh V. Recent developments of imidazo [1, 2-a] pyridine analogues as antituberculosis agents. RSC Med Chem. 2023;14(4):644–657. doi:10.1039/D3MD00019B
30. Yates TA, Tomlinson LA, Bhaskaran K, et al. Lansoprazole use and tuberculosis incidence in the United Kingdom Clinical Practice Research Datalink: a population based cohort. PLoS Med. 2017;14(11):e1002457. doi:10.1371/journal.pmed.1002457
31. Hartkoorn RC, Sala C, Neres J, et al. Towards a new tuberculosis drug: pyridomycin - nature’s isoniazid. EMBO Mol Med. 2012;4(10):1032–1042. doi:10.1002/emmm.201201689
32. Almasirad A, Samiee-Sadr S, Shafiee A. Synthesis and Antimycobacterial Activity of 2-(Phenylthio) benzoylarylhydrazone Derivatives. Iran J Pharm Res. 2011;10(4):727–731.
33. Matsa R, Makam P, Sethi G, et al. Pyridine appended 2-hydrazinylthiazole derivatives: design, synthesis, in vitro and in silico antimycobacterial studies. RSC Adv. 2022;12(29):18333–18346. doi:10.1039/D2RA02163C
34. Scarim CB, Pavan FR. Thiazole, triazole, thio-and semicarbazone derivatives-Promising moieties for drug development for the treatment of tuberculosis. Eur J Med Chem Rep. 2021;1:100002. doi:10.1016/j.ejmcr.2021.100002
35. Ebaid MS, Ibrahim HA, Kassem AF, Sabt A. Recent studies on protein kinase signaling inhibitors based on thiazoles: review to date. RSC Adv. 2024;14(50):36989–37018. doi:10.1039/D4RA05601A
36. Kalita T, Choudhury A, Shakya A, Ghosh SK, Singh UP, Bhat HR. A review on synthetic thiazole derivatives as an antimalarial agent. Curr Drug Discov Technol. 2024;21(5):10–42. doi:10.2174/0115701638276379231223101625
37. Helal MHM, Salem MA, El-Gaby MSA, Aljahdali M. Synthesis and biological evaluation of some novel thiazole compounds as potential anti-inflammatory agents. Eur J Med Chem. 2013;65:517–526. doi:10.1016/j.ejmech.2013.04.005
38. Sabt A, Abdelrahman MT, Abdelraof M, Rashdan HR. Investigation of novel mucorales fungal inhibitors: synthesis, in‐silico study and anti‐fungal potency of novel class of coumarin‐6‐sulfonamides‐thiazole and thiadiazole hybrids. ChemistrySelect. 2022;7(17):e202200691. doi:10.1002/slct.202200691
39. Mokale SN, Sanap PT, Shinde DB. Synthesis and hypolipidemic activity of novel 2-(4-(2-substituted aminothiazole-4-yl) phenoxy) acetic acid derivatives. Eur J Med Chem. 2010;45:3096–3100. doi:10.1016/j.ejmech.2010.03.043
40. Luckner SR, Machutta CA, Tonge PJ, Kisker C. Crystal structures of Mycobacterium tuberculosis KasA show mode of action within cell wall biosynthesis and its inhibition by thiolactomycin. Structure. 2009;17:1004–1013. doi:10.1016/j.str.2009.04.012
41. Ranjbar S, Haridas V, Nambu A, et al. Cytoplasmic RNA sensor pathways and nitazoxanide broadly inhibit intracellular Mycobacterium tuberculosis growth. iScience. 2019;22:299–313. doi:10.1016/j.isci.2019.11.001
42. Fox LM, Saravolatz LD. Nitazoxanide: a new thiazolide antiparasitic agent. Clin Infect Dis. 2005;40:1173–1180. doi:10.1086/428839
43. Harausz EP, Chervenak KA, Good CE, et al. Activity of nitazoxanide and tizoxanide against Mycobacterium tuberculosis in vitro and in whole blood culture. Tuberculosis. 2016;98:92–96. doi:10.1016/j.tube.2016.03.002
44. Shaikh SA, Labhade SR, Kale RR, et al. Thiadiazole-thiazole derivatives as potent anti-tubercular agents: synthesis, biological evaluation, and In silico docking studies. Eur J Med Chem Rep. 2024;12:100183. doi:10.1016/j.ejmcr.2024.100183
45. Makam P, Kannan T. 2-Aminothiazole derivatives as antimycobacterial agents: synthesis, characterization, in vitro and in silico studies. Eur J Med Chem. 2014;87:643–656. doi:10.1016/j.ejmech.2014.09.086
46. Corey EJ, Enders D. Synthetic routes to polyfunctional molecules via metallated N, N-dimethylhydrazones. Tetrahedron Lett. 1976;17(1):11–14. doi:10.1016/S0040-4039(00)71309-8
47. Batran RZ, Sabt A, Khedr MA, Allayeh AK, Pannecouque C, Kassem AF. 4-Phenylcoumarin derivatives as new HIV-1 NNRTIs: design, synthesis, biological activities, and computational studies. Bioorg Chem. 2023;141:106918. doi:10.1016/j.bioorg.2023.106918
48. Sabt A, Abdelraof M, Hamissa MF, Noamaan MA. Antibacterial activity of quinoline‐based derivatives against methicillin‐resistant Staphylococcus aureus and Pseudomonas aeruginosa: design, synthesis, DFT and molecular dynamic simulations. Chem Biodivers. 2023;20(11):e202300804. doi:10.1002/cbdv.202300804
49. Verma G, Marella A, Shaquiquzzaman M, Akhtar M, Ali MR, Alam MM. A review exploring biological activities of hydrazones. J Pharm Bioallied Sci. 2014;6(2):69–80. doi:10.4103/0975-7406.129170
50. Wang Y, Cao D, Liu G. Application of antimicrobial drugs in Mycobacterium tuberculosis and research progress. Microb Pathogenesis. 2025;206:107794. doi:10.1016/j.micpath.2025.107794
51. Batran RZ, Sabt A, Dziadek J, Kassem AF. Design, synthesis and computational studies of new azaheterocyclic coumarin derivatives as anti-Mycobacterium tuberculosis agents targeting enoyl acyl carrier protein reductase (InhA). RSC Adv. 2024;14(30):21763–21777. doi:10.1039/D4RA02746A
52. Kassem AF, Sabt A, Korycka-Machala M, et al. New coumarin linked thiazole derivatives as antimycobacterial agents: design, synthesis, enoyl acyl carrier protein reductase (InhA) inhibition and molecular modeling. Bioorg Chem. 2024;150:107511. doi:10.1016/j.bioorg.2024.107511
53. Sabt A, Abdulla MH, Ebaid MS, et al. Identification of 2-(N-aryl-1, 2, 3-triazol-4-yl) quinoline derivatives as antitubercular agents endowed with InhA inhibitory activity. Front Chem. 2024;12:1424017. doi:10.3389/fchem.2024.1424017
54. Khaleel EF, Sabt A, Korycka-Machala M, et al. Identification of new anti-mycobacterial agents based on quinoline-isatin hybrids targeting enoyl acyl carrier protein reductase (InhA). Bioorg Chem. 2024;144:107138. doi:10.1016/j.bioorg.2024.107138
55. Abu-Melha S. Pyridyl thiosemicarbazide: synthesis, crystal structure, DFT/B3LYP, molecular docking studies and its biological investigations. Chem Cent J. 2018;12:1–7. doi:10.1186/s13065-018-0469-3
56. Makam P, Kankanala R, Prakash A, Kannan T. 2-(2-Hydrazinyl)thiazole derivatives: design, synthesis and in vitro antimycobacterial studies. Eur J Med Chem. 2013;69:564–576. doi:10.1016/j.ejmech.2013.08.054
57. Franzblau SG, Witzig RS, McLaughlin JC, et al. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J Clin Microbiol. 1998;36:362–366. doi:10.1128/JCM.36.2.362-366.1998
58. Korycka-Machała M, Kawka M, Lach J, et al. 2,4- Disubstituted pyridine derivatives are effective against intracellular and biofilm-forming tubercle bacilli. Front Pharmacol. 2022;13:1004632. doi:10.3389/fphar.2022.1004632
59. Korycka-Machała M, Viljoen A, Pawełczyk J, et al. 1 H-Benzo [d] Imidazole derivatives affect MmpL3 in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2019;63(10):10–128. doi:10.1128/AAC.00441-19
60. Korycka-Machała M, Brzostek A, Dziadek B, et al. Evaluation of the mycobactericidal effect of thio-functionalized carbohydrate derivatives. Molecules. 2017;22(5):812. doi:10.3390/molecules22050812
61. Korycka-Machała M, Pawełczyk J, Borówka P, et al. PPE51 Is Involved in the Uptake of Disaccharides by Mycobacterium tuberculosis. Cells. 2020;9(3):603. doi:10.3390/cells9030603
62. Sabbah M, Mendes V, Vistal RG, et al. Fragment-based design of Mycobacterium tuberculosis InhA inhibitors. J Med Chem. 2020;63:4749–4761. doi:10.1021/acs.jmedchem.0c00007
63. He X, Alian A, Stroud R, Ortiz de Montellano PR. Pyrrolidine carboxamides as a novel class of inhibitors of enoyl acyl carrier protein reductase from Mycobacterium tuberculosis. J Med Chem. 2006;49(21):6308–6323. doi:10.1021/jm060715y
64. Ha NX, Le CH. In silico molecular docking and ADMET study of Isodon coetsa phytochemicals targeting TNF‐α in inflammation‐mediated diseases. Vietnam J Chem. 2024;62(3):387–393. doi:10.1002/vjch.202300187
65. Eberhardt J, Santos-Martins D, Tillack AF, Forli S. AutoDock Vina 1.2. 0: new docking methods, expanded force field, and python bindings. J Chem Inf Model. 2021;61(8):3891–3898. doi:10.1021/acs.jcim.1c00203
66. Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. doi:10.1002/jcc.20291
67. Johansen MD, Herrmann JL, Kremer L. Nontuberculous mycobacteria and the rise of Mycobacterium abscessus. Nat Rev Microbiol. 2020;18(7):392–407.
68. Bekier A, Kawka M, Lach J, et al. Imidazole-thiosemicarbazide derivatives as potent anti-Mycobacterium tuberculosis compounds with antibiofilm activity. Cells. 2021;10:3476. doi:10.3390/cells10123476
69. Kawka M, Brzostek A, Dzitko K, et al. Mycobacterium tuberculosis binds human serum amyloid A, and the interaction modulates the colonization of human macrophages and the transcriptional response of the pathogen. Cells. 2021;10(5):1264. doi:10.3390/cells10051264
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.