Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
Intronic Variants in OCT1 are Associated with All-Cause and Cardiovascular Mortality in Metformin Users with Type 2 Diabetes
Authors Schweighofer N, Genser B, Maerz W, Kleber ME ![]() , Trummer O, Pieber TR
, Trummer O, Pieber TR ![]() , Obermayer-Pietsch B
, Obermayer-Pietsch B
Received 22 October 2019
Accepted for publication 1 May 2020
Published 18 June 2020 Volume 2020:13 Pages 2069—2080
DOI https://doi.org/10.2147/DMSO.S235663
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Antonio Brunetti
Natascha Schweighofer,1,2 Bernd Genser,3,4 Winfried Maerz,5,6 Marcus E Kleber,5 Olivia Trummer,1 Thomas R Pieber,1,2 Barbara Obermayer-Pietsch1,2
1Department of Internal Medicine, Division of Endocrinology and Diabetology, Medical University Graz, Graz, Austria; 2CBmed GmbH, Center for Biomarker Research in Medicine, Graz, Austria; 3BG Stats Consulting, Vienna, Austria; 4Institute of Public Health, Social and Preventive Medicine, Medical Faculty of Mannheim, University of Heidelberg, Mannheim, Germany; 5Medical Clinic V (Nephrology, Hypertensiology, Rheumatology, Endocrinology, Diabetology), Medical Faculty Mannheim, University of Heidelberg, Mannheim, Germany; 6SynLaboratory Academy, SynLaboratory Holding Deutschland GmbH, Mannheim and Augsburg, Germany
Correspondence: Barbara Obermayer-Pietsch
Department of Internal Medicine, Division of Endocrinology and Diabetology, Medical University Graz, Auenbruggerplatz 15, Graz A-8036, Austria
Tel +43 316 385 80253
Fax +43 316 385 13428
Email [email protected]
Purpose: Organic cation transporters (Octs) use cations like endogenous compounds, toxins, and drugs, such as metformin, as substrates. Therefore, these proteins determine the pharmacokinetics and -dynamics of metformin and thus its efficacy. Of note, metformin is today the most commonly used pharmaceutical in the treatment of type 2 diabetes (T2DM) with nevertheless a great variability in clinical response, which attributes to genetic variances. The aim of this study was to determine the influence of intronic OCT1 SNPs on prevalence of all-cause and cardiovascular death.
Patients and Methods: Genotypes of 27 intronic SNPs in OCT1 were investigated in the LURIC study, a prospective cohort of 3316 participants scheduled for coronary angiography. We investigated whether these variants were associated with all-cause and cardiovascular death in 73 individuals with T2DM under metformin therapy, in individuals without diabetes, individuals with T2DM and individuals with T2DM without metformin therapy.
Results: In a multivariate Cox regression analysis adjusted for classical cardiovascular risk factors, 4 intronic OCT1 SNPs were significantly associated with all-cause and cardiovascular mortality in individuals with T2DM on metformin therapy.
Conclusion: According to their OCT1 genotype, some individuals with T2DM on metformin therapy might be prone to an increased risk of cardiovascular death.
Keywords: organic cation transporter 1, SNP, T2DM, cardiovascular death, metformin
Introduction
According to WHO data collected in 2014, cardiovascular diseases (CVDs) are globally the primary cause of death. Data obtained in the year 2016 showed that at least 17.9 million of people died from CVDs representing 31% of all deaths worldwide.1 In Europe, more than half of all deaths are caused by CVDs.
Individuals with type 2 diabetes mellitus (T2DM) show significantly increased cardiovascular morbidity and all-cause mortality compared to subjects without diabetes.2,3 In T2DM, coronary artery disease (CAD) and stroke increase 2.4-fold and the risk of heart failure increases 2.8-fold due to diabetic vascular disease.4
Metformin is the recommended first-line therapy and hence the most frequently prescribed drug in T2DM treatment. A recent review by Griffin and colleagues targeted the influence of metformin therapy on cardiovascular diseases and showed that all outcomes, except the risk of stroke, were improved by the use of metformin; however, none of these associations achieved statistical significance.5
Metformin reduces glucose absorption in the gastrointestinal tract and suppresses hepatic gluconeogenesis by inhibiting the mitochondrial respiratory chain complex6,7 I: leading to a decrease in gluconeogenetic enzymes,8 II: via AMPK to an increased fatty acid uptake and beta-oxidation9 and III: inhibition of mitochondrial glycerophosphate dehydrogenase.10 AMPK activation can also occur independent of AMP, complex I and mitochondria.11 In the gastrointestinal tract metformin changes the microbiota composition.11 As seen in mice, metformin increases the number of Lactobacillus species, which in turn leads to a normalization of sodium–glucose cotransporter-1 (SGLT1) expression in the host which is changed in T2DM. Via the induction of GLP-1 secretion, SGLT1 decreases glucose production.12–14
In addition to its effects on glycemic control, it exerts favourable effects on surrogate parameters like body mass index (BMI) and waist circumference and reduces micro- and macrovascular complications.15 In the last 6 years, increasing evidence points to the effectiveness of metformin in the treatment of cancer.16 Metformin not only proved to be useful in the treatment of T2DM but also of pre-diabetes, type 1 diabetes mellitus, polycystic ovary syndrome and gestational diabetes. Positive effects of metformin were also seen in congestive heart failure, chronic liver and kidney disease, multiple sclerosis, or non-alcoholic fatty liver disease (reviewed in17). This drug not only might be used in the future for treatment of further disorders but also in anti-aging therapy. Current research to examine the potential of metformin on slowing the progress of age-related and age-dependent diseases in elderly individuals is ongoing.18
Metformin needs membrane transporters to penetrate organs and cross cell membranes due to its low hydrophobicity. Since metformin is not metabolized in the body, transport proteins regulating its gastrointestinal and hepatic uptake and renal elimination are particularly important in determining metformin pharmacokinetics and -dynamics. Organic cation transporter family members (Oct) are involved in the transport of small organic cations, including drugs, endogenous compounds or toxic substances with distinct molecular structures.19 Metformin is a substrate of the gastrointestinal and liver expressed Oct1, which is mainly responsible for the uptake.20,21 Of note, there is a reproducible heritability of glycemic response to metformin, up to 34%, based on genome-wide complex trait analyses. This suggests an important influence of genetic variants on the variance in glycemic response to metformin.22 The effects of coding SNPs in OCT1 on uptake, and thus efficacy of metformin, have extensively been investigated by a variety of studies.23–25 Recent work done in the field has begun to look more closely onto intronic SNPs in this region, since, on the one hand, genome-wide association studies (GWAS) targeting type 1 and type 2 diabetes showed top hits located in intronic regions of various genes26,27 and, on the other hand, metformin pharmacokinetics and -dynamics have been linked to genetic variants in transcription factors.28 Thus, coding as well as non-coding SNPs in OCT 1 play an important role in inter-patient difference of metformin efficacy.29–34 The investigated SNPs might therefore be important for metformin use in a variety of diseases.
The aim of this study is to determine the influence of intronic SNPs in one of the Oct transporter genes (OCT1), on critical outcomes in a large European cardiovascular risk cohort, such as the prevalence of all-cause and cardiovascular death where endpoint data were available. We investigate, whether this effect is metformin-dependent and thus relevant to metformin-users only or might be a more generalized effect.
Patients and Methods
Participants, Study Description, and Definition of Comorbidities
Data were obtained from the Ludwigshafen Risk and Cardiovascular Health (LURIC) study, a prospective cohort study designed to evaluate the determinants of cardiovascular health.35–37 3316 Caucasian subjects, aged 62.6 ± 10 years, referred for coronary angiography between July 1997 and January 2000, were recruited at a coronary care tertiary referral center (Herzzentrum Ludwigshafen, Germany). Participants with acute illness (except for acute coronary syndrome), non-cardiac chronic disease, or malignant neoplasms within the past 5 years were excluded. Written informed consent from each participant and the study approval by the institutional review board at the Ärztekammer Rheinland-Pfalz were obtained. The study was conducted in accordance with the Declaration of Helsinki. More detailed information about subjects in the study, examinations, recruitment and comorbidities have been previously described.35,38
Of note, during the recruitment phase of the LURIC study, metformin was not the first-line therapy option in type 2 diabetes therapy, due to the occurrence of several cases of lactic acidosis and consecutive safety concerns, leading to only 73 metformin users in this study. In the follow-up period (median 9.9 years), 894 (27%) of the study participants died. During follow-up no patient was lost. Local registries were used to gain information on mortality. Classification of death due to cardiovascular or non-cardiovascular causes was done by the use of death certificates. Classification of causes of death was done by two physicians who reviewed death certificates and hospital records without knowledge of study participants’ baseline characteristics. Cardiovascular deaths included sudden cardiac death (SCD), fatal myocardial infarction, deaths due to heart failure, death after intervention to treat CAD, stroke, and other deaths due to heart disease. SCD was defined as a sudden unexpected death either within 1 h of onset of symptoms or within 24 h of having been observed alive and without symptoms.
The presence of a visible luminal narrowing (>20% stenosis) in at least 1 of 15 coronary segments in coronary imaging was used to define coronary artery disease (CAD) according to the classification of the American Heart Association.35 Hypertension was defined as a systolic and/or diastolic blood pressure ≥140 and/or ≥90 mmHg or a significant history of hypertension. The glomerular filtration rate was estimated by using the 2012 CKD-EPI eGFRcreatcys equation.39 Pre-diabetes and diabetes were determined according to the American Diabetes Association (ADA) criteria.40 Impaired fasting glycemia (IFG) was determined by plasma glucose concentrations between 5.6 and 6.9 mM, and fasting type 2 diabetes mellitus was determined by plasma glucose concentrations ≥7.0 mM or HbA1c levels ≥6.5%. Based on a 2 h post-oral glucose tolerance test (oGTT), impaired glucose tolerance (IGT) was diagnosed by plasma glucose concentrations between 7.8 and 11.0 mM, and 2 h post-load type 2 diabetes mellitus by plasma glucose concentrations ≥11.1 mM. Individuals who required antidiabetic medication (ie oral antidiabetic and/or insulin use for control of glycemia) were also defined as diabetic41. Number of subjects per group is shown in Figure 1. Individuals included in this analysis either belonged to the group of individuals without diabetes (no diabetes: ND) or to the group of individuals with type 2 diabetes mellitus (T2DM), both groups together are referred to as all individuals (all). The latter was divided into metformin users (metformin users with type 2 diabetes: MUT) and non-metformin users (non-metformin users with type 2 diabetes: NMUT). Number of individuals in each group is described in the flow chart (Figure 1).
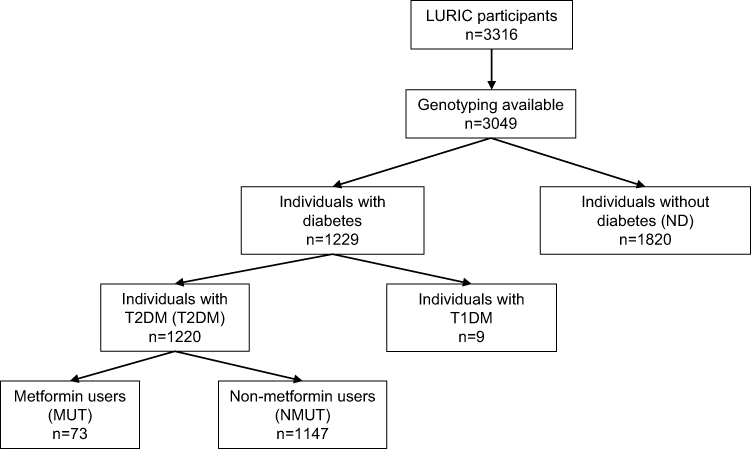 |
Figure 1 Flow chart of subjects per investigated group. Abbreviations: T2DM, type 2 diabetes mellitus; T1DM, type 1 diabetes mellitus. |
SNP Selection and Analysis of Functional Consequences
Based on a minor allele frequency (MAF) >0.01 in a central European population, we selected 34 non-coding, intronic SNPs in the transporter gene OCT1 (gene SLC22A1 and 4 kb upstream of the first translational start site). Linkage disequilibrium (LD) analysis was performed with the LDlink tool of the NIH National cancer institute (RRID: SCR_011403, https://analysistools.nci.nih.gov/LDlink/) and was additionally checked in HaploReg v4.1 (RRID: SCR_006796).42,43 The investigated SNPs were not in LD with any coding SNPs in OCT1.
OCT1 genotyping data were available from 3061 individuals (92.3% of the entire cohort) due to technical reasons. OCT1 SNPs were imputed in pre-existing genotyping data and an in silico analysis was performed.
Analysis of cis or trans regulation of gene expression was determined by database search (GTEx Portal V8 (RRID: SCR_001618) or HaploReg v4.1 (RRID: SCR_006796).42,43 Changes in transcription factor binding sites and sites of epigenetic modification were determined via HaploReg v4.1.
The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 02/05/2020 and/or dbGaP accession number phs000424.vN.pN on 02/05/2020.
Statistical Analysis
Associations between the non-coding SNPs in OCT1 and changes in all-cause and cardiovascular mortality were analyzed using multivariate Cox regression. Hazard ratios (HR) with 95% CIs for the mortality categories all-cause death and cardiovascular death were calculated using Cox proportional hazards regression models, which enabled adjustment for potential confounding parameters. In these analyses, an additive model was calculated, the unadjusted model describes the crude association, the adjusted model was adjusted for sex, BMI, systolic blood pressure, hypertension, lipid parameters, C-reactive protein (CRP), sodium levels, cortisol, cystatin c, NT-pro-BNP, arterial fibrillation, left ventricular hypertrophy, smoking and coronary artery disease stages, respectively.
To determine the impact of glycemic control on the SNP effects on CVD, we adjusted in a third model additionally for HbA1c and HOMA-IR. HOMA-IR was calculated as follows: (fasting glucose [mmol/L] × fasting insulin [U/L])/22.5. Changes in HR are given per minor allele present and are referred to the homozygous major allele of each SNP, respectively.
Associations between investigated SNPs and mortality categories were determined for all LURIC participants. Subclassification of individuals with type 2 diabetes in metformin users, non-metformin users and comparison with subjects without diabetes should indicate whether the effect is metformin dependent or not.
Statistical significance was defined as p<0.05. Statistical analyses were done using the statistical software package STATA, StataCorp. 2017. Stata Statistical Software: Release 15. College Station, TX: StataCorp LLC.
Results
Baseline Characteristics of the Study Cohort
The baseline characteristics of the study cohort are given in Table 1.
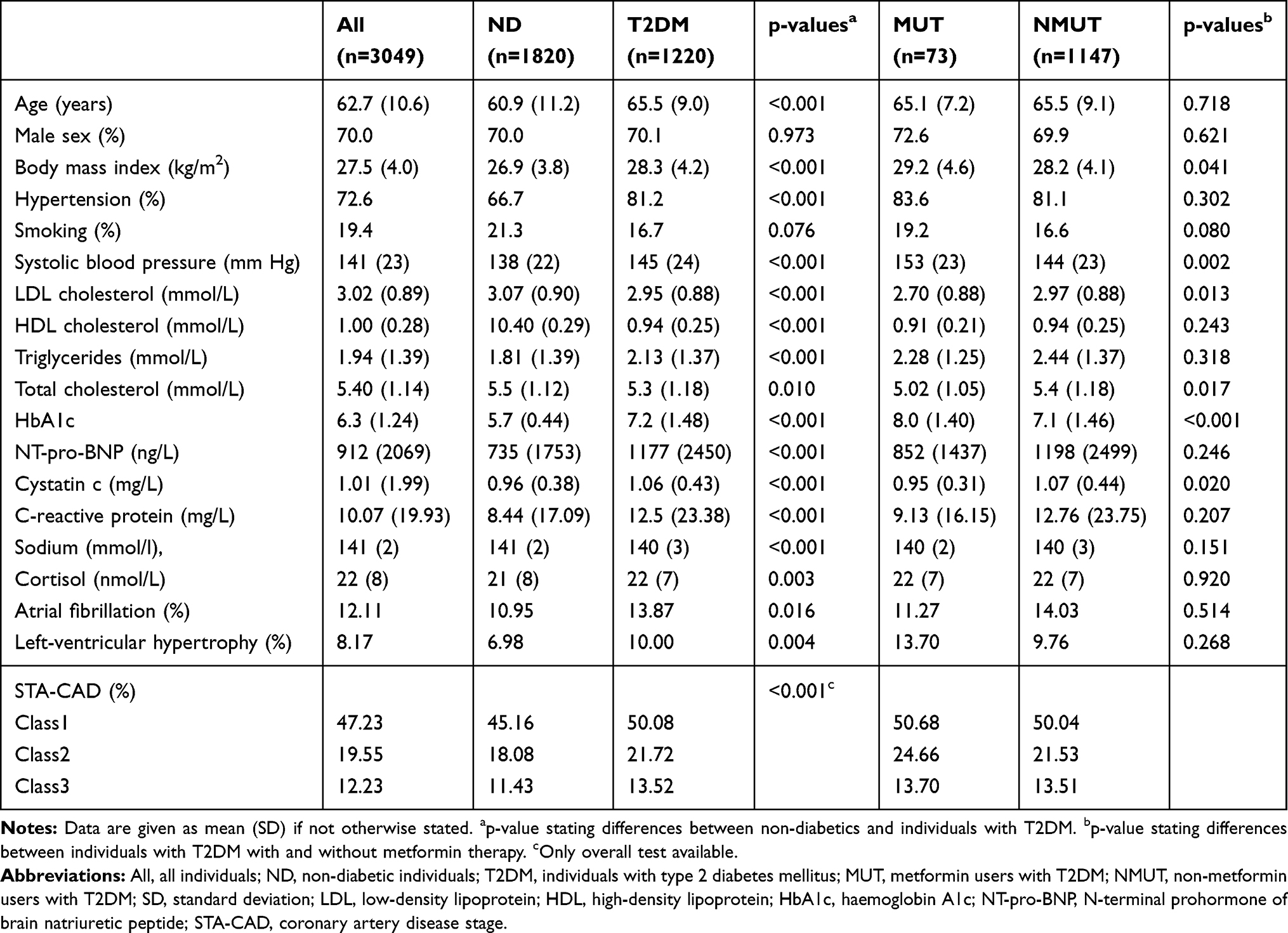 |
Table 1 Baseline Characteristics of the Study Participants |
Data on Mortality of the Investigated Cohort and Subgroups
Data on all-cause and cardiovascular death of all subjects and the investigated subgroups are given in Table 2. Data on all-cause and cardiovascular death per genotype in all subjects and metformin users with T2DM are given in Table 3.
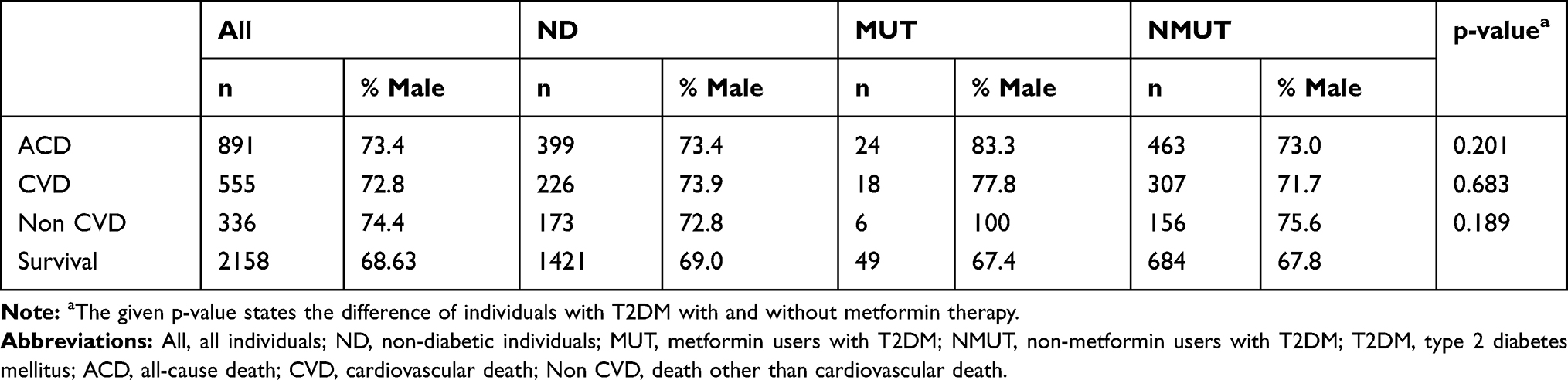 |
Table 2 Mortality Data of the Investigated Subgroups |
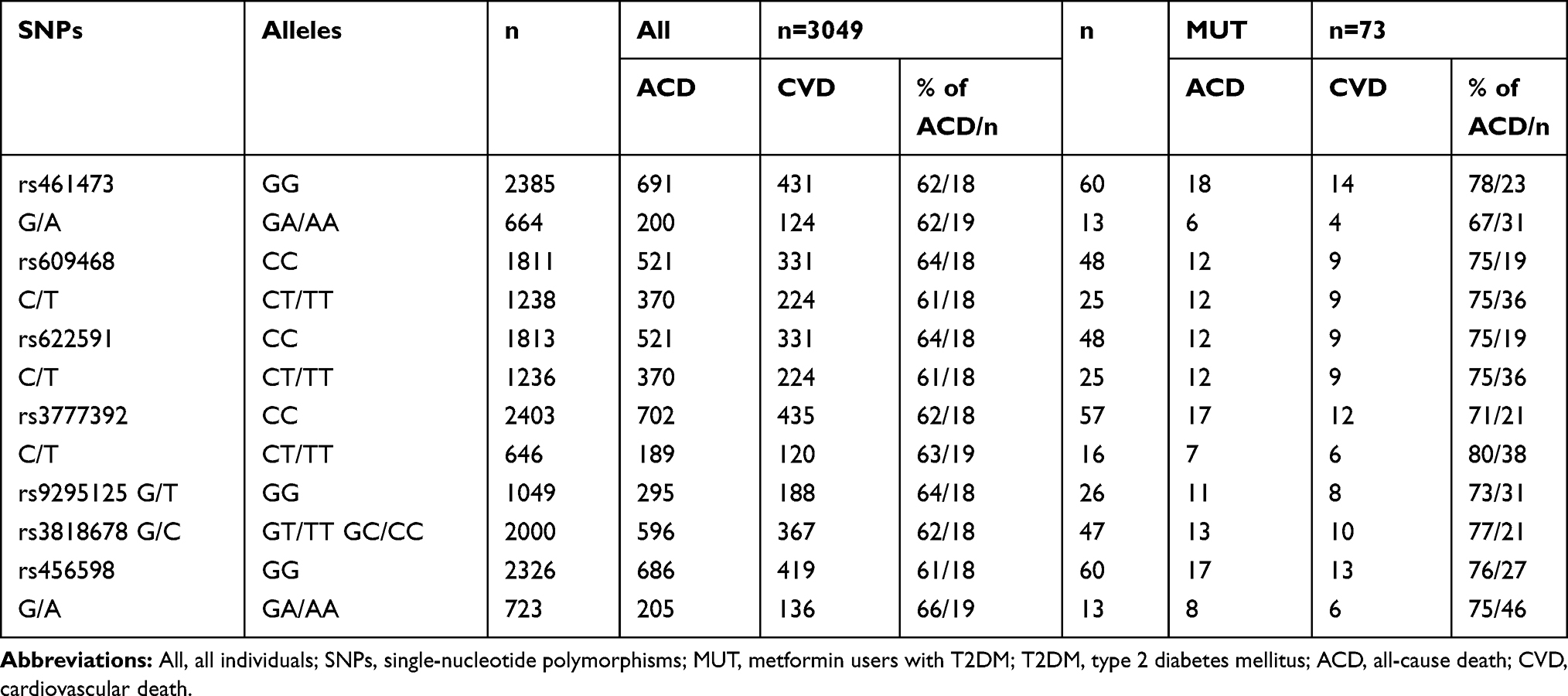 |
Table 3 Mortality Data per Genotype in All Individuals and Individuals with T2DM Using Metformin |
Minor Allele Frequency of the SNPs
The minor allele frequency of the intronic OCT1 SNPs rs461473, rs609468, rs622591, rs3777392, rs9295125/rs3818678 and rs456598 were 0.118, 0.229, 0.228, 0.114, 0.412 and 0.127 respectively in our cohort.
Association of OCT1 SNPs with All-Cause and Cardiovascular Death in Individuals with Type 2 Diabetes with and Without Metformin Therapy and Non-Diabetic Individuals
Only in metformin users with T2DM, SNP rs461473 was significantly associated with all-cause as well as cardiovascular death even after adjustment (see the 'Patients and Methods’ section). Each copy of the minor A allele was significantly associated with an increase in HR for all-cause and cardiovascular death (see Table 4 for details). No association with either all-cause or cardiovascular death was seen in non-metformin users with T2DM and non-diabetic subjects (see Tables 5 and 6 for details).
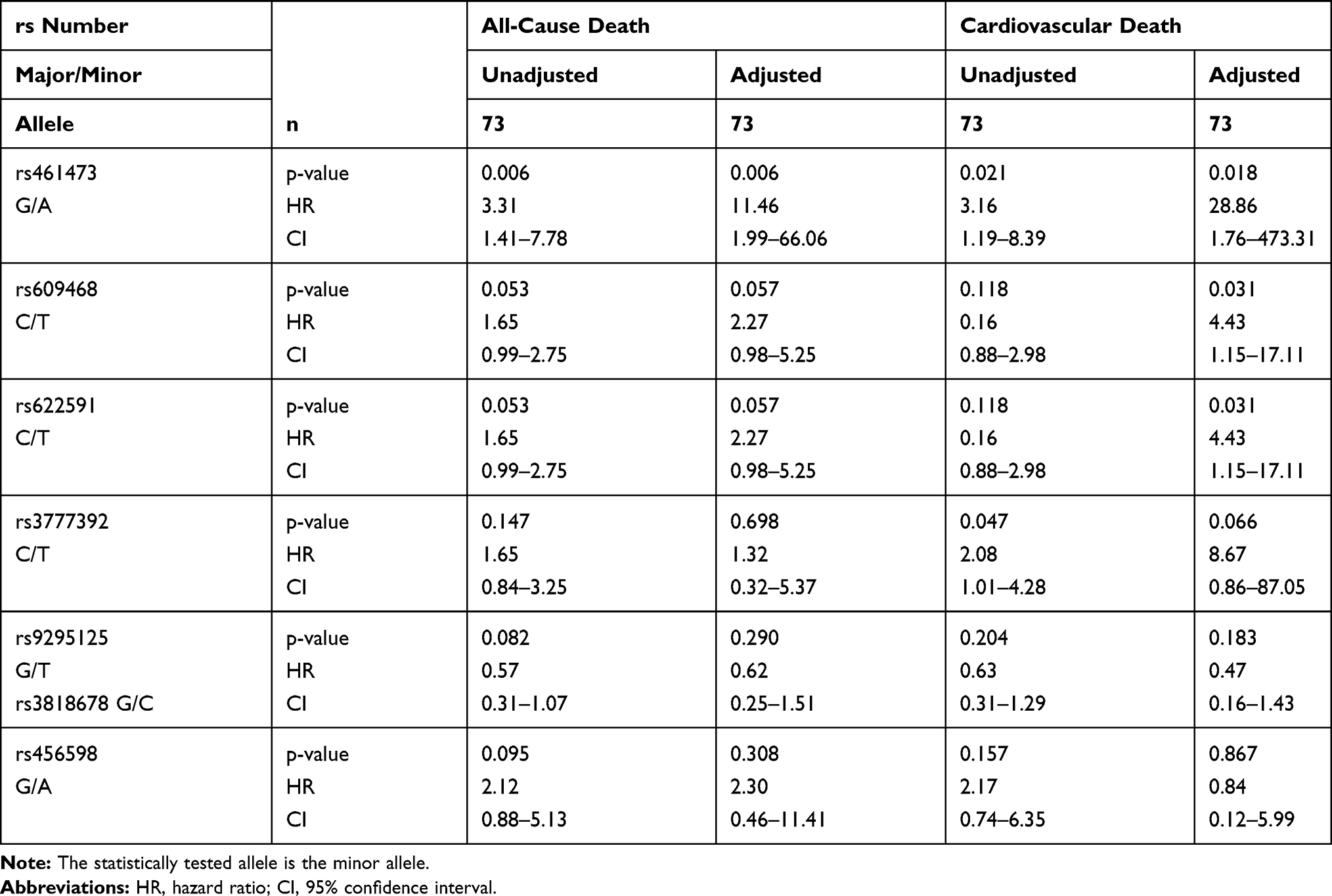 |
Table 4 Association of Intronic OCT1 SNPs with All-Cause and Cardiovascular Death in Metformin Users with T2DM (MUT) |
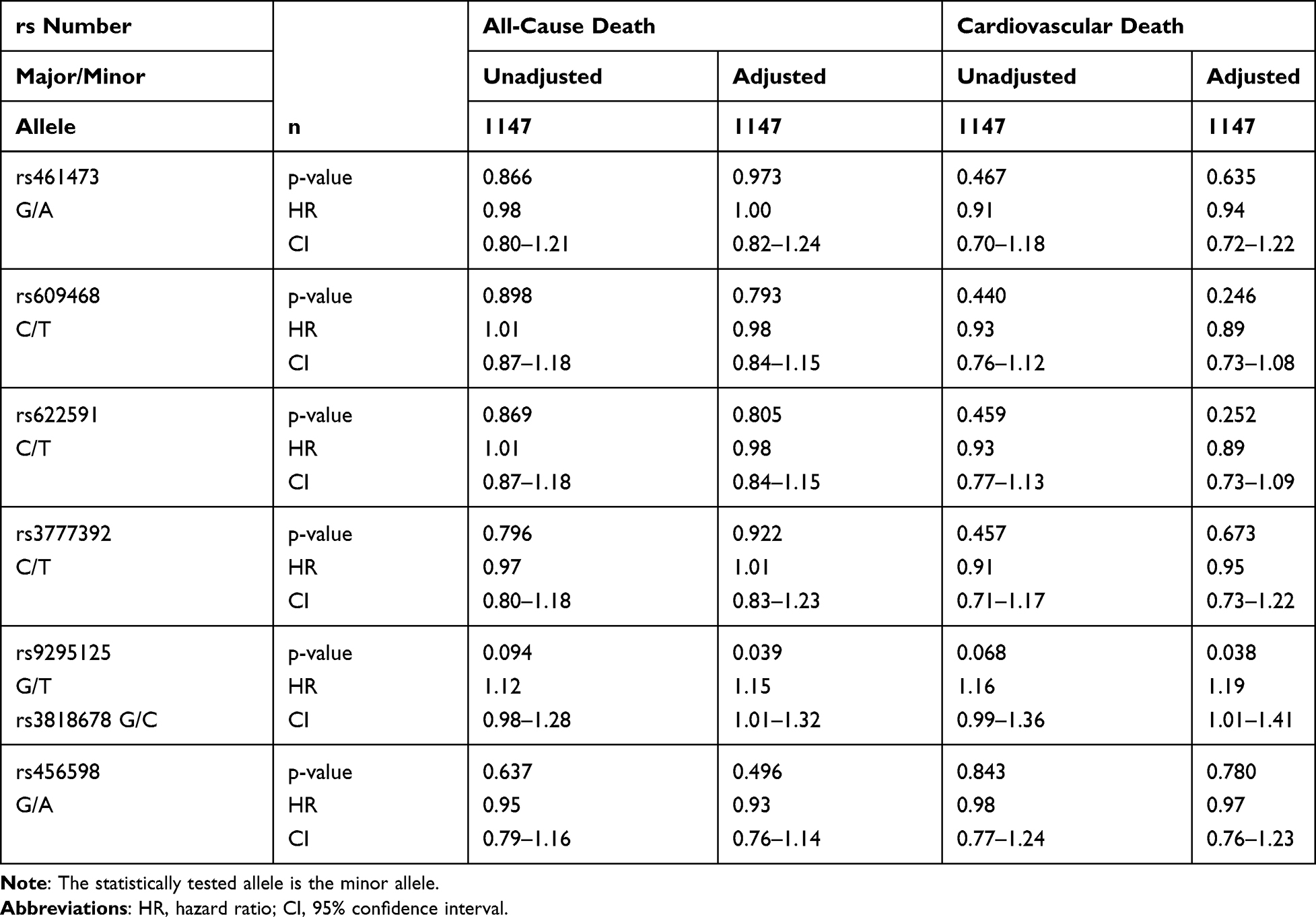 |
Table 5 Association of Intronic OCT1 SNPs with All-Cause and Cardiovascular Death in Non-Metformin Users with T2DM (NMUT) |
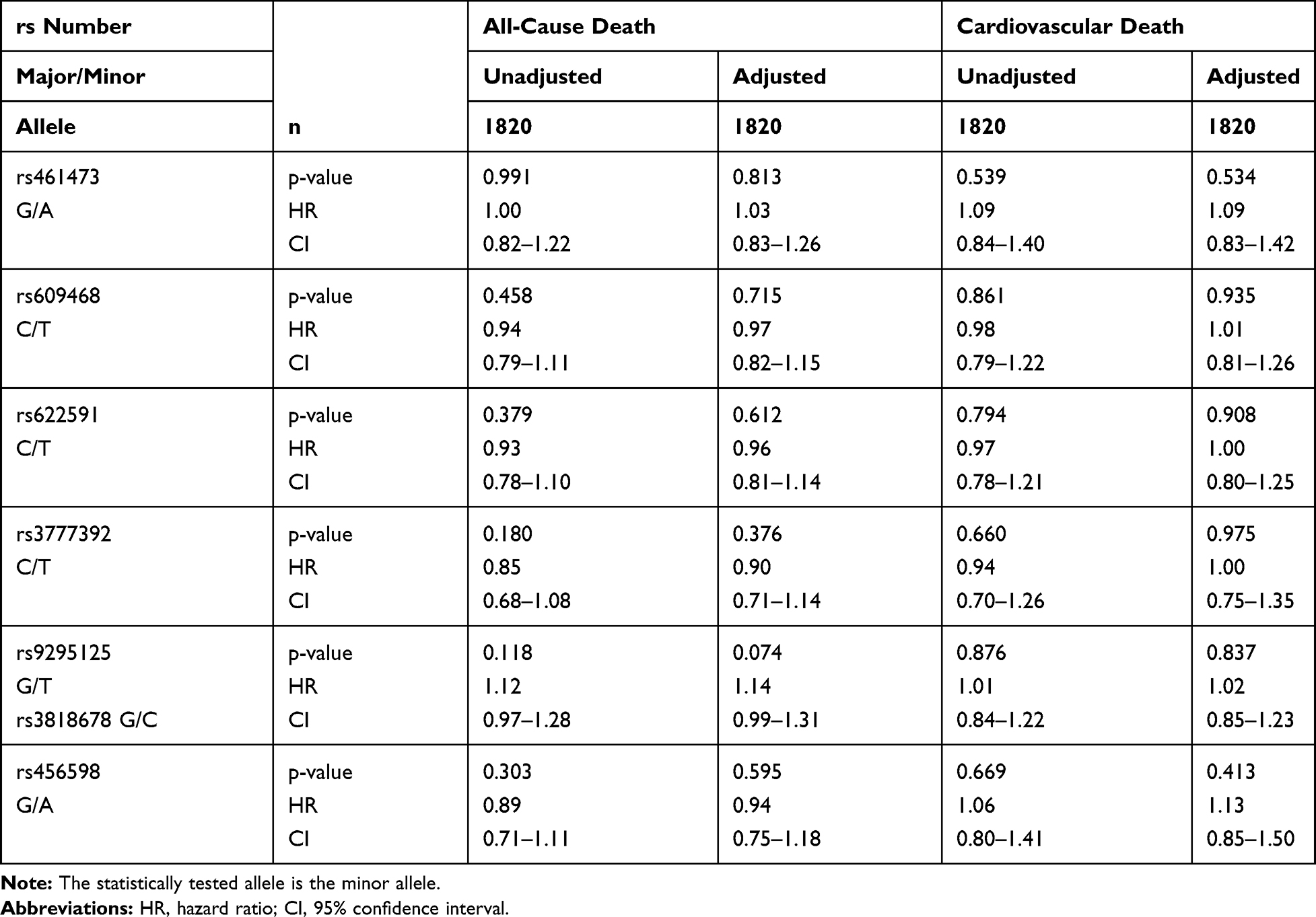 |
Table 6 Association of Intronic OCT1 SNPs with All-Cause and Cardiovascular Death in Non-Diabetic Patients (ND) |
SNPs rs609468 and rs622591 were borderline associated with all-cause death and only after adjustment significantly associated with cardiovascular death in metformin users with T2DM. Each copy of the minor T allele was associated with an increased HR, respectively. No associations with either all-cause or cardiovascular death were seen in non-metformin users with T2DM and non-diabetic subjects (see Tables 5 and 6 for details).
SNP rs3777392 was significantly associated with cardiovascular death in the unadjusted model, the association was only seen by trend after adjustment in metformin users with T2DM. None of the mentioned associations were seen in non-metformin users with T2DM or non-diabetics (see Tables 4–6 for details).
SNPs rs9295125, rs3818678 and rs456598 showed an association with all-cause death in metformin users with T2DM only in the unadjusted model, which disappeared after adjustment (see Table 4). The minor alleles of SNPs rs9295125 and rs3818678 showed a by trend association with all-cause death in the unadjusted model. Each copy of the minor allele was associated with a decrease in HR for all-cause death. In individuals with T2DM who were non-metformin users, both SNPs were by trend associated with all-cause and cardiovascular death in the unadjusted model, which was significant only after adjustment (see Table 5 for details). In this patient subgroup, the minor allele was associated with an increase in HR. In non-diabetic subjects, both SNPs showed an association with all-cause death only in the adjusted model. As in non-metformin users with T2DM, the minor allele of both SNPs was associated with an increased HR for all-cause death (see Table 5 for details). An overview of the influence of each minor allele of the investigated SNPs on the hazard ratio of cardiovascular risk is given in Figure 2.
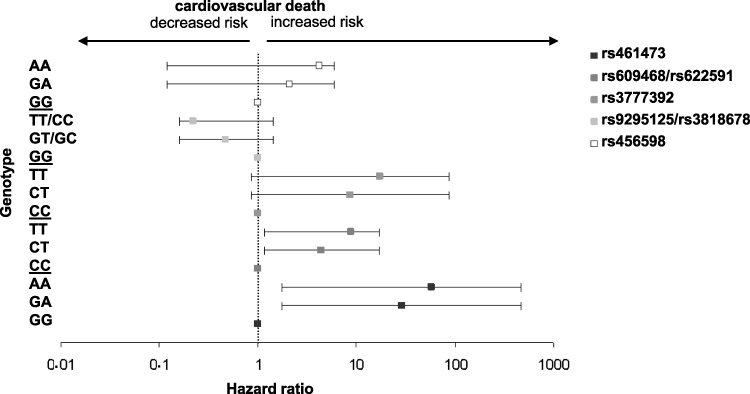 |
Figure 2 Hazard ratios for cardiovascular death per OCT1 genotype in diabetic metformin users. Squares: hazard ratios calculated with an additive cox proportional hazards regression model adjusted for confounders, error bars: 95% confidence interval, different shades of grey represent different intronic OCT1 SNPs or SNP combinations. Homozygosity for the major allele is set as 1. |
Impact of Glycemic Control on the Influence of Intronic OCT1 SNPs on All-Cause and Cardiovascular Death
To determine whether glycemic control may affect the described influence of intronic OCT1 SNPs on all-cause and cardiovascular death in metformin users with T2DM we included HbA1c and HOMA-IR in our analysis. Rs461473, rs609468 and rs622591 were still significantly associated with an increased risk of all-cause death per minor allele. An association with an increased risk of cardiovascular death per minor allele was still seen for rs609468 and rs622591 (both p=0.012) and rs461473 (p=0.061), rs1777392 (p=0.070) and rs9295125/rs3818678 (p=0.077). For details see Table 7.
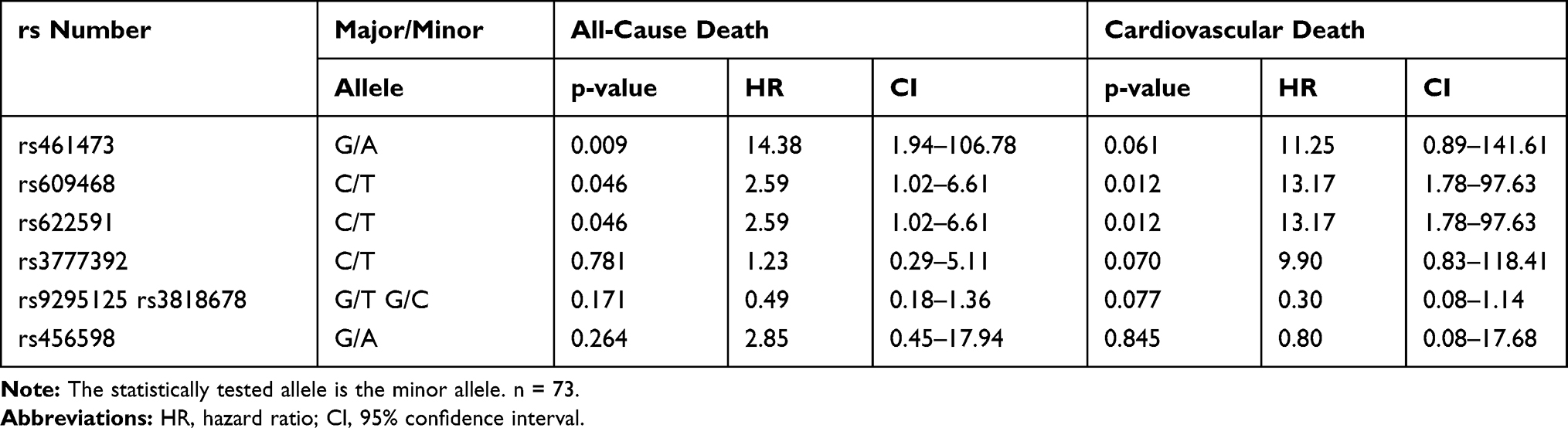 |
Table 7 Association of Intronic OCT1 SNPs with All-Cause and Cardiovascular Death in Metformin Users with T2DM (MUT) After Adjustment for Glycemic Control |
Prediction of Functional Consequences
Functional consequences of SNPs were determined by database search in HaploReg v4.1 and GETx Portal V8.
QTL Results
Rs461473, rs609468, rs622591, rs9295125, rs3818678 and rs456598 correlate with SLC22A1 expression. Rs9295125 correlates with expression of RP3-393E18.2, a large intergenic non-coding RNA locus, rs456598 with SOD2 expression and the presence of metabolites of the tryptophan and acylcarnitine metabolism.
Epigenomic Information
According to the chromatin 25-states model using 12 imputed marks, rs461473 creates a poised promoter in fat tissue, a primary H3K27ac possible enhancer in rectal mucosa and pancreatic islets and an active transcription start site in liver. It creates an active enhancer in the right atrium, duodenum mucosa, liver pancreas and an active promoter in the liver. In the liver it inactivates an enhancer and a promoter. Rs609468 creates an active enhancer in aorta, liver and an active promoter in the spleen. Rs622591 creates an active enhancer in the liver. Rs9295125 creates an active enhancer in fat, aorta and liver and rs3818678 in aorta, stomach smooth muscle, placenta and liver. Rs465698 generates an active enhancer in the right ventricle and lung and an active promoter in liver and spleen, whereas an inactive enhancer in the right ventricle is generated.
Changes in Transcription Factor Binding Motifs
Rs461473 and rs609468 do not lead to changes in transcription factor binding sites. All other SNPs change various transcription factor binding sites: rs622591 changes Dbx1, Hoxa4, Ifr, Mef2, Zfp105, rs9295125 Foxo, Pax-8, Pou1f1 and Rox11, rs3818678 DMRT5, Foxo and TCF4 and rs456598 AIRE binding sites.
Discussion
In this study, we investigated the effect of intronic OCT1 SNPs on all-cause and cardiovascular death in 3040 Caucasians with increased cardiovascular risk. We identified 4 SNPs (rs461473, rs609468, rs622591, rs3777392), which were significantly associated with an increased risk per minor allele for all-cause and/or cardiovascular death in 73 individuals with T2DM on metformin therapy. This effect varied between 4.4 and 28.9 fold increased risk of cardiovascular death per minor allele in metformin users with T2DM. After adjustment for parameters of glycemic control, rs609468 and rs622591 were still significantly associated with an increased risk per minor allele for cardiovascular death, implicating that glycemic control does not impact their influence on CVD risk. Rs461473, rs3777392 and rs9295125 showed an association by trend with cardiovascular death after adjustment for parameters HbA1c and HOMA-IR, suggesting that poor glycemic control in our cohort is partly, but not completely, responsible for the association detected.
Intronic OCT1 SNPs might be influencing gene expression by either acting in cis (on the OCT gene cluster), in trans or even by modification of enhancers (influencing other “metformin” transporters or proteins determining metformin efficacy). The alteration of OCT gene expression in “cis” by changing transcription factor binding sites (as predicted for rs622591, rs3777392, rs9295125, rs3818678 and rs456598) would not only directly impact the transport of metformin but also the transport of physiological substrates. The transport of metformin determines its concentrations in blood, hence in end organs, and in the gastrointestinal tract. The first determines the capacity of metformin to inhibit hepatic gluconeogenesis by inhibition of the mitochondrial respiratory chain complex.6,7 The second changes the microbiota composition in the gastrointestinal tract which might be achieved via inhibition of bacterial complex I homologues.44 Alterations in microbiota on the one hand decrease gut permeability45 and alter on the other hand the profile of bacterial products generated. Bacterial products per se might contribute to the effects of metformin on glucose metabolism11 or have direct effects on inflammation like indole,46 butyrate47 and small chain fatty acids.48 Since inflammation-mediated processes might be one of the main mechanisms the gut microbiota influences T2DM development and progression as well as CVD risk, metformin acts on both by decreasing systemic inflammation. This might be one of the mechanism rs622591 influences the risk of CVD, since Becker and colleagues could not see any effect of its genotypes on HbA1c levels.32
An alteration of OCT gene expression also affects the transport of their natural substrates like neurotransmitters, polyamines, prostaglandins and thiamine. All these substances are linked to glucose and lipid metabolism as well as cardiovascular disease either in humans or animal models49–62 and might thus be responsible for the observed increased cardiovascular risk. Christensen and colleagues could not detect any change of plasma metformin concentrations in individuals with different rs461473 alleles rather implicating a mechanism other than cis-regulation.22 This might include trans-regulatory processes like creating or destroying enhancers or via non-coding RNAs. Trans regulation may affect other transport proteins using metformin as a substrate63 like the plasma monoamine transporter (PMAT), the multidrug toxin and extrusion (MATE 1), and MATE 2, again influencing the metformin concentration in gut and thus the gut microbiota Trans regulation may also affect the expression of mediators of metformin action or efficacy like AMPK,64 LBK1,65 SRR and BDNF. Another candidate gene is SLC2A2, encoding the metformin target GLUT2 transporter.66 Rs456598 and rs3777392, ie affect SOD2 gene expression, which is associated with microvascular complications of diabetic ischemia.67 Intronic SNPs might also influence epigenetic regulation and thus the accessibility of chromatin as predicted for some of the SNPs.
Based on the results of this study, it might be of interest to monitor several individuals with T2DM currently using metformin, which might have an increased risk of cardiovascular incidents according to their OCT1 genotype. Since metformin is also used for a number of other indications than type 2 diabetes therapy, these findings might also concern other disease groups.
Major limitations of this study are the relatively small sample size of metformin users with T2DM due to lack of safety data for metformin ahead and during the recruitment period and the fact that we performed the analysis without direct access to sample material to measure additional parameters to the given ones. We did not select functional variants in this study, because their effect on metformin efficacy in our patient groups has already been widely investigated. Furthermore, statistical power to investigate some of these functional OCT1 SNPs known to be associated with decreased metformin efficacy was too low due to a very low allele frequency in our cohort. The selection of intronic SNPs might be another limitation of this study. In contrast to coding SNPs, which directly affect transporter efficacy by changing the protein sequence and thus the ability to transport its substrates, intronic SNPs may change the expression on the genes in their vicinity, or far from their location, enhancing further the complexity of possible ways of action.
Since these data were generated in a cohort with a small number of metformin users it is necessary to replicate our findings in a cohort reflecting presently usage of metformin as first-line therapy in type 2 diabetes treatment and to further include coding SNPs in OCT1 for the analysis of complex intertwining between metformin, glycemic control and thereafter cardiovascular outcomes.
Conclusion
We were able to show in a small number of persons with type 2 diabetes mellitus on metformin therapy a susceptibility to an increased risk of cardiovascular death according to their OCT1 genotype. This unexpected effect was only partly due to impaired glycemic control implicating other pleiotropic effects of metformin. This finding might also be interesting for potential metformin users in other indications than diabetes mellitus.
Acknowledgments
We specially thank our colleague Christoph W. Haudum for his assistance in data analysis and statistical analysis.
Disclosure
Dr Natascha Schweighofer reports grants from the Austrian Federal Government within the COMET K1 Centre Program, Land Steiermark and Land Wien, during the conduct of the study; and she is an employee in CBmed GmbH, Center for Biomarker Research in Medicine, outside the submitted work. Dr Marcus E Kleber reports personal fees from Bayer, outside the submitted work. Prof. Dr. Thomas R Pieber reports grants, personal fees from AstraZeneca, grants, personal fees from Novo Nordisk, personal fees from Adocia, personal fees from Arecor, personal fees from Sanofi, personal fees from Roche Diagnostics, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Mendis S, Armstrong T, Bettcher D, et al. Global Status Report on Noncommunicable Diseases 2014. World Health Organisation. 2014. ISBN 9789241564854.
2. Martín-Timón I. Type 2 diabetes and cardiovascular disease: have all risk factors the same strength? World J Diabetes. 2014;5(4):444. doi:10.4239/wjd.v5.i4.444
3. Gu K, Cowie CC, Harris MI. Diabetes and decline in heart disease mortality in US adults. J Am Med Assoc. 1999;281(14):1291–1297. doi:10.1001/jama.281.14.1291
4. Diabetes Drafting Group. Prevalence of small vessel and large vessel disease in diabetic patients from 14 centres - the World Health Organisation multinational study of vascular disease in diabetics. Diabetologia. 1985;28(8):615–640. doi:10.1007/BF00290267
5. Griffin SJ, Leaver JK, Irving GJ. Impact of metformin on cardiovascular disease: a meta-analysis of randomised trials among people with type 2 diabetes. Diabetologia. 2017;60(9):1620–1629. doi:10.1007/s00125-017-4337-9
6. El-Mir MY, Nogueira V, Fontaine E, Avéret N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275(1):223–228. doi:10.1074/jbc.275.1.223
7. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(3):607–614. doi:10.1042/0264-6021:3480607
8. Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494(7436):256–260. doi:10.1038/nature11808
9. Fullerton MD, Galic S, Marcinko K, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med. 2013;19(12):1649–1654. doi:10.1038/nm.3372
10. Madiraju AK, Erion DM, Rahimi Y, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510(7506):542–546. doi:10.1038/nature13270
11. Prattichizzo F, Giuliani A, Mensa E, et al. Pleiotropic effects of metformin: shaping the microbiome to manage type 2 diabetes and postpone ageing. Ageing Res Rev. 2018;48:87–98. doi:10.1016/j.arr.2018.10.003
12. Fiorentino TV, Suraci E, Arcidiacono GP, et al. Duodenal sodium/glucose cotransporter 1 expression under fasting conditions is associated with postload hyperglycemia. J Clin Endocrinol Metab. 2017;102(11):3979–3989. doi:10.1210/jc.2017-00348
13. Bauer PV, Duca FA, Waise TMZ, et al. Metformin alters upper small intestinal microbiota that impact a glucose-SGLT1-sensing glucoregulatory pathway. Cell Metab. 2018;27(1):101–117.e5. doi:10.1016/j.cmet.2017.09.019
14. Gorboulev V, Rehman S, Albert CM, et al. Assay conditions influence affinities of rat organic cation transporter 1: analysis of mutagenesis in the modeled outward-facing cleft by measuring effects of substrates and inhibitors on initial uptake. Mol Pharmacol. 2018;93(4):402–415. doi:10.1124/mol.117.110767
15. Papanas N, Maltezos E, Mikhailidis DP. Metformin: diamonds are forever. Expert Opin Pharmacother. 2009;10(15):2395–2397. doi:10.1517/14656560903176453
16. Heckman-Stoddard BM, DeCensi A, Sahasrabuddhe VV, Ford LG. Repurposing metformin for the prevention of cancer and cancer recurrence. Diabetologia. 2017;60(9):1639–1647. doi:10.1007/s00125-017-4372-6
17. Romero R, Erez O, Hüttemann M, et al. Metformin, the aspirin of the 21st century: its role in gestational diabetes mellitus, prevention of preeclampsia and cancer, and the promotion of longevity. Am J Obstet Gynecol. 2017;217(3):282–302. doi:10.1016/j.ajog.2017.06.003
18. Valencia WM, Palacio A, Tamariz L, Florez H. Metformin and ageing: improving ageing outcomes beyond glycaemic control. Diabetologia. 2017;60(9):1630–1638. doi:10.1007/s00125-017-4349-5
19. Sakata T, Anzai N, Kimura T, et al. Functional analysis of human organic cation transporter OCT3 (SLC22A3) polymorphisms. J Pharmacol Sci. 2010;113(3):263–266. doi:10.1254/jphs.09331SC
20. Wang DS, Jonker JW, Kato Y, Kusuhara H, Schinkel AH, Sugiyama Y. Involvement of organic cation transporter 1 in hepatic and intestinal distribution of metformin. J Pharmacol Exp Ther. 2002;302(2):510–515. doi:10.1124/jpet.102.034140
21. Dujic T, Causevic A, Bego T, et al. Organic cation transporter 1 variants and gastrointestinal side effects of metformin in patients with type 2 diabetes. Diabet Med. 2016;33(4):511–514. doi:10.1111/dme.13040
22. Christensen MMH, Brasch-Andersen C, Green H, et al. The pharmacogenetics of metformin and its impact on plasma metformin steady-state levels and glycosylated hemoglobin A1c. Pharmacogenet Genomics. 2011;21(12):837–850. doi:10.1097/FPC.0b013e32834c0010
23. Tzvetkov MV, Vormfelde SV, Balen D, et al. The effects of genetic polymorphisms in the organic cation transporters OCT1, OCT2, and OCT3 on the renal clearance of metformin. Clin Pharmacol Ther. 2009;86(3):299–306. doi:10.1038/clpt.2009.92
24. Becker ML, Visser LE, Van Schaik RHN, Hofman A, Uitterlinden AG, Stricker BHC. Interaction between polymorphisms in the OCT1 and MATE1 transporter and metformin response. Pharmacogenet Genomics. 2010;20(1):38–44. doi:10.1097/FPC.0b013e328333bb11
25. Bradfield JP, Qu HQ, Wang K, et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet. 2011;7(9):e1002293. doi:10.1371/journal.pgen.1002293
26. Saxena R, Voight BF, Lyssenko V, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316(5829):1331–1336. doi:10.1126/science.1142358
27. Goswami S, Yee SW, Stocker S, et al. Genetic variants in transcription factors are associated with the pharmacokinetics and pharmacodynamics of metformin. Clin Pharmacol Ther. 2014;96(3):370–379. doi:10.1038/clpt.2014.109
28. Reitman ML, Schadt EE. Pharmacogenetics of metformin response: a step in the path toward personalized medicine. J Clin Invest. 2007;117(5):1226–1229. doi:10.1172/JCI32133
29. Shikata E, Yamamoto R, Takane H, et al. Human organic cation transporter (OCT1 and OCT2) gene polymorphisms and therapeutic effects of metformin. J Hum Genet. 2007;52(2):117–122. doi:10.1007/s10038-006-0087-0
30. Shu Y, Sheardown SA, Brown C, et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest. 2007;117(5):1422–1431. doi:10.1172/JCI30558
31. Zhou K, Donnelly LA, Kimber CH, et al. Reduced-function SLC22A1 polymorphisms encoding organic cation transporter 1 and glycemic response to metformin: a GoDARTS study. Diabetes. 2009;58(6):1434–1439. doi:10.2337/db08-0896
32. Becker ML, Visser LE, van Schaik RHN, Hofman A, Uitterlinden AG, Stricker BHC. Genetic variation in the organic cation transporter 1 is associated with metformin response in patients with diabetes mellitus. Pharmacogenomics J. 2009;9(4):242–247. doi:10.1038/tpj.2009.15
33. Dujic T, Zhou K, Yee SW, et al. Variants in pharmacokinetic transporters and glycemic response to metformin: a metgen meta-analysis. Clin Pharmacol Ther. 2017;101(6):763–772. doi:10.1002/cpt.567
34. Schweighofer N, Lerchbaum E, Trummer O, Schwetz V, Pieber T, Obermayer-Pietsch B. Metformin resistance alleles in polycystic ovary syndrome: pattern and association with glucose metabolism. Pharmacogenomics. 2014;15(3):305–317. doi:10.2217/pgs.13.223
35. Winkelmann BR, März W, Boehm BO, et al. Rationale and design of the LURIC study - a resource for functional genomics, pharmacogenomics and long-term prognosis of cardiovascular disease. Pharmacogenomics. 2001;2(1 SUPPL. 1):S1–S73. doi:10.1517/14622416.2.1.S1
36. Pilz S, März W, Wellnitz B, et al. Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J Clin Endocrinol Metab. 2008;93(10):3927–3935. doi:10.1210/jc.2008-0784
37. Thomas GN, Hartaigh BÓ, Bosch JA, et al. Vitamin D levels predict all-cause and cardiovascular disease mortality in subjects with the metabolic syndrome: the Ludwigshafen Risk and Cardiovascular health (LURIC) study. Diabetes Care. 2012;35(5):1158–1164. doi:10.2337/dc11-1714
38. Brandenburg VM, Kleber ME, Vervloet MG, et al. Fibroblast growth factor 23 (FGF23) and mortality: the Ludwigshafen risk and cardiovascular health study. Atherosclerosis. 2014;237(1):53–59. doi:10.1016/j.atherosclerosis.2014.08.037
39. Inker LA, Schmid CH, Tighiouart H, et al. Estimating glomerular filtration rate from serum creatinine and cystatin C. N Engl J Med. 2012;367(1):20–29. doi:10.1056/NEJMoa1114248
40. American Diabetes Association. Summary of revisions to the 2011 clinical practice recommendations. Diabetes Care. 2011;34(Suppl 1).
41. O’Hartaigh B, Neil Thomas G, Silbernagel G, et al. Association of 25-hydroxyvitamin D with type 2 diabetes among patients undergoing coronary angiography: cross-sectional findings from the LUdwigshafen Risk and Cardiovascular Health (LURIC) study. Clin Endocrinol (Oxf). 2013;79(2):192–198. doi:10.1111/cen.12024
42. Ward LD, Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016;44(D1):D877–D881. doi:10.1093/nar/gkv1340
43. Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40(D1):D930–D934. doi:10.1093/nar/gkr917
44. Friedrich T, Scheide D. The respiratory complex I of bacteria, archaea and eukarya and its module common with membrane-bound multisubunit hydrogenases. FEBS Lett. 2000;479(1–2):1–5. doi:10.1016/s0014-5793(00)01867-6
45. Chelakkot C, Choi Y, Kim DK, et al. Akkermansia muciniphila-derived extracellular vesicles influence gut permeability through the regulation of tight junctions. Exp Mol Med. 2018;50(2):e450–11. doi:10.1038/emm.2017.282
46. Beaumont M, Neyrinck AM, Olivares M, et al. The gut microbiota metabolite indole alleviates liver inflammation in mice. FASEB J Off Publ Fed Am Soc Exp Biol. 2018:fj201800544. doi:10.1096/fj.201800544.
47. Wang X, He G, Peng Y, Zhong W, Wang Y, Zhang B. Sodium butyrate alleviates adipocyte inflammation by inhibiting NLRP3 pathway. Sci Rep. 2015;5(1):12676. doi:10.1038/srep12676
48. Vinolo MAR, Rodrigues HG, Nachbar RT, Curi R. Regulation of inflammation by short chain fatty acids. Nutrients. 2011;3(10):858–876. doi:10.3390/nu3100858
49. Turgan N, Ersöz B, Çoker C, Hamulu F, Yilmaz C, Elmaci S. Glucose metabolism and catecholamine responses during physical exercise in non-insulin-dependent diabetes. Clin Chem Lab Med. 1996;34(9):683–690. doi:10.1515/cclm.1996.34.9.683
50. Guigas B, de Leeuw van Weenen JE, van Leeuwen N, et al. Sex-specific effects of naturally occurring variants in the dopamine receptor D2 locus on insulin secretion and type 2 diabetes susceptibility. Diabet Med. 2014;31(8):1001–1008. doi:10.1111/dme.12464
51. Straznicky NE, Grima MT, Lambert EA, et al. Arterial norepinephrine concentration is inversely and independently associated with insulin clearance in obese individuals with metabolic syndrome. J Clin Endocrinol Metab. 2015;100(4):1544–1550. doi:10.1210/jc.2014-3796
52. Pellinger TK, Dumke BR, Halliwill JR. Effect of H1- and H2-histamine receptor blockade on postexercise insulin sensitivity. Physiol Rep. 2013;1(2). doi:10.1002/phy2.33
53. Kim K, Oh CM, Ohara-Imaizumi M, et al. Functional role of serotonin in insulin secretion in a diet-induced insulin-resistant state. Endocrinology. 2015;156(2):444–452. doi:10.1210/en.2014-1687
54. Ilcol YO, Cansev M, Yilmaz MS, Hamurtekin E, Ulus IH. Peripheral administration of CDP-choline and its cholinergic metabolites increases serum insulin: muscarinic and nicotinic acetylcholine receptors are both involved in their actions. Neurosci Lett. 2008;431(1):71–76. doi:10.1016/j.neulet.2007.11.024
55. Pienaar PR, Micklesfield LK, Levitt NS, et al. Insulin resistance is associated with lower acetylcholine-induced microvascular reactivity in nondiabetic women. Metab Syndr Relat Disord. 2014;12(3):178–184. doi:10.1089/met.2013.0126
56. Sala-Rabanal M, Li DC, Dake GR, et al. Polyamine transport by the polyspecific organic cation transporters OCT1, OCT2, and OCT3. Mol Pharm. 2013;10(4):1450–1458. doi:10.1021/mp400024d
57. Lin Y, Zhang X, Wang L, et al. Polyamine depletion attenuates isoproterenol-induced hypertrophy and endoplasmic reticulum stress in cardiomyocytes. Cell Physiol Biochem. 2014;34(5):1455–1465. doi:10.1159/000366350
58. Mastracci TL, Robertson MA, Mirmira RG, Anderson RM. Polyamine biosynthesis is critical for growth and differentiation of the pancreas. Sci Rep. 2015;5(1). doi:10.1038/srep13269
59. Suzuki J, Ogawa M, Watanabe R, et al. Roles of prostaglandin E2 in cardiovascular diseases. Int Heart J. 2011;52(5):266–269. doi:10.1536/ihj.52.266
60. Docanto MM, Ham S, Corbould A, Brown KA. Obesity-associated inflammatory cytokines and prostaglandin E2 stimulate glucose transporter mRNA expression and glucose uptake in primary human adipose stromal cells. J Interf Cytokine Res. 2015;35(8):600–605. doi:10.1089/jir.2014.0194
61. Nakhjavani M, Ghazizadeh Z, Aghajaninargesi A, et al. Prostaglandin F2 alpha plasma concentration predicts glycemic control and oxidation status in patients with type 2 diabetes mellitus. Clin Lab. 2014;60(12):2073–2080. doi:10.7754/Clin.Lab.2014.140405
62. Liang X, Yee SW, Chien HC, et al. Organic cation transporter 1 (OCT1) modulates multiple cardiometabolic traits through effects on hepatic thiamine content. PLoS Biol. 2018;16(4):e2002907. doi:10.1371/journal.pbio.2002907
63. He L, Wondisford FE. Metformin action: concentrations matter. Cell Metab. 2015;21(2):159–162. doi:10.1016/j.cmet.2015.01.003
64. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60(9):1577–1585. doi:10.1007/s00125-017-4342-z
65. Lizcano JM, Göransson O, Toth R, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004;23(4):833–843. doi:10.1038/sj.emboj.7600110
66. Krishan S, Richardson DR, Sahni S. Adenosine monophosphate -activated kinase and its key role in catabolism: structure, regulation, biological activity, and pharmacological activation. Mol Pharmacol. 2015;87(3):363–377. doi:10.1124/mol.114.095810
67. Tian C, Fang S, Du X, Jia C. Association of the C47T polymorphism in SOD2 with diabetes mellitus and diabetic microvascular complications: a meta-analysis. Diabetologia. 2011;54(4):803–811. doi:10.1007/s00125-010-2004-5
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.