Back to Archived Journals » Research and Reports in Neonatology » Volume 5
Intrauterine hypoxia: clinical consequences and therapeutic perspectives
Authors Thompson L, Crimmins S, Telugu B, Turan S
Received 30 April 2015
Accepted for publication 21 June 2015
Published 15 September 2015 Volume 2015:5 Pages 79—89
DOI https://doi.org/10.2147/RRN.S57990
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Robert Schelonka
Loren P Thompson,1 Sarah Crimmins,1 Bhanu P Telugu,2 Shifa Turan1
1Department of Obstetrics, Gynecology and Reproductive Sciences, University of Maryland School of Medicine, Baltimore, MD, USA; 2Department of Animal Sciences, University of Maryland, College Park, MD, USA
Abstract: Intrauterine hypoxia is a significant clinical challenge in obstetrics that affects both the pregnant mother and fetus. Intrauterine hypoxia can occur in pregnant women living at high altitude and/or with cardiovascular disease. In addition, placental hypoxia can be generated by altered placental development and spiral artery remodeling leading to placental insufficiency and dysfunction. Both conditions can impact normal maternal cardiovascular homeostasis leading to preeclampsia and/or impair transfer of O2/nutrient supply resulting in fetal growth restriction. This review discusses the mechanisms underlying altered placental vessel remodeling, maternal and fetal consequences, patient management, and potential future therapies for improving these conditions.
Keywords: fetal growth restriction, oxidative stress, extravillous trophoblast invasion, Doppler ultrasound, pulsatility index, preeclampsia
Introduction
Embryogenesis, fetal growth, and survival depend on optimal maternal health and normal placental development. Maternal exposure to a persistently hypoxic environment may lead to abnormal placenta development and negatively impact fetal growth.1 The consequences of abnormal trophoblast invasion and altered remodeling of the maternal vasculature can result in preeclampsia, as well as intrauterine fetal growth restriction (FGR), asphyxia, multiorgan failure, premature delivery, and perinatal demise. This review will discuss the mechanisms associated with placental hypoxia leading to maternal complications and FGR. It will discuss the clinical consequences of intrauterine hypoxia and how to identify patients that present with placental pathophysiology. We will also discuss the current therapeutic approaches for management of the mother and fetus exposed to intrauterine hypoxia, as well as drug treatments currently under investigation that hope to provide future potential therapies.
Normal placental development: key terms
In humans, cytotrophoblasts (CTBs), the precursor population of the placenta, give rise to two distinct placental subtypes, the multinucleated syncytiotrophoblast (STB) and a migratory extravillous trophoblast (EVT) population. The STBs mediate an initial breach of the uterine epithelium and implantation within the uterine wall. Following implantation, the fetal trophoblasts undergo a period of rapid growth to establish the maternal–fetal interface (ie, the “junctional zone”). The junctional zone can be anatomically categorized into the “basal plate”, that is shed along with the placenta at birth, and “placental bed”, that remains attached to the uterus. The STBs, along with CTBs, form the “floating villi”, and are the primary site of a maternal–fetal exchange. The villi that attach to the basal plate are called “anchoring villi” and contain a proliferating population of precursor EVTs called the “cell columns” that anchor the villi to the basal plate.2–5
Role of oxygen in placental development
Oxygen (O2) is one of the key regulators in trophoblast differentiation, proliferation, and migration of trophoblasts in normal placental development.2 In humans, the initial invasion of trophoblasts during the first trimester of pregnancy occurs in relatively low partial pressure of O2 of 18–40 mmHg measured in the intervillous space6 and endometrial tissue7,8 which increases to 60–80 mmHg after spiral artery remodeling and increased placental perfusion.6,8,9 The placenta, unlike the fetus, undergoes proliferation and growth within a low O2 environment. Low O2 stimulates the expression and stability of transcription factors such as hypoxia-inducible factors (HIFs). HIF1α and HIF2α proteins can each form a complex with the constitutive HIF1β isoform, which then binds to consensus DNA recognition sequences promoting activation of gene expression of angiogenic factors such as vascular endothelial growth factor (VEGF), placental growth factor (PLGF), and angiopoietins 1 and 2.10–12 The hypoxia-related gene expression of angiogenic factors contributes to blood vessel growth within the endometrial tissue.13 In addition, hormones such as insulin, progesterone, estrogen, gonadotropin releasing hormone, and human chorionic gonadotropin and its hyperglycosylated isoform14 are important in the regulation of angiogenesis and trophoblast differentiation.2 During the initial hypoxia phase, the EVTs within the cell column undergo key changes including a change in the integrin profile, become migratory, and invade the decidual interstitium (interstitial EVT) and maternal spiral arteries (endovascular EVT). The EVTs initially plug the spiral artery lumen, thereby maintaining a low O2 environment.15 Subsequently, the endothelial cells within the spiral arteries are replaced by EVTs, the vascular smooth cells undergo apoptosis, and the lumen diameter increases, creating low resistance, high capacitance vessels that increase placental perfusion.8,16–18
The role of low O2 in EVT behavior remains a bit unclear. Initial studies15,19 have demonstrated that low O2 promotes trophoblast proliferation, whereas high O2 induces differentiation of trophoblasts. Using isolated cultured CTBs, proliferation was increased in 2% O2 compared to 21% O2, while invasion was inhibited,15,19,20 suggesting low O2 conditions inhibit endovascular invasion. However, studies have also shown that a hypoxic environment may promote invasion of EVTs.21 Graham et al22 reported increased invasion through Matrigel of cultured HTR-8/SVneo cells (an immortalized trophoblast cell line) in 1% O2 compared to 20% O2 and proposed that hypoxia is a factor that promotes invasion by EVTs similar to that for other invasive cell types such as carcinoma cells.14 Therefore, the role of O2 in regulating EVT invasion may depend on the conditions of study (in vitro versus in vivo), cell types studied, and gestational age from which the placental cells are derived.2,21,23 On the other hand, while trophoblast behaviors (ie, proliferation and invasion) are sensitive to O2, the effect of sustained placental hypoxia, in vivo, on EVT proliferation/invasion in the endometrium may differ from those occurring in in vitro conditions.
Normal endothelial cell function may also play a role in endovascular invasion of EVTs. For example, expression of endothelial cell adhesion molecules such as VE-cadherin, VCAM-1, and αβ integrins may be important in the endothelial cell–trophoblast interaction and spiral artery remodeling since CTBs have been shown to express similar endothelial cell surface receptors.24–26 In preeclampsia, the expression of endothelial cell markers such as VE-cadherin, VCAM-1 and α,β-integrins in EVTs has been shown to be reduced, suggesting that the endothelial cell surface markers are necessary for endovascular invasion by EVTs.25,26 However, other studies do not support expression of endothelial cell markers on EVTs as a mechanism contributing to endovascular invasion,27–30 suggesting the role of endothelial cell receptors remains incompletely understood. Overall, if EVTs have not established adequate occupation of the decidual tissue and spiral arteries, the consequences may be superficial trophoblast invasion and/or suboptimal vessel remodeling, respectively.31,32
Causes/consequences of intrauterine hypoxia
Hypoxia is defined as a reduction in O2 supply relative to the O2 demand of the tissue.1 Intrauterine hypoxia refers to a relative deficiency of partial pressure of O2 in maternal, placental, or fetal compartments as a result of compromised O2 supply/demand balance. Placental O2 varies over the course of pregnancy as O2 delivery and metabolic demand increases with both placental and/or fetal development.
Intrauterine hypoxia can be caused by living at high altitude.28 Pregnant women of high- compared to low-altitude populations have reduced uterine blood flow and placental perfusion33 and FGR.28 In addition, pregnant women with pulmonary hypertension and cyanotic heart disease have reduced cardiac output, which negatively impacts O2 delivery to the placenta.29,34 Placental oxygenation may be reduced from inadequate O2 delivery, placental insufficiency, and/or abnormal spiral artery remodeling.35,36 Regardless of the cause, the consequences of placenta hypoxia/ischemia are inadequate fetal oxygenation and placental pathology that contribute to a range of maternal symptoms of hypertension, systemic inflammation, and immune responses.37
Role of oxidative stress in placental hypoxia
In placentas of preeclamptic women, increased oxidative stress is associated with a defective trophoblast invasion.38 Oxidative stress occurs with excess generation of reactive oxygen species beyond the antioxidant capacity of the cell. This may occur under conditions of hypoxia/reoxygenation injury or even hypoxia alone when incomplete reduction of O2 generates excess electrons in the mitochondrial respiratory chain.21,38–40 Both increased levels of reactive oxygen species (superoxide anions, hydrogen peroxides, and hydroxyl radicals) and decreased expression of antioxidant enzyme systems (eg, superoxide dismutase, catalase, glutathione peroxidase, and/or peroxiredoxins) contribute to oxidative stress.41 In addition, superoxide anions interact with nitric oxide to form peroxynitrite, which disrupts membrane and enzyme function. Further, protein nitration by peroxynitrite has several functional consequences in the placenta that include altered signal transduction, immunogenicity of proteins, and enzyme activity.42 Oxidized lipids, proinflammatory cytokines (eg, interleukin-6 [IL-6], tumor necrosis factor-α [TNF-α]), as well as reactive oxygen species (superoxide anions) are increased in the hypoxic placenta43 where the disruptive effects on trophoblast invasion may contribute to altered spiral artery remodeling, along with tissue inflammation, apoptosis of the trophoblasts, and shedding of surface membrane microparticles from STBs44 into the maternal circulation.24 The occurrences of reduced endovascular invasion, altered spiral artery remodeling, and membrane fragment shedding are considered causative to the development of preeclampsia.29
Clinical consequences of intrauterine hypoxia
Clinical consequences of intrauterine hypoxia associated with maternal, placental, and fetal conditions may differ in outcomes between the mother and fetus. We can classify intrauterine hypoxia into two categories: 1) pre-placental hypoxia, where the mother and fetus are both hypoxic (ie, high altitude exposure, cyanotic maternal heart diseases); and 2) uteroplacental hypoxia, where the mother’s oxygenation is normal but the uteroplacental circulation is impaired. As a result, both maternal and fetal consequences can occur.
Maternal consequences
Pregnancy complications associated with intrauterine hypoxia and disruption of normal spiral artery remodeling play a key role in the occurrence of the development of preeclampsia. Preeclampsia is defined as new-onset hypertension after 20 weeks gestation with new-onset proteinuria or other features (Table 1).
 | Table 1 Diagnostic criteria for preeclampsia |
Hypertension is defined by elevations of systolic blood pressure (BP) >140 mmHg and diastolic BP >90 mmHg on two occasions greater than 4 hours apart. Preeclampsia is classified by features that include: 1) systolic BP >160 mmHg or diastolic BP >110 mmHg on two occasions at least 4 hours apart; 2) thrombocytopenia (platelet count <100,000/μL); 3) impaired circulating liver transaminase levels (twice the upper limit of normal concentration); 4) new development of renal insufficiency (serum creatinine >1.1 mg/dL); 5) pulmonary edema; and/or 6) new-onset cerebral or visual disturbances.45
Preeclampsia is secondary to altered endothelial function in several organ systems. For example, endothelial dysfunction of the hepatic circulation contributes to the onset of HELLP (hemolysis, elevated liver enzymes, and low platelet count) syndrome.46 Dysfunction of the endothelium of the cerebral circulation leads to neurological symptoms or even eclampsia.47 Glomerular endotheliosis contributes to preeclamptic symptoms via increased edema formation and eventual proteinuria.48 Lastly, low serum albumin levels and edema formation in the lower extremities and lungs are associated with microangiopathic hemolytic anemia and hyperpermeability of the systemic vascular endothelium in preeclampsia.49 In addition, preeclampsia restricts blood flow to the placenta, which contributes to a fourfold increased risk for FGR.50
Signs and symptoms of cerebral edema in preeclamptic women include headache, tinnitus, visual disorders, and brisk tendon reflexes. Other symptoms include oliguria (secondary to acute renal failure), uterine contractions or vaginal bleedings (secondary to placental abruption), and vomiting and epigastric pain (secondary to HELLP syndrome). Blood tests should include a complete blood count with platelets, and serum levels of haptoglobin, lactate dehydrogenase, bilirubin, and liver enzymes (ie, aspartate transaminase and alanine transaminase) to identify HELLP syndrome. Proteinuria and increased levels of electrolytes, urea, and creatinine are diagnostic for acute renal failure. Maternal mortality is caused by intracranial hemorrhage, liver failure, eclampsia, and/or lung complications.51
Fetal consequences
Uteroplacental hypoxia impacts the fetus and causes FGR. Abnormal placental vascular development decreases normal placental blood supply, contributing to reduced O2/nutrient delivery to the fetus. Optimal fetal growth is dependent on the transfer of O2 and nutrients across the placenta with glucose and amino acids as the primary nutrients for growth and maturation.52 Of the amount of O2/nutrient supply delivered to the placenta via the uterine artery, ~40% of O2 and 70% of glucose are consumed by the placenta53 and the remaining is available for fetal transfer. With mild placental insufficiency, the placental mass is maintained while nutrient transfer to the fetus is reduced.54
The growth-restricted fetus compensates for placental insufficiency via hematologic and hemodynamic responses. There is an increase in erythropoiesis, nucleated red blood cells, and hematocrit55 related to the degree of in utero hypoxia.56 In FGR, immune dysfunction occurs, platelet counts decline, and there can be increased abnormal villous vasculature. This is correlated with the severity of placental insufficiency40 along with platelet activation/aggregation and a tenfold increase in thrombocytopenia.57,58 The immune dysfunction manifests as decline in fetal white blood cell counts, neutrophils, monocytes, and lymphocytes.59
Hemodynamic responses to hypoxic stress include redistribution of cardiac output to critical organs of survival. For example, blood flow in the hypoxic fetus is shunted to the brain, myocardium, and upper body and away from the kidneys, gastrointestinal tract, and lower extremities. Vasoconstriction of the lower extremities increases right ventricular afterload.60,61 At the same time, cerebral vasodilation is protective against hypoxic brain injury and decreases left ventricular afterload. If the hypoxia persists, fetal distress emerges since the hemodynamic changes eventually become inadequate to maintain appropriate oxygenation. Neurodevelopment is negatively impacted by fetal hypoxia.62 Long-term (up to age 6 years) cognitive and neurological abnormalities have been demonstrated in FGR infants.63 In the late stages of fetal distress, prior to death, atrial pressure waves are transmitted into the ductus venosus (DV) and umbilical vein (UmbV) as detected by Doppler waveforms. If the fetus is unable to compensate, fetal demise will occur. The rates of stillbirth are much higher in the setting of FGR compared to normal-growth fetuses.40,64 Neonates who survive have an increased risk of arterial hypertension and cardiovascular disease later in life.65,66
Imaging of the growth-restricted fetus
Doppler ultrasound (US) has been widely used in the clinical setting to detect and understand the origin of the uteroplacental hypoxia. Once gestational age is confirmed, Doppler US is used to estimate fetal weight. An abdominal circumference of less than the tenth percentile indicates FGR. Once FGR is confirmed, inherent fetal causes of growth restriction must be excluded. Both karyotype abnormalities, as well as evidence of fetal infection, can be detected by US. For example, infants with trisomy 13, 18, or 21 all have US markers that are evaluated on US. In trisomy 18, 90% of fetuses have FGR with sonographic findings such as rocker bottom feet, low-set ears, and ventriculomegaly. A majority of these infants will also have a cardiac defect.67 If any marker of chromosomal abnormalities or suspicion of infection is demonstrated, invasive fetal testing via amniocentesis is offered to determine the karyotype or provide evidence of intrauterine infection.
After the exclusion of abnormal karyotype or infection, placental insufficiency is the presumed diagnosis of FGR. As early as 12–14 weeks gestation, Doppler US can provide information about the uteroplacental unit. The presence of diastolic notching of the uterine arteries at 12–14 weeks suggests incomplete trophoblastic invasion of the spiral arteries.68 This is manifest as an elevated uteroplacental vascular resistance presumably due to reduced spiral artery remodeling in the maternal compartment.69,70
Arterial and venous Doppler US of the fetal circulation is performed to assess abnormal villous branching and fetal status. The pulsatility index (PI) is a measure of variability in the blood velocity in a vessel, which is the difference between the peak systolic and minimum diastolic velocities divided by the mean velocity during the cardiac cycle. Abnormal villous formation or progressive vascular occlusion can be measured by an elevation in the fetal umbilical artery (UmbA) Doppler waveform, indicative of increased vascular resistance. A decrease in the UmbA end-diastolic velocity is detected with a reduction of normal development of the fetal villous vasculature.70 The occurrence of absent or reversed end-diastolic flow in the UmbA is indicative of ~60% abnormal villous vasculature.71,72 The degree of hypoxia and acidemia is proportional to the severity of the UmbA Doppler abnormality.73 A reduction in the Doppler PI of the anterior or middle cerebral artery, referred to as “brain sparing”,74 indicates an increase in right ventricular afterload and is evident of a maladaptive response to intrauterine hypoxia.
Doppler waveforms of the DV and UmbV can be utilized to evaluate the alterations in cardiac preload associated with intrauterine hypoxia. A failure to maintain an appropriate cardiac preload75 can be identified by an absence or reversal of forward flow during atrial systole in the DV or UmbV Doppler waveform. Pulsatile waveforms in the UmbV or reversed flow in the DV are poor prognostic signs and correlate with birth acidemia and perinatal demise.76,77 Renal perfusion declines with intrauterine hypoxia in the compromised fetus, leading to a decline in amniotic fluid volume.
Normal fetal behavior develops sequentially starting with body movements and progressive maturation of fetal breathing. As the nervous system develops, autonomic reflexes override the intrinsic cardiac activity and regulate fetal heart rate characteristics. With fetal maturation, there is a decreased baseline heart rate and increased heart rate variability with episodic accelerations that are coupled to fetal movement. This is normally accomplished by 28 weeks gestation.78 Intrauterine hypoxia can delay the maturation profile of the nervous system and is associated with a decline in global fetal movement.79 With worsening degrees of hypoxia, fetal breathing declines, followed by a decrease in gross body movements and tone, which are eventually lost in an acidemic environment.79,80 Maturation profile of the nervous system is assessed with a biophysical profile score (BPS). This is an US surveillance of fetal activity that can be used to evaluate fetal movement, tone, amniotic fluid volume, breathing, and heart rate activity.81 Scoring is based on a 10-point scale with an acceptable score of either 8/10 or 10/10. A score of 6/10 with normal fluid is considered equivocal and a 6/10 with abnormal amniotic fluid volume is considered abnormal. A score that is <6/10 is considered abnormal and delivery should be considered. An abnormal BPS helps identify a pattern of deterioration that includes a decline in heart rate accelerations, followed by decreased fetal breathing movements, amniotic fluid volume, and body movements and tone.80
Clinical management of the pregnant mother
Clinical management depends on both the fetal and maternal status. Pregnancy complications associated with intrauterine hypoxia are managed with close antenatal Doppler US surveillance in order to time intervention and determine timing between exams. In the presence of intrauterine hypoxia, FGR is of major concern and management should be directed toward establishing the fetal status. Both fetal Doppler US and biophysical profiles are the best testing modalities for identifying or classifying the degree of FGR. Doppler surveillance, initiated after 24 weeks gestation, is used in cases of early onset FGR with intrauterine hypoxia. Altered placental development is identified with progressive worsening of the UmbA PI. This is followed by Doppler indices identifying fetal brain sparing and cardiovascular compromise (ie, abnormal DV). Changes in Doppler waveforms precede changes in fetal movement. The deterioration of the composite BPS should initiate prompt delivery. The decisions that lead to delivery take into consideration the prenatal and postnatal risks to the fetus. Prior to 28 weeks gestation in utero, the fetus gains a 2% increased chance of survival per day.82 Therefore, it is essential to use integrated testing modalities to maximize the chance of survival.
Integrated fetal surveillance (ie, the combination of BPS and Doppler US) can be utilized in order to determine monitoring intervals. The timing of follow-up can be based on specific Doppler abnormalities. If UmbA PI is increased but still has a normal positive end-diastolic flow, together with normal amniotic fluid and BPS, weekly or biweekly Doppler US are performed. With the onset of brain sparing, both BPS and Doppler US are repeated weekly. In the presence of significant blood flow redistribution, when there is absent or reverse diastolic flow in the UmbA, fetal hypoxemia is common with possible asphyxia. In these circumstances, delivery is initiated with fetuses more than 34 weeks gestation. At less than 32 weeks gestation, antenatal steroids are administered and vaginal delivery suggested, unless there is a maternal contraindication.
If significant flow redistribution is present, along with an increased DV PI, then signs of hypoxemia are likely associated with acidemia and asphyxia. Under these conditions, management would proceed to vaginal delivery after 32 weeks. If the fetus is less than 32 weeks, steroid administration for lung development is suggested with daily Doppler US and BPS testing until the completion of steroid treatment.
Fetal decompensation will be considered by the presence of absent or reverse UmbA end-diastolic flow, brain sparing, and absent or reverse DV A-wave flow (absent or reverse A-wave in D: indicates cardiac compromise and cardiac failure). In this situation, there is a high mortality rate and stillbirth can occur, irrespective of intervention. Therefore, immediate delivery is suggested at a tertiary center with the highest level of neonatal intensive unit care.83
The management algorithm in Table 2 is based on the ability to perform arterial and venous Doppler, as well as a full five-component BPS. This is the typical management approach we practice at the University of Maryland Medical Center, Baltimore, for preterm growth-restricted fetuses (unless otherwise indicated).
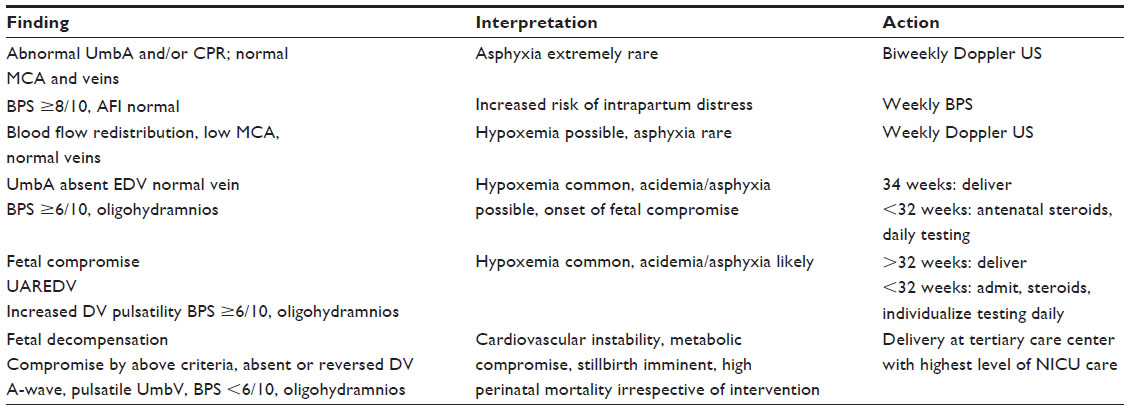 | Table 2 The management algorithm for pregnancies complicated by FGR |
Clinical management for pregnancy complicated with preeclampsia will depend on the maternal status. In this circumstance, the mother is monitored for signs and symptoms of worsening hypertension or preeclampsia. Delivery is the only curative treatment once preeclampsia is diagnosed. The timing for delivery is based on two interrelated factors: 1) the gestational age at diagnosis; and 2) the severity of preeclampsia. Preeclampsia, without severe symptoms, can be managed without delivery until 37 weeks or until severe features develop. The latter are identified in Table 1 for the mother and as Doppler waveform abnormalities in the fetus in Table 2. The maternal indications for severity include: eclampsia, HELLP syndrome, abruptio placentae, severe uncontrolled hypertension, pulmonary edema, or oliguria.84
The timing of delivery in preeclampsia with only severe hypertension (>160 mmHg systolic or >110 mmHg diastolic BP) is dependent on gestational age. Between 24 and 34 weeks, individuals who develop severe hypertension may be observed for the onset of the additional symptoms outlined above without delivery. If additional symptoms are developed then immediate delivery should be undertaken. In pregnancies progressing to 34–37 weeks gestation, conditions of severe hypertension with or without other symptoms demand delivery. After 37 weeks, there is no value of continuing the pregnancy, regardless of the severity of preeclampsia.45
Current therapies
The current therapies of treatment are aimed at the underlying etiology of the intrauterine hypoxia. If FGR is secondary to placental insufficiency, the therapeutic interventions are limited and the best of treatment for fetuses is delivery. Between 24 and 34 weeks gestation, the mother is given one course of antenatal corticosteroids (12 mg betamethasone intramuscular, two doses every 24 hours) in order to reduce neonatal morbidity and mortality. Corticosteroid stimulation of gene expression and physiologic functions result in maturation of lungs and other tissues.85 The course of corticosteroids has been proven to improve neonatal lung development, as well as reduce neonatal death, necrotizing enterocolitis, and interventricular hemorrhage.86
If maternal preeclampsia is the underlying cause of FGR, the mother is treated in two ways. First, antihypertensive therapy is used to control BP in order to prevent pulmonary edema and/or cerebral hemorrhage. Effective treatment should normalize systolic BP to less than 160 mmHg and diastolic BP to less than 110 mmHg.87 Second, magnesium sulfate is used to prevent seizures and normalize BP by mechanisms that are multifactorial but remain unclear. Magnesium sulfate may act as a vasodilator to decrease peripheral vascular resistance and protect the blood–brain barrier, limiting cerebral edema formation.88 By treating preeclampsia with antihypertensive medications and magnesium sulfate, the direct benefit to the fetus is pregnancy prolongation. Each day gained by treatment in utero increases fetal survival and intact survival by 1%–2% up to 28 weeks gestation.82
Low-dose aspirin (81 mg each night) is also given to the patient in the presence of preeclampsia and FGR. A meta-analysis of 34 randomized controlled trials determined that if low-dose aspirin was started at 16 weeks or earlier, the incidence of FGR is reduced by 16.3% in controls and 7.3% in the treatment group. The risk of preeclampsia with low-dose aspirin is reduced from 21.3% to 7.3%.89 In pregnancies without history of preeclampsia or FGR, the uterine artery Doppler US can be used in the first trimester to screen for patients at risk of developing preeclampsia, FGR, and perinatal death.69 The uterine artery Doppler alone has a poor positive predictive value for preeclampsia (12.1%) and FGR (21.2%) in the first trimester. When used, however, with first trimester serum screening, maternal characteristics, and history, the detection rates rise to 83.8% with 5% false-positive rate.90,91
Potential future therapies
Intensive research over the past several decades has investigated the molecular mechanisms associated with the etiology of preeclampsia. Placental hypoxia/ischemia has been linked to preeclampsia by measurement of increased antiangiogenic factors (soluble fms tyrosine kinase 1 [sFLT1], soluble endoglin),92 proinflammatory cytokines (IL-6, TNF-α),93 and oxidative stress markers (malondialdehyde, lipid hydroperoxide, oxidized low-density lipoprotein)94 in maternal serum as signaling molecules that contribute to hypertension, renal dysfunction, and vascular inflammation.95,96 Currently, there are limited drug therapies that can target the specific synthetic pathways of these factors or their actions and alleviate preeclamptic symptoms. Pharmacological agents and approaches are discussed in the following paragraphs as potential future clinical treatments against placental insufficiency and/or preeclampsia.
Reducing sFLT1 levels
Preeclampsia is characterized by widespread endothelial dysfunction, hypertension, and proteinuria,29 and accompanied by elevated maternal levels of sFLT1, all of which can contribute to glomerular endotheliosis, hypertension, and reduced VEGF and PLGF levels. Increases in VEGF/PLGF levels in excess of sFLT1 would be expected to reduce the negative impact of elevated sFLT1 on endothelial function and enhance the angiogenic action in preeclampsia. In a pilot study, multiple extracorporeal therapy treatments of a dextran sulfate cellulose apheresis was used to lower maternal sFLT1 levels in three pregnant women with very preterm preeclampsia.97 This reduced sFLT1 levels to 25%–35% pretreatment levels with each treatment and was accompanied by concomitant reductions in BP and proteinuria, as well as being well tolerated by both mother and baby.
TNFα antagonists
TNFα has been shown to be elevated in preeclamptic patients.98 Clinical trials for TNFα antagonists are ongoing to evaluate possible teratogenic effects on the newborn (etanercept and adalimumab) and to collect information on pregnancy outcomes (ie, normal and abnormal live births with infliximab) during pregnancy. Etanercept will be administered to pregnant women in the Organization of Teratology Information Services (OTIS) Autoimmune Diseases in Pregnancy Project (NLM identifier: NCT00116272) and incidence rates of birth defects will be measured. Infliximab is administered in a second trial entitled Analysis of Birth Outcomes of Swedish, Danish and Finnish Women Exposed to Remicade With Inflammatory Bowel Disease, Rheumatoid Arthritis, Psoriatic Arthritis, Ankylosing Spondylitis, and Psoriasis (NLM identifier: NCT00658827) given to women with these diseases. Adalimumab is administered in the OTIS Autoimmune Diseases in Pregnancy Project (NLM identifier: NCT01086059) to pregnant women with Crohn’s disease or rheumatoid arthritis.37 After a systematic literature review of 50 studies on pregnant patients administered anti-TNFα therapy (ie, antibodies against TNFα), it was concluded that although the therapy benefited in optimizing maternal disease control during pregnancy, it must be weighed against the potential negative effects on the developing fetus (ie, increased rate of still birth, preterm birth, and low birth weight) until definitive safety to the fetus has been confirmed.99 As further understanding of anti-TNFα therapy is advanced and tolerability studies identify favorable outcomes, it is anticipated that TNFα inhibitors may be a potential therapeutic approach for preventive treatment for the preeclamptic patient in the future.37
Antioxidants
Antioxidants have also been considered as a therapeutic strategy against conditions associated with oxidative stress. Administration of vitamin E and C supplements to women with risk factors for preeclamptic women showed a reduction in the ratio of plasminogen activator inhibitor-1 to plasminogen activator inhibitor-2, a biomarker of endothelial dysfunction, and 8-epi prostaglandin F2α, a marker of lipid peroxidation.100 Yet in a randomized control trial, when the study was powered to identify differences in the incidence of preeclampsia with antioxidant treatment, there was no difference in the number of women who developed preeclampsia,101 nor was there an improvement in the prevention of preeclampsia in either high-risk102 or lower-risk women.103,104 Despite the lack of evidence for antioxidants in preventing preeclampsia, antioxidant supplementation continues to be investigated as a therapeutic strategy against oxidative stress in pregnancy.101,105
Statins
Statins play an important role in the treatment of hypercholesterolemia by inhibiting the enzyme HMG-CoA reductase in the liver106 and may have potential as a therapeutic in preeclampsia.107 Statins have been shown to increase the expression of heme oxygenase-1 and inhibit sFLT1 release induced by cytokines in placental explants.108 Because of its ability to increase PLGF and reduce the effect of sFLT1, and its inability to cross the placenta,109,110 pravastatin is being tested in a clinical trial (Prevastatin for Prevention of Preeclampsia; NLM identifier: NCT01717586) for its potential to reduce symptoms of early-onset preeclampsia.
Peptide-based therapies
Recently, peptide-based therapies for targeting signaling factors such as sFLT1, proinflammatory cytokines, and reactive oxygen species, important in the generation of preeclampsia, are being considered to ameliorate the consequences of endothelial dysfunction and end-organ damage. Polymeric carrier proteins bound separately to VEGF, NF-ºB (regulatory protein of inflammatory pathway), and nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2) docking sequence111 have been developed, which are expected to reduce sFLT1 levels, inhibit the generation of proinflammatory cytokines, and reduce NOX2 (ie, superoxide anion generation) activity, respectively. Because they are bound to a high molecular weight protein, their actions would be limited to the maternal circulation and not access the fetus. Polymeric carrier proteins are currently under investigation as an experimental approach as a future therapeutic delivery system for the management of preeclampsia.
Pharmacogenetics
Recent pharmacogenetics approaches for individualizing drug therapy are underway to optimize the effectiveness of drug treatment, which varies among patients. Several polymorphisms of specific genes associated with pathophysiological mechanisms reveal an underlying cause of disparities in the efficacy of drug action.112 Only recently has this novel approach of targeting specific genes been considered for the development of a pharmacological treatment of preeclampsia.113 For example, polymorphisms of the CYP3A family, which metabolizes nifedipine, a calcium channel blocker of uterine smooth muscle and effective tocolytic in preterm labor therapy, may negatively impact the concentration levels in the maternal blood114,115 since the pharmacokinetics of nifedipine is influenced by the CYP3A5 genotype.115 Recent efforts have identified that polymorphisms of the eNOS (endothelial nitric oxide synthase) gene alter the effectiveness of antihypertensive therapy in preeclamptic women,105,116 as well as the effectiveness of statins in preeclampsia.117 Thus, several examples now identify that polymorphisms in selected genes may alter drug efficacy. As we advance our knowledge in the causes of preeclampsia and other pregnancy disorders, we may be able to develop personalized therapies that target the patients’ individual symptoms.
Conclusion
Intrauterine hypoxia is one of the most significant clinical challenges facing obstetric practice and can be generated under conditions of placental insufficiency, high-altitude environments, and exposure to toxic substances. Preeclampsia is a maternal consequence that is associated with placental dysfunction and reduced spiral artery development and impacts the health of both the mother and fetus. Understanding the underlying mechanisms associated with the placental dysfunction associated with intrauterine hypoxia is key toward improving therapeutic strategies for clinical management. Future therapies in managing preeclampsia and other placental disorders are expected to improve as we increase our understanding of the factors that regulate trophoblast invasion of the placenta and its spiral arteries, as well as how the regulatory mechanisms are altered under conditions of placental hypoxia.
Disclosure
The authors report no conflicts of interest in this work.
References
Herrera EA, Krause B, Ebensperger G, et al. The placental pursuit for an adequate oxidant balance between mother and the fetus. Front Pharmacol. 2014;5:149. | |
Ji L, Brkic J, Liu M, Fu G, Peng C, Wang YL. Placental trophoblast cell differentiation: physiological regulation and pathological relevance to preeclampsia. Mol Aspects Med. 2013;34:981–1023. | |
Kaufmann P, Black S, Huppertz B. Endovascular trophobast invasion: implications for the pathogenesis of intrauterine growth retardation and preeclampsia. Biol Reprod. 2003;69:1–7. | |
Lyall F. Primary and remodeling of human placenta bed spiral arteries during pregnancy – a review. Placenta. 2005;26 Suppl A:S31–S36. | |
Knöfler M. Critical growth factors and signaling pathways controlling human trophoblast invasion. Int J Dev Biol. 2010;54(2–3):269–280. | |
Rooth G, Sjostedt S, Caligara F. Hydrogen concentration, carbon dioxide tension and acid base balance in blood of human umbilical cord and intervillious space of placenta. Arch Dis Child. 1961;36:278–285. | |
Jauniaux E, Watson A, Ozturk O, Quick D, Burton G. In-vivo measurement of intrauterine gases and acid-base values early in human pregnancy. Hum Reprod. 1999;14(11):2901–2904. | |
Rodesch F, Simon P, Donner C, Jauniaux E. Oxygen measurement in endometrial and trophoblastic tissues during early pregnancy. Obstet Gynecol. 1992;80(2):283–285. | |
Jauniaux E, Poston L, Burton GJ. Placental-related diseases of pregnancy: Involvement of oxidative stress and implications in human evolution. Hum Reprod Update. 2006;12:747–755. | |
Prahbakar NR, Semenza GL. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-induced factors 1 and 2. Physiol Rev. 2012;92:967–1003. | |
Kaelin WG Jr, Ratcliffe PJ. Oxygen Sensing by metazoan: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. | |
Iyer NV, Kotch LE, Agani F, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–162. | |
Ahmed A, Dunk C, Ahmad S, Khaliq A. Regulation of placental vascular endothelial growth factor (VEGF) and placenta growth factor (PIGF) and soluble Flt-1 by oxygen – a review. Placenta. 2000;21 Suppl A:S16–S24. | |
Cole La. Hyperglycosylated hCG, a review. Placenta. 2010;31(8):653–664. | |
Genbacev O, Zhou Y, Ludlow JW, Fisher SJ. Regulation of human placental development by oxygen tension. Science. 1997;277(5332):1669–1672. | |
Robertson WB, Brosens I, Dixon G. Maternal uterine vascular lesions in the hypertensive complications of pregnancy. Perspect Nephrol Hypertens. 1976;5:115–127. | |
Hirano H, Imai Y, Ito H. Spiral artery of placenta: development and pathology-immunohistocehmical microscopical, and electro-microscopic study. Kobe J Med Sci. 2002;48:13–23. | |
Robertson WB, Manning PJ. Elastic tissue in uterine blood vessels. J Pathol. 1974;112(4):237–243. | |
Genbacev O, Joslin R, Damsky CH, Polliotti BM, Fisher SJ. Hypoxia alters early gestation human cytotrophoblast differentiation/invasion in vitro and models the placental defects that occur in preeclampsia. J Clin Invest. 1996;97(2):540–550. | |
Caniggia I, Mostachfi H, Winter J, et al. Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFbeta(3). J Clin Invest. 2000;105: 577–587. | |
Burton GJ. Oxygen, the Janus gas; its effects on human placental development and function. J Anat. 2009;215:27–35. | |
Graham CH, Postovit LM, Park H, Canning MT, Fitzpatrick TE. Adriana and Luisa Castellucci award lecture 1999: role of oxygen in the regulation of trophoblast gene expression and invasion. Placenta. 2000;21:443–450. | |
Graham CH, Lala PK. Mechanisms of placental invasion of the uterus and their control. Biochem Cell Biol. 1992;70(10–11):867–874. | |
Myatt L, Webster RP. Vascular biology of preeclampsia. J Thomb Haemost. 2009;7:375–384. | |
Zhou Y, Fisher SJ, Janatpour M, et al. Human cytotrophoblasts adopt a vascular phenotype as they differentiate. A strategy for successful endovascular invasion? J Clin Invest. 1997;99:2139–2151. | |
Zhou Y, Damksy CH, Fisher SJ. Preeclampsia is associated with failure of human cytotrophoblasts to mimic a vascular phenotype. One cause of defective endovascular invasion in this syndrome? J Clin Invest. 1997;99:2152–2164. | |
Lyall F, Bulmer JN, Duffie E, Cousins F, Theriault A, Robson SC. Human trophoblast invasion and spiral artery transformation: the role of PECAM-1 in normal pregnancy, preeclampsia, and fetal growth restriction. Am J Pathol. 2001;158:1713–1721. | |
Moore LG, Shriver M, Bernis L, et al. Maternal adaptation to high-altitude pregnancy: an experiment of nature – a review. Placenta. 2004;25 Suppl A:S60–S71. | |
Roberts JM, Taylor RN, Musci TJ, Rodgers GM, Hubel CA, Mclaughlin MK. Preeclampsia: an endothelial cell disorder. Am J Obstet Gynecol. 1989;161:1200–1204. | |
Divers MJ, Bulmer JN, Miller D, Lilford RJ. Beta 1 integrins in third trimester human placentae: no differential expression in pathological pregnancy. Placenta. 1995;16:245–260. | |
Redman CW, Sargent IL. Latest advances in understanding preeclampsia. Science. 2005;308(5728):1592–1594. | |
Redman CW, Sargent IL. Placental stress and pre-eclampsia: a revised view. Placenta. 2009;30 Suppl A:S38–S42. | |
Zamudio S, Plamer SK, Droma T, Stamm E, Coffin C, Moore LG. Effect of altitude on uterine artery blood flow during pregnancy. J Appl Physiol (1985). 1995;79:7–14. | |
Roberts JM, Taylor RN, Goldfien A. Clinical and biochemical evidence of endothelial cell dysfunction in the pregnancy syndrome preeclampsia. Am J Hypertens. 1991;4:700–708. | |
Murray AJ. Oxygen delivery and fetal-placental growth: beyond a question of supply and demand? Placenta. 2012;33 Suppl 2:e16–e22. | |
Schneider H. Oxygenation of the placental-fetal unit in humans. Respir Physiol Neurobiol. 2011;178:51–58. | |
George EM. New approaches for managing preeclampsia: clues from clinical and basic research. Clin Ther. 2014;36:1873–1881. | |
Wang Y, Walsh SW. Increased superoxide generation is associated with decreased superoxide dismutase activity and mRNA expression in placental trophoblast cells in pre-eclampsia. Placenta. 2001; 22(2–3):206–212. | |
Guzy RD, Hoyos B, Robin E, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and celullar oxygen sensing. Cell Metab. 2005;1:401–408. | |
Wilcox CR, Trudinger BJ. Fetal platelet consumption: a feature of placental insufficiency. Obstet Gynecol. 1991:616–621. | |
Myatt L. Review: Reactive oxygen and nitrogen species and functional adaptation of the placenta. Placenta. 2010;31 Suppl:S66–S69. | |
Webster RP, Roberts VH, Myatt L. Protein nitration in placenta – functional significance. Placenta. 2008;29(12):985–994. | |
Laresgoiti-Servitje E, Gomez-Lopez N. The pathophysiology of preelcampsia involves altered levels of angiogenic factors promoted by hypoxia and autoantibody-mediated mechanisms. Biol Reprod. 2012;87(2):36. | |
Huppertz B, Kingdom J, Caniggia I, et al. Hypoxia favours necrotic versus apoptitic shedding of placental syncytiotrophoblast into the maternal circulation. Placenta. 2003;24:181–190. | |
American College of Obstetricians and Gynecologists; Task Force on Hypertension in Pregnancy. Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet Gynecol. 2013;122(5):1122–1131. | |
Sibai BM, Ramadan MK, Usta I, Salama M, Mercer BM, Friedman SA. Maternal morbidity and mortality in 442 pregnancies with hemolysis, elevated liver enzymes, and low platelets (HELLP syndrome). Am J Obstet Gynecol. 1993;169:1000–1006. | |
Postma IR, Slager S, Kremer HP, de Groot JC, Zeeman GG. Long-term consequences of the posterior reversible encephalopathy syndrome in eclampsia and preeclampsia: a review of the obstetric and nonobstetric literature. Obstet Gynecol Surv. 2014;69(5):287–300. | |
Sircar M, Thadhani R, Karumanchi SA. Pathogenesis of Preeclampsia. Curr Opin Nephrol Hypertens. 2015;24(2):131–138. | |
Bombrys AE, Barton JR, Habli M, Sibai BM. Expectant management of severe preeclampsia at 27(0/7) to 33(6/7) weeks’ gestation: maternal and perinatal outcomes according to gestational age by weeks at onset of expectant management. Am J Perinatol. 2009;26(6):441–446. | |
Odegård RA, Vatten LJ, Nilsen ST, Salvesen KA, Austgulen R. Preeclampsia and fetal growth. Obstet Gynecol. 2000;96:950–955. | |
Haddad B, Deis S, Goffinet F, Paniel BJ, Cabrol D, Sibai BM. Maternal and perinatal outcomes during expectant management of 239 severe preclamptic women between 24 and 33 weeks’ gestation. Am J Obstet Gynecol. 2004;190(6):1590–1595; discussion 1595–1597. | |
Reece EA, Wiznitzer A, Le E, Homko CJ, Behrman H, Spencer EM. The relation between human fetal growth and fetal blood levels of insulin-like growth factors I and II, their binding proteins, and receptors. Obstet Gynecol. 1994;84:88–95. | |
Meschia G, Battaglia FC, Hay WW, Sparks JW. Utilization of substrates by the ovine placenta in vivo. Fed Proc. 1980;39:245–249. | |
Rigano S, Bozzo M, Ferrazzi E, Bellatti M, Battagli FC, Galan HL. Early and persistent reduction in umbilical vein blood flow in the growth-restricted fetus: a longitudinal study. Am J Obstet Gynecol. 2001;185:834–838. | |
Levy R, Smith SD, Chandler K, Sadovsky Y, Nelson DM. Apoptosis in human cultured trophoblasts is enhanced by hypoxia and diminshed by epidermal growth factor. Am J Physiol Cell Physiol. 2000;278: C982–C988. | |
Nishi H, Nakada T, Kyo S, Inoue M, Shay JW, Isaka K. Hypoxia-induced factor 1 mediates upregulation of telomerase (hTERT). Mol Cell Biol. 2004;24(13):6076–6083. | |
Trudinger B, Song JZ, Wu ZH, Wang J. Placental insufficiency is characterized by platelet activation in the fetus. Obstet Gynecol. 2003;101:975–981. | |
Baschat AA, Gembruch U, Reiss I, Cortner L, Weiner CP, Harman CR. Absent umbilical artery end-diastolic veolicty in growth-restricted fetuses: a risk factor for neonatal thrombocytopenia. Obstet Gynecol. 2000;96:162–166. | |
Davies N, Snijder R, Nicolaides KH. Intra-uterine starvation and fetal leucocyte count. Fetal Diagn Ther. 1991;6:107–112. | |
al-Ghazali W, Chita SK, Chapman MG, Allan LD. Evidence of redistribution of cardiac output n asymmetrical growth retardation. Br J Obstet Gynaecol. 1989;96(6):697–704. | |
Rizzo G, Arduini D, Romanini C. Doppler echocardiographic assessment of fetal cardiac function. Ultrasound Obstet Gynecol. 1992;2(6):434–445. | |
Soothill PW, Nicolaides KH, Campbell S. Prenatal asphyxia, hyperlacticaemia, hypoglycemia, and erythroblastosis in growth retarded fetusues. Br Med J (Clin Res Ed). 1987;294(6579):1051–1053. | |
Mccarton CM, Wallace IF, Divon M, Vaughan HG Jr. Cognitive and neurologic development of premature, small for gestational age infant through age 6: comparison by birth weight and gestational age. Pediatrics. 1996;98(6 Pt 1):1167–1178. | |
Gardosi J, Madurasinghe V, Williams M, Malik A, Francis F. Maternal and fetal risk factors for stillbirth:population based study. BMJ. 2013;346:f108. | |
Barker DJ. The fetal origins of adult hypertension. J Hypertens Suppl. 1992;10(7):S39–S44. | |
Bonamy AK, Bendito A, Martin H, Andolf E, Sedin G, Norman M. Preterm birth contributes to increased vascular resistance and higher blood pressure in adolescent girls. Pediatr Res. 2005;58(5):845–849. | |
Hsiao CC, Tsao LY, Chen HN, Chiu HY, Chang WC. Changing clinical presentations and survival pattern in trisomy 18. Pediatr Neonatol. 2009;50:147–151. | |
Harrington K, Carpenter RG, Goldfrad C, Campbell S. Transvaginal Doppler ultrasound of the uteroplacental circulation in the early prediction of pre-eclampsia and interuine growth retardation. Br J Obstet Gynaecol. 1997;104:674–681. | |
Papageorghiou AT, Yu CK, Nicolaides KH. The role of uterine artery Doppler in predicting adverse pregnancy outcome. Best Pract Res Clin Obstet Gynaecol. 2004;18(3):383–396. | |
Morrow RJ, Adamson SL, Bull SB, Ritchie JW. Effect of placental embolization on the umbilical artery velocity waveform in fetal sheep. Am J Obstet Gynecol. 1989;161:1055–1060. | |
Baschat AA, Hecher K. Fetal growth restriction due to placental disease. Semin Perinatol. 2004;28(1):67–80. | |
Nicolaides KH, Bilardo CM, Soothill PW, Campbell S . Absence of end diastolic frequencies in umbilical artery: a sign of fetal hypoxia and acidosis. BMJ. 1988;297(6655):1026–1027. | |
Weiner CP. The relationship between the umbilical artery systolic/diastolic ratio and umbilical blood gas measurements in specimens obtained by cordocentesis. Am J Obstet Gynecol. 1990;162: 1198–1202. | |
Gramellini D, Folli MC, Raboni S, Vadora E, Merialdi A. Cerebral-umbilical Doppler ratio as a predictor of adverse perinatal outcome. Obstet Gynecol. 1992;79:416–420. | |
Hecher K, Campbell S, Doyle P, Harrington K, Nicolaides K. Assessment of fetal compromise by Doppler ultrasound investigation of the fetal circulation. Arterial, intracardiac, and venous blood flow studies. Circulation. 1995;91:129–138. | |
Carvalho FH, Moron AF, Mattar R, et al. Ductus venosus Doppler velocimetry in the prediction of acidemia at birth: which is the best parameter? Prenat Diagn. 2005;25(13):1212–1216. | |
Schwarze A, Gembruch U, Krapp M, Katalinic A, Germer U, Axt-Fliedner R. Qualitative venous Doppler flow waveform analysis in preterm intrauterine growth-restricted fetuses with ARED flow in the umbilical artery – correlation with short-term outcome. Ultrasound Obstet Gynecol. 2005;25(6):573–579. | |
Manning FA. Fetal biophysical profile. Obstet Gynecol Clin North Am. 1999;26:557–577, v. | |
Ribbert LS, Nicolaides KH, Visser GH. Prediction of fetal acidaemia in intrauterine growth retardation: comparison of quantified fetal activity with biophysical profile score. Br J Obstet Gynaecol. 1993;100(7):653–656. | |
Vintzileos AM, Femiling AD, Scorza WE, et al. Relationship between fetal biophysical activity and umbilical cord blood gas values. Am J Obstet Gynecol. 1991;165:707–713. | |
Crimmins S, Desai A, Block-Abraham D, Berg C, Gembruch U, Baschat AA. A Comparison of Doppler and biophysical findings between liveborn and stillborn growth-restricted fetuses. Am J Obstet Gynecol. 2014;211(6):e1–e10. | |
Baschat AA, Cosmi E, Bilardo CM, et al. Predictors of neonatal outcome in early-onset placental dysfunction. Obstet Gynecol. 2007;109: 253–261. | |
Turan S, Miller J, Baschat AA. Integrated testing and management in fetal growth restriction. Semin Perinatal. 2008;32:194–200. | |
Belghiti J, Kayem G, Tsatsaris V, Goffinet F, Sibai BM, Haddad B. Benefits and risks of expectant management of severe preeclampsia at less than 26 weeks gestation: the impact of gestational age and severe fetal growth restriction. Am J Obstet Gynecol. 2011;205:465. e1–e6. | |
Bonanno C, Wapner RJ. Antenatal corticosteroid treatment: what’s happened since Drs Liggins and Howie? Am J Obstet Gynecol. 2009; 200:448–457. | |
Roberts D, Dalziel S. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst Rev. 2006;(3):CD004454. | |
Cunningham FG. Severe preeclampsia and eclampsia: systolic hypertension is also important. Obstet Gynecol. 2005;105(2):237–238. | |
Roberts JM. Endothelial dysfunction in preelclampsia. Semin Reprod Endocrinol. 1998;16:5–15. | |
Bujold E, Roberge S, Lacasse Y, et al. Prevention of preeclampsia and interuterine growth restriction with aspirin started in early pregnancy: a meta-analysis. Obstet Gynecol. 2010;116(2 Pt 1):402–414. | |
Poon LC, Stratieva V, Piras S, Piri S, Nicolaides KH. Hypertensive disorders in pregnancy: combined screening by uterine artery Doppler, blood pressure and serum PAPP-A at 11–13 weeks. Prenat Diagn. 2010;30(3):216–223. | |
Vainio M, Kuhansuu E, Iso-Mustajärvi M, Mäenpää J. Low dose Acetylsalicylic acid in prevention of pregnancy-induced hypertension and interuterine growth retardation in women with bilateral uterine artery notches. BJOG. 2002;109(2):161–167. | |
Ahmed A, Ramma W. Unravelling the theories of pre-eclampsia: are the protective pathways the new paradigm? Br J Pharmacol. 2015;172: 1574–1586. | |
Redman CW, Sacks GP, Sargent IL. Preeclampsia: an excessive maternal inflammatory response to pregnancy. Am J Obstet Gynecol. 1999;180:499–506. | |
Genc H, Uzun H, Benian A, et al. Evaluation of oxidative stress markers in first trimester for assessment of preeclampsia risk. Arch Gynecol Obstet. 2011;284:1367–1373. | |
Burton GJ, Jauniaux E. Placental oxidative stress: from miscarriage to preeclampsia. J Soc Gynecol Invest. 2004;11:342–352. | |
Hubel CA. Oxidative stress in the pathogenesis of preeclampsia. Proc Soc Exp Biol Med. 1999;222:222–235. | |
Thadhani R, Kisner T, Hagmann H, et al. Pilot study of extracorporeal removal of soluble fms-like tyrosine kinase 1 in preeclampsia. Circulation. 2011;124:940–950. | |
Kupferminc MJ, Peaceman AM, Wigton TR, Rehnberg KA, Socol ML. Tumor Necrosis factor-alpha is elevated in plasma and amniotic fluid of patients with severe preeclampsia. Am J Obstet Gynecol. 1994;170: 1752–1757. | |
Marchioni RM, Lichtenstein GR. Tumor necrosis factor-α inhibitor therapy and fetal risk: a systemic literature review. World J Gastroenterol. 2013;19:2591–2602. | |
Poston L, Igosheva N, Mistry HD, et al. Role of oxidative stress and antioxidant supplementation in pregnancy disorders. Am J Clin Nutr. 2011;94(6 Suppl):1980S–1985S. | |
Poston L, Briley AL, Seed PT, Kelly FJ, Shennan AH; Vitamins in Pre-eclampsia (VIP) Trial Consortium. Vitamin C and vitamin E in pregnant women at risk for pre-eclampsia (VIP trial): randomized placebo-controlled trial. Lancet. 2006;367:1145–1154. | |
Villar JK, Purwar M, Merialdi M, et al. World Health Organization mulitcentre randonised trial of supplementation with vitamins C and E among pregnant women at high risk for pre-eclampsia in population of low nutritional status from developing countries. BJOG. 2009;116:780–788. | |
Rumbold AR, Crowther CA, Haslam RR, Dekker GA, Robinson JS; ACTS Study Group. Vitamins C and E and the risks of preeclampsia and prenatal complications. N Eng J Med. 2006;354:1796–1806. | |
Roberts JM, Myatt L, Spong CY, et al. Vitamins C and E to prevent complications of pregnancy-associated hypertension. N Eng J Med. 2010;362:1282–1291. | |
Ilekis JV, Reddy UM, Roberts JM. Preeclampsia – a pressing problem: an executive summary of a National Institute of Child Health and Human Development workshop. Reprod Sci. 2007;14(6):508–523. | |
Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29:431–438. | |
Ramma W, Ahmed A. Therapeutic potential of statins and the induction of heme oxygenase-1 in preeclampsia. J Reprod Immunol. 2014;101–102:153–160. | |
Cudmore M, Ahmad S, Al-Ani B, et al. Negative regulation of soluble Flt-1 and soluble endoglin release by heme oxygenase-1. Circulation. 2007;115:1789–1797. | |
Mctaggart F, Buckett L, Davidson R, et al. Preclinical and clinical pharmacology of Rosuvastatin, a new 3-hydroxy-3-methylglutaryl coezyme A reductase inhibitor. Am J Cardiol. 2001;87: 28B–32B. | |
Hatanaka T. Clinical pharmacokinetics of pravastatin: mechanism of pharmacokinetics events. Clin Pharmacokinet. 2000;39(397–412). | |
Bidwell GL 3rd, George EM. Maternally sequestered therapeutic polypeptides- a new approach for the management of preeclampsia. Front Pharmacol. 2014;5:201. | |
Haas DM. Pharmacogenetics and individualizing drug treatment during pregnancy. Pharmacogenomics. 2014;15(1):69–78. | |
Luizon MR, Sandrim VC. Pharmacogenomic approaches that may guide preeclampsia therapy. Pharmacogenomics. 2013;14(6):591–593. | |
Haas DM, Quinney SK, McCormick CL, Jones DR, Renbarger JL. A pilot study of the impact of genotype on nifedipine pharmacokinetics when used as a tocolytic. J Matern Fetal Neonatal Medicine. 2012; 25(4):419–423. | |
Haas DM, Quinney SK, Clay JM, et al. Nifedipine pharmacokinetics are influenced by CYP3A5 genotype when used as a preterm labor tocolytic. Am J Perinatol. 2013;30(4):275–281. | |
Sandrim VC, Palei AC, Luizon MR, Izidoro-Toledo TC, Cavalli RC, Tanus-Santos JE. eNOS haplotypes affect the responsiveness to antihypertensive therapy in preeclampsia but not in gestational hypertension. Pharmacogenomics J. 2010;10(1):40–45. | |
Ahmed A. New insights into the etiology of preeclampsia: identification of key elusive factors for the vascular complications. Thromb Res. 2011;127 Suppl 3:S72–S75. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.