Back to Journals » International Medical Case Reports Journal » Volume 12
Intramedullary spinal cord involvement: a rare presentation of sarcoidosis
Authors Padooru KR ![]() , Sen M
, Sen M
Received 14 April 2019
Accepted for publication 1 June 2019
Published 2 July 2019 Volume 2019:12 Pages 199—203
DOI https://doi.org/10.2147/IMCRJ.S212229
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ronald Prineas
Keerthi Reddy Padooru,1 Mitali Sen2
1Department of Internal Medicine, Morehouse School of Medicine, Atlanta, GA, USA; 2Department of Rheumatology, Emory University School of Medicine, Atlanta, GA, USA
Abstract: Sarcoidosis is a systemic granulomatous disease of uncertain etiology, which predominantly affects the lungs, eyes, lymph nodes, and skin. Nervous system involvement (neurosarcoidosis) occurs in approximately 5–10% of patients, but spinal cord involvement is very rare, affecting <1% of patients. We present a rare case of intramedullary sarcoidosis.
Keywords: spinal sarcoidosis, intramedullary lesion, neurosarcoidosis
Introduction
Neurosarcoidosis is relatively rare, and sole spinal cord disease is extremely rare. Intramedullary spinal sarcoidosis, as observed in the patient described in this report, is seen in less than 1% of cases of sarcoidosis.1 To date, very few cases have been described in the literature as the initial manifestation of spinal cord sarcoidosis without overt systemic complaints. We present the case of a 44-year-old African American man who had an intramedullary expansile patchy spinal cord lesion in the cervical and thoracic segments, which is very suggestive of spinal sarcoidosis.
Case presentation
The authors confirm that written informed consent has been provided by the patient to have the case details and any accompanying images published. Institutional approval is not required for publishing case details.
A 42-year-old African American man with a past medical history significant for uncontrolled type 2 diabetes mellitus and hypertension was transferred from an outside facility owing to acute numbness and a tingling sensation in the legs, with progression to difficulty in walking over the course of 2–3 weeks. His symptoms started with neck pain and stiffness. Shortly afterwards, his legs began to feel heavy, then he developed a sensation of numbness and tingling. He described his problem as “trying to search to find the floor with my feet”. It progressed to the point where he could not walk independently, following which he approached the emergency department. He also had a loss of fine motor skills, so that he was no longer able to type on his phone. Around the same time, he noticed a sensation of bladder fullness despite urination, but no issues with defecation.
A review of systems was negative for lymphadenopathy, fever, weight and appetite changes, cough, shortness of breath, and chest pain. He denied any known allergies or exposure to molds, chemicals, or asbestos. He also denied a history of tuberculosis (TB) exposure. Family history was not significant. He works as a police officer. He uses alcohol occasionally, but denied smoking or recreational drug use. His vital signs were within normal limits. Cardiology, head and neck, skin, gastrointestinal, and pulmonary examinations were normal. He was alert, and oriented to time, place, and person. On neurological examination, his language was fluent and he had a normal attention span. Cranial nerve examination was normal. Sensation for pinprick, temperature, and light touch was abnormal from sensory level T5–T6 and below. He had normal bulk and tone. Strength was 5/5 (Medical Research Council grade) throughout except in the left deltoid and iliopsoas, where it was 4+/5. Reflexes were graded 2+ in the bilateral upper and lower extremities. A gait examination could not be completed as he was not able to stand up for long without falling. Neurogenic bladder requiring a Foley catheter, and proprioceptive defects in the distal lower extremities, were present.
As the initial part of the work-up, magnetic resonance imaging (MRI) of the brain along with the cervical and thoracic spine was performed with and without contrast. Brain MRI showed no abnormal intracranial mass or enhancement. A heterogeneous expansile intramedullary spinal cord lesion centered at C5–T3 was seen, with nodular foci of enhancement at C3 and C4, along with moderate spinal canal stenosis (Figure 1), and extensive edema from the cervical medullary junction to T7 (Figure 2), with formation of a syrinx at C7 level. Cerebrospinal fluid (CSF) was clear and colorless with 2 white blood cells/mm3, 4 red blood cells/mm3, glucose 3.4 mmol/L, and slightly elevated protein at 0.56 g/L. CSF cultures and cytometry were negative. Initially, the plan was to obtain a spinal cord biopsy, but having noticed incidental bilateral hilar lymphadenopathy on thoracic spinal imaging, computed tomography (CT) of the chest was performed. With definitive hilar adenopathy, the spinal cord biopsy was cancelled and a transbronchial biopsy of the lymph nodes was obtained. The biopsy was significant for numerous non-necrotizing epithelioid granulomas in the background of small lymphocytes, consistent with granulomatous lymphadenitis (Figure 3). Bronchoalveolar lavage cultures were negative. Flow cytometry of blood and peripheral lymph nodes was normal. In view of the patient’s history, the presence of multiple non-caseating granulomas in the biopsy, along with patchy and nodular lesions of the spine and the lack of infectious symptoms, his presentation was felt to be consistent with a new diagnosis of sarcoidosis with involvement of the cervical and thoracic spine. The diagnosis was discussed among the medicine, rheumatology, neurology, and neurosurgery departments. Eye examination and echocardiogram revealed no evidence of sarcoid involvement. Hepatitis serology and QuantiFERON gold testing were normal. His laboratory tests were negative for HIV and anti-nuclear antibody, significant for low vitamin D levels (44.93 nmol/L), normal for vitamin B12 and thyroid stimulating hormone levels, and normal for CSF angiotensin-converting enzyme (ACE) (31 U/L). Aquaporin-4-IgG was negative, ruling out neuromyelitis optica spectrum disorders. Myelin oligodendrocyte glycoprotein IgG was not tested.
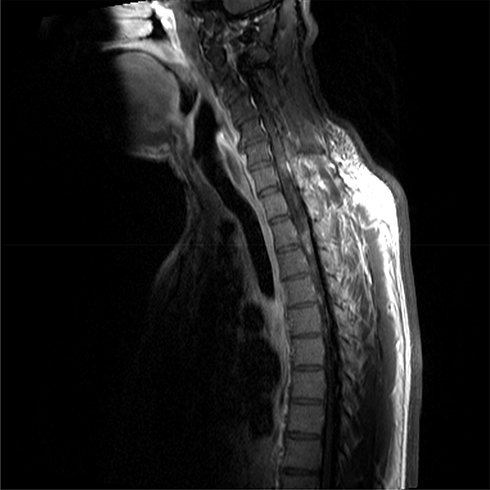 |
Figure 1 T1 sagittal magnetic resonance imaging: patchy and expansile intramedullary spinal cord lesion. |
 |
Figure 2 Short tau inversion recovery magnetic resonance imaging: edema in the cervical cord. |
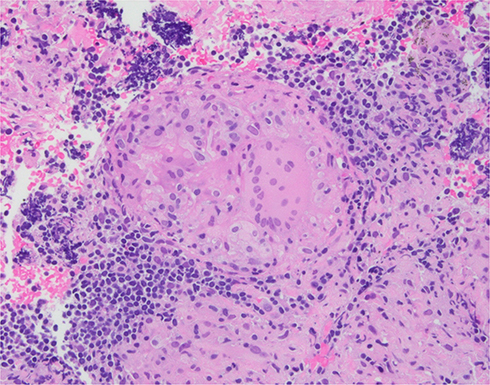 |
Figure 3 Lymph-node biopsy: non-caseating granuloma. |
Treatment was initiated with 60 mg intravenous methylprednisolone every 12 hours. He developed a Foley catheter-related urinary tract infection, treated with ceftriaxone, after which he was started on infliximab 5 mg/kg for one dose and subsequently discharged on oral prednisone 30 mg twice daily. During his hospital stay, he had mild improvement in sensation up to the bilateral thighs, although he continued to suffer proprioceptive deficits in the distal lower extremities. His left arm strength recovered. He was discharged to acute rehabilitation for 2 weeks. On follow-up at 2 months, he reported improvement in mobility with physical therapy. He could stand on his own when supporting himself with his upper extremities, and was able to take a few steps unassisted with a walker. However, there was not much improvement in lower extremity sensory loss and he still felt as if there were a tight band across his chest. He still suffered from paresthesia in his hands, but with normal strength. The neurogenic bladder was persistent, requiring intermittent self-catheterization and bethanechol.
Discussion
Sarcoidosis is an idiopathic granulomatous multisystem disorder, which primarily affects the lungs, lymph nodes, eyes, and skin. However, it is also known to affect other organs, including joints, liver, heart, spleen, gastrointestinal tract, nervous system, paranasal sinuses, parotid, breast, tongue, and skeletal muscle, which accounts for its myriad of symptoms and presentations.2 Nervous system involvement is seen in 5–15% of cases, with 6–8% of these cases having spinal cord involvement; isolated spinal cord involvement is noticed in less than 0.5% of patients.3,4 Postmortem studies found an increasing frequency of neurosarcoidosis cases, only half of which were diagnosed prior to autopsy. Spinal sarcoidosis is underrecognized or misdiagnosed as it resembles other longitudinally extensive types of myelitis, such as neuromyelitis optica spectrum disorder, although this can be differentiated by the presence of aquaporin-4-IgG antibodies.5 Sarcoidosis is more prevalent in African Americans than in Caucasians of European heritage and Asians.2 In a study conducted in Olmsted county in Minnesota, USA, the incidence of sarcoidosis was 11 per 100,000 per year in mainly white people of northern European ancestry.6 In the Optum insurance database, African Americans had an incidence of 17.8 per 100,000.6 In the majority of case reports in the literature, sarcoidosis occurs between 20 and 45 years of age; however, in more recent studies, the peak age was reported to be close to 30–55 years old.6 Women are more commonly affected than men.7 Sarcoidosis most commonly affects cranial nerves, with the seventh nerve, followed by the second nerve, being mostly frequently involved.4 Prognosis is good for clinical manifestations such as cranial and peripheral neuropathy, cognitive difficulty, and seizures, but a bad prognosis is reported for patients with parenchymal brain lesions, meningeal disease, and myopathy.4 The clinical presentation of spinal sarcoidosis varies depending on extradural, intradural, or intramedullary involvement.4 Cervical and thoracic segments are most commonly affected.4 Clinical features include radiculopathy, autonomic dysfunction, cauda equina syndrome, neurogenic bladder, paraparesis, and tetraparesis. Myelopathic features are noticed caudal to the spinal lesion, whereas radiculopathy is seen at the level of the lesion.4 Prognosis can be favorable with early recognition and if treatment is initiated, particularly if spinal cord atrophy has not occurred.4
Modified Zajicek criteria are used for the diagnosis of neurosarcoidosis:8
- Definitive: clinical presentations should prompt diagnosis along with positive neural biopsy.
- Probable: evidence of central nervous system inflammation on imaging and positive extraneural biopsy with exclusion of other diseases.
- Possible: clinical presentation prompting the diagnosis with exclusion of other diseases without histopathological confirmation.
Our patient had probable spinal sarcoidosis according to these criteria. MRI with and without contrast is more sensitive than CT, particularly in depicting the number of lesions and extent of involvement, although it can be normal in patients who are receiving steroids.4 CT with contrast is suggested in those with contraindications to MRI.4 Spinal sarcoidosis can have various manifestations. Most common is leptomeningeal enhancement, followed by pachymeningeal and then intramedullary enhancing lesions.9 MRI findings in spinal sarcoidosis can be divided into four stages: 1) leptomeningeal linear enhancement; 2) intramedullary enhancing lesions with cord enlargement due to spread of inflammation to parenchyma through the perivascular spaces; 3) normal size cord with focal or multifocal enhancement; and 4) spinal atrophy and no enhancement.10 Our patient had stage 2 and 3 disease with multifocal patchy enhancement and cord edema. Diagnosis is challenging when spinal sarcoidosis presents without overt systemic complaints, as observed in our patient.10 Patients with neurosarcoidosis also have a high frequency of pulmonary involvement, and hence chest X-rays can be taken as part of the initial work-up.4 Our patient had normal chest X-ray but incidental hilar adenopathy on thoracic spine CT, which was confirmed on a dedicated chest CT. The sensitivity of ACE levels is low (24–55%), although specificity is high (94–95%).4 ACE levels are found to be not elevated in some case series of spinal sarcoidosis patients.9 Levels were normal in both serum and CSF analyses in our patient. The diagnosis of neurosarcoidosis is not definitive as biopsy of the nervous system is invasive and should be avoided. We confirmed this diagnosis by means of paratracheal lymph-node biopsy. Alternative diagnosis with neural biopsy is needed if patients fail to improve with standard therapy or if the likelihood of their having spinal sarcoidosis is low, even with established extraneural disease.4 Many patients have cervical stenosis, leading to a consideration of spondylotic myelopathy, and thus they are subjected to decompression surgery with no improvement.11 Central nervous system lesions may be small, granulomas are not well formed, and biopsy may produce negative results.3
For asymptomatic individuals incidentally identified on imaging, prednisone or hydroxychloroquine can be given. For symptomatic cases, initial treatment with corticosteroids 1 mg/kg/day can be tried. Severe disabling cases can be treated with pulse-dose intravenous methylprednisolone followed by slow tapering based on the individual patient’s response over 6–12 months.4,8 If no response is seen and symptoms are severe, administration of infliximab or cyclophosphamide is tried. For mild to moderate cases, mycophenolate mofetil, methotrexate, and azathioprine can be tried. Severe resistant cases may need radiotherapy and surgery.4 In a 2018 meta-analysis of neurosarcoidosis (brain and spine) outcomes, partial or complete remission occurred in 59%, disease remained quiescent in 24%, and progression occurred in 6% of treated patients.8 Our patient showed a good response to steroids and infliximab infusion. His motor function recovered to baseline, with improvement in proprioception in the proximal lower extremities and stable persistent distal lower extremity deficits during follow-up.
Conclusion
General physicians must have a high index of suspicion to include sarcoidosis as an important differential for patients of African American descent who are aged 20–50 years and present with spinal cord lesions. Early identification and treatment with steroids can prevent further progression of the disease.
Acknowledgments
No funding was received for this work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Saadi A, Rajashekara S. Intramedullary spinal neurosarcoidosis. Radiol Case Rep. 2015;7(4):739. doi:10.2484/rcr.v7i4.739
2. Kidd DP. Sarcoidosis of the central nervous system: clinical features, imaging, and CSF results. J Neurol. 2018;265:1906–1915. doi:10.1007/s00415-018-8928-2
3. Kasliwal MK, Harbhajanka A, Nag S, O’Toole JE. Isolated spinal neurosarcoidosis: an enigmatic intramedullary spinal cord pathology-case report and review of the literature. J Craniovertebr Junction Spine. 2013;4(2):76–81. doi:10.4103/0974-8237.128536
4. Terushkin V, Stern BJ, Judson MA, et al. Neurosarcoidosis: presentations and management. Neurologist. 2010;16(1):2–15. doi:10.1097/NRL.0b013e3181c92a72
5. Flanagan EP, Kaufmann TJ, Krecke KN, et al. Discriminating long myelitis of neuromyelitis optica from sarcoidosis. Ann Neurol. 2016;79:437–447. doi:10.1002/ana.24582
6. Arkema EV, Cozier YC. Epidemiology of sarcoidosis: current findings and future directions. Ther Adv Chronic Dis. 2018;9(11):227–240. doi:10.1177/2040622318790197
7. Radwan W, Lucke-Wold B, Robadi IA, Gyure K, Roberts T, Bhatia S. Neurosarcoidosis: unusual presentations and considerations for diagnosis and management. Postgrad Med J. 2016;93(1101):401–405. doi:10.1136/postgradmedj-2016-134475
8. Owen CI, Jabeen F, Bhattacharjee A. Application of the modified Zajicek criteria to diagnose probable spinal cord neurosarcoidosis. Clin Case Rep. 2018;6(9):1718–1722. doi:10.1002/ccr3.1712
9. Sakushima K, Yabe I, Nakano F, et al. Clinical features of spinal cord sarcoidosis: analysis of 17 neurosarcoidosis patients. J Neurol. 2011;258(12):2163–2167. doi:10.1007/s00415-011-6080-3
10. Soni N, Bathla G, Pillenahalli Maheshwarappa R. Imaging findings in spinal sarcoidosis: a report of 18 cases and review of the current literature. Neuroradiol J. 2019:32(1):17–28. doi:10.1177/1971400918806634.
11. Kwon DH, Lee SH, Kim ES, Eoh W. Intramedullary sarcoidosis presenting with delayed spinal cord swelling after cervical laminoplasty for compressive cervical myelopathy. J Korean Neurosurg Soc. 2014;56(5):436–440. doi:10.3340/jkns.2014.56.5.436
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.