Back to Journals » International Journal of Nanomedicine » Volume 12
Intracellular “activated” two-photon photodynamic therapy by fluorescent conveyor and photosensitizer co-encapsulating pH-responsive micelles against breast cancer
Authors Luo L, Zhong H, Liu S, Deng L, Luo Y, Zhang Q, Zhu Y, Tian Y, Sun Y, Tian X
Received 25 April 2017
Accepted for publication 30 June 2017
Published 21 July 2017 Volume 2017:12 Pages 5189—5201
DOI https://doi.org/10.2147/IJN.S140345
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Lei Luo,1 Hong Zhong,1 Shuang Liu,1 Lidong Deng,1 Yonghuang Luo,1 Qiong Zhang,2 Yingzhong Zhu,2 Yupeng Tian,2 Yuan Sun,3 Xiaohe Tian4
1College of Pharmaceutical Sciences, Southwest University, Chongqing, People’s Republic of China; 2Department of Chemistry, Key Laboratory of Functional Inorganic Material Chemistry of Anhui Province, Anhui University, Hefei, People’s Republic of China; 3Department of Chemistry and Biochemistry, The Ohio State University, Columbus, OH, USA; 4School of Life Science, Anhui University, Hefei, People’s Republic of China
Abstract: The application of photodynamic therapy (PDT) for the diagnosis and treatment of cancer is hindered by the intrinsic defects of the currently available photosensitizers (PSs), such as poor water solubility and limited light-penetration depth. In this study, pH-responsive polymeric micelles that co-encapsulate therapeutic PSs and organooxotin two-photon compounds were applied for two-photon PDT (TP-PDT) against breast cancer. The TP-PDT effect of the drug-loaded micelles was “activated” when the micelles turned into aggregates at a triggering pH level. The in vitro therapeutic effect was evaluated on 4T1 murine breast cancer cells by viability assays, real-time morphology collapsing, and reactive oxygen species determination. Time-dependent ex vivo organ distribution and in vivo anticancer efficacy results suggested that the drug carriers could accumulate in tumors and suppress tumor growth by TP-PDT. The delivery system could enhance the solubility and distribution of PSs and, if administered along with a tissue-penetrating prolonged light source, could thus have good potential for cancer therapy.
Keywords: photodynamic therapy, two-photon absorbance, drug delivery, pH responsiveness, cancer therapy
Introduction
Photodynamic therapy (PDT) is a noninvasive treatment for cancer that combines light, photosensitizers (PSs), and reactive oxygen species (ROS) for therapeutic effect.1–3 However, most of the PSs applied in clinical settings belong to the porphyrin family, and these compounds are often limited by their poor water solubility. The extended delocalized aromatic π electron system of PS molecules leads to aggregation, which subsequently inhibits ROS generation.4 To improve the solubility of PSs, various polymeric micelles have been used as drug carriers.5 Meanwhile, micellar drug delivery systems can enhance drug accumulation in tumor tissues by the enhanced permeability and retention (EPR) effect.6 However, a certain extent of PS aggregation happens inside the micelles and thus restrains the PDT effect.7 Therefore, designing a proper carrier for PSs still remains a challenge.
Furthermore, porphyrins show absorption spectra with a characteristic peak in the Soret band (~400 nm) and several weak absorption peaks in the visible range (Q-band, ~660 nm).8 The visible light used for activation within these absorption bands penetrates poorly through biological tissues, which limits PDT therapeutic application and damages normal tissues. To overcome this problem, near-infrared (NIR) light is used to increase the tissue-penetration depth. NIR-excitable PSs and lanthanide-doped upconversion nanoparticles (UCNPs) are the two main approaches. However, the former are difficult to synthesize, and the introduced rare-earth UCNPs are currently undergoing biocompatibility evaluation.9 Another method is the indirect excitation of PSs with NIR light by two-photon absorption (TPA). TPA is a nonlinear optical process, in which two photons are absorbed simultaneously at a wavelength that is practically in the NIR region to promote a molecule to the excited state corresponding to the combined energy of the two photons.10 However, conventional PSs typically possess a low TPA cross-section; for example, most porphyrins have only 1–10 GM (1 GM =10−50 cm4·s/photon) in the visible region.11 Compounds with a large TPA cross-section have been reported and used as donors to upconvert NIR light to excite PSs.12–14 This nonlinear optical process is referred to as Förster resonance energy transfer or fluorescence resonance energy transfer (FRET). FRET occurs when the absorption spectra of the acceptor (the PS) overlaps with the upconverted emission spectra of the donor and, normally, if the distance between the donor and the acceptor is <10 nm.
Recently, we described the design, synthesis, and full characterization of a novel triphenylamine cyanoacetic acid-based two-photon absorbing compound (LTPA, Scheme 1C).15 LTPA is an organooxotin compound exhibiting a large TPA cross-section (~2,500 GM) in the NIR region (800–900 nm). Herein, we present a strategy of co-encapsulating both hydrophobic LTPA and PS pyropheophorbide a (PPa, Scheme 1A) in pH-responsive micelles for two-photon PDT (TP-PDT) against breast cancer. A pH-responsive diblock polymer, esterified poly (2-hydroxypropyl methacrylate)-co-poly(2-diisopropylamino) ethyl methacrylate (PSHPMA-PDPA, Scheme 1B), was reported for incorporation in an artificial biomineral;16 however, its pH-responsive property and drug-loading capability have not yet been fully exploited. PSHPMA-PDPA forms micelles at both acidic and alkalic pH levels and aggregates at weakly acidic pH levels because of the opposite charges of the two ionizing blocks. The TP-PDT effect of PSs is inhibited in the micelles and “activated” in the polymer aggregates at pH 6.8. The morphology, size distribution, and zeta potential of cargo-loaded micelles have been characterized by dynamic light scattering (DLS) and scanning electron microscopy (SEM). We also report a series of in vitro studies including light on/off cell viability assays, cell apoptosis evaluation, two-photon laser scanning confocal microscope (TP-LSCM)-based cell morphology destruction, and ROS level detection. In addition, in vivo studies including anticancer efficacy and biodistribution have been performed on mice bearing xenograft tumors. The concept of this work is illustrated in Scheme 2.
 | Scheme 1 Molecular structures of (A) pyropheophorbide a, a photosensitizer, (B) PSHPMA70-PDPA50, and (C) LTPA, a two-photon absorbing compound. |
 | Scheme 2 Schematic illustration of PSHPMA-PDPA as a pH-responsive drug carrier that is activated in the cell for TP-PDT when the micelles are transformed into aggregates. |
Materials and methods
Materials
Please refer to the supplementary data for details. The PSHPMA-PDPA diblock copolymer was prepared as described in the literature.16
Preparation of micelles
Empty micelles (EM), encapsulated-PPa micelles (PM), encapsulated-LTPA micelles (LTPAM), and co-encapsulated-PPa/LTPA copolymer micelles (PLTPAM) were prepared by the thin-film hydration method.17,18 PSHPMA70-PDPA50 polymer (10 mg) and the drug (1 mg of PPa and/or LTPA, 1:1 w/w when co-encapsulated) were dissolved in 3 mL of methanol and 2 mL of chloroform, respectively. A drug-loaded polymer film was formed by solvent evaporation in a vacuum oven for 10 min. A round-bottomed flask containing the film was placed in a vacuum oven for further 2 h to remove the solvent residues. The thin film was then hydrated by adding 1 mL of 0.01 M PBS, and the solution was stirred at room temperature for 6 h to form micelles. The solution was filtered through a 0.22 μm membrane filter to remove unloaded hydrophobic drugs. The final solutions (PPa =1.87 mM and LTPA =0.63 mM) were stored at 4°C before use.
Characterization of micelles
The size and zeta potentials of the micelles at different pH levels were measured with a Zeta-sizer (Malvern Instruments, Malvern, UK) at a wavelength of 633 nm at 25°C. The intensity of the scattered light was detected at a scattering angle of 90°. Each measurement was performed in triplicate. The morphologies of micelles were characterized by S-4800 SEM (Hitachi, Tokyo, Japan). The sample (0.1 mg/mL) was prepared on a single-crystal silicon wafer and coated with gold prior to examination. The ultraviolet/visible (UV/Vis) absorption and emission spectra of micelles were obtained with a UV-6100 spectrophotometer (Shanghai Mapada Instruments, Shanghai Shi, China) and an F-7000 fluorescence spectrophotometer (Shimadzu, Kyoto, Japan), respectively.
TPA cross-section determination
The TPA cross-sections (δ) of the samples were obtained by the two-photon-excited fluorescence method with a femtosecond laser pulse and a Ti:Sapphire system (680–1,080 nm, 80 MHz, 140 fs, Chameleon II) as the light source. The concentration of the LTPAM solution was 1.0×10−14 mol/L. Thus, the δ values of the sample were determined by the following equation:
|
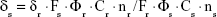
In this equation, the subscript s is the sample; subscript r, the reference molecule; F, the integrated area of the detected two-photon-induced fluorescence signal; Φ, the fluorescence quantum yield; C, the concentration of the solution; and n, the refractive index of the solution. The δ value of reference is taken from the literature.19
In vitro release studies
The in vitro release behaviors of PPa and LTPA from PLTPAM were assessed by a dialysis method. The pH-sensitive release properties were determined in PBS at 37°C (pH =6.3 and pH =7.4, respectively). At pH 7.4, 5 mL of PLTPAM solution at a drug concentration of 1 mg/mL was placed in a dialysis bag (molecular weight cut-off =3,500 Da), and the bag was immersed in 100 mL of 0.01 M PBS with stirring at 37°C for 36 h. An aliquot was withdrawn at set time points and passed through a 0.22 μm membrane filter before quantification. The cumulative release of PPa and LTPA was calculated based on the absorbance at the corresponding maximum absorption wavelengths. To investigate the drug release from polymer aggregates, an equivalent PLTPAM solution was also placed in a dialysis bag, and the bag was immersed in 100 mL of 0.01 M PBS (pH 6.5) with stirring at 37°C for 36 h. An aliquot was withdrawn at set time points and the pH level was adjusted back to 7.4 with NaOH. The added volume of base solution was recorded for the calculations.
The amount of PPa was calculated based on the calibration curve at a wavelength of 710 nm (Figure S1A). After deducting absorbance of PPa at 400 nm, LTPA was calculated according to the standard curve at a wavelength of 400 nm (Figure S1B). Calibration curves were determined by a UV/vis spectrometer UV6100 (Shanghai Mapada Instruments, Shanghai Shi, China).
Determination of singlet oxygen (1O2)
The generation of 1O2 from PPa in various solutions was quantitatively evaluated by detecting the chemical oxidation of anthracene-9,10-dipropionic acid disodium salt (ADPA), which is an amphiphilic singlet-oxygen indicator. The absorption of ADPA at 400 nm should be quenched in the presence of 1O2.20 Testing samples were first mixed with 20 μM ADPA in a PBS solution (pH =7.4) and then transferred into a quartz microcuvette (1 cm pathlength, 1 mm width, and 0.5 cm3 solution volume) before exposure to a laser beam (Xian Midriver Optoelectronics Technology, Xian, China) of specific excitation wavelength (660 and 808 nm laser for one- and two-photon excitations, respectively).
Cellular uptake and in vitro PDT assays
A ZEISS 710 TP-LSCM (CarlZeiss, Oberkochen, Germany) was employed for live-cell imaging experiments. For cellular uptake of PLTPAM, 4T1 cells were incubated with PLTPAM (5.1 μM PPa and 1.3 μM LTPA) for 2 h. PPa (5.1 μM) and LTPA (1.3 μM) in dimethylsulfoxide (DMSO) were also incubated individually for 2 h for comparison. The commercial cell dyes 4,6-diamidino-2-phenylindole (DAPI) and Syto9 were used for staining, and the cells were incubated at 37°C (5% CO2) for 3 h. The uptake of the live cells was monitored for 30 min after PLTPAM was added to the culture plate. Lambda stack scanning was performed by treating the cells with PLTPAM and LTPAM for 2 h, and the scanning range was set from 396 to 716 nm with an excitation wavelength of 808 nm. The level of intracellular ROS was quantified by using a Reactive Oxygen Species Assay Kit with 2′,7′-dichlorofluorescein diacetate; the fluorescence was captured under a confocal microscope (excitation wavelength [Ex] =485 nm, emission wavelength [Em] =535 nm), and the relative levels of ROS were analyzed by an image processing software ImageJ. Endocytosis inhibitor treatment was required to maintain the serum-free conditions throughout the experiment. Cells were treated with the six inhibitors chlorpromazine, chloroquine, nocodazole, 2-deoxy-D-glucose, colchicine, and NH4Cl at the suggested concentrations for 30 min before the addition of PLTPAM.21,22
In vitro cytotoxicity
All the cells involved in this paper were supplied by KeyGEN Ltd. The murine breast cancer cells (4T1) and the human embryonic lung fibroblasts (HELF) cells were grown in Dulbecco’s Modified Eagle’s Medium with high glucose, cultured in a humidified incubator at 37°C (5% CO2/95% air atmosphere) with 10% fetal bovine serum and 1% antibiotics (streptomycin, 100 U/mL, plus penicillin). The cells were harvested with 0.02% ethylenediaminetetraacetate and 0.025% trypsin and rinsed. The resultant cell suspension was used in the following experiments. A dark cytotoxicity assay (light off) and a laser-mediated cytotoxicity assay (light on) were applied individually. Free PPa and LTPA samples used in the cell experiments were diluted from a freshly prepared 10 mM stock solution in DMSO. The final DMSO concentrations in cellular experiments were below 0.1% (v/v).
For the light-off assays, the relative cytotoxicities of samples against different cell lines (4T1 and HELF cells) were evaluated in vitro with a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. For 4T1 cells, the cells were seeded in 96-well plates at a density of 5×103 cells/well in 100 μL of complete medium and incubated overnight to settle down. Three concentrations (1, 10, and 20 μM) of PSHPMA70-PDPA50, free PPa, and PM were incubated with the cells for 24 h, and PBS was applied as a control. Seven concentrations of free LTPA, LTPAM, and PLTPAM ranging from 0.01 to 10 μM (concentration of LTPA) were incubated with the cells for 24 h, and PBS was applied as a control. For HELF cells, 0.1, 1, and 10 μM of free LTPA, LTPAM, and PLTPAM (concentration of LTPA) were incubated with the cells for 24 h, and PBS was applied as a control. MTT solution (5 mg/mL, 10 μL) was added into each well, and the entire medium was removed after 4 h of incubation. This was followed by the addition of 150 μL of DMSO to each well. The absorbance was measured by a UV/Vis plate reader at a wavelength of 570 nm with a 690 nm background subtraction. The cell viability was calculated with the following formula: viable cells% = (ODsample/ODcontrol) ×100%, in which ODsample and ODcontrol represent the absorbance of the sample and control (PBS) wells, respectively.
For the light-on group, the cells were incubated with PLTPAM (0.3 μM PPa and 0.1 μM LTPA) for 24 h and then dosed with 808 nm laser light (0.3 W/cm2) for four intensities 0, 18, 90, and 180 J/cm2. The MTT assay was carried out similar to that with the light-off group. The IC50 of light-on group was performed by using an 808 nm laser beam to irradiate the PLTPAM-treated cells and incubated for 24 h. Same concentration of PLTPAM-treated cells in dark were used as positive control, PBS-treated cells with same light dosage were applied as negative controls for light-on group and PBS-treated cells without light exposing were used as negative controls for light-off group.
Annexin V/propidium iodide (PI) apoptosis detection assays
The apoptotic cell populations induced by PLTPAM were assessed with an Annexin V–fluorescein isothiocyanate (FITC) and PI kit. 4T1 cells were seeded in 6-well plates (1×106 cells/well) overnight. The PLTPAM + Laser group was assessed by incubating PLTPAM (0.55 μM PPa and 0.15 μM LTPA) with cells and then dosing with an 808 nm laser beam for 10 min. The PLTPAM (0.55 μM PPa and 0.15 μM LTPA) group was assessed in the same way except without light dosing. Cells were then harvested and Annexin V–FITC and PI were added to the cellular suspensions. The mixtures were analyzed by using a Calibur fluorescence-activated cell sort flow cytometer (Becton–Dickinson, Franklin Lakes, NJ, USA). The populations of dead, late apoptotic (necrosis), early apoptotic, and viable cells are represented in the results with the names P2-Q1, P2-Q2, P2-Q3, and P2-Q4, respectively.
Animal experiments
Female BALB/c mice (20±2 g, 6–8 weeks old) were selected for tumor models. The animals were purchased from the Experimental Animal Center, Chongqing University of Medicine in China, and were housed in a controlled-temperature room with regular alternating cycles of light and darkness. The animal experiments were performed in compliance with China’s guidelines for the care and use of laboratory animals and were approved by the experimental animal ethics committee of the School of Pharmaceutical Sciences and School of Chinese Medicine, Southwest University.
Ex vivo fluorescence imaging
The biodistribution of the micelles was determined by using 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine (DiR) (Ex =748 nm, Em =790 nm) as a fluorescent probe. DiR-loaded micelles were prepared by the thin-film hydration method mentioned earlier and were injected into mice by the tail vein (n=3 per group). At set time intervals after administration, mice were sacrificed. The heart, liver, spleen, lungs, and tumors were excised and observed with the Fx Pro In Vivo Image System (Bruker Corporation, Karlsruhe, Germany). The mean fluorescence intensity was calculated by using Analysis Workstation software.
In vivo anticancer activity
The in vivo anticancer activity of drug-loaded micelles was also evaluated on a 4T1 tumor-bearing murine breast tumor model. When the tumor volume reached 50–100 mm3, the mice were randomized into four groups: group 1, PBS – injection of PBS solution via the tail vein; group 2, laser only; group 3, PLTPAM – injection of PLTPAM intravenously without laser exposure; group 4, PLTPAM + laser – injection of PLTPAM via the tail vein and irradiation with an 808 nm laser at a power intensity of 0.3 W/cm2 for 30 min (540 J/cm2) after injection for 4 h, the PPa and LTPA doses being 5 and 2 mg/kg, respectively. The tumor size was measured with a caliper every day for 20 d, and the tumor volume was calculated with the following equation: V = (d2×l)/2, where d and l were the width and length of the tumor, respectively. In addition, the body weights and survival rates of the mice were recorded.
Histology examination
For histology examinations, tumor tissues from control and treated mice were harvested, fixed in 10% neutral buffered formalin, processed routinely into paraffin, sectioned into 4 μm thick slices, stained with H&E, and examined with a digital microscope.
Results
Synthesis of PSHPMA70-PDPA50 diblock copolymers
The synthesis of PSHPMA70-PDPA50 was performed in accordance with a previously published paper.16 1H NMR spectroscopy showed that PSHPMA70-PDPA50 was fully esterified (Figure S2). After esterification and purification of the polymer, 1H NMR spectroscopy indicated that there is no change in the proton signal due to the PDPA block. The high degree of esterification was determined by comparison of the 4 protons from the succinic acid groups at 3.05 ppm with the 12 methyl protons from the DPA residues at 1.05 ppm. Tetrahydrofuran gel permeation chromatography (GPC) analysis showed that both the PHPMA70 homopolymer and the PHPMA70-PDPA50 diblock copolymer precursor have relatively narrow molecular weight distributions, and the molecular weights increased linearly as expected. There were no “shoulder” or “tailing” problems observed in these curves, which indicated that no significant combination occurred under these conditions and chain growth was well controlled during atom transfer radical polymerization. There was no GPC protocol to characterize the PSHPMA70-PDPA50 diblock copolymer. However, because the esterification was carried out under relatively mild conditions, the low molecular weight distribution is expected to be maintained (Figure S3).
Characterization of the copolymer micelles
The characterization of the micelles was in agreement with previous data from the literature. The carboxyl and tertiary amine groups that exist on the PSHPMA and PDPA blocks, respectively, can be charged at different pH values. The positive-to-negative overturned zeta potential demonstrated the micelle core–shell reversing process as the pH level was modulated from 2 to 10 (Figure 1E). The PDPA blocks assembled as the core and were surrounded by a PSHPMA corona if the micelles assembled in PBS solution (0.01 M) above pH 7.4. The core–shell components were “reversed” if the micelles were formed at a pH value <5.5. Aggregates were formed between pH 5.5 and 7 as a result of the opposite-charge attraction between the blocks, and pH 6.5 was the polymer isoelectric point. The particle sizes were varied suddenly and the DLS measurements were unable to detect the particle sizes between pH 5.5 and 7 (Figure 1D, shadow box). The morphologies of PSHPMA70-PDPA50 at pH 2.0, 6.5, and 7.4 were captured with SEM (Figure 1A–C).
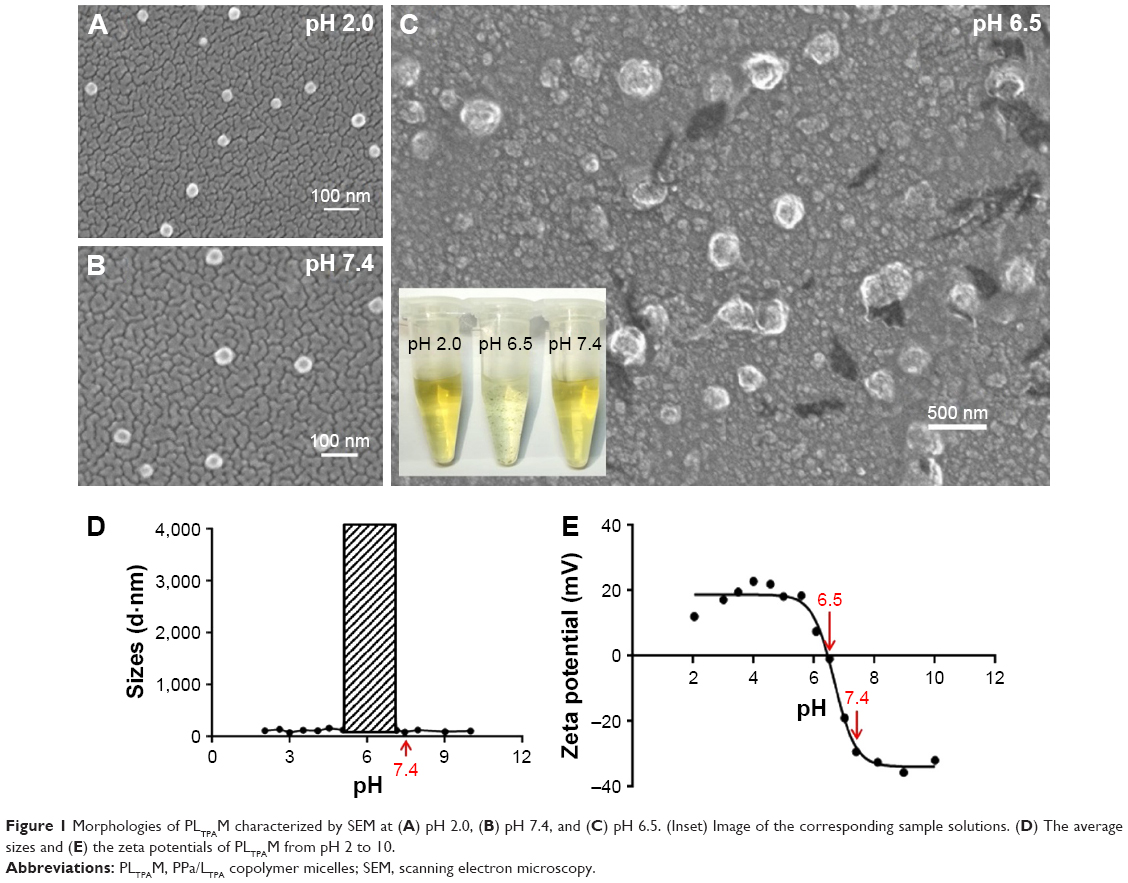 | Figure 1 Morphologies of PLTPAM characterized by SEM at (A) pH 2.0, (B) pH 7.4, and (C) pH 6.5. (Inset) Image of the corresponding sample solutions. (D) The average sizes and (E) the zeta potentials of PLTPAM from pH 2 to 10. |
The thin-film hydration method was applied to prepare EM, LTPAM, PM, and PLTPAM. The average size of EM at pH 7.4 was 55.3±4.2 nm. LTPAM, PM, and PLTPAM showed average sizes of 58.1±2.1, 59.1±3.1, and 63.7±4.8 nm, respectively (Figure S4).
In order to illustrate the co-encapsulation of LTPA and PPa, the UV/Vis absorption and one-photon fluorescence emission spectra of LTPAM, PM, and PLTPAM were investigated. The peaks at 490 and 670 nm on the emission spectrum of PLTPAM were evidence of the inclusion of both compounds (Figure 2B). This feature was further corroborated by the observation of the characteristic individual absorption peaks of PPa and LTPA at 710 and 400 nm (Figure 2A).
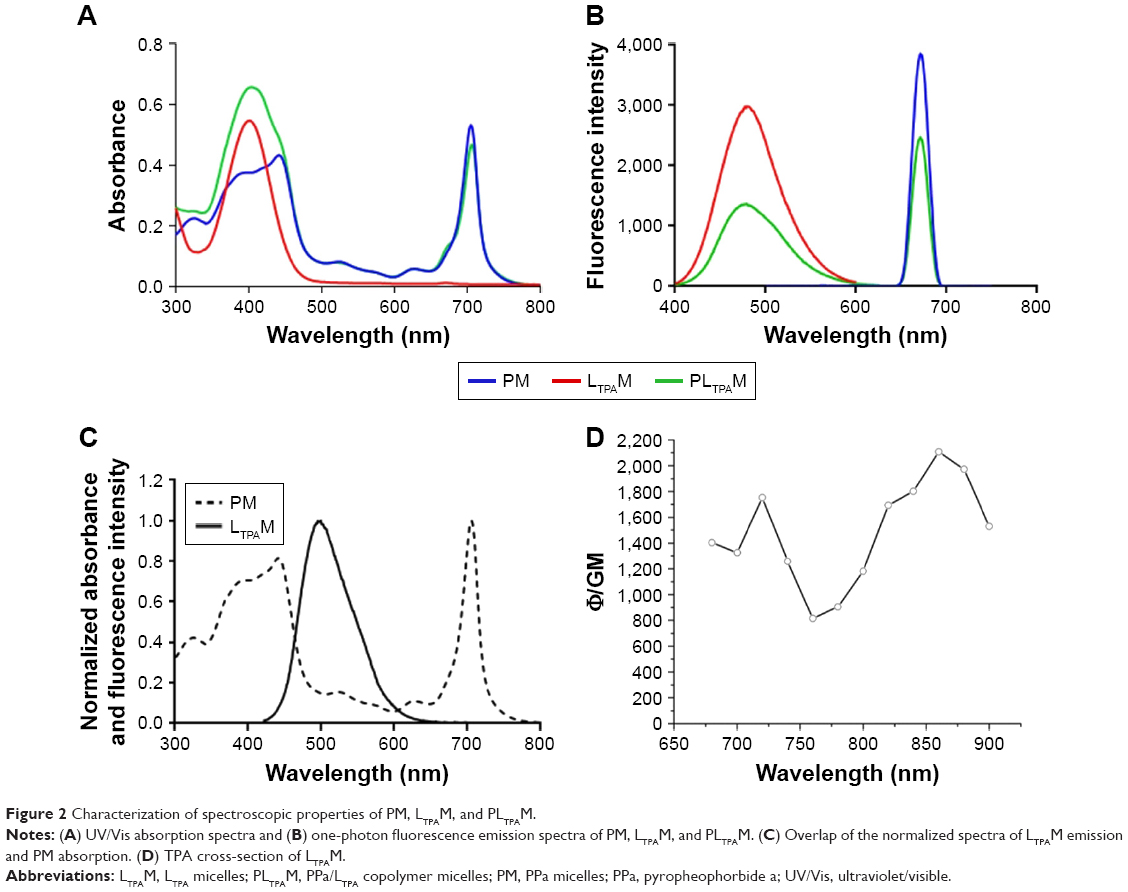 | Figure 2 Characterization of spectroscopic properties of PM, LTPAM, and PLTPAM. |
For the purpose of indirectly exciting PPa with NIR light, a FRET process is required. According to the results, the emission spectrum of LTPA partially overlapped with the Soret band of PPa (Figure 2C). Meanwhile, the TPA cross-section of LTPA in the micelles was around 1,200 GM at 808 nm; this property might come from the formation of a π-conjugated system to enable electronic distribution in the LTPA molecules (Figure 2D). Both results were conducive to the occurrence of FRET.
A dialysis method was applied to evaluate the in vitro release profile of PLTPAM at pH 7.4. The cumulative release of both PPa and LTPA within 24 h was around 40% (Figure 3A, inset). At pH 6.5, samples were acquired at certain time points from the dialysis bag and then the pH level was adjusted back to pH 7.4 to transform the aggregates into soluble micelles. Approximately 40% of PPa and LTPA had been released from the aggregates after dialysis for 24 h (Figure 3A). For the control groups, modulation of the pH level and filtration were performed, and around 10% of the encapsulated compounds were released after this process had been repeated nine times. Furthermore, PLTPAM at pH 7.4 generated much less singlet oxygen (1O2) than the PLTPAM aggregates at pH 6.5 after irradiation with an 808 nm laser (Figure 3B and Figure S5).
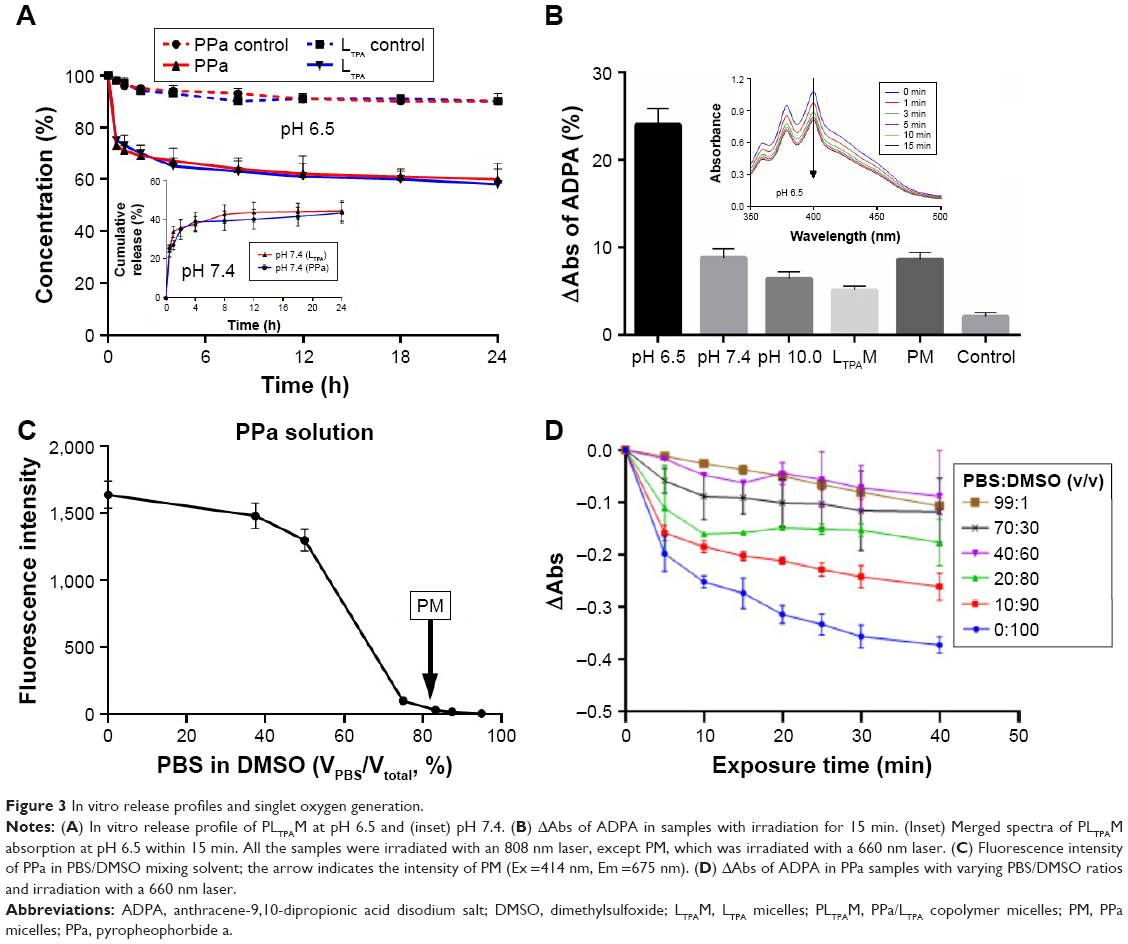 | Figure 3 In vitro release profiles and singlet oxygen generation. |
It was also noted that encapsulated-PM hardly generated any 1O2 with 660 nm laser irradiation. This could be a result of partial aggregation of the hydrophobic PS molecules inside the micelles. The inhibition of 1O2 generation and the fluorescence decay of PPa in micelles were investigated. Equal amounts of PPa were dissolved into a series of PBS/DMSO mixing solutions (Figure 3C). The fluorescence was quenched as the PBS proportion was increased, and >90% of the fluorescence was quenched if the PBS content was 70%. PM showed very limited fluorescence (28.65±3.18 au), which was equal to that of PPa in a solution with 80% PBS. Meanwhile, 1O2 generation of PPa was inhibited if the water proportion of the mixing solution was increased (Figure 3D).
The polymer:cargo ratio was optimized according to the values of encapsulation efficiency (EE% wt) and loading capacity (LC% wt), which were determined by using variable concentrations of cargo and polymers for encapsulation. To pursue economical and efficient encapsulation, an optimal polymer:cargo ratio of 10:1 was selected for the remaining study based on the results (Figure S6).
Cellular uptake and in vitro PDT assays
4T1 murine breast cancer cells were used for cell-based analysis including two-photon microscopy imaging, viability assays, and flow cytometry. Both LTPA and PPa were taken up by the cells after 2 h and primarily distributed in the cytosol (Figure 4A, left panel). Furthermore, co-localization analysis through a scatter plot of the Pearson’s correlation coefficient (r=0.762) indicated an overlap between the donor LTPA and the acceptor PPa (Figure 4A, right panel). The real-time cellular uptake of PLTPAM showed a simultaneous increase in the LTPA and PPa fluorescence intensities (Figure 4B). By contrast, free LTPA and PPa were both taken up by cells slowly, which further emphasized the importance of the micelles in assisting the cellular internalization of the two compounds (Figure S7). The cellular uptake mechanism of PLTPAM was systematically investigated by changing the incubation temperature (4°C and 25°C) and studying the inhibition with well-documented endocytosis and active transport inhibitors.21,22 A significant decrease in fluorescence intensity strongly suggested that both cellular-active transport and endocytosis contribute to the cellular entry of PLTPAM (Figure S8).
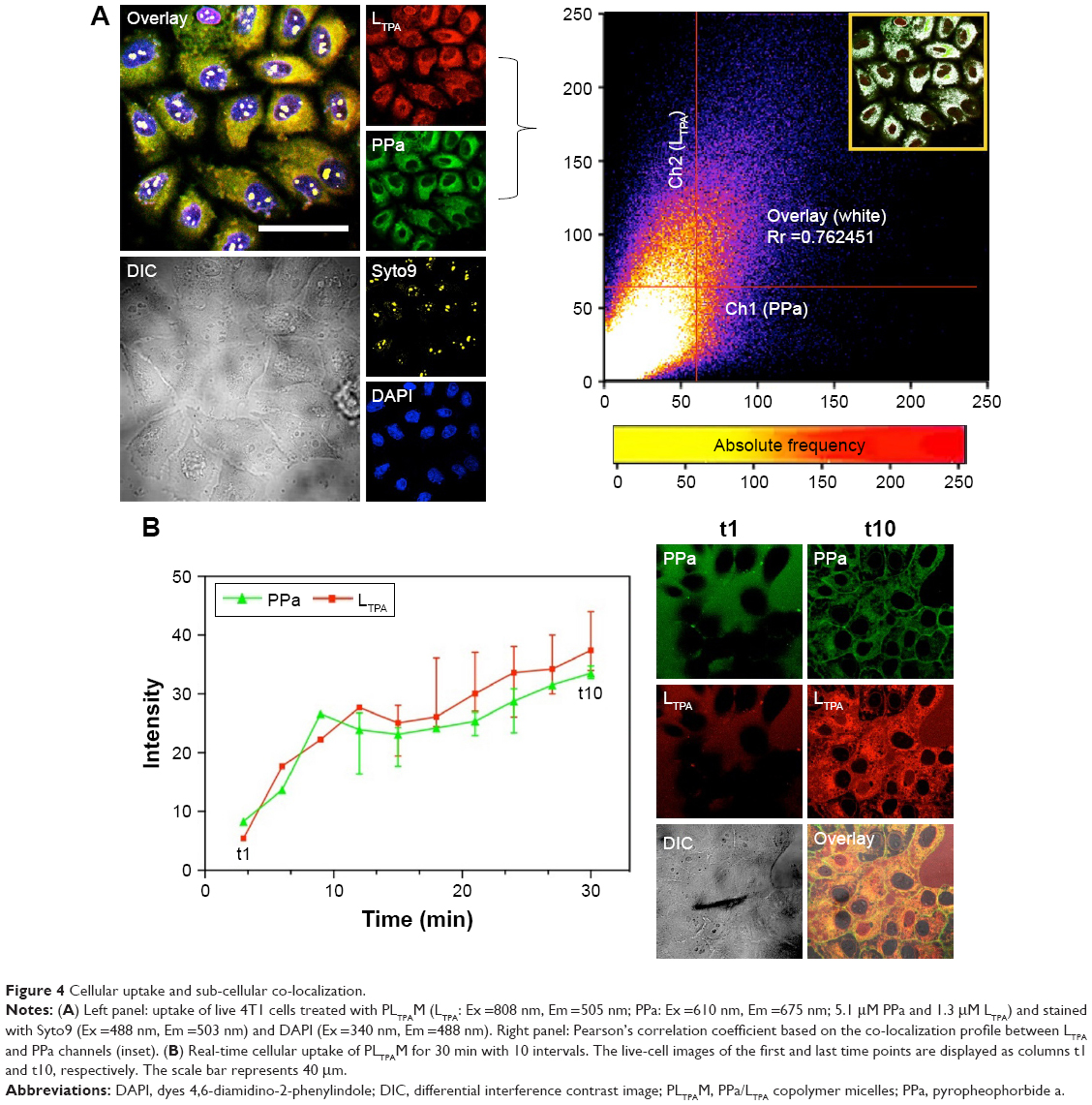 | Figure 4 Cellular uptake and sub-cellular co-localization. |
The TP-PDT effect was then induced by irradiating the cells with a laser under a TP-LSCM. A significant change in the cell morphology was observed (Figure 5A), and a video of the cell morphology collapsing is provided in the supplementary data. These changes are associated with cell apoptosis/necrosis caused by ROS generation, as further proven by the ROS-specific fluorescent probe. The cellular response to oxidative stress of the cells treated with PLTPAM showed an enhanced signal of green fluorescence, which indicated an increase in ROS generation after irradiation with an 808 nm laser (Figure S9).
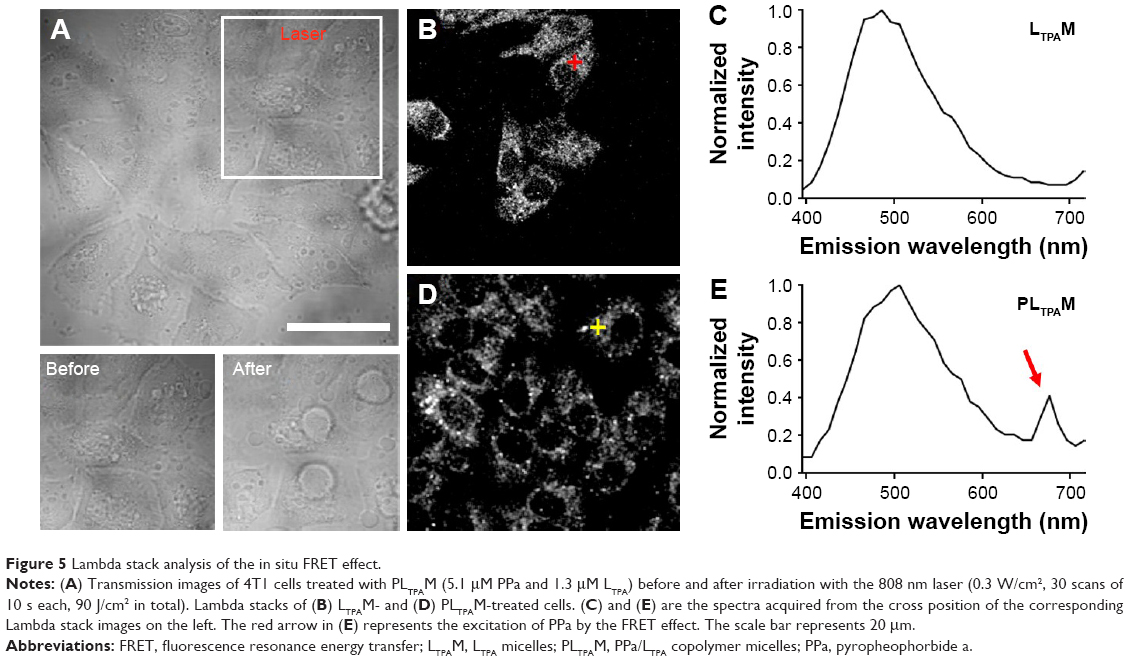 | Figure 5 Lambda stack analysis of the in situ FRET effect. |
To prove that these morphological changes and the ROS generation result from the NIR laser excitation of LTPA and transfer energy to PPa, the emission spectra of both PLTPAM and LTPAM locally on cells were recorded by using Lambda stack scanning from 396 to 716 nm, which allowed the collection of the emission spectra (Ex =808 nm). The red and yellow crosses were randomly selected, and the corresponding spectra represented the fluorescence spectra at each point (Figure 5B and D). The results revealed the characteristic peak of PPa in the spectrum of PLTPAM at 676 nm, which points to the intracellular activation of FRET-based TP-PDT (Figure 5C and E).
Subsequently, we tested light-off cell inactivation with 4T1 murine breast cancer cells. No significant cytotoxicity (viability >70%) was shown up to 20 μM PPa doses. The polymer itself did not show any obvious cytotoxicity with doses up to 20 μM (Figure 6A). Compounds from the organooxotin class have been reported to possess anticancer activity,23 and LTPA could reduce the viability of A549 and MDA-MB-231 cells from our previous research.15 The cytotoxicity of LTPA was also observed in 4T1 murine breast cancer cells, and the micelles can further increase its cytotoxicity (Table 1 and Figure S10A). The cytotoxicity of LTPA on normal cells was studied with human embryo lung fibroblast cells. PLTPAM showed a 50% inhibition rate with dosing at 10 μM, which was 100 times higher than the half-maximal inhibitory concentration (IC50) on 4T1 cells (Figure 7).
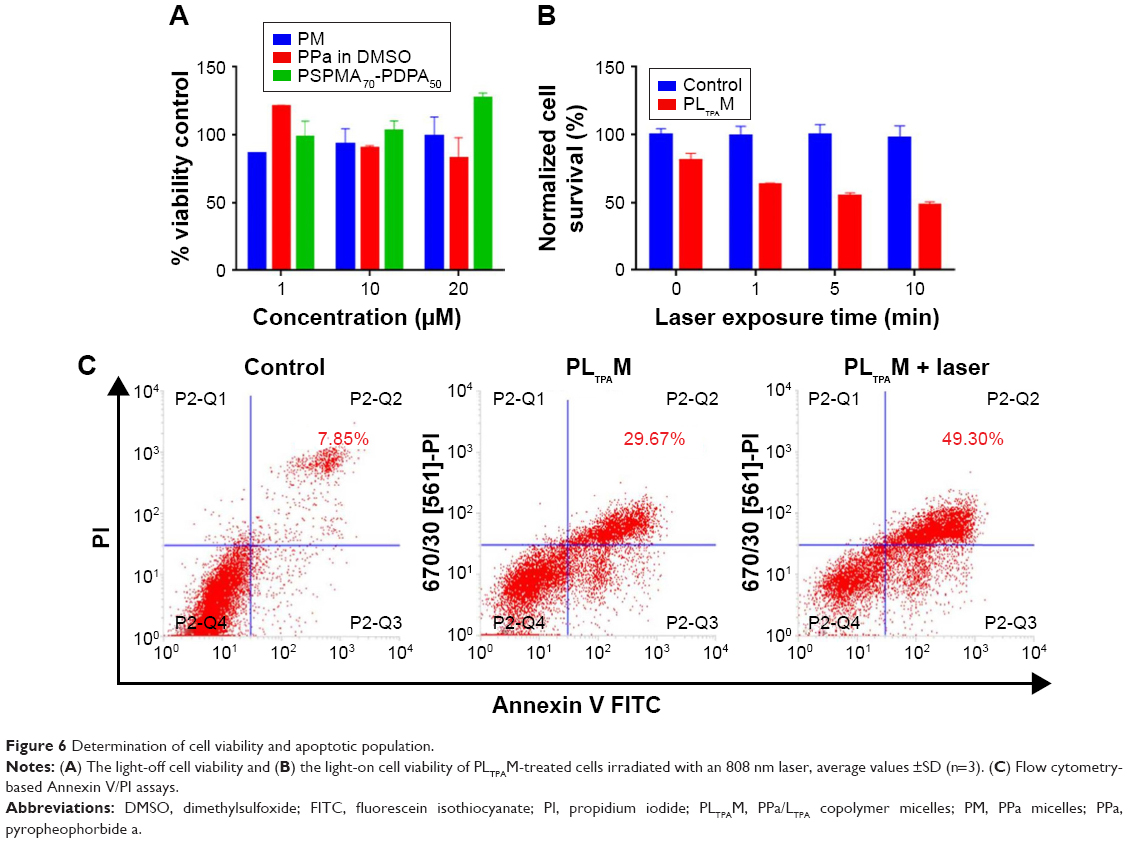 | Figure 6 Determination of cell viability and apoptotic population. |
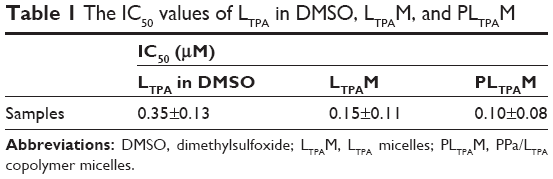 | Table 1 The IC50 values of LTPA in DMSO, LTPAM, and PLTPAM |
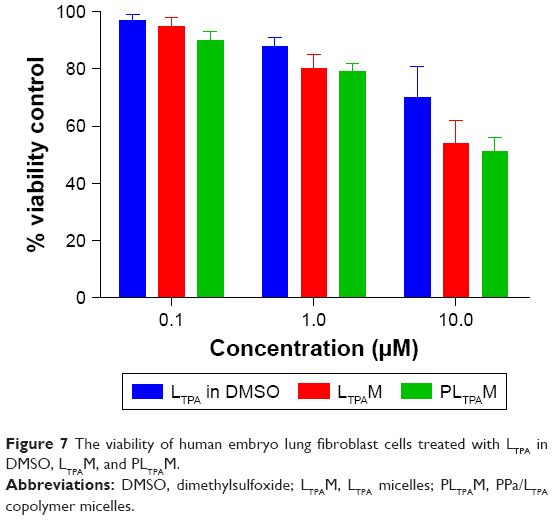 | Figure 7 The viability of human embryo lung fibroblast cells treated with LTPA in DMSO, LTPAM, and PLTPAM. |
Light-on cell inactivation was measured under the same conditions as those for the light-off group but with irradiation with an 808 nm laser at irradiation periods of 0, 1, 5, and 10 min, which correspond to a power density of 0, 18, 90, and 180 J/cm2. The cell viability decreased to 45% after dosing with light for 10 min, whereas the cell viability of the control group remained at around 95% (Figure 6B). Furthermore, the PLTPAM-treated cells dosing with laser showed an IC50 of 0.06±0.05 μM and, in the contrary, IC50 in the dark was 0.10±0.04 μM (Figure S10B). PDT has been proved to be associated with cancer cell apoptosis.24 An Annexin V/PI apoptosis detection assay was used to verify whether the TP-PDT-mediated decrease in cell viability was related to late apoptosis (necrosis). Cells treated with PLTPAM (0.55 μM PPa and 0.15 μM LTPA) showed an obvious increase in the late apoptotic cell population (29.67%) compared with the blank sample population (7.85%; Figure 6C). The late apoptotic cell population was approximately doubled with irradiation for 10 min. This result illustrated the combined anticancer effect of both TP-PDT and LTPA.
In vivo anticancer evaluation
Mice subcutaneously bearing 4T1 murine breast cancer were used for ex vivo and in vivo assays. The ex vivo fluorescence imaging study was carried out to investigate the tumor targeting efficiency and guide time point for light dosing. Due to the faint signal of PPa on ex vivo organs, a hydrophobic probe that can be excited by NIR light, DiR, was encapsulated into the micelles. The fluorescence accumulation in the tumors reached a maximum at around 6 h post injection (Figure 8A), and the light dosing time was therefore set at 4 h after injection. The micelles were largely distributed in excised livers and spleens, which could be a result of the PSHPMA polymer being recognized by the reticuloendothelial system (Figure 8B).
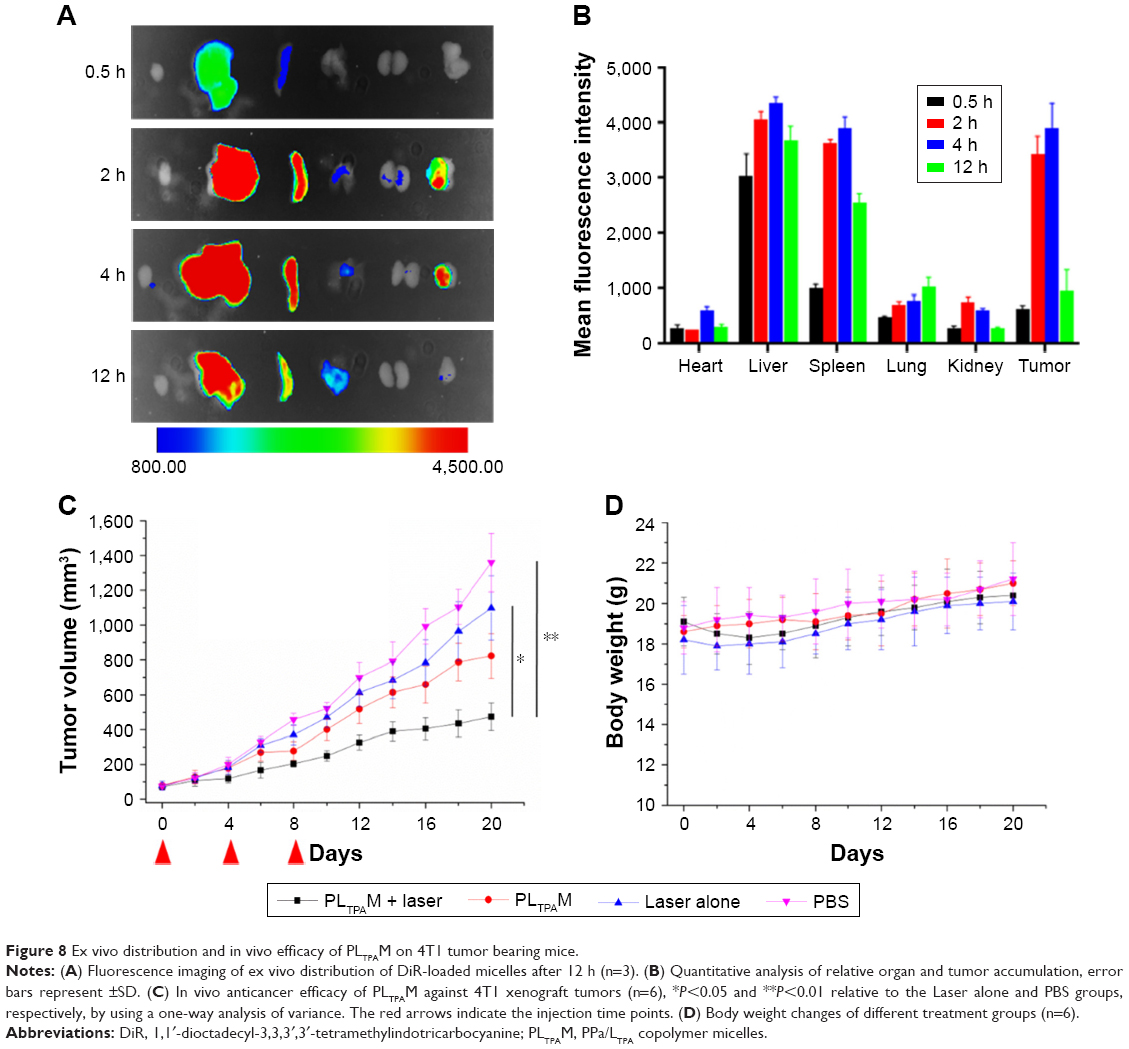 | Figure 8 Ex vivo distribution and in vivo efficacy of PLTPAM on 4T1 tumor bearing mice. |
To evaluate the in vivo anticancer efficacy, PLTPAM were intravenously injected into mice bearing 4T1 tumors and was followed with 30 min NIR laser irradiation (540 J/cm2). The PLTPAM + Laser group slowed down the growth of the tumor, with a final mean tumor size of 474±80 mm3 after 20 d (Figure 8C). The average tumor volume of the PBS group (1,359±168 mm3) was larger than the volume of the laser group (1,099±184 mm3) at the 20th day, which indicated that the laser exposure could provide an additional therapeutic effect. No significant body weight differences were observed for the mice between various treatments (Figure 8D), which suggested low toxicity of PLTPAM after injection. Histological examination results (Figure S11), obtained by H&E staining, revealed significant cell death in the tumor tissues of mice treated with PLTPAM and accompanying laser irradiation.
Discussion
We have prepared pH-sensitive micelles (PSHPMA70-PDPA50) co-encapsulating a photosensitizer (PPa) and a two-photon absorbing conveyor (LTPA) for NIR-laser-activated TP-PDT against breast cancer. The TP-PDT of PLTPAM could be activated at weakly acidic pH levels when the micelles were transformed into aggregates. Moreover, this effect could be turned on intracellularly and generated therapeutic ROS. PLTPAM was able to accumulate in tumors and effectively slowed down 4T1 breast cancer in vivo.
The pH-responsive property was derived from ionization of the carboxyl groups and tertiary amine groups that are present on the PSHPMA and PDPA blocks, respectively. At physiological pH value, the morphology of the co-encapsulated micelles was intact (Figure 1B), which could benefit the delivery of inclusions. Such a small particle size could accumulate preferentially at the tumor by the EPR effect, which would enable a drug delivery system with a decreased drug concentration in healthy tissues and reduced side effects.25,26 Aggregates were formed between pH 5.5 and pH 7 by the opposite-charge attraction of both blocks (Figure 1D).
Our in vitro release results demonstrated that the cargoes wrapped in the micelles were maintained in the polymer aggregates instead of being quickly released at the trigger pH value (Figure 3A). It was proved that both the fluorescence and 1O2 generation of PPa in the micelles imparted a certain extent of inhibition. PPa probably aggregated in the micelles through a π–π stacking owing to their rigid planar structure. Although the core of the micelles is generally regarded as an “oily bulk”, the micelles could exchange solute with the surrounding hydrophilic environment and possess slight water permeability.27 The large-sized and non-ionized aggregates could produce a more hydrophobic inner area than the micelles. Thus, the TP-PDT effect could be “activated” in the aggregates while PPa exists in the molecular state. These pH-dependent properties would be of benefit for specific therapies, to avoid activation in normal organs.
The cellular uptake results showed that the micelles enhanced internalization of both compounds and that the intracellular localizations of LTPA and PPa were favorably overlapped. This provided the required short distance for FRET to occur (Figure 4A). Meanwhile, the intracellular LTPA and PPa concentrations increased simultaneously, which demonstrated the concurrent delivery of both cargoes by LTPAM to enable efficient energy transfer (Figure 4B). Intracellular FRET was proved to occur by a combination of the results of MTT assays, cell morphology collapsing, and ROS determination.
We also reported the TP-PDT and LTPA combined therapy both in vitro and in vivo. LTPA showed cytotoxicity to cancer cells and realized in vivo TP-PDT. A normal cell cytotoxicity assay was performed with human embryo lung fibroblast cells and LTPA showed a much higher IC50 value on the normal cell line than the 4T1 cells. Mice administered with LTPA were alive without obvious body weight changes after 20 d. Investigation on the mechanism and metabolism of LTPA and screening for nontoxic, highly FRET-efficient, and rapidly metabolizable two-photon compounds are the focus of ongoing work in our group.
In the in vivo anticancer assays, the PLTPAM group effectively slowed down tumor growth. Such an effect could be a combination of the anticancer effect of LTPA, penetration-enhanced TP-PDT, and possibly PDT-induced immunotherapy. In conclusion, our study indicates an important role for TP-PDT against cancer and suggests that pH-responsive micelles could provide a safe and effective delivery strategy.
Acknowledgments
We acknowledge financial support from the National Natural Science Foundation of China (Nos 81503009, 51432001, 21501001, 21602003, 51672002, and 51472002) and the Chongqing Science and Technology Commission (Nos cstc2015shmszx80018 and cstc2016jcyjA0550).
Disclosure
The authors report no conflicts of interest in this work.
References
Nyman ES, Hynninen PH. Research advances in the use of tetrapyrrolic photosensitizers for photodynamic therapy. J Photochem Photobiol B. 2004;73(1–2):1–28. | ||
Chang JE, Oak C-H, Sung N, Jheon S. The potential application of photodynamic therapy in drug-resistant tuberculosis. J Photochem Photobiol B. 2015;150:60–65. | ||
Spring BQ, Rizvi I, Xu N, Hasan T. The role of photodynamic therapy in overcoming cancer drug resistance. Photochem Photobiol Sci. 2015;14(8):1476–1491. | ||
Sibani SA, Mccarron PA, Woolfson AD, Donnelly RF. Photosensitizer delivery for photodynamic therapy. Part 2: systemic carrier platforms. Expert Opin Drug Deliv. 2008;5(11):1241–1254. | ||
Díaz-Moscoso A. Soft versus hard nanoparticles in the delivery of aromatic macrocycles for photodynamic therapy of cancer. Int J Med Biomed Res. 2012;1(1):12–23. | ||
Lim CK, Heo J, Shin S, et al. Nanophotosensitizers toward advanced photodynamic therapy of cancer. Cancer Lett. 2013;334(2):176–187. | ||
Nakagishi Y, Morimoto Y, Kawauchi S, et al. Polymeric micelle encapsulating dendrimer-based phthalocyanine for enhanced photodynamic therapy. Cancer Res. 2006;66. | ||
Stables GI, Ash DV. Photodynamic therapy. Cancer Treat Rev. 1995;21(4):311–323. | ||
Gnach A, Lipinski T, Bednarkiewicz A, Rybka J, Capobianco JA. Upconverting nanoparticles: assessing the toxicity. Chem Soc Rev. 2015;44(6):1561–1584. | ||
Kachynski AV, Pliss A, Kuzmin AN, et al. Photodynamic therapy by in situ nonlinear photon conversion. Nat Photonics. 2014;8(6):455–461. | ||
Kruk M, Karotki A, Drobizhev M, Kuzmitsky V, Gael V, Rebane A. Two-photon absorption of tetraphenylporphin free base. J Lumin. 2003;105(1):45–55. | ||
Sapsford KE, Berti L, Medintz IL. Materials for fluorescence resonance energy transfer analysis: beyond traditional donor–acceptor combinations. Angew Chem Int Ed Engl. 2006;45(28):4562–4588. | ||
Zhang K, Tang X, Zhang J, et al. PEG-PLGA copolymers: their structure and structure-influenced drug delivery applications. J Control Release. 2014;183:77–86. | ||
Wang SZ, Zhang JG, Chen HQ, Wang L. An optical FRET inhibition sensor for serum ferritin based on Mn2+-doped NaYF4:Yb, Tm NIR luminescence up-conversion nanoparticles. J Lumin. 2015;168:82–87. | ||
Zhao X, Liu J, Wang H, et al. Synthesis, crystal structures and two-photon absorption properties of triphenylamine cyanoacetic acid derivative and its organooxotin complexes. Dalton Trans. 2015;44(2):701–709. | ||
Kim YY, Ganesan K, Yang P, et al. An artificial biomineral formed by incorporation of copolymer micelles in calcite crystals. Nat Mater. 2011;10(11):890–896. | ||
Chen YW, Chen J, Liu YJ, Luo XL, Li SM. Micelles formed by self-assembling of low molecular weight phosphorylcholine-containing poly(L-lactide). Polym Adv Technol. 2012;23(10):1357–1361. | ||
Song ZM, Zhu W, Liu N, Yang F, Feng R. Linolenic acid-modified PEG-PCL micelles for curcumin delivery. Int J Pharm. 2014;471(1–2):312–321. | ||
Albota MA, Xu C, Webb WW. Two-photon fluorescence excitation cross sections of biomolecular probes from 690 to 960 nm. Appl Opt. 1998;37(31):7352–7356. | ||
Yoon HK, Lou X, Chen YC, Lee YEK, Yoon E, Kopelman R. Nanophotosensitizers engineered to generate a tunable mix of reactive oxygen species, for optimizing photodynamic therapy, using a microfluidic device. Chem Mater. 2014;26(4):1592–1600. | ||
Ziegler HK, Unanue ER. Decrease in macrophage antigen catabolism caused by ammonia and chloroquine is associated with inhibition of antigen presentation to T-cells. Proc Natl Acad Sci U S A. 1982;79(1):175–178. | ||
Puckett CA, Barton JK. Mechanism of cellular uptake of a ruthenium polypyridyl complex. Biochemistry. 2008;47(45):11711–11716. | ||
Lippert B. Book review: metal complexes in cancer chemotherapy. Angew Chem Int Ed Engl. 1994;33(15–16):1672–1673. | ||
Sui B, Yue X, Kim B, Belfield KD. Near-IR two-photon fluorescent sensor for K+ imaging in live cells. ACS Appl Mater Interfaces. 2015;7(32):17565–17568. | ||
Maeda H. Macromolecular therapeutics in cancer treatment: the EPR effect and beyond. J Control Release. 2012;164(2):138–144. | ||
Biswas S, Kumari P, Lakhani PM, Ghosh B. Recent advances in polymeric micelles for anti-cancer drug delivery. Eur J Pharm Sci. 2016;83:184–202. | ||
Maibaum L, Dinner AR, Chandler D. Micelle formation and the hydrophobic effect. J Phys Chem B. 2004;108(21):6778–6781. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.