Back to Journals » Therapeutics and Clinical Risk Management » Volume 12
Intestinal permeability in a patient with liver cirrhosis
Authors Aguirre Valadez JM, Rivera-Espinosa L ![]() , Méndez-Guerrero O, Chávez-Pacheco JL
, Méndez-Guerrero O, Chávez-Pacheco JL ![]() , García Juárez I, Torre A
, García Juárez I, Torre A ![]()
Received 27 June 2016
Accepted for publication 7 August 2016
Published 18 November 2016 Volume 2016:12 Pages 1729—1748
DOI https://doi.org/10.2147/TCRM.S115902
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Jonathan Manuel Aguirre Valadez,1 Liliana Rivera-Espinosa,2 Osvely Méndez-Guerrero,1 Juan Luis Chávez-Pacheco,2 Ignacio García Juárez,1 Aldo Torre1
1Department of Gastroenterology, Instituto Nacional de Ciencias Médicas y Nutrición”Salvador Zubirán, 2Pharmacology Laboratory, Instituto Nacional de Pediatría, Mexico City, Mexico
Abstract: Liver cirrhosis is a worldwide public health problem, and patients with this disease are at high risk of developing complications, bacterial translocation from the intestinal lumen to the mesenteric nodes, and systemic circulation, resulting in the development of severe complications related to high mortality rate. The intestinal barrier is a structure with a physical and biochemical activity to maintain balance between the external environment, including bacteria and their products, and the internal environment. Patients with liver cirrhosis develop a series of alterations in different components of the intestinal barrier directly associated with the severity of liver disease that finally increased intestinal permeability. A “leaky gut” is an effect produced by damaged intestinal barrier; alterations in the function of tight junction proteins are related to bacterial translocation and their products. Instead, increasing serum proinflammatory cytokines and hemodynamics modification, which results in the appearance of complications of liver cirrhosis such as hepatic encephalopathy, variceal hemorrhage, bacterial spontaneous peritonitis, and hepatorenal syndrome. The intestinal microbiota plays a fundamental role in maintaining the proper function of the intestinal barrier; bacterial overgrowth and dysbiosis are two phenomena often present in people with liver cirrhosis favoring bacterial translocation. Increased intestinal permeability has an important role in the genesis of these complications, and treating it could be the base for prevention and partial treatment of these complications.
Keywords: liver cirrhosis, hepatic encephalopathy, spontaneous bacterial peritonitis, variceal hemorrhage, bacterial translocation, intestinal permeability
Introduction
Liver cirrhosis is a worldwide public health problem, and patients with this disease are at high risk of developing complications associated with bacterial infections, particularly spontaneous bacterial peritonitis (SBP) and hepatic encephalopathy (HE).1–5
There is a very close link between the liver and the gastrointestinal tract; the liver is constantly exposed to nutrients, toxins, food-derived antigens, microbial products, and gastrointestinal tract microorganisms.
Susceptibility to the development of bacterial infections in this population is due to various abnormalities in defense mechanisms, including deficient bactericidal capacity and opsonization, abnormal monocytic cell activity, decreased reticuloendothelial phagocytic potential, deficient chemotaxis, decreased complement levels, increased intestinal transit time resulting in bacterial overgrowth, and an increase in intestinal permeability leading to bacterial translocation.
In liver cirrhosis, bacterial translocation is a common denominator in the genesis of several of its complications: upper gastrointestinal bleeding, HE, and SBP.
Increased intestinal permeability may precede and promote translocation of bacteria (migration of microbes or their products into mesenteric lymph nodes),6 endotoxins (such as lipopolysaccharides), and pathogen-associated molecular patterns (PAMPs) into the portal venous system and extraintestinal sites; their presence has been reported in animal models of cirrhosis with ascites (45%–78%) versus normal animals (0%–4%) and in patients with cirrhosis undergoing laparotomy, particularly in those with the greatest liver function compromise.7
Bacterial translocation leads to a systemic inflammatory response with subsequent increases in portal hypertension, exacerbating the characteristic hyperdynamic circulation in these patients, all negatively impacting liver function.6,8
Intestinal bacterial overgrowth plays an important role in the pathogenesis of bacterial translocation in cirrhotic patients; this hypothesis has been supported by the results of clinical and experimental investigation, in which growth inhibition of intestinal Gram-negative aerobic flora decreases the incidence of SBP in these patients.9 This practice is recommended in the international clinical practice guidelines to prevent the development and recurrence of SBP10 and the incidence of intestinal bacterial overgrowth, bacterial translocation, and bacterial peritonitis in animal models.
Another key event in bacterial translocation and overgrowth is the delay in intestinal transit time in patients with cirrhosis and experimental cirrhosis models; some prokinetic drugs such as cisapride have been shown to decrease bacterial translocation and overgrowth in patients with liver cirrhosis.11 This effect has also been described with the use of nonselective beta-blockers (propranolol) in animal models of cirrhosis, accelerating intestinal transit time and decreasing bacterial translocation.12
Likewise, there are structural and functional changes in the intestinal barrier that may be secondary to vascular stasis resulting from portal hypertension and that can lead to increased intestinal permeability to bacterial migration and their products. Structural changes such as mucosal congestion and edema have been observed in cirrhotic patients in whom broadening of intercellular spaces has also been described.13
In this population, there is also recent evidence of probable susceptibility to the development of increased intestinal permeability. Patients with the nucleotide-binding oligomerization domain containing 2 (NOD2) and Toll-like receptor-2 (TLR2) are also at greater risk of developing SBP.14,15
Intestinal barrier
A defensive function is also necessary to prevent the passage of potentially noxious substances, such as pathogenic microorganisms, antigens, and proinflammatory factors, from the intestinal lumen into the internal milieu and that may simultaneously allow selective passage of substances favoring the development of the intestinal immune system; these functions depend on the intestinal barrier.16
The intestinal barrier is a physical and functional separation between the environment and the organism’s interior. It is formed by a mucinous component secreted by intestinal epithelial cells and by the intestinal epithelium per se, creating an intercellular junction layer that allows selective passage of substances. The gastrointestinal epithelium constitutes the longest interphase with the external environment and allows nutrient absorption while also acting as a physical barrier to proinflammatory molecules such as pathogens, toxins, and antigens. In the intestinal lumen, bacteria and antigens are degraded by gastric acid and pancreatic juice and promote a microenvironment fostering commensalism and the generation of antibacterial products.12,17–19 The most important components of the intestinal barrier are hereby described.
Intestinal mucosa
The intestinal epithelium is covered by a microclimate consisting of a nonagitated mucus layer 100 μm in thickness, rich in mucin, water, and glycocalyx, mainly secreted by goblet cells with hydrophobic and tensioactive properties, which prevent the adhesion of enteric bacteria to the intestinal epithelium.19 Externally, there is a layer of agitated mucus composed of mucin and antimicrobial substances; the thickness and composition of the mucus layer vary depending on its location in the intestinal tract, but it is most viscous in the distal colon.20 Although the mucus layer prevents some organisms and large molecules such as food particles to directly access the epithelium, it can only slightly mitigate the flow of small molecules, ions, and water. The epithelial cell membrane is an efficient barrier against most hydrophilic solutes, but it would not be an appropriate barrier should the space between individual cells were not sealed by a series of intercellular junctions.20
Cell types
Various cell types constitute the intestinal barrier’s integrity. In order of frequency, we find absorptive cells or enterocytes, goblet cells, Paneth cells, enteroendocrine cells, and pluripotent stem cells (Figure 1).21
 | Figure 1 Components of the intestinal barrier. It is composed of intestinal microbiota, a mucin layer with IgA immunoglobulin (and other defense proteins) covering the mixed columnar epithelial cells (enterocytes, globet cells, enterochromaffin cells and intestinal stem cells). Subepithelial region containing the lamina propria, the enteric nervous system, connective tissue and muscle layers. Intraepithelial lymphocytes are above, underlying basement membrane. There are many differents immune cells, like macrophages, dendritic cells, plasma cells, lymphocytes and in some cases, neutrophils, and organized lymphoid tissue like lymphoid structures compound as the Peyer’s patch (terminal ileum), containing M cells, dendritic cells and lymphocytes. |
Enterocytes or absorptive cells
These are characterized by a great abundance of microvilli on their apical surface, tightly distributed and parallel to each other, forming a structure known as a “brush border”. Each enterocyte has a mean of 3,000 microvilli, translating into a very large absorptive surface; enterocytes are key elements to the maintenance of the barrier’s physical integrity. They also play a role in the development of immunologic activity since they express receptors implicated in the innate immune response,22 and they act as nonprofessional antigen-presenting cells and release several cytokines and chemokines such as thymic stromal lymphopoietin, transforming growth factor-β1,23 interleukin-25 (IL-25),24 B cell proliferation stimulating factor (APRIL), and B-cell activating factor,25,26 recruiting and activating leukocytes and regulating the local immune response.
Intestinal microbiota
The term intestinal microbiota refers to the group of microorganisms residing in the human intestine that establish a symbiotic relationship with host. Most intestinal bacteria belong to ten phyla; five phyla represent the majority of bacteria that comprise the gut microbiota: 90% are Firmicutes (200 genera) and Bacteroidetes (25 genera). Other phyla are Actinobacteria, Proteobacteria, Verrucomicrobia, Fusobacteria, and Cyanobacteria.27
Microbiota functions can be grouped into three large areas: protective, trophic, and metabolic, and all play a role in the maintenance of epithelial cell integrity; they decrease colonization by pathogens by producing antimicrobial substances, modify the pH, stimulate mucus secretion, and hence contribute to the baseline conservation of the host’s defenses in a stationary state (Figure 1).27
Intercellular junction proteins
Epithelial cells foster selective permeability in the following two ways: one is transcellular and the other is paracellular. The first (transcellular) involves nutrient absorption via transporters or channels located on the apical or basolateral membrane, while the paracellular mechanism is associated with transport in the intercellular space between adjacent epithelial cells; it is regulated by apical union complexes and formed by tight junctions (TJ), anchoring junctions, and communicating junctions (GAP; Table 1).28–30 Two types of intestinal permeability have been described: mediated by pores (characterized by its highly selective capacity in terms of particle size and charge) and by “leakage” (characterized by its low selective capacity).
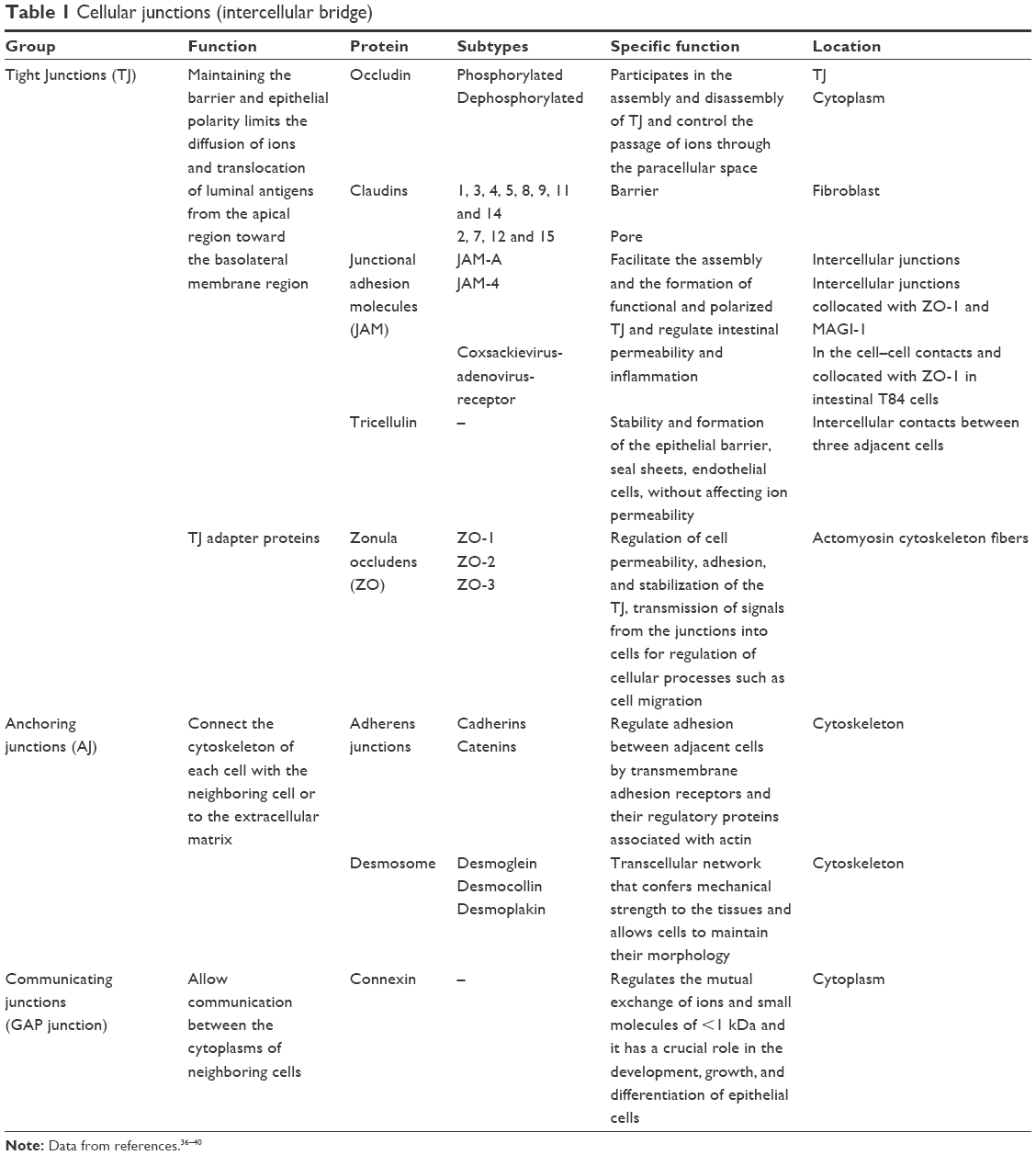 | Table 1 Cellular junctions (intercellular bridge) |
The paracellular pathway is associated with transport through the intercellular space between adjacent epithelial cells; it is regulated by apical junctional complexes and conformed by TJ and adhesion junctions. The latter, in along with desmosomes, create strong links between epithelial cells and foster intercellular communication but do not determine paracellular permeability.31 TJ not only determine paracellular selective permeability to solutes and create the barrier to noxious molecules but also form permeable pores to ions, solutes, and water. TJ are complexes formed by multiple proteins such as claudins, occludin, and zonula occludens 1 and a broad spectrum of cytosolic proteins. Modifications to the TJ barrier and paracellular permeability are dynamically regulated by several extracellular stimuli, closely linked to our health or disease susceptibility (Table 1).32
Injury to the TJ-mediated barrier and the consequent increase in paracellular permeability and luminal passage of proinflammatory proteins may lead to activation of the immune system associated with mucosa, a sustained inflammatory process and perpetuating tissue injury. Experimentally, the TJ-mediated barrier is evaluated by measuring the transepithelial electrical resistance and the paracellular passage of small molecules such as mannitol, dextran, and inulin.32–37
TJ regulation
TJ regulation has been studied in physiological conditions, in cell culture, and in animal models. TJ barrier dysfunction mediated by cytokines leads to immune activation and tissue inflammation, which may be an important factor triggering and/or in the development of several intestinal and systemic diseases.30
- Interferon gamma: this cytokine is mainly secreted by T-cells and natural killer cells. It is a proinflammatory cytokine associated with increased paracellular intestinal permeability due to TJ protein redistribution; induces TJ internalization and a subsequent increase in intestinal permeability.
- TNF-α: this is a proinflammatory protein preferentially produced by T-cells and macrophages. This factor is known to induce apoptosis and inflammation of intestinal epithelial cells while also interferes with the intestinal TJ barrier through different mechanisms. TNF-α alters the TJ intestinal barrier by rearranging the cytoskeleton and TJ expression.30 In enterocytes decreases occludin mRNA expression but increases that of claudin 2 and 8.38
- IL-4: it is mainly secreted by T-cells, basophils, and mastocytes, and plays a significant role in the humoral and adaptive immune responses. The mechanism through which IL-4 alters TJ remains unclear, but it has been shown to decrease the epithelial electric resistance and increase the flow of dextran.30
- IL-6: this is a cytokine with pleiotropic activities, whose expression is important in host defense against a great number of pathogens. IL-6 selectively increases intestinal permeability to cations but not to macromolecules, by increasing the pores formed by claudin 2.
- IL-10: this protein is primarily produced by regulatory T-cells, monocytes, macrophages, and dendritic cells. It appears that IL-10 has a protective effect on the TJ barrier since it is an antagonist to some of the effects of TNF-α. In intestinal cells, IL-10 prevents the effects of interferon gamma-mediated increase in mannitol flow. Injury from parenteral nutrition in animal models is associated with decreased mucosal IL-10 levels.30
Intestinal immunity
The intestinal barrier does not totally impede the passage of antigens and their penetration is restricted by local immune mechanisms. In particular, the subepithelial lamina propria contains a large number of antigen-presenting dendritic cells that sample the environment and process commensal and pathogenic bacteria; they subsequently migrate to mesenteric lymph nodes or Peyer patches and prime virgin T-lymphocytes. Live enteric bacteria have been found in the liver although contained by Kupffer cells, suggesting that the liver is a second barrier limiting and depurating commensal or pathogenic bacteria if the first line of defense mechanisms have been overwhelmed.39
Another fundamental component of the intestinal barrier is constituted by intestinal macrophages (CX3CR1) that through TLRs can detect and subsequently activate innate lymphoid tissue secreting IL-22 that in turn promotes the repair and maintenance of the epithelium’s integrity (Figure 1).40
Effects of liver cirrhosis on the intestinal barrier
Inflammation and TJ in patients with liver cirrhosis
Recent studies have shown that patients with high Child–Pugh–Turcotte scores have increased serum TNF-α levels.15,41 Other cytokines and the presence of necrosis may lead to short- and long-term liver decompensation. In cirrhosis, one of the main contributors to TJ abnormalities is the increased production of TNF-α by monocytes in mesenteric lymph nodes,42 favoring the decreased expression of proteins such as occludin but an increase in claudin 2 and 8.
A recent study by Assimakopoulos et al43 revealed altered intestinal TJ protein expression in patients with liver cirrhosis as a pathogenic mechanism of increased intestinal permeability; they demonstrated that occludin and claudin 1 expression is decreased in the intestinal epithelium of these patients. According to their immunohistochemistry results, this downregulation was more significant in decompensated cirrhosis (CHILD B and C). A specific intestinal pattern was observed in the case of occludin, with a gradual decrease in the expression of crypts in villi.
The presence of activated CD33+CD14+ intestinal macrophages (nitric oxide for producing cells: TREM-1 iNOS), which in other diseases such as necrotizing enterocolitis are associated with intestinal mucosal repair,44 has been recently found in duodenal epithelium in patients with decompensated cirrhosis; they were associated with increased serum levels of lipopolysaccharide, nitric oxide, IL-8, IL-6, and claudin levels, decreased duodenal resistance and increased intestinal permeability.45
The chemoattractant cytokine CX3CR1, plays an important role in modulating inflammatory responses, including monocyte homeostasis and macrophage phenotype and function.46 In a nonalcoholic steatohepatitis mouse model, CX3CR1 was shown to limit the progression of liver inflammation and early glucose intolerance by maintaining the intestinal barrier’s integrity; this effect hinged on the intestinal microbiota’s composition and the receptor acts as a gatekeeper in diet-induced steatohepatitis, maintaining intestinal permeability.40
Intestinal mucus in the patient with liver cirrhosis
Mucin secretion is affected by transcription factors, such as nuclear factor-κB, growth factors, lipopolysaccharides, the presence of microorganisms, and proinflammatory cytokines.
Increased mucins MUC2 and MUC3 mRNA expression has been reported in the ileum of rats with cirrhosis compared with rats without cirrhosis.47 Intestinal mucus modulates bacterial adhesion to the intestinal surface.
Intestinal microbiota in the patient with liver cirrhosis
Two pathological processes of intestinal microbiota have been described in patients with liver cirrhosis, fostering an intestinal and systemic inflammatory state: small intestinal bacterial overgrowth (SIBO) and bacterial dysbiosis, two phenomena that are not mutually exclusive.48
Small intestinal bacterial overgrowth
SIBO is defined as excessive bacterial growth in the small intestine and the constellation of symptoms to which it leads, but there is no clear consensus on this entity’s definition. Tests such as proximal jejunal aspirate culture have historically defined SIBO as bacterial growing at ≥105 colony forming units, and noninvasive tests such as breath tests with different carbohydrates have also been used for diagnosis.49 Factors fostering SIBO development in the liver cirrhosis population include decreased gastric acid production, alterations in gastrointestinal motility (increased intestinal transit time), decreased intestinal biliary acid availability as well as decreased peptides with antimicrobial activity.50
SIBO appears to play an important role in the pathogenesis of bacterial translocation; it has been frequently associated with the severity of liver disease, particularly in patients with a history of SBP and/or HE.51 Clinical and experimental research has shown that inhibition of intestinal Gram-negative aerobic flora growth decreases the incidence of SBP in patients with cirrhosis.10
Intestinal dysbiosis in the patient with liver cirrhosis
Dysbiosis refers to the altered diversity in bacterial families in the gastrointestinal tract, resulting from a decrease in commensal microbiota; dysbiosis has been described in the study of the stool, saliva, and colonic mucosa of patients with liver cirrhosis and it appears to correlate with disease severity, systemic inflammation, and the number of hospitalizations.52–54
Changes in diet and intestinal inflammation may promote intestinal dysbiosis; for example, a high-fat diet changes the ratios from Firmicutes and Proteobacterias to Proteobacterias and Bacteroides, thus activating the inflammasome and leading to the development of steatohepatitis in a mouse model.55
Abnormalities in intestinal microbiota have also been shown in patients with HE when compared with patients with hepatic cirrhosis and no HE and healthy individuals; certain bacterial families appear to be associated with HE (Alcaligenaceae, Porphyromonadaceae, and Enterobacteriaceae), cognitive dysfunction, and inflammation in these patients.56
Intestinal transit
Another important factor in bacterial translocation and bacterial overgrowth is the delay in intestinal transit time observed in the cirrhotic patient and in experimental cirrhosis models. Patients with decompensated cirrhosis have a slower transit time compared with patients with compensated cirrhosis and healthy individuals.50 Moreover, some prokinetic drugs such as cisapride have been shown to decrease bacterial translocation and overgrowth in cirrhotic patients.11 This effect has also been described with the use of nonselective beta-blockers (propranolol) in cirrhosis animal models, and they also accelerated their intestinal transit time and decreased bacterial translocation.12
Intestinal wall structural changes
Portal hypertension-induced vascular stasis and that may lead to an increase in intestinal permeability. Structural changes such as mucosal congestion and edema have been observed in cirrhotic patients in whom broadening of intercellular spaces has also been described.13
There is also recent evidence of probable susceptibility in this population to increased intestinal permeability, since patients with the NOD2 and TLR2 polymorphisms are at greater risk of developing SBP and have greater mortality than patients with minor alleles, associated with increased intestinal permeability and a greater presence of markers derived from bacterial products.14,15
Intestinal permeability in the patient with liver cirrhosis
Alteration in intestinal permeability (increase) is a common finding in patients with liver injury, particularly in patients with liver cirrhosis although it has also been described in different entities associated with chronic liver disease such as alcohol-induced injury, nonalcohol fatty liver disease, and HCV-mediated injury.57 In patients with liver cirrhosis, alterations in intestinal permeability correlate with the degree of liver injury whereby Child–Pugh C patients and those with ascites or a positive history of SBP have increased intestinal permeability to the passage of molecules58 although patients with compensated cirrhosis have been shown to have increased colonic permeability but not gastroduodenal or intestinal.59 However, other authors have been unable to reproduce these results.60
Consequences of increased intestinal permeability
Increased intestinal permeability may precede and promote translocation of bacteria,6 endotoxins (such as lipopolysaccharides), and pathogen-associated molecules into the portal venous system and extraintestinal sites.
Bacterial translocation
There are at least four mechanisms underlying bacterial translocation: intestinal bacterial overgrowth, bacterial dysbiosis, immune system abnormalities, and increased intestinal permeability. The gastrointestinal tract is an actively immune organ that essentially includes all leukocyte types involved in the immune response. Changes in the local and systemic immune system are clinically relevant since they foster bacterial translocation in cirrhosis. Patients with cirrhosis have immunologic abnormalities that facilitate the development of infections and bacterial translocation. In summary, these four previously mentioned factors play a pivotal role in the pathogenesis of bacterial translocation in cirrhosis and also explain part of the high prevalence in cirrhosis compared with other clinical scenarios in which only some of these factors are actively involved.
Experimental studies have also suggested that bacterial translocation is associated with deterioration in the hemodynamics of patients with cirrhosis.61–63
Liver injury
Intestinal inflammation and bacterial translocation impact directly the progression to liver fibrosis and decompensation of preexisting chronic liver damage.
The products of bacterial metabolism may be hepatotoxic substances (phenols, ethanol, acetaldehyde, ammonia, and benzodiazepines) that are metabolized in the liver via the portal circulation.48 Moreover, the lipopolysaccharides released by Gram-negative bacteria attach to the lipopolysaccharide-binding protein that activates Kupffer cells by binding to CD14. The association between CD14 and TLR4 on the cell surface triggers the inflammation cascade, suggesting that microbiota may directly mediate inflammatory processes in the liver.
The persistent liver inflammation generated by an aggressive stimulus (ie, alcohol, virus, fat deposits, etc) plus the inflammation resulting from the translocation of bacteria or their products play an important role in the development of liver fibrosis through the activation of TLR2, a receptor for Gram-positive bacterial products that promotes an inflammatory cascade mediated by the monocytes in the intestinal lamina propria after its activation via the tumor necrosis factor receptor type I (TNFR1). A study of knock-out mice for the TLR2 gene (TLR2−/−) with cholestatic liver injury revealed deceased bacterial translocation into the mesenteric lymph nodes and less endotoxemia compared with wild mice; furthermore, TNFRI−/− mice are protected against the development of liver fibrosis due to decreased extracellular matrix deposits.64 Decreasing the levels of serum lipopolysaccharides with nonabsorbable antibiotics (neomycin) attenuated liver fibrosis development in a nonalcoholic steatohepatitis animal model, by decreasing intestinal permeability as a result of increased TJ expression and decreased TRL4 activity and hepatic stellate cell activation.65
Acute-on-chronic liver failure refers to a condition that develops in patients with chronic liver disease leading to organ failure and associated with high short-term mortality rates; it is a distinct entity from liver decompensation or acute liver failure.66 The most frequent precipitating factor is infection, particularly SBP, pneumonia, and urinary tract infection.67
Variceal hemorrhage
Patients with cirrhosis and upper gastrointestinal bleeding are at increased risk of acquiring bacterial infections (25%–65%), particularly SBP during the first 7 days after bleeding; furthermore, bacterial infections increase the risk of early rebleeding.68 Patients with liver cirrhosis and increased intestinal permeability, increased lipopolysaccharide binding protein, and elevated IL-6 levels represent a population highly prone to develop variceal hemorrhage.15
Infectious processes have been described to increase portal pressure and changes in homeostasis, and the administration of prophylactic antibiotic therapy appears to have a beneficial role in terms of controlling hemorrhage and preventing rebleeding. Current international guidelines actually recommend the use of prophylactic antibiotics in patients with cirrhosis and gastrointestinal bleeding, regardless of the presence or lack of ascites.37,69 A recent meta-analysis of 12 studies comparing a population of 1,241 patients with cirrhosis and gastrointestinal bleeding determined that the use of prophylactic antibiotics significantly decreased the incidence of bacterial infections, the incidence of rebleeding, the duration of hospitalization and mortality.70
Infections, SBP
Bacteremia and SBP are the main consequences of bacterial translocation in patients with cirrhosis.71 Patients with decompensated cirrhosis and high-risk alleles for the NOD2 associated with functional alterations of the intestinal wall are at increased risk of developing SBP and bacteriascites.72,73
Almost half of cirrhotic patients are infected at hospital admission or develop infections during their hospitalization.71 Inhospital mortality is greater in patients who develop bacterial infections than in those who do not.74 The prognosis of bacterial peritonitis has improved in the past few decades due to early diagnosis, better established criteria, and the timely use of antibiotic therapy.10
Bacterial DNA translocation in patients with ascites and portal hypertension worsens systemic circulation, leading to subsequent exacerbation of peripheral vasodilation; this has been related to an increase in the inflammatory status, characterized by elevated TNF-α levels.62 Worsening of hyperdynamic circulation in this inflammatory state due to bacterial translocation has been suggested to play a role in the development of portal hypertension complications, particularly the hepatorenal syndrome. Recent experimental studies have shown that in the cirrhotic population, the expression of TLR4 and proinflammatory cytokines (TNF-α) are increased in the kidney and more prone to inflammatory insult in the presence of bacterial translocation.75
The presence of bacterial DNA in refractory ascites showed that translocation was associated with abnormalities in cardiovascular and kidney function, as well as a greater risk of hepatorenal syndrome and death; the use of antibiotics like rifaximin improves systemic hemodynamics alterations and renal function.76,77
One-third of patients with SBP with antibiotics develop kidney failure that may be transient, permanent, or rapidly progressive in 25%, 33%, and 42% of cases, respectively.78,79
International associations recommend that primary prophylaxis of SBP be administered to cirrhotics with a low protein concentration in ascitic fluid (<1.5 g/dL), as well as to patients with renal dysfunction (serum creatinine ≥1.2 mg/dL, BUN ≥25 mg/dL, or serum sodium ≤130 mEq/L) or severe liver dysfunction (Child–Pugh–Turcotte ≥9 and serum bilirubin ≥3 mg/dL), and those with a previous episode of SBP. The antibiotics of choice are norfloxacin, ciprofloxacin, and trimethoprim–sulfamethoxazole.10
Hepatic encephalopathy
In the last decade, several studies have suggested a synergistic effect of inflammation and infection on the pathogenesis of HE.80,81
HE is usually associated with signs of the systemic inflammatory response syndrome.82 A prospective study proved the association between systemic inflammatory response syndrome or infection and the development of severe HE, independently of ammonia levels.83 Systemic inflammation associated with bacterial translocation may play a role in the pathogenesis of HE. SIBO has also been associated with bacterial translocation and an increased prevalence of HE in liver cirrhosis.84
Bajaj et al52 demonstrated an association between certain intestinal bacterial families, altered cognition, and IL-17/IL-23-mediated inflammation in patients with HE.
Analytic methods to determine intestinal permeability
Intestinal permeability refers to the property that allows the exchange of solutes and fluid between the intestinal lumen and tissues.
Several tools have been developed to measure it, both in vivo and ex vivo. Although there are accessible and applicable methods to measure permeability in humans, result interpretation may be complex. Various authors still debate the clinical significance of increased intestinal permeability and even the normal values of intestinal permeability settings (Table 2). These differences might be explained by several factors such as intestinal transit time, surface area, kidney function, different liver disease entities, time evolution of the disease, comorbidities, the type of test used to measure intestinal permeability (testing with isotopes is considered the gold standard test), and the considered cutoff limits (Table 2).
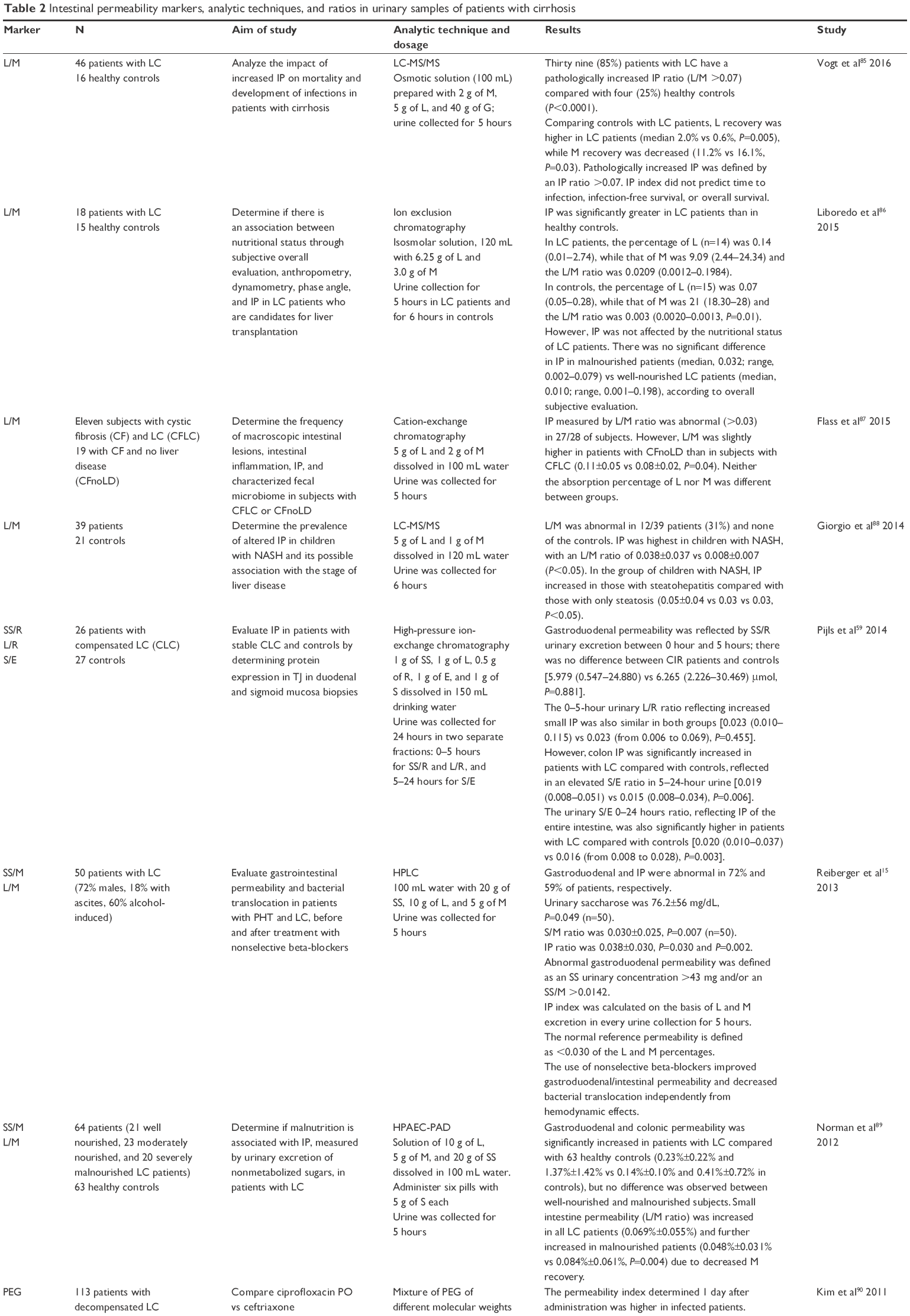 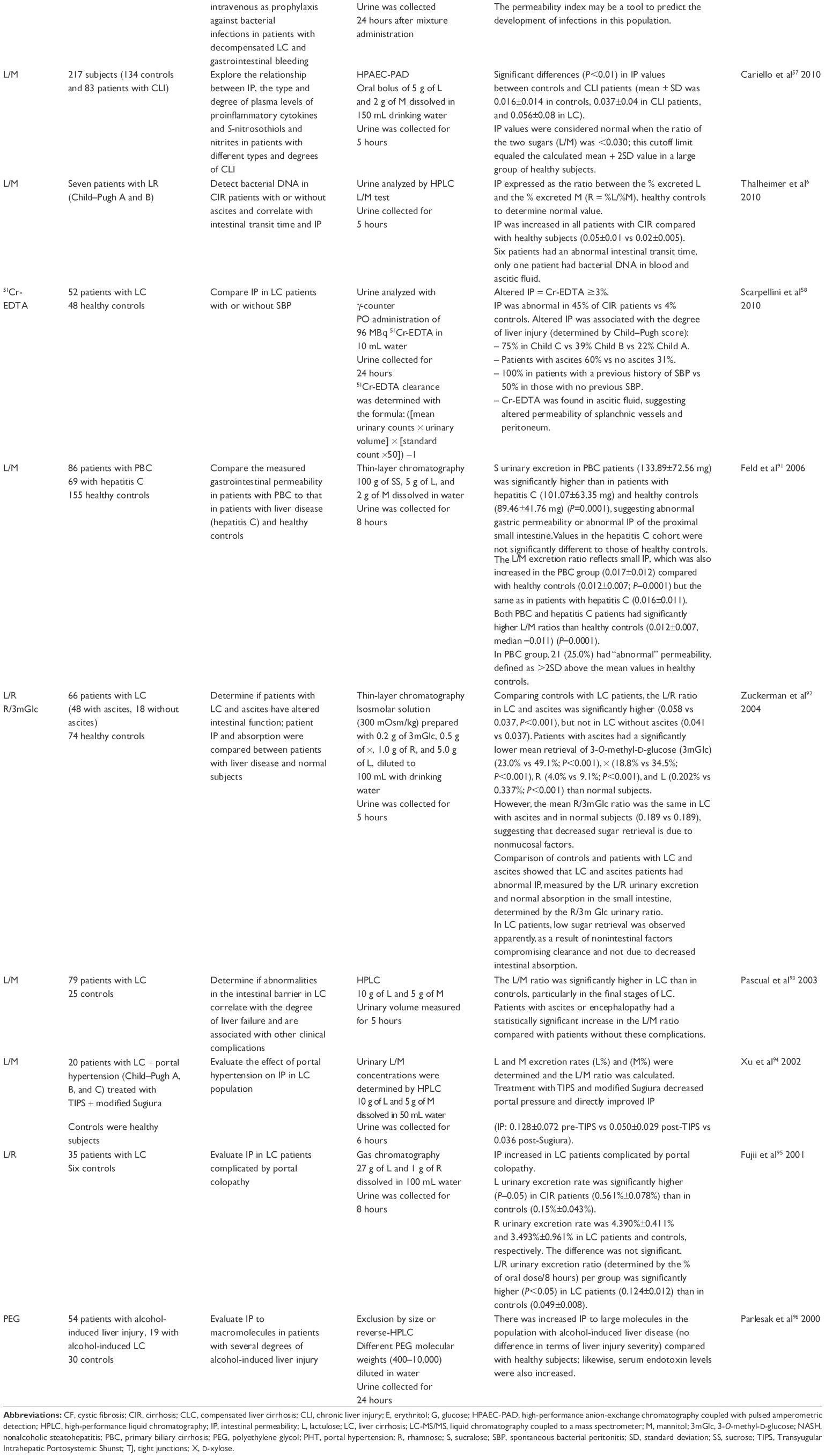 | Table 2 Intestinal permeability markers, analytic techniques, and ratios in urinary samples of patients with cirrhosis |
Studies using sugar as a differential marker in absorption tests, combining an oligosaccharide and a monosaccharide, such as in lactulose/mannitol (L/M) and saccharose/mannitol, revealed increased intestinal permeability in patients with liver cirrhosis.6,15,57–59,85–96
Intestinal permeability can be measured with the administration of nondigestible markers that cross the mucosal barrier by passive diffusion.97 These markers have been observed to pass through the intestinal lumen to the extraintestinal space and can be detected in blood, urine, tissues, and other organs. The most commonly used markers are some saccharides, radioisotopes such as Cr-EDTA58 and polyethylene glycols.96,98
In the 1970s, oligosaccharides were introduced for intestinal permeability evaluation.99 Throughout the world, saccharides with different molecular weights, absorption routes, and levels of urinary excretion have been used as parameters to determine intestinal permeability.100–104
Several saccharide quantification methods have been developed to determine changes in intestinal and gastroduodenal permeability, since sugars, such as lactulose, saccharose, mannitol, xylose, and rhamnose, among others, are known to provide this information.98,101,105–110
Mannitol is one of the most studied monosaccharides. It is absorbed in healthy small intestinal mucosa, travels via the transcellular aqueous pores, the hydrophilic region of the cell membrane, ultimately reflecting the absorption of small molecules.106 Lactulose and saccharose, which are absorbed through the cell wall union complexes, that is, the TJ and extrusion zones of intervillous spaces, reflecting permeability to large molecules.104,107
Mannitol is currently considered a marker of the mucosal absorption zone, while lactulose absorption is a marker of TJ integrity in the intestinal mucosa.11,12 The L/M ratio is an index evaluating intestinal permeability; when it increases, one can conclude that intestinal permeability is also increased. However, lactulose and mannitol are degraded by colonic bacteria and are of no use in evaluating colonic permeability. Lactulose is large in size and can only cross the barrier if there is intestinal epithelial injury, so it is considered a marker of epithelial integrity. Furthermore, mannitol is one-third the size of lactulose; it crosses the intestinal epithelium through pores and it is used to determine the surface area. In patients with liver cirrhosis and in other diseases compromising the intestinal wall, intestinal permeability is increased.13,111
The saccharose/mannitol ratio is used as an index of gastroduodenal permeability.112 Saccharose is an ideal marker to evaluate gastric permeability and it has been evaluated in endoscopic studies analyzing mucosal injury after the ingestion of acetylsalicylic acid,113 alcohol use,114 and other diseases.115 In the liver cirrhosis population, gastric permeability to saccharose has been demonstrated in patients with portal hypertension and it correlates with the severity of gastric lesions (urinary retrieval ≥0.15%); gastric permeability to saccharose returned to normal after the administration of nonselective beta-blockers.116,117
The quantitative analysis of sugars is difficult since they are highly polar compounds, as well as hydrophilic, thermosensitive, their structure lacks chromophores, their pKa’s are >10, and they are not easily ionized due to their low acidity and volatility.
However, saccharides have been identified and quantified with several extraction techniques and different detection instruments. High-pressure liquid chromatography is the most often used instrument, coupled to various types of detectors such as ultraviolet or visible UV light,118 fluorescence, electrochemical, evaporative light scattering detector,119 refraction index detector,120 charged aerosol detector,121 and masses.104,108–110
Additionally, gas chromatography–mass spectrometry122,123 in which some mono- and disaccharides are derivatized for quantification is used; other methods include high-performance anion-exchange chromatography with pulsed amperometric detection,124,125 capillary electrophoresis, among others.
Due to the complexity of sugar quantification, derivatization has been used as an alternative for their measurement. Some examples of compounds used for derivatization are 1-phenyl-3-methyl-5-pyrazolone (1-(2-naphthyl)-3-methyl-5-pyrazolone)126 and p-aminobenzoic ethyl ester that react by reducing carbohydrates127 for ultraviolet detection; in gas chromatography, trimethylsilyl or acetate derivatives123,128 are used before analysis since saccharides need to become volatile and stable derivatives. Quantification by fluorescence, benzamidine has been used as a reducing agent129 and 8-aminopyrenesulfonic acid for derivatization.130
Additionally, in general, sugar extraction from the biological matrix (urine) is through direct precipitation and filtration in order to clean the sample104,108–110 although solid-phase extraction has also been used; its disadvantage is its high cost compared with precipitation,131 so numerous assays have been performed to obtain the optimal quantification conditions.
Most techniques developed for sugar determination have been applied to the study of food products and drinks, but they are also of use in studies of intestinal permeability in patients with diseases such as cystic fibrosis, Crohn’s disease, malnutrition, pancreatitis, colon cancer, and cirrhosis.89,104,108–110,132–134
Several studies on intestinal permeability in cirrhotic patients have been conducted by many authors; Table 2 shows some of these and the different quantification methods that have been applied as well as results on the sugar ratios reflecting permeability.
There are multiple results on the L/M quantification ratio and they are generally expressed as the percentage of excreted lactulose over the percentage of excreted mannitol. The normal proposed value is ≤0.03,57 and a ratio of >0.03 reflects increased small intestine permeability, and higher ratios are associated with even greater permeability.
Measures to diminish increased intestinal permeability in the liver cirrhosis population
Intestinal decontamination
Selective intestinal decontamination refers to a use of antibiotics to modify and select intestinal microbiota; it is common practice in patients with cirrhosis in the management of certain complications (HE, variceal hemorrhage, prophylaxis of bacterial peritonitis, etc) and it is recommended in the international guidelines on the management of each of these complications.10,69,135
Rifaximin is an antibiotic with luminal activity and minimal systemic absorption; it is a broad-spectrum antibiotic against Gram-positive bacteria and its risk of fostering resistance has been reported to be low. Rifaximin in conjunction with lactulose is effective in the prevention of overt HE recurrence.135 A study published by Vlachongiannakos et al136 reported that the use of rifaximin was associated with a significant decrease endotoxemia and simultaneously, portal venous gradient in a population of decompensated alcoholic-related cirrhosis. Rifaximin has also been associated with improvement in cognitive function as well as in endotoxemia in patients with minimal HE; Bajaj et al137 proved that rifaximin modified bacterial networks and their metabolites, fostering the generation of beneficial metabolites (saturated and unsaturated fatty acids) that ameliorate cognitive function.
Probiotics
The use of probiotics has been associated with decreased intestinal permeability and decreased bacterial translocation and endotoxemia in animal models and clinical studies.138 Patients eligible for liver transplants presented with increased intestinal permeability compared with healthy controls, a 30-day treatment with S. boulardii did not improve this intestinal permeability or the severity scores.139
Better trials are needed to establish that probiotics effectively improve intestinal permeability and their clinical effects; a recent meta-analysis was unable to confirm that probiotics were effective in the management of HE.140
Increased gastrointestinal motility
The use of prokinetics has been evaluated as a measure to decrease bacterial translocation. A study conducted in humans and rats showed that cisapride (5-HT4 agonist) decreased SIBO and bacterial translocation.11 Moreover, a study of rats with liver cirrhosis analyzed bacterial translocation, SIBO, and intestinal transit time before and after cisapride or placebo administration: rats on cisapride had lower rates of bacterial translocation, endotoxin translocation, and intestinal bacterial overgrowth that appeared to depend on an increase in intestinal transit time and improved intestinal permeability.141 The use of combined norfloxacin and cisapride versus cisapride alone decreases the incidence of SBP in patients with liver cirrhosis and ascites at high risk of developing SBP (alcohol-induced cirrhosis, low albumin levels in ascitic fluid, gastrointestinal hemorrhage, and low serum albumin).142
In an animal model,12 the administration of propranolol to rats with liver cirrhosis was evaluated and compared with placebo. The rats on propranolol had a lower portal pressure (20.9±4 mmHg vs 17.2±4 mmHg), a lower intestinal transit time (0.23±0.1 vs 0.44±0.1), lower rates of SIBO (67% vs 15%), and bacterial translocation (58% vs 15%).
Other strategies
Studies in rats have shown that the duodenal administration of a 15% alcohol solution or the application of red wine to the duodenal intraluminal surface increases intestinal permeability, while the application of melatonin can prevent this effect of alcohol.143
Diets that improve intestinal saturated fatty acid levels may decrease alcohol-induced liver injury by stabilizing the intestinal barrier and preserving intestinal eubiosis; this is suggested in a study conducted in humans and mice by Chen et al.144
The use of propranolol in patients with liver cirrhosis not only decreased the portal venous pressure gradient but also intestinal permeability when tested with the three-sugar test; it also decreased bacterial translocation and serum IL-6 levels, an effect that was not limited to hemodynamically responding patients.15,117
Úbeda et al145 showed that administration of obeticholic acid (a potent farnesoid X receptor agonist) for 2 weeks to cirrhotic rats with ascites decreased bacterial translocation, increased the expression of antimicrobial peptides in the ileum, and also increased TJ protein expression (zonulin 1 and occludin); it also decreased the degree of liver fibrosis and normalized the expression of inflammatory cytokines and TLR4. In an experimental model of cholestatic liver injury, obeticholic acid improved intestinal barrier function at the ileal level and increased the expression of claudin 1 and occludin; it also led to a significant decrease in bacterial translocation by attenuating the degree of intestinal permeability.146
A new potential therapeutic target may be the inhibition of microRNA-155 (miRNA-155) expression. Mice that are deficient in miRNA-155 were protected from alcohol-induced intestinal inflammation and they showed no elevation in endotoxin levels, suggesting that miRNA-155 may play a role in the maintenance of epithelial integrity after alcohol ingestion.147
Conclusion
Patients with liver cirrhosis deteriorate as a result of several complications secondary to the presence of bacterial translocation and their products from the intestinal lumen into the systemic circulation. Identification of the mechanisms implicated in this increase in intestinal permeability will promote the development of therapeutic targets that may prevent decompensation or death in this population. The usefulness and limitations of selective decontamination should be more clearly defined. Further research including innate immune responses against structural components of microbes may open new possibilities to manage this alteration. It remains to define the true impact of short- and long-term use of beta-blockers in the management of intestinal permeability, with a cautious use in people with advanced liver disease and refractory ascites.
There are other promising therapeutic strategies such as the use of obeticholic acid and miRNAs, which are still in evaluation. Studies attempting to standardize the available methods to evaluate intestinal permeability in the liver cirrhosis population are lacking; the different results might be explained by several factors such as intestinal transit time, surface area, kidney function, different liver disease entities, time evolution of the disease, comorbidities, the type of test used to measure intestinal permeability, and the considered cutoff limits.
Disclosure
The authors report no conflicts of interest in this work.
References
Ginès P, Schrier RW. Renal failure in cirrhosis. N Engl J Med. 2009;13:1279–1290. | ||
Munoz SJ. Hepatic encephalopathy. Med Clin North Am. 2008;92(4):795–812. | ||
Olson J, Wendon JA, Kramer DJ, et al. Intensive care of the patient with cirrhosis. Hepatology. 2011;54(5):1864–1872. | ||
Seki E, Schnabl B. Role of innate immunity and the microbiota in liver fibrosis: crosstalk between the liver and gut. J Physiol. 2012;590(3):447–458. | ||
Pleguezuelo M, Benitez JM, Jurado J. Diagnosis and management of bacterial infections in decompensated cirrhosis. World J Hepatol. 2013;27:6–25. | ||
Thalheimer U, De Lorio F, Capra F, et al. Altered intestinal function precedes the appearance of bacterial DNA in serum and ascites in patients with cirrhosis: a pilot study. Eur J Gastroenterol Hepatol. 2010;22(10):1228–1234. | ||
Cirera I, Beuer TM, Navasa M. Bacterial translocation of enteric organism in patients with cirrhosis. J Hepatol. 2001;34:32–37. | ||
Goulis J, Armonis A, Patch D, Sabin C, Greenslade L, Burroughs AK. Bacterial infection is independently associated with failure to control bleeding in cirrhotic patients with gastrointestinal hemorrhage. Hepatology. 1998;27(5):1207–1212. | ||
Gines P, Rimola A, Planas R, et al. Norfloxacin prevents spontaneous bacterial peritonitis recurrence in cirrhosis: results of a double blind, placebo-controlled trial. Hepatology. 1990;12:716–724. | ||
Runyon BA. Management of adult patients with ascites due to cirrhosis: update 2012. Hepatology. 2009;492087:107. | ||
Pardo A, Bartoli R, Lorenzo Zúñiga V, et al. Effect of cisapride on intestinal bacterial overgrowth and bacterial translocation in cirrhosis. Hepatology. 2000;31:858–863. | ||
Pérez Páramo M, Muñoz J, Albillos A, et al. Effect of propranolol on factors promoting bacterial translocation in cirrhotic rats with ascites. Hepatology. 2000;31:43–48. | ||
Such J, Guardiola JV, de Juan J, et al. Ultrastructural characteristics of distal duodenum mucosa in patients with cirrhosis. Eur J Gastroenterol Hepatol. 2002;14(4):371–376. | ||
Nischalke HD, Berger C, Aldenhoff K, et al. Toll like receptor (TLR2) promoter and intron 2 polymorphism are associated with increased risk for spontaneous bacterial peritonitis in liver cirrhosis. J Hepatol. 2011;55:1010–1016. | ||
Reiberger T, Ferlitsch A, Payer BA, et al. Non selective betablocker therapy decreases intestinal permeability and serum levels of LBP and IL6 in patients with cirrhosis. J Hepatol. 2013;58:911–921. | ||
Turner J. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9(11):799–809. | ||
Chang CS, Chen GH, Lien HC, Yeh HZ. Small intestine dysmotility and bacterial overgrowth in cirrhotic patients with spontaneous bacterial peritonitis. Hepatology. 1998;28(5):1187–1190. | ||
Tilg H, Wilmer A, Vogel W, et al. Serum levels of cytokines in chronic liver diseases. Gastroenterology. 1992;103(1):264–274. | ||
Qin X, Caputo FJ, Xu DZ, Deitch EA. Hydrophobicity of mucosal surface and its relationship to gut barrier function. Shock. 2008;29(3):372–376. | ||
Swidsinski A, Sydora BC, Doerffel Y, et al. Viscosity gradient within the mucus layer determines the mucosal barrier function and the spatial organization of the intestinal microbiota. Inflamm Bowel Dis. 2007;13(8):963–970. | ||
Salvo-Romero E, Alonso Cotoner C, Pardo-Camacho C, et al. The intestinal barrier function and its involvement in digestive disease. Rev Esp Enferm Dig. 2015;107:686–696. | ||
Pott J, Hornef M. Innate immune signalling at the intestinal epithelium in homeostasis and disease. EMBO Rep. 2012;13(8):684–698. | ||
Zeuthen L, Fink L, Frokiaer H. Epithelial cells prime the immune response to an array of gut-derived commensals towards a tolerogenic phenotype through distinct actions of thymic stromal lymphopoietin and transforming growth factor-beta. Immunology. 2008;123(2):197–208. | ||
Zaph C, Du Y, Saenz S, et al. Commensal-dependent expression of IL-25 regulates the IL-23-IL-17 axis in the intestine. J Exp Med. 2008;205(10):2191–2198. | ||
Xu W, He B, Chiu A, et al. Epithelial cells trigger frontline immunoglobulin class switching through a pathway regulated by the inhibitor SLPI. Nat Immunol. 2007;8(3):294–303. | ||
He B, Xu W, Santini P, et al. Intestinal bacteria trigger T cell-independent immunoglobulin A(2) class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity. 2007;26(6):812–826. | ||
Marchesi JR, Adams DH, Fava F, et al. The gut microbiota and host health: a new clinical frontier. Gut. 2015;0:1–10. | ||
Odenwald M, Turner G. Intestinal permeability defects: is it time to treat? Clin Gastroenterol Hepatol. 2013;11(9):1075–1083. | ||
Caricilli AM, Castoldi A, Câmara NO, et al. Intestinal barrier: a gentlemen’s agreement between microbiota and immunity. World J Gastrointest Pathophysiol. 2014;5(1):18–32. | ||
Suzuki T. Regulation of intestinal epithelial permeability by tight junctions. Cell Mol Life Sci. 2013;70(4):631–659. | ||
Baum B, Georgiou M. Dynamic of adherens junction in epitelial establishment, maintenance, and remodeling. J Cell Biol. 2011;192:907–917. | ||
Balda M, Matter K. Tight junctions at a glance. J Cell Sci. 2008;121:3677–3682. | ||
Schulzke J, Fromm M. Tight junctions: molecular structure meets function. Ann N Y Acad Sci. 2009;1165:1–6. | ||
Rao R. Occludin phosphorylation in regulation of epithelial tight junctions. Ann N Y Acad Sci. 2009;1165:62–68. | ||
Hartsock A, Nelson W. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778(3):660–669. | ||
Liu Y, Nusrat A, Schnell F, et al. Human junction adhesion molecule regulates tight junction resealing in epithelia. J Cell Sci. 2000;113(pt 13):2363–2374. | ||
Ikenouchi J, Furuse M, Furuse K, Sasaki H, Tsukita S, Tsukita S. Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol. 2005;171(6):939–945. | ||
Ye D, Guo S, Al-Sadi R, Ma TY. Micro RNA regulation of intestinal epithelial tight junction permeability. Gastroenterology. 2011;141(4):1323–1333. | ||
Balmer ML, Slack E, de Gottardi A, et al. The liver may act as a firewall mediating mutusalism between the host and its gut comensal microbiota. Sci Transl Med. 2014;6:237–266. | ||
Schneider KM, Beighs V, Heymann F, et al. CX3CR1 is a gatekeeper for intestinal barrier integrity in mice: Limiting steatohepatitis by maintaining intestinal homeostasis. Hepatology. 2015;62(5):1405–1416. | ||
Frazier TH, DiBaise JK, McClain CJ. Gut microbiota, intestinal permeability, obesity-induced inflammation, and liver injury. JPEN J Parenter Enteral Nutr. 2011;35(5 suppl):14S–20S. | ||
Genescà J, Martí R, Rojo F, et al. Increased tumor necrosis factor alpha production in mesenteric lymph nodes of cirrhotic patients with ascites. Gut. 2003;52:1054–1059. | ||
Assimakopoulos SF, Tsamandas AC, Tsiaoussis GI, et al. Altered intestinal tight junctions’ expression in patients with liver cirrhosis: a pathogenetic mechanism of intestinal hyperpermeability. Eur J Clin Invest. 2012;42(4):439–446. | ||
Anad RJ, Dai S, Rippel C, et al. Activated macrophages inhibit enterocyte gap junctions via the release of nitric oxide. Am J Physiol Gastrointest Liver Physiol. 2008;294(1):G109–G119. | ||
Plessis JD, Vanheel H, Janseen CEI, et al. Activated intestinal macrophages in patients with cirrhosis release NO and IL-6 that may disrupt intestinal barrier function. J Hepatol. 2013;58(6):1125–1132. | ||
Medina-Contreras O, Greem D, Laur O, et al. CX3CR1 regulates intestinal macrophages homeostasis, bacterial translocation, and colitogenic Th17 responses in mice. J Clin Invest. 2011;121:4787–4795. | ||
Xie YR, Liu SL, Liu X, et al. Intestinal microbiota and innate immunity-realted gene alteration in cirrhotic rats with liver transplantation. Transplant Proc. 2011;43:3973–3979. | ||
Abu-Shanab A, Quigley EM. The role of the gut microbiota in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2010;7(12):691–701. | ||
Rezaie A, Pimentel M, Rao SS. How to test and treat small intestinal overgrowth: an evidence-based approach. Curr Gastroenterol Rep. 2016;18:8. | ||
Roland BC, Garcia-Tsao G, Ciarleglio MM, et al. Decompensated cirrhotics have slower intestinal transit time as compared with compensated cirrhotics and healthy controls. J Clin Gastroenterol. 2013;47:888–893. | ||
Chang CS, Chen GH, Lien HC, et al. Small intestine dysmotility and bacterial overgrowth versus antimicrobial capacity in patients with spontaneous bacterial peritonitis. Scand J Gastroenterol. 2001;36:92–96. | ||
Bajaj JS, Hylemon PB, Ridlon JM, et al. Colonic mucosal microbiome differs from stool microbiome in cirrhosis and hepatic encephalopathy and is linked to cognition and inflammation. Am J Physiol Gastrointest Liver Physiol. 2012;303(6):G675–G685. | ||
Bajaj JS, Heuman DM, Hylemon PB, et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol. 2014;60(5):940–947. | ||
Bajaj JS, Betrapally ND, Hylemon PH, et al. Salivary microbiota reflects changes in gut microbiota in cirrhosis with hepatic encephalopathy. Hepatology. 2015;62(4):1260–1271. | ||
Henao Mejía J, Elinav E, Jin C, et al. Inflammasome mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. | ||
Bajaj JS, Ridlon JM, Hylemon PB, et al. Linkage of gut microbiome with cognition in hepatic encephalopathy. Am J Physiol Gastrointest Physiol. 2012;302(1):G168–G175. | ||
Cariello R, Federico A, Sapone A, et al. Intestinal permeability in patients with chronic liver diseases: its relationship with a etiology and the entity of liver damage. Dig Liv Dis. 2010;42:200–204. | ||
Scarpellini E, Valenza V, Gabrielli M, et al. Intestinal permeability in cirrhotic patients with and without spontaneous bacterial peritonitis: is the ring closed? Am J Gastroenterol. 2010;105(2):323–327. | ||
Pijls KE, Koek GH, Elamin EE, de Vries H, Masclee AA, Jonkers DM. Large intestine permeability is increased in patients with compensated liver cirrhosis. Am J Physiol Gastrointest Liver Physiol. 2014;306(2):G147–G153. | ||
Kalaitzakis E, Johansson JE, Bjarnason I, Björnsson E. Intestinal permeability in cirrhotic patients with and without ascites. Scand J Gastroenterol. 2006;41(3):326–330. | ||
Wiest R, Cadelina G, Milstien S, McCuskey RS, Garcia-Tsao G, Groszmann RJ. Bacterial translocation up-regulates GTP-cyclohydrolase I in mesenteric vasculature of cirrhotic rats. Hepatology. 2003;38(6):1508–1515. | ||
Bellot P, Garcia-Pagan JC, Frances R, et al. Bacterial DNA translocation is associated with systemic circulatory abnormalities and intrahepatic endothelial dysfunction inpatients with cirrhosis. Hepatology. 2010;52(6):2044–2052. | ||
Albillos A, de la Hera A, Gonzalez M, et al. Increased lipopolysaccharide binding protein in cirrhotic patients with marked immune and hemodynamic derangement. Hepatology. 2003;37(1):208–217. | ||
Hartmann P, Haimerl M, Mazagova M, et al. Toll like receptor 2-mediated intestinal injury and enteric tumor necrosis factor receptor I contribute to liver fibrosis in mice. Gastroenterology. 2012;143:1330–1340. | ||
Douhara A, Moriya K, Yoshiji H, et al. Reduction of endotoxin attenuates liver fibrosis through suppression of hepatic stellate cell activation and remission of intestinal permeability in a rat non-alcoholic steatohepatitis model. Mol Med Rep. 2015;11(3):1693–1700. | ||
Moreau R, Jalan R, Gines P, et al. Acute on chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology. 2013;144:1426–1437. | ||
Gusto T, Felleiter P, Pickkers P, et al; EPIC II Group of Investigators. Impact of infection on the prognosis of critically ill cirrhotic patients: results from a large worldwide study. Liver Int. 2014;34(10):1496–1503. | ||
Bernard B, Cadranel JF, Valla D, Escolano S, Jarlier V, Opolon P. Prognostic significance of bacterial infection in bleeding cirrhotic patients: a prospective study. Gastroenterology. 1995;108(6):1828–1834. | ||
Franchis R, Baveno VI Faculty. Expanding consensus in portal hypertension Report of the Baveno VI Consensus Workshop: stratifying risk and individualizing care for portal hypertension. J Hepatol. 2015;63:743–752. | ||
Chavez T, Barrientos-Gutierrez T, Tellez AF, et al. Meta-analysis: antibiotic prophylaxis for cirrhotic patients with upper gastrointestinal bleeding – an update Cochrane review. Aliment Pharmacol Ther. 2011;34:509–518. | ||
Guarner C, Soriano G. Spontaneous bacterial peritonitis. Semin Liver Dis. 1997;17(3):203–217. | ||
Appenrodt B, Grünhage F, Gentemann MG, et al. Nucleotide binding oligomerization domain containing 2 (NOD2) variants are genetic risk factors for death and spontaneous bacetrial peritonitis in liver cirrhosis. Hepatology. 2010;51:1327–1333. | ||
Bruns T, Peter J, Reuken PA, et al. NOD 2 gene variants are a risk factor for culture positive spontaneous bacterial peritonitis and monomicrobial bacterascites in cirrhosis. Liver Int. 2012;32:223–230. | ||
Caly WR, Strauss E. A prospective study of bacterial infections in patients with cirrhosis. J Hepatol. 1993;18:353–358. | ||
Shah N, Dhar D, El Zahraa MF, et al. Prevention of acute kidney injury in a rodent model of cirrhosis follow in selective gut decontamination is associated with reduce renal TLR4 expression. J Hepatol. 2012;56:1047–1053. | ||
Angeli P, Brun P, Cesari M, et al. Prevalence of bacterial DNA in patients with cirrhosis and refractory ascites and its role in the development of cardiac and renal dysfunctions. Hepatology. 2010;52:342A. | ||
Kalambokis GN, Mouzaki A, Rodi M, et al. Rifaximin improves systemic hemodynamics and renal function inpatients with alcohol-related cirrhosis and ascites. Clin Gastroenterol Hepatol. 2012;10:815–818. | ||
Fernandez J, Navasa M, Planas R, et al. Primary prophylaxis of spontaneous bacterial peritonitis delays hepatorenal syndrome and improves survival in cirrhosis. Gastroenterology. 2007;133:818–824. | ||
Follo A, Llovet JM, Navasa M, et al. Renal impairment after spontaneous bacterial peritonitis in cirrhosis: incidence, clinical course, predictive factors and prognosis. Hepatology. 1994;20:1495–1501. | ||
Ramachandran A, Prabhu R, Thomas S, Reddy JB, Pulimood A, Balasubramanian KA. Intestinal mucosal alterations in experimental cirrhosis in the rat: role of oxygen free radicals. Hepatology. 2002;35:622–629. | ||
Guarner C, Soriano G, Tomas A, et al. Increased serum nitrite and nitrate levels in patients with cirrhosis: relationship to endotoxemia. Hepatology. 1993;18:1139–1143. | ||
Blei AT. Infection, inflammation and hepatic encephalopathy, synergism redefined. J Hepatol. 2004;40:327–330. | ||
Shawcross DL, Sharifi Y, Canavan JB, et al. Infection and systemic inflammation, not amonia, are associated with grade 3/4 hepatic encephalopathy, but nor mortality in cirrhosis. J Hepatol. 2011;54:640–649. | ||
Jun DW, Kim KT, Lee OY, et al. Association between small intestinal bacterial overgrowth and periphereal bacterial DNA in cirrhotic patients. Dig Dis Sci. 2010;55:1465–1471. | ||
Vogt A, Reuken PA, Stengel S, Stallmach A, Bruns T. Dual-sugar tests of small intestinal permeability are poor predictors of bacterial infections and mortality in cirrhosis: a prospective study. World J Gastroenterol. 2016;22(11):3275–3284. | ||
Liboredo JC, Vilela EG, Ferrari Mde L, et al. Nutrition status and intestinal permeability in patients eligible for liver transplantation. JPEN J Parenter Enteral Nut. 2015;39:163–170. | ||
Flass T, Tong S, Frank DN, et al. Intestinal lesions are associated with altered intestinal microbiome and are more frequent in children and young adults with cystic fibrosis and cirrhosis. PLoS One. 2015;10(2):e0116967. | ||
Giorgio V, Miele L, Principessa L, et al. Intestinal permeability is increased in children with non-alcoholic fatty liver disease, and correlates with liver disease severity. Dig Liver Dis. 2014;46:556–560. | ||
Norman K, Pirlich M, Schulzke JD, et al. Increased intestinal permeability in malnourished patients with liver cirrhosis. Eur J Clin Nutr. 2012;66(10):1116–1119. | ||
Kim BI, Kim HJ, Park JH, et al. Increased intestinal permeability as a predictor of bacterial infections in patients with decompensated liver cirrhosis and hemorrhage. J Gastroenterol Hepatol. 2011;26(3):550–557. | ||
Feld JJ, Meddings J, Heathcote EJ. Abnormal intestinal permeability in primary biliary cirrhosis. Dig Dis Sci. 2006;51(9):1607–1613. | ||
Zuckerman MJ, Menzies IS, Ho H, et al. Assessment of intestinal permeability and absorption in cirrhotic patients with ascites using combined sugar probes. Dig Dis Sci. 2004;49(4):621–626. | ||
Pascual S, Such J, Esteban A, et al. Intestinal permeability is increased in patients with advanced cirrhosis. Hepatogastroenterology. 2003;50(53):1482–1486. | ||
Xu WH, Wu XJ, Li JS. Influence of portal pressure change on intestinal permeability in patients with portal hypertension. Hepatobiliary Pancreat Dis Int. 2002;1(4):510–514. | ||
Fujii T, Seki T, Maruoka M, et al. Lactulose-L-rhamnose intestinal permeability test in patients with liver cirrhosis. Hepatol Res. 2001;19: 158–169. | ||
Parlesak A, Schäfer C, Schütz T, et al. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol. 2000;32(5):742–747. | ||
Sun Z, Wang X, Andersson R. Role of intestinal permeability in monitoring mucosal barrier function. History, methodology, and significance of pathophysiology. Dig Surg. 1998;15(5):386–397. | ||
Lee S, Son SC, Han MJ, et al. Increased intestinal macromolecular permeability and urine nitrite excretion associated with liver cirrhosis with ascites. World J Gastroenterol. 2008;14(24):3884–3890. | ||
Menzies IS. Absorption of intact oligosaccharide in health and disease. Biochem Soc Trans. 1974;1:1042–1046. | ||
Van Elburg RM, Uil JJ, Mulder CJ, Heymans HS. Intestinal permeability in patients with coeliac disease and relatives of patients with coeliac disease. Gut. 1993;34(3):354–357. | ||
Dastych M, Dastych M Jr, Novotná H, Cíhalová J. Lactulose/mannitol test and specificity, sensitivity, and area under curve of intestinal permeability parameters in patients with liver cirrhosis and Crohn’s disease. Dig Dis Sci. 2008;53(10):2789–2792. | ||
Celli M, D’Eufemia P, Dommarco R, et al. Rapid gas-chromatographic assay of lactulose and mannitol for estimating intestinal permeability. Clin Chem. 1995;41(5):752–756. | ||
Miki K, Butler R, Moore D, Davidson G. Rapid and simultaneous quantification of rhamnose, mannitol and lactulose in urine by HPLC for estimating intestinal permeability in pediatric practice. Clin Chem. 1996;42(1):71–75. | ||
Lostia AM, Lionetto L, Principessa L, et al. A liquid chromatography/mass spectrometry method for the evaluation of intestinal permeability. Clin Biochem. 2008;41(10–11):887–892. | ||
van Nieuwenhoven MA, Geerling BJ, Deutz NE, Brouns F, Brummer RJ. The sensitivity of the lactulose/rhamnose gut permeability test. Eur J Clin Invest. 1999;29(2):160–165. | ||
Di Leo V, Venturi C, Baragiotta A, et al. Gastroduodenal and intestinal permeability in primary biliary cirrhosis. Eur J Gastroenterol Hepatol. 2003;15(9):967–973. | ||
Hessels J, Snoeyink EJ, Platenkamp AJ, et al. Assessment of intestinal permeability: enzymatic determination of urinary mannitol, raffinose, sucrose and lactose on Hitachi analyzer. Clin Chem Lab Med. 2003;41(1):33–38. | ||
Camilleri M, Nadeau A, Lamsam J, et al. Understanding measurements of intestinal permeability in healthy humans with urine lactulose and mannitol excretion. Neurogastroenterol Motil. 2010;22(1):e15–e26. | ||
Kubica P, Kot-Wasik A, Wasik A, Namieśnik J, Landowski P. Modern approach for determination of lactulose, mannitol and sucrose in human urine using HPLC-MS/MS for the studies of intestinal and upper digestive tract permeability. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;907:34–40. | ||
Lee GO, Kosek P, Lima AA, et al. Lactulose: mannitol diagnostic test by HPLC and LC-MSMS platforms: considerations for field studies of intestinal barrier function and environmental enteropathy. J Pediatr Gastroenterol Nutr. 2014;59(4):544–550. | ||
Murphy MS, Eastham EJ, Nelson R, Pearson AD, Laker MF. Intestinal permeability in Crohn’s disease. Arch Dis Child. 1989;64(3):321–325. | ||
Sutherland LR, Verhoef M, Wallace JL, et al. A simple, non invasive marker of gastric damage: sucrose permeability. Lancet. 1994;343:998–1000. | ||
Rabassa AA, Goodgame R, Sutton FM, Ou CN, Rognerud C, Graham DY. Effects of aspirin and Helicobacter pylori on the gastroduodenal mucosal permeability to sucrose. Gut. 1996;39(2):159–163. | ||
Kershavarzian A, Fields JZ, Vaeth J, et al. The differing effects of acute and chronic on gastric and intestinal permeability. Am J Gastroenterol. 1994;89:2205–2211. | ||
Vogelsang H, Oberhuber G, Wyatt J. Lymphocytic gastritis and gastric permeability in patients with celiac disease. Gastroenterology. 1997;111:73–77. | ||
Giofré MR, Meduri G, Pallio S, et al. Gastric permeability to sucrose is increased in portal hypertensive gastropathy. Eur J Gastroenterol Hepatol. 2000;12(5):529–533. | ||
Senzolo M, Fries W, Buda A, et al. Oral propranolol decreases intestinal permeability in patients with cirrhosis: another protective mechanism against bleeding? Am J Gastroenterol. 2009;104(12):3115–3116. | ||
Lv Y, Yang X, Zhao Y, et al. Separation and quantification of component monosaccharides of the tea polysaccharides from Gynostemma pentaphyllum by HPLC with indirect UV detection. Food Chem. 2009;112:742–746. | ||
Cao Y, Wang Y, Chen X, et al. Study on sugar profile of rice during ageing by capillary electrophoresis with electrochemical detection. Food Chem. 2004;86:131–136. | ||
Chavez-Servin JL, Castellote AI, Lopez-Sabater MC. Analysis of mono- and disaccharides in milk-based formulae by high-performance liquid chromatography with refractive index detection. J Chromatogr A. 2004;1043(2):211–215. | ||
Hutchinson JP, Remenyi T, Nesterenko P, et al. Investigation of polar organic solvents compatible with corona charged aerosol detection and their use for the determination of sugars by hydrophilic interaction liquid chromatography. Anal Chim Acta. 2012;750:199–206. | ||
Martínez-Augustin O, Boza JJ, Romera JM, Gil A. A rapid gas-liquid chromatography method for the determination of lactulose and mannitol in urine: clinical application in studies of intestinal permeability. Clin Biochem. 1995;28(4):401–405. | ||
Ruiz-Matute AI, Hernández-Hernández O, Rodríguez-Sánchez S, Sanz ML, Martínez-Castro I. Derivatization of carbohydrates for GC and GC-MS analyses. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879(17–18):1226–1240. | ||
Pico J, Martínez MM, Martín MT, Gómez M. Quantification of sugars in wheat flours with an HPAEC-PAD method. Food Chem. 2015;173:674–681. | ||
Gangola MP, Jaiswal S, Khedikar YP, Chibbar RN. A reliable and rapid method for soluble sugars and RFO analysis in chickpea using HPAEC-PAD and its comparison with HPLC-RI. Food Chem. 2014;154:127–133. | ||
You J, Sheng X, Ding C, et al. Detection of carbohydrates using new labeling reagent 1-(2-naphthyl)-3-methyl-5-pyrazolone by capillary zone electrophoresis with absorbance (UV). Anal Chim Acta. 2008;609(1):66–75. | ||
Blanco Gomis D, Muro Tamayo D, Mangas Alonso JJ. Evolution of sugars in cider brandy aged in oak barrels: a contribution to its characterization. J Agric Food Chem. 2003;51(4):923–926. | ||
Medeiros PM, Simoneit BR. Analysis of sugars in environmental samples by gas chromatography-mass spectrometry. J Chromatogr A. 2007;1141(2):271–278. | ||
Kakita H, Kamishima H, Komiya K, Kato Y. Simultaneous analysis of monosaccharides and oligosaccharides by high-performance liquid chromatography with postcolumn fluorescence derivatization. J Chromatogr A. 2002;961(1):77–82. | ||
Račaitytė K, Kiessig S, Kálmán F. Application of capillary zone electrophoresis and reversed-phase high-performance liquid chromatography in the biopharmaceutical industry for the quantitative analysis of the monosaccharides released from a highly glycosylated therapeutic protein. J Chromatogr A. 2005;1079(1–2):354–365. | ||
Marsilio R, D’Antiga L, Zancan L, Dussini N, Zacchello F. Simultaneous HPLC determination with light-scattering detection of lactulose and mannitol in studies of intestinal permeability in pediatrics. Clin Chem. 1998;44(8 pt 1):1685–1691. | ||
Flick JA, Schnaar RL, Perman JA. Thin-layer chromatographic determination of urinary excretion of lactulose, simplified and applied to cystic fibrosis patients. Clin Chem. 1987;33(7):1211–1212. | ||
Vilela EG, Torres HO, Ferrari ML, Lima AS, Cunha AS. Gut permeability to lactulose and mannitol differs in treated Crohn’s disease and celiac disease patients and healthy subjects. Braz J Med Biol Res. 2008;41(12):1105–1109. | ||
Sukkar SG, Schenone E, Foppiani L, Nobile MT. Experimental assessment of chemotherapy-induced early intestinal damage in colon cancer the lactulose-mannitol permeability test. Tumori. 2004;90(5):461–463. | ||
Vilstrup H, Amodio P, Bajaj J, et al. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver. J Hepatol. 2014;61(3):642–659. | ||
Vlachongiannakos J, Saveriadis AS, Viazis N, et al. Intestinal decontamination improves liver haemodynamics in patients with alcohol related decompensated cirrhosis. Aliment Pharmacol Ther. 2009;29:992–999. | ||
Bajaj JS, Heuman DM, Sanyal AJ, et al. Modulation of the metabiome by rifaximin in patients with cirrhosis and minimal hepatic encephalopathy. PLoS One. 2013;8(4):e60042. | ||
Lata J, Jurankova J, Kopacova M, Vitek P. Probiotics in hepatology. World J Gastroenterol. 2011;17(24):2890–2896. | ||
Liboredo JC, Ferrari Mde L, Vilela EG, et al. The effect of Saccharomyces boulardii in patients eligible for liver transplantation. Nutr Hosp. 2014;31:778–784. | ||
McGee RG, Bakens A, Wiley K, Riordan SM, Webster AC. Probiotics for patients with hepatic encephalopathy. Cochrane Database Syst Rev. 2011;(11):CD008716. | ||
Zhang SC, Wang W, Ren WY, He BM, Zhou K, Zhu WN. Effect of cisapride on intestinal bacterial and endotoxin translocation in cirrhosis. World J Gastroenterol. 2003;9(3):534–538. | ||
Sandhu BS, Gupta R, Sharma J, Singh J, Murthy NS, Sarin SK. Norfloxacin and cisapride combination decreases the incidence of spontaneous bacterial peritonitis in cirrhotic ascites. J Gastroenterol Hepatol. 2005;20(4):599–605. | ||
Sommansson A, Saudi WS, Nylander O, Sjöblom M. Melatonin inhibits alcohol-induced increases in duodenal mucosal permeability in rats in vivo. Am J Physiol Gatsrointest Liver Physiol. 2013;305(1):G95–G105. | ||
Chen P, Torralba M, Tan J, et al. Supplementation of saturated long-chain fatty acids maintains intestinal eubiosis and reduces ethanol-induced liver injury in mice. Gastroenterology. 2015;148(1):203–214. | ||
Úbeda M, Lario M, Muñoz L, et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J Hepatol. 2016;64(5):1049–1057. | ||
Verbeke L, Farre R, Verbinnen B, et al. The FXR agonist obeticholic acid prevents gut barrier dysfunction and bacterial translocation in cholestatic rats. Am J Pathol. 2015;185(2):409–419. | ||
Lippai D, Bala S, Catalano D, et al. MircoRNA-155 deficiency prevents alcohol induced serum endotoxin increase and small bowel inflammation in mice. Alcohol Clin Exp Res. 2014;38:2217–2224. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.