Back to Archived Journals » International Journal of Interferon, Cytokine and Mediator Research » Volume 8
Interleukin-8 in gastrointestinal inflammation and malignancy: induction and clinical consequences
Authors Cotton JA, Platnich JM, Muruve DA, Jijon H, Buret AG, Beck PL
Received 17 June 2015
Accepted for publication 24 July 2015
Published 10 May 2016 Volume 2016:8 Pages 13—34
DOI https://doi.org/10.2147/IJICMR.S63682
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Randall Davis
James A Cotton,1 Jaye M Platnich,1 Daniel A Muruve,1 Humberto B Jijon,1 Andre G Buret,2 Paul L Beck,1
1Department of Medicine, 2Department of Biological Sciences, University of Calgary, Calgary, AB, Canada
Abstract: Owing to its immense surface area, the mucosal surface of the gastrointestinal (GI) tract is continually subject to numerous endogenous and exogenous antigens capable of inducing an inflammatory response in the appropriate setting. In certain individuals, these inflammatory responses may also become dysregulated and result in the development of chronic GI inflammation, such as inflammatory bowel disease. Furthermore, gastric and colonic malignant processes are extremely common and, to date, still have high mortality rates. As a result, continuing research has focused on elucidating potential molecular mechanisms involved in these processes in an attempt to understand the pathophysiology of these diseases and, potentially, aid in the development of novel therapeutics. Interleukin-8 (CXCL8) is a small molecular weight chemokine identified almost 30 years ago. Ongoing research has demonstrated that this factor contributes to a multitude of pathophysiological processes within the GI tract, including chronic GI inflammatory states, such as inflammatory bowel disease, and gastric and colonic carcinomas. This review highlights the role of CXCL8 in GI inflammation and malignancy. It describes molecular mechanisms involved in promoting and restraining CXCL8 production in GI cells and how CXCL8 contributes to the pathophysiology of a variety of GI inflammatory processes. Finally, this review describes a variety of mechanisms by which CXCL8–CXCL1/2 signaling may contribute to GI malignancy.
Keywords: CXCL8, inflammatory bowel disease, IBD, colon cancer, IL-8, Crohn’s disease, ulcerative colitis
Introduction
The gastrointestinal (GI) tract represents the largest mucosal surface in contact with the external environment and, as such, is exposed to numerous endogenous and exogenous antigens capable of inducing an inflammatory response in the appropriate setting. In addition, many malignant processes occur within the GI tract, and continual exposure to chronic inflammation increases the likelihood of these processes.1,2 Inflammatory bowel disease (IBD) is subdivided into two main entities: Crohn’s disease and ulcerative colitis (UC). These diseases are chronic, relapsing, and remitting disorders of the GI tract that result from a combination of genetic, environmental, and immune factors. The incidence and prevalence of Crohn’s disease and UC appears to be increasing worldwide, and both represent significant economic burdens to health care systems and countries.3,4 Similarly, colorectal and gastric cancers are two of the most commonly identified malignancies and leading causes of mortality worldwide.5,6 To date, our understanding of these disease processes remains incomplete, and resistance to standard therapeutics has been reported for patients with IBD and gastric or colorectal cancer (CRC). The mechanisms underlying this resistance remain unclear.7,8 As such, ongoing research has focused on elucidating the molecular mechanisms involved in the development of GI inflammatory and malignant processes in the hopes of identifying novel therapeutic targets.
Nearly 30 years ago, three separate research groups simultaneously described a small molecular weight protein secreted by stimulated human mononuclear cells that induced polymorphonuclear cell (PMN) chemotaxis, degranulation, and superoxide and hydrogen peroxide (H2O2) production.9–11 As such, the compound plays a critical role in the body’s antimicrobial defense mechanisms. This product was subsequently named interleukin-8 and now often abbreviated as IL-8 or CXCL8 with both terms being used interchangeably.12,13 Research has subsequently demonstrated that CXCL8 is involved in a variety of processes apart from PMN activation, and its expression has been observed in a multitude of acute and chronic inflammatory processes, including many GI disease states. Its expression has been reported in a variety of resident and invading cell types within the GI tract, including monocytes, macrophages, endothelial cells, epithelial cells, fibroblasts, and neutrophils. Indeed, CXCL8 expression occurs acutely during GI infection caused by bacteria, viruses, and parasites, while elevated levels of CXCL8 are also reported in patients with chronic GI inflammation. In addition, sustained CXCL8 expression is reported in tumorous GI epithelial cell lines and in mucosal biopsy tissues collected from patients with gastric cancer or CRC. As a result, the expression or dysregulation of CXCL8 has been postulated to contribute to the pathophysiology of various disease states, and furthermore, targeting its expression may be of therapeutic benefit. In addition, the measurement of CXCL8 has been proposed as a potential diagnostic or prognostic marker for several GI disease states. The following review provides a summary of our current understanding of how CXCL8 expression contributes to the pathophysiology of various GI disease states.
Classification as a CXCL family chemokine
The primary amino acid sequence of interleukin-8 contains two N-terminal cysteine residues separated by a single amino acid that promotes formation of two disulfide bonds in higher-level amino acid structures.14,15 The presence of this sequence results in its assignment to the CXCL family of chemokines, hence the abbreviation CXCL8. Furthermore, the primary amino acid sequence of CXCL8 contains a glutamate–leucine–arginine (ELR+) sequence proximal to the CXC motif that is essential for binding to its two receptors;16–18 deletion or modification of this motif attenuates CXCL8 receptor binding.19,20 The presence of these motifs designates CXCL8 as an ELR+ CXC chemokine. The CXCL8 gene is located on chromosome 4q12-13 in close approximation to other ELR+ CXC chemokines that perform similar functions, including CXCL1 (growth-related oncogene α; GROα), CXCL2 (GROβ), CXCL3 (GROγ), CXCL5 (epithelial-derived neutrophil-activating peptide 78; ENA-78), and CXCL7 (neutrophil-activating peptide 2; NAP-2).15,21,22 Like several genes in this cluster, the CXCL8 gene contains four exons, three introns, and single CAT- and TATA-like structures.23 Together, these genes share ~30%–50% amino acid homology.15 As mentioned, CXCL8 primarily binds two receptors that are responsible for the majority of activated intracellular signaling events and are now designated as CXCR1 and CXCR2.24,25 Characterization of these receptors has also led to their identification on a variety of cell types, including endothelial cells, epithelial cells, fibroblasts, and neutrophils.26,27 This suggests that CXCL8 has a multitude of effects that extend beyond PMN activation and chemotaxis. Moreover, although CXCR1 and CXCR2 are highly homologous, differences in intracellular signaling cascades and biological processes activated following CXCL8 binding have been described.28 Molecular studies also indicate that only CXCL6 and CXCL8 are capable of binding CXCR1, while CXCR2 is capable of binding CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, and CXCL8.29,30 Therefore, although CXCL8 may structurally resemble other proinflammatory mediators of the ELR+ CXCL family, the differences in receptor–ligand binding suggest a, potentially, nonredundant role for this factor in a variety of biological or pathological processes. See Tables 1 and 2 for chemokine family members related to CXCL8 and ELR+ CXCL members.
 | Table 1 CXCL chemokine family related to CXCL8 |
 | Table 2 ELR+ CXCL chemokine family members related to CXCL8 |
Molecular signaling cascades and CXCL8 expression
An abundance of research examining the molecular mechanisms of CXCL8 mRNA transcription has been performed and aided in the study of how this chemokine contributes to disease in various organ systems, including the GI tract. Initial experiments using in vitro intestinal epithelial monolayers have demonstrated that GI CXCL8 mRNA transcription is regulated via the cooperation of inducible and constitutively expressed transcription factors, whereby the constitutively expressed transcription factors activator protein-1 (AP-1) and cis-regulatory enhancer binding protein-like factor cooperate with inducible nuclear factor κB (NF-κB) transcription factors to induce expression of CXCL8 mRNA.31–35 These factors bind cis-acting elements within the −94 to −71 region of the CXCL8 promoter to induce mRNA transcription.35,36 The NF-κB pathway has received much attention for its role in CXCL8 gene transcription, and numerous studies have demonstrated aberrant activation of this pathway during GI disease states.
The NF-κB pathway promotes transcription of a variety of anti- and proinflammatory mediators, including CXCL8, and is subdivided into a classical and alternative pathway based on activating stimuli and composition of transcription factor subunits. NF-κB transcription factors are homo- or heterodimers of five different subunits: p50/p105, p52/p100, p65 (RelA), RelB, or c-Rel.37 In the absence of proinflammatory stimuli, these transcription factors are bound to inhibitor of κB (IκB) proteins and retained within the cytosol so as to prevent their nuclear translocation and subsequent induction of gene transcription.37,38 Activation of the classical NF-κB pathway occurs in response to a variety of host- or pathogen-derived proinflammatory stimuli that bind different receptor families, including toll-like receptors (TLRs),39 nucleotide oligomerization domain-containing protein 1 and 2,40 interleukin-1 receptor (IL-1R),41 and tumor necrosis factor α (TNFα) receptor.42,43 Activation of the NF-κB pathway is also modulated by microtubule-associated serine/threonine protein kinase-3 (MAST3), and alteration of this pathway has been shown to affect CXCL8 secretion.44 Moreover, CXCL8 expression and activation of the NF-κB pathway has been reported to occur in response to reactive oxygen species (ROS), hypoxia, and acidosis.45 It should be noted, however, that these experiments have not all been performed within GI experimental models, and additional experiments are required to confirm the relevance of these observations in the GI tract. Upstream signaling cascades from these various receptors converge and phosphorylate residues on the IκB kinase (IKK) complex to activate the classical NF-κB pathway; this complex is composed of two kinase subunits, IKKα and IKKβ, and a regulatory subunit IKKγ/NEMO.46 Activation of the classical pathway results in phosphorylation of Ser177 and Ser181 residues on IKKβ and downstream phosphorylation, ubiquitination, and proteasomal degradation of IκB proteins.47,48 Degradation of IκB proteins permits the nuclear translocation of p65-containing NF-κB transcription factors that subsequently induce CXCL8 mRNA transcription.49,50
In the GI tract, CXCL8 mRNA transcription occurs following activation of the classical NF-κB pathway. The upstream signaling mechanisms involved in NF-κB p65-induced CXCL8 mRNA transcription may be cell specific. In vitro experiments using gastric epithelial cells and monocytic THP-1 cells exposed to Helicobacter pylori demonstrated that these two cell lines utilize different upstream signaling cascades for CXCL8 mRNA transcription.51 Stimulation of TLR2 is also implicated in H. pylori-induced E-cadherin disruptions, which have a well-established association with the development of gastric cancer. Indeed, recent findings have established that H. pylori infection is associated with elevated serum levels of an 80 kDa E-cadherin ectodomain in human patients. This was thought to arise via a heat-labile H. pylori surface component that activated the host protease calpain to cleave E-cadherin in human gastric cells, independently of the virulence factors CagA and VacA.52 Whether and how these pathways interact with CXCL8 signaling in gastric epithelial cells have yet to be assessed. Regardless, numerous in vitro experiments have shown that pathogen- or host-derived proinflammatory mediators promote NF-κB p65 nuclear accumulation and subsequent transcription and translation of CXCL8 within GI cell lines.53–58 Moreover, inhibition of the classical NF-κB pathway in in vitro intestinal epithelial monolayers reduces CXCL8 mRNA production.31 Similarly, mice infected with the attaching and effacing pathogen Citrobacter rodentium display elevated levels of activated NF-κB transcription factors and a paralleled increase in the CXCL8-related chemokines CXCL1 (KC) and CXCL2 (MIP-2) that were attenuated following NF-κB inhibition.59 In separate experiments, NF-κB p65 subunit inhibition ameliorated 2,4,6-trinitrobenzenesulfonic acid-induced colitis in vivo; however, these experiments did not analyze expression of CXCL8-related chemokines.60 These in vivo and in vitro experimental observations are further corroborated with data collected from human patients. Mucosal biopsy tissues collected from patients with H. pylori-induced gastritis display elevated levels of nuclear NF-κB p65 and increased levels of CXCL8 that parallel an increase in PMN tissue infiltration.61–66 Similarly, both activated NF-κB p65 and elevated CXCL8 have been observed in inflamed mucosal biopsy specimens collected from IBD patients, and increased expression of activated NF-κB p65 and CXCL8 directly correlated with indices of inflammation.60,67–70 Elevated levels of activated NF-κB p65 have also been reported in gastric and CRC lesions;71–73 however, association studies examining increased nuclear accumulation NF-κB p65 and elevated CXCL8 have not been performed. Collectively, these studies demonstrate the importance of the classical NF-κB pathway in contributing to CXCL8 production within the GI tract. Furthermore, they highlight the importance of this pathway in contributing to CXCL8-mediated GI disease. See Figure 1A and B for details on factors impacting CXCL8 and possible outcomes in inflammation and neoplasia. For details on mouse orthologs and ELR+ CXCL chemokine expression in mice see Table 1 and Figure 2 for details.
 | Figure 1 An overview of CXCL8 gene regulation and the signalling cascades downstream of CXCL8-mediated CXCR1/2 activation. |
 | Figure 2 CXC ELR+ chemokines in mice. |
Restraining NF-κB activation and CXCL8 production
Estimates suggest that the human GI tract contains ~1014 microorganisms with an estimated collective biomass of more than 1 kg.74–76 As a result, cells within the GI tract are constantly exposed to a multitude of potentially noxious and/or inflammatory stimuli capable of activating the NF-κB pathway and promoting transcription of various proinflammatory cytokines, including CXCL8. Despite this, the GI tract and its microbiota have evolved to exist in a state of equilibrium.77,78 A key mechanism that limits inflammation to a low level is the secretion of a mucus blanket which, in health, separates the microbiota from the host epithelium. In addition, researchers have described a variety of mechanisms employed by GI tissues aimed at attenuating activation of the NF-κB pathway and transcription of proinflammatory genes, such as CXCL8. One of the most basic mechanisms employed by the GI tract to ensure homeostasis with the gut microbiota is via the downregulation of receptor molecules capable of activating the classical NF-κB pathway on epithelial cells lying in close apposition to the microbiota. Experiments have demonstrated that in vitro intestinal epithelial monolayers are hyporesponsive to TLR2 and TLR4 stimulation due to downregulation of these receptors. Notwithstanding, these cells were capable of activating the classical NF-κB pathway and inducing CXCL8 mRNA transcription following transgenic expression of TLR2 or TLR4 and administration of their respective ligands.79,80 Importantly, experiments have demonstrated that administration of proinflammatory interferon γ or TNFα to in vitro intestinal monolayers restores TLR4 expression and NF-κB-mediated CXCL8 production induced via lipopolysaccharide.81 These experiments parallel observations of mucosal biopsy tissues collected from IBD patients, whereby areas of active inflammation were associated with increased TLR2 and TLR4 expression.82–84 Another mechanism employed by GI tissue to maintain homeostasis with resident microbiota is the preferential expression of receptors on epithelial surfaces separated from the gut microbiota. For example, the flagellin receptor TLR5 is primarily expressed on the basolateral surface of intestinal epithelial monolayers to ensure TLR5-mediated induction of the NF-κB pathway, and subsequent CXCL8 expression only occurs following disruption of monolayer integrity, such as during infection with Salmonella typhimurium.85,86 Similarly, TLR9 ligation activates distinctive receptor-signaling cascades within intestinal epithelial cells depending on the intestinal epithelial surface, whereby only basolateral TLR9 ligation resulted in NF-κB activation and CXCL8 secretion.87 These processes may be actively altered by enteropathogens, as illustrated in the Campylobacter jejuni-induced disruption of protective apical TLR9, which in turn primes the gut for heightened levels of CXCL8 and inflammation upon exposure to proinflammatory stimuli.88 Collectively, these results demonstrate that the GI epithelium, at least partially, regulates CXCL8 transcription via the NF-κB pathway through reduced expression and/or localization of TLRs.
In order to restrain inflammatory responses, the GI tract also expresses molecules that negatively regulate TLR/IL-1R proinflammatory signaling cascades, such as single IgG IL-1-related receptor (SIGIRR) and Toll-interacting protein (TOLLIP).80,89–91 These inhibitory proteins are essential for attenuating GI CXCL8 responses and associated with a variety of clinical GI disease states. SIGIRR and TOLLIP expressions are downregulated in mucosal biopsy tissues collected from areas of active inflammation in IBD patients.92 These molecules are also downregulated in immature GI tissues collected from patients with necrotizing enterocolitis and are associated with a concomitant increase in CXCL8 expression.93 Decreased TOLLIP mRNA has been found in association with increased levels of TLR mRNA in colon cancer and H. pylori-induced gastritis.94,95 Interestingly, decreased TOLLIP mRNA and increased TLR mRNA are observed as progression from H. pylori-induced gastritis to adenocarcinoma occurred.94 These observations may help, at least partially, explain experimental data suggesting that CXCL8 is the most upregulated gene in in vitro gastric epithelial cells exposed to H. pylori.96 Clinical observations suggesting these inhibitory proteins attenuate CXCL8 production are supported experimentally. The genetic ablation of TOLLIP and SIGIRR in vitro results in enhanced NF-κB responses and increased CXCL8 mRNA transcription, while their overexpression leads to blunted CXCL8 expression.90 Epithelial SIGIRR deficiency in vivo increases susceptibility to dextran sulfate sodium (DSS)-induced colitis, enhances expression of CXCL8-related chemokines, and increases tumorigenesis rates following DSS + azoxymethane treatment.97 Similarly, genetic deletion of TOLLIP in vivo enhances susceptibility to experimental models of colitis and leads to upregulated NF-κB activity and expression of CXCL8-related cytokines.98 Collectively, these results demonstrate that the GI tract expresses molecules capable of dampening CXCL8 expression, and absence of these molecules can result in pathological CXCL8 expression. This pathological expression of CXCL8 may contribute to the development of chronic GI inflammatory disease states or malignancies.
CXCL8 gene polymorphisms
Although the CXCL8 promoter region is essential to mRNA transcription, gene polymorphism studies indicate that other regions within the gene may contribute to CXCL8 production and, potentially, susceptibility to certain GI disease states. For example, multiple polymorphisms within the CXCL8 gene are thought to influence susceptibility to the development of irritable bowel syndrome.99 A collection of research also suggests that polymorphisms at position −251 of the CXCL8 gene enhance susceptibility to a variety of GI disease states. Individuals with adenosine residues at position −251 (−251A) of the CXCL8 gene are thought to be at increased risk of infection from Clostridium difficile, enteroaggregative Esherichia coli, and H. pylori, and following infection, have elevated inflammatory responses and/or more clinically significant disease.100–109 It is currently unknown whether these polymorphisms enhance susceptibility to other GI pathogens. Moreover, −251A polymorphisms are thought to increase susceptibility to the development of gastric cancer and IBD.110–114 It is worth noting that the reproducibility of some of the above studies has been questioned. As a result, it has been postulated that the presence of these mutations may influence the development of GI disease only within certain populations.115 It is unknown whether other CXCL8 gene polymorphisms contribute to the development of certain GI diseases. Research to date examining the relationship between CXCL8 gene polymorphisms and GI disease has focused on the −251 region, and additional research is required to clarify whether polymorphisms at other sites also factor into susceptibility to GI inflammation or cancer. Other CXCL8 gene polymorphisms are associated with a variety of inflammatory and malignant states, including osteoarthritis,116 ovarian cancer,117 and systemic lupus erythematosus nephritis.118 Additionally, it is currently unknown how −251A polymorphisms affect CXCL8 transcription. To date, experiments have only demonstrated that lipopolysaccharide stimulation of peripheral blood cells collected from patients with −251A polymorphisms results in enhanced CXCL8 production;119 the mechanism via which this occurs remains obscure.
Interleukin-8 protein
CXCL8 is synthesized as a 99 amino acid precursor and secreted following proteolytic processing of leader sequences. These sequences are variable, approximately 20 amino acid residues in length, and cleaved by a variety of extracellular enzymes. The variability associated with the cleavage site results in the generation of an assortment of CXCL8 isoforms.57 The most commonly reported active CXCL8 isoforms contain 77 (CXCL8 1-77) or 72 (CXCL8 6-77) amino acids. CXCL8 6-77 is primarily secreted by activated macrophages and monocytes and has potent effects on PMNs.62,105 It is compared to CXCL8 1-77 largely derived from endothelial cells and fibroblasts.120,121 Active CXCL8 isoforms can be further processed via a myriad of factors that further modulate CXCL8 potency. Importantly, the ELR motif must remain intact for its stimulatory effect on PMNs, while CXCL8 1-77 can be further processed by thrombin into the more potent isoform CXCL8 6-77.122 Moreover, PMN-derived cathepsin G, elastase, matrix metalloproteinase-9 (MMP-9; gelatinase B), and proteinase-3 are capable of proteolytically processing CXCL8 1-77 into more potent isoforms.123,124 Therefore, PMNs may induce a positive feedback loop that further potentiates the recruitment of additional PMNs. It has recently been proposed that modification of CXCL8 via various factors be divided into three distinct groups. The first group involves isoforms of CXCL8 generated by cleavage with aminopeptidases, which yields isoforms of 75–79 amino acids in length with intermediate activity toward PMNs. The second group includes CXCL8 isoforms of 69–72 amino acids in length that are generated by proteolytic cleavage and are highly potent PMN chemoattractants. Finally, the third group is generated via modification of arginine to citrulline residues and is a weak PMN chemoattractant.125 Although citrullination of CXCL8 dampens PMN tissue responses, it may enhance CXCL8’s ability to mobilize PMNs within the blood stream.126 Collectively, these studies demonstrate that CXCL8 exists in a multitude of isoforms that are subject to modification by the surrounding host tissue. It remains largely unknown how proteolytic modifications to CXCL8 affect various inflammatory or malignant GI disease states. See Figure 1A and B and Tables 3 and 4 for a review of a CXCL8 signaling and in promoting inflammation and neoplasia.
 | Table 3 The role of CXCL8 in promoting inflammation |
 | Table 4 Role of CXCL8 in promoting tumorigenesis and malignancy in gastric and intestinal carcinomas |
MMP-9-mediated proteolytic processing
Several proteases capable of processing CXCL8 have been shown to be elevated in GI disease states. MMP-9 is associated with a variety of GI disease states, and it has been shown experimentally that intestinal epithelial MMP-9 expression aggravates experimental colitis and delays wound healing.127 Expression of this protease is also elevated in many patients with active IBD and colorectal or gastric cancer, and heightened expression is associated with poor cancer disease prognosis.128–138 Additionally, H. pylori infection is associated with elevated MMP-9 expression in an NF-κB-dependent manner,139–141 and pathogen eradication results in attenuated MMP-9 expression.142 High expression of MMP-9 and CXCL8-related chemokines has been observed in animals infected with C. rodentium143 and in vivo models of colitis-associated colon cancer.144 In addition, serum MMP-9 and CXCL8 are concomitantly elevated in patients with stages II–IV CRC,145 and elevated MMP-9 expression is associated with increased CXCR1 and CXCR2 in gastric biopsy tissues collected from patients with gastric cancer.146 Although both MMP-9 and CXCL8 are co-expressed in several GI disease states, it remains unknown whether proteolytic processing of CXCL8 by MMP-9 contributes to disease pathogenesis. However, in vivo studies have demonstrated that manipulation of MMP-9 alters susceptibility to experimental colitis, as inhibition of MMP-9 activity in mice is associated with decreased PMN accumulation and decreased expression of the CXCL8-related chemokine CXCL5.147 Overexpression of intestinal epithelial MMP-9 in vivo enhanced animal susceptibility to DSS- or S. typhimurium-induced colitis, as was evidenced by increased intestinal PMN accumulation and CXCL1 expression.148,149 Collectively, these results demonstrate that MMP-9 is capable of modifying GI disease states, but additional research is required to examine whether MMP-9-mediated proteolytic processing of CXCL8 contributes to various GI disease states.
CXCL8 contributes to the tissue recruitment of neutrophils
Bone marrow egression and extravasation
A multitude of research findings have established that CXCL8 plays numerous roles in the recruitment of PMNs into GI tissues. CXCL8 has been shown to contribute to PMN bone marrow egression, extravasation, and tissue migration. Although not all the experimental studies have been conducted in GI tissues, the following provides a framework for describing how CXCL8 contributes to the recruitment of PMNs in GI tissues. The initial recruitment of PMNs requires their mobilization from the bone marrow into the circulation. During homeostasis, PMNs are largely retained within the bone marrow and only ~2% are found in circulation.150 As in vivo experiments have shown, the retention of PMNs in the bone marrow involves antagonism between CXCR2 and CXCR4, whereby the CXCR4 ligand, CXCL12 (stroma cell-derived factor 1; SDF-1), retains PMNs within the bone marrow by enhancing receptor–ligand interactions between PMN integrins and surface receptors on bone marrow endothelial and stromal cells.151–153 During an acute inflammatory response, including GI inflammation, the release of granulocyte colony-stimulating factor decreases CXCL12 expression, while simultaneously increasing the expression of CXCR2 ligands, including CXCL8, by endothelial cells to promote PMN bone marrow egression.154 This has been demonstrated during in vivo studies showing that intravenous administration of CXCL8 induces rapid mobilization of PMNs and their progenitor cells from the bone marrow into the bloodstream.155,156
Following this, PMNs exit the vasculature and enter tissues via postcapillary venules in a series of highly characterized steps.157–159 Briefly, PMN extravasation is subdivided into five overlapping steps: tethering, rolling, firm adhesion, crawling, and transmigration.157 Exposure to proinflammatory stimuli induces the release of a multitude of proinflammatory mediators, such as histamines, leukotrienes, and cytokines. These mediators are released from tissue-resident immune and epithelial cells that induce expression of P- and E-selectin molecules on the luminal surfaces of endothelial cells. These molecules bind glycosylated ligands, such as P-selectin glycoprotein 1 (PSGL-1), on the surface of PMNs to induce PMN tethering and, subsequently, PMN rolling.160 Firm adhesion of PMNs to the vascular endothelium occurs as rolling PMNs contact ELR+ PMN chemokines, such as CXCL8. Endothelial cells are capable of transcytosing CXCL8, produced by various cell types, to the luminal surface where it is subsequently bound to heparan sulfate;28,161 this ensures that secreted chemokines are immobilized and able to induce firm adhesion and migration in circulating PMNs.162,163 Binding of CXCL8 to its receptor triggers a signaling cascade that enhances the binding affinity and avidity of PMN integrin molecules, such as CD11a/CD18 and CD11b/CD18. This enables them to strongly bind endothelial surface ligands, such as intercellular adhesion molecules, and undergo firm adhesion.160,164,165 Following this, PMNs crawl across the surface of the endothelium and, subsequently, preferentially migrate into host tissues via tricellular endothelial cell corners.166,167 Collectively, these results demonstrate that CXCL8 participates in PMN bone marrow egression and extravasation during inflammatory responses, including those in the GI tract.
PMN tissue migration
CXCL8 is also involved in directing extravasated PMNs through GI tissues to an infectious or inflammatory site. Following extravasation, PMNs migrate through multiple and sequential chemotactic gradients within host tissues.168 These gradients are classified into a hierarchy of intermediate and end-target chemoattractants. End-target chemoattractants are produced immediately at the inflammatory site and are the final chemotactic gradients PMNs will migrate through; these are host- or microbially-derived factors located in the immediate vicinity of an infectious or inflammatory site. Examples include bacterial formylated peptides and the complement fragment C5a. Intermediate chemoattractants are host-derived factors that direct PMNs toward end-target chemoattractants and are produced at distances farther from the infectious or inflammatory site. End-target chemoattractant signaling cascades override those generated from intermediate chemoattractants. Therefore, intracellular signaling cascades from intermediate chemoattractants, such as CXCL8, via the p38 MAPK pathway are superseded by phosphatidylinositol-3-kinase (PI3K) signaling following PMN receptor binding of end-target chemoattractants.169 For example, S. typhimurium infection of the intestinal epithelium drives expression of CXCL8 and the arachidonic acid metabolite lipoxin A4; CXCL8 promotes PMN recruitment to the basolateral membrane, and lipoxin A4 induces PMN transepithelial migration.170–172 Taken together, these results demonstrate that CXCL8 plays an important role in directing extravasated PMNs toward infectious or inflammatory sites until its signal is overridden by end-target chemoattractant signals.
CXCL8 aids in the activation of neutrophil antimicrobial machinery
Respiratory burst
Following directed chemotaxis to an inflammatory or infectious site, PMNs utilize a variety of mechanisms to clear host tissues of invading or translocated microbes, including the respiratory burst. This process is essential to host defense against a variety of infectious organisms, as has been demonstrated in patients with chronic granulomatous disease. Individuals with chronic granulomatous disease are highly susceptible to a variety of catalase-positive bacterial and fungal infections, due to mutations in genes essential for the respiratory burst.173,174 Similarly, genetic deletion of factors essential for the respiratory burst enhances susceptibility to in vivo infection.175–178 The respiratory burst involves rapid utilization of oxygen, increased glucose consumption, and the release of ROS.179 In PMNs, production of ROS involves the assembly of the multicomponent enzymatic nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase (NOX2) complex. This complex is composed of several subunits distributed throughout the cytosol and cell membranes of inactive PMNs. The flavocytochrome b558 subunit, composed of gp91phox/Nox2 and p22phox, is located on the plasma membrane and within specific granules, while the other components (p67phox, p47phox, p40phox, and the small G-protein Rac2) are cytosolic proteins.180 Assembly and activation of the NOX2 system on plasma and phagosomal membranes occur following PMN exposure to and phagocytosis of microbes.181–183 This process initially results in the production of superoxide anion radicals (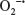 ) by the NADPH-oxidase (NOX) system, whereby is subsequently converted to other oxygen radicals, such as H2O2.184
) by the NADPH-oxidase (NOX) system, whereby is subsequently converted to other oxygen radicals, such as H2O2.184
CXCL8 itself does not induce NOX2 activity and, therefore, overt production of ROS. This has been shown experimentally in mice transgenically engineered to continually express human intestinal epithelial CXCL8; these animals show extensive PMN tissue infiltration, but lack apparent histological signs of mucosal injury that could be, potentially, induced by ROS.185 Rather, CXCL8 pretreatment has been shown to enhance ROS production when PMNs are subsequently treated with formylated bacterial peptides (such as formyl-methionyl-leucyl phenylalanine [fMLF]), phorbol 12-myristate 13-acetate, or platelet activating factor.186,187 This appears to occur via the CXCR1 receptor.188,189 Importantly, this CXCL8-mediated priming of NOX2 enhances the microbicidal activity of PMNs.190 Ongoing research suggests that CXCL8-mediated priming of NOX2 activity in PMNs may occur via multiple mechanisms. PMNs pretreated with CXCL8 have elevated surface expression of the fMLF receptors, which is postulated to contribute to enhanced fMLF-induced ROS production.191 Separately, it has been demonstrated that increased intracellular Ca2+ is required for activation of the NOX2 system, potentially via activation of phospholipase A2.192–194 Accordingly, CXCL8 pretreatment has been shown to increase intracellular Ca2+.195 Finally, the phosphorylation of the p47phox and p67phox subunits of the NOX2 system has been suggested to be essential for its activation.196–198 CXCL8 pretreatment has also been shown to induce the pre-assembly of the NOX2 system within PMN lipid rafts via phosphorylation of p47phox and p67phox.199 Collectively, these results demonstrate that CXCL8 plays a critical role in the production of ROS within PMNs.
It is well known that chronic GI inflammatory states are capable of progressing to various GI cancers.200 Although the NOX2 system is vital in microbial elimination, the production of ROS also results in bystander damage to host tissues that can predispose to the development of cancer. PMN-derived oxidants are found in excess in mucosal biopsy tissues collected from areas of active inflammation in patients with IBD201 and gastric biopsies collected from patients with chronic H. pylori infection.202,203 Furthermore, ROS production has been shown to contribute to intestinal epithelial damage in patients with IBD.204 This ongoing damage during chronic inflammatory states triggers a multitude of changes within host tissues that can result in the development of cancer. Indeed, tissues collected from patients with either H. pylori infection or IBD have increased rates of DNA damage, elevated mutation levels, and defective DNA mismatch repair systems.205–209 Furthermore, biopsies collected from areas of active inflammation in patients with UC demonstrated higher mutation rates in the tumor suppressor p53 gene; this was attributed to an increase in ROS production.210 Currently, it remains unknown whether CXCL8-mediated priming of the NOX2 NADPH-oxidase system is involved in promoting the progression of chronic GI inflammation to malignancy, and additional studies would be required to confirm these hypotheses.
Degranulation
PMN granules are storage organelles containing a plethora of noxious antimicrobial compounds and divided into three subsets based on their contents, order of production, and release within host tissues.180 Primary granules (also known as azurophilic granules) are positive for myeloperoxidase and, therefore, have a critical role in the potentiation of the respiratory burst.211 Furthermore, these granules contain a bevy of antimicrobial compounds such as defensins, lysozyme, and bactericidal/permeability-increasing protein and three serine proteases: cathepsin G, neutrophil elastase, and proteinase 3.212 These are the first granules formed during PMN differentiation.213 Secondary granules (also known as specific granules) contain lactoferrin and a variety of antimicrobial compounds, including neutrophil gelatinase-associated lipocalin, cathelicidin, and lysozyme.212 These are formed after primary granules.213 Finally, tertiary granules (also referred to as gelatinase granules) contain a large number of metalloproteinases;212 these are the last granules formed during PMN differentiation.213 The utilization of these granules by PMNs occurs in an order opposite to their production during differentiation. Degranulation of PMN tertiary granules occurs during migration through the basement membrane. It has been postulated that metalloproteinases degrade components of this structure to ease PMN migration,214,215 while degranulation of primary and secondary granules occurs once PMNs have arrived at the inflammatory site.
The initial identification and characterization of CXCL8 as a PMN-activating compound demonstrated its ability to induce PMN degranulation.10,216–218 Follow-up experiments demonstrated that CXCL8 was capable of inducing PMN degranulation of primary, secondary, and tertiary granules via CXCR1 and CXCR2.188,219,220 Interestingly, tertiary granule exocytosis and release of MMP-9 was reported to occur downstream of CXCR2.221 Additionally, the release of antimicrobial peptides also serves as a chemoattractant for T-cells.222,223 CXCL8 is subject to modification via numerous proteases elevated in several infectious, inflammatory, or cancerous states of the GI tract, where elevated CXCL8 has been concomitantly reported. In these states, PMN degranulation products represent an abundant source of proteases capable of proteolytically processing CXCL8. PMN elastase is increased in fecal samples collected from patients with CD and UC,224 while separate reports have indicated that its antagonists, elafin and secretory leukocyte protease inhibitor (SLPI), are significantly decreased.225–227 Elevated PMN elastase is reported in patients with CRC,228 while H. pylori infection results in suppression of SLPI.229 As such, the increased expression of proteases coupled with decreased antiprotease expression may lead to heightened proteolytic activity within GI tissues and, as a result, enhanced processing of CXCL8. Additional research is required to investigate this hypothesis. To date, it has been shown that exogenous administration of elafin and SLPI reduces inflammation in in vivo models of colitis, whereby a decrease in PMN tissue infiltration and expression of CXCL8-related chemokines was observed.226,227 However, little research has examined whether the protease:antiprotease imbalance results in modifications to CXCL8 that contribute to pathology associated with IBD, various cancers, or other GI disease states.
Interestingly, certain GI pathogens have been shown to release proteases that proteolytically process CXCL8 and alter their effects on PMNs. Entamoeba histolytica cysteine protease 2 cleaves CXCL8, and this product is more effective at inducing PMN chemotaxis.230 In contrast, Giardia duodenalis cathepsin B cysteine proteases were shown to degrade CXCL8 and attenuate CXCL8-induced PMN chemotaxis.231 This research may explain how in vivo G. duodenalis infections were capable of attenuating granulocyte infiltration induced by intrarectal instillation of C. difficile TcdA/TcdB or reducing CXCL chemokine expression from ex vivo inflamed intestinal mucosal biopsy tissues.232 It remains unknown whether other GI pathogens produce proteases or factors capable of modifying CXCL8 and whether this could, potentially, affect the pathogenesis of infection.
Involvement of CXCL8 in malignant processes
Over expression of CXCL8 and CXCR1/2 has been described in numerous inflammatory and neoplastic disease states. The link between chronic inflammation and neoplasia has been well described, and cancer-associated inflammation, due to changes in tumor microenvironment or changes within the tumor cell, can be associated with increased tumor growth, metastasis, resistance to therapy, all of which impact prognosis and survival (see Figure 1A and B and Tables 3 and 4 for a review a CXCL8 signaling and in promoting inflammation and neoplasia).
CXCL8 induces angiogenesis
Angiogenesis, or the formation of new blood vessels, involves a sequential sequence of events beginning with basement membrane proteolysis, endothelial cell proliferation and chemotaxis, and finally, organization and maturation of tubular structures. This process has long been recognized as an integral hallmark in cancer development,233 and disrupting angiogenesis has been proposed as an effective therapeutic target for a variety of cancers.234 Indeed, angiogenesis is essential to providing the primary neoplasm with an adequate blood supply and contributes to cancer metastasis.235,236 The induction of angiogenesis occurs early during the development of multistage cancers and has been observed in histological specimens collected from premalignant, noninvasive lesions such as dysplasias and in situ carcinomas.237 It is thought that tumor progression induces activation of variety of angiogenic switches that promote surrounding vasculature to produce new vessels.238 These newly formed blood vessels tend to be heterogeneous in size and structure, are poorly formed, and have inadequate function.239–241 The angiogenic switches responsible for new blood vessel formation have been proposed to occur through direct and indirect mechanisms. Oncogenes within tumor cells may directly induce expression of angiogenic factors. Alternatively, these angiogenic factors may be produced indirectly by infiltrating inflammatory cells, including macrophages, PMNs, mast cells, and myeloid progenitor cells.242 Indeed, individuals with chronic inflammatory disorders are at increased risk of developing cancer, which may be, at least in part, due to expression of angiogenic factors produced by infiltrating inflammatory cells.243 For example, it has been well established that chronic inflammation with H. pylori infection is associated with increased risk of gastric cancer.244 Similarly, individuals with IBD are at increased risk of colon cancer via a variety of mechanisms that remain incompletely understood.1
CXCL8 and related chemokines have been demonstrated to have oncogenic properties capable of inducing angiogenesis;245–247 this occurs independently of their ability to induce PMN chemotaxis248–250 and results via activation of CXCR2.251–253 Indeed, mucosal biopsy tissues collected from patients with gastric carcinomas have increased CXCL8 levels that parallel increased tumor vascularity.254 Furthermore, one study has suggested that the survival rate of patients with gastric carcinomas was significantly reduced in patients with tumors expressing high levels of CXCL8.255 A variety of signaling molecules produced during tumor metabolism may induce CXCL8 secretion and, resultantly, promote tumor angiogenesis. For example, the production of lactate has been shown to induce autocrine CXCL8 production and thereby promote tumor angiogenesis in an in vivo model of CRC.256 Experimentally, increased CXCL8 expression in vivo enhanced blood vessel formation, while genetic deletion of CXCR2 blunted this effect.257 Research has now demonstrated that CXCL8 contributes to angiogenesis via multiple mechanisms, including inducing expression of vascular endothelial growth factor258 as well as MMP-2 and MMP-9 within endothelial cells; these two factors are involved in basement degradation during angiogenesis.249 In patients with CRC, a concomitant increase in serum CXCL8 and MMP-9 is observed in patients with stage II disease or higher.145 CXCL8 is also capable of inducing endothelial cell proliferation and capillary tube reorganization in a concentration-dependent manner, whereby lower rather than higher concentrations are more effective at inducing these effects.259 Finally, CXCL8 has also been shown to inhibit endothelial cell apoptosis via increasing expression of antiapoptotic proteins, such as Bcl-XL and Bcl-Xs.249,260 Similarly, in in vivo models of CRC, the transgenic expression of CXCL8 in animals results in enhanced tumor burdens that were, at least partially, due to increased tumor angiogenesis.261 CXCL8 and/or CXCL1/2 have also been shown to promote angiogenesis in several other cancers, including melanoma, pancreatic, prostate, and non-small-cell lung cancer.262
CXCL8 induces epithelial-to- mesenchymal transition, migration, and invasion
Tumor epithelial-to-mesenchymal transition (EMT) is a process involving the dedifferentiation of epithelial cells into an invasive mesenchymal phenotype. This process involves the loss of epithelial cell characteristics, including a loss in cell polarity, cell–cell contacts, epithelial surface markers, and a concomitant increase in mesenchymal properties. The properties include increased expression of mesenchymal-associated proteins, enhanced cell motility, and metastatic potential.263 Indeed, in vitro experiments have demonstrated that gastric epithelial cell lines treated with exogenous CXCL8 increased expression of the epidermal growth factor receptor, MMP-9, and vascular endothelial growth factor; at the same time, this treatment decreased E-cadherin expression and increased the invasive potential of these cells.264 Clinical research has indicated that CXCL8 levels are positively associated with the extent of tissue invasion and metastasis. In patients with CRC, elevated levels of CXCL8 have been positively associated with tumor size, level of infiltration, and liver metastases.265,266 Heightened expression of both CXCL8 receptors in gastric mucosal biopsy tissues has been associated with gastric cancer metastasis.146 CXCL8 tissue expression levels have been reported to increase as colonic adenocarcinoma cells become less differentiated, while serum levels have been shown to increase in proportion to the pathological staging of CRC tumors and distant metastases.267 These positive correlations between CXCL8 and tissue invasion and metastasis are further supported by in vitro experiments, whereby knockdown of CXCR1 and CXCR2 proteins inhibited the migratory capability of gastric epithelial cells.146 Similarly, overexpression of CXCL8 in in vitro colonic epithelial cell lines enhanced cellular proliferation, migration, and invasive potential of these cells.268 Studies in prostate cancer found that CXCL8 enhances tumor cell migration and invasion via activation of Src and FAK.269 Moreover, in vitro experiments using a variety of colonic epithelial cell lines have demonstrated that expression of CXCL8 parallels metastatic potential, whereby increased expression of CXCL8 and its receptors is found in cell lines with higher invasive potential.270 Indeed, CXCL8 and its receptors are downstream targets for several EMT transcription factors.271,272 The overexpression of CXCL8 by in vitro CRC cell lines has also been shown to act as an autocrine growth factor.273 Furthermore, CXCL8 may also confer chemoresistance, as pretreatment of in vitro colonic and gastric epithelial cells with CXCL8 resulted in enhanced cell survival to oxaliplatin treatment.268,274,275 Moreover, CXCL8 treatment has been shown to reduce in vitro epithelial sensitivity to apoptosis.276 Importantly, inhibition of CXCR2 signaling in intestinal epithelial cells has been shown to increase sensitivity to oxaliplatin.277 Based on the above data, it has been proposed that targeting the CXCL8 pathway represents a potential therapeutic target for gastric cancer and CRC.278
Myeloid-derived suppressor cells
Immature myeloid cells are a heterogeneous group of cells and comprise numerous subpopulations, including myeloid-derived suppressor cells (MDSCs). Experimental evidence has shown that these cells are not fully differentiated and, therefore, express surface markers for both granulocytic and monocytic cell types.279 Ongoing research has implicated MDSCs in the pathophysiology of numerous cancers.280 In patients with CRC, MDSC populations are elevated both within cancerous tissue and serum; moreover, the level of elevation positively correlates with the cancer stage and extent of metastasis.281,282 A complete discussion of the research surrounding MDSCs is beyond the scope of this review. However, ongoing research has demonstrated that MDSCs participate in the progression of cancer via a number of mechanisms, including via the suppression of antitumor immune responses280,283 and promotion of angiogenesis.242 As a result, targeting MDSCs has been proposed as a potential chemotherapeutic target.284 Experimental research has just begun to elucidate the role that CXCL8 plays in recruiting MDSCs. In in vivo models of colon cancer, mice transgenically engineered to express human CXCL8 displayed increased infiltration of immature myeloid cells into dysplastic and surrounding tissues; this ectopic expression of CXCL8 was associated with enhanced angiogenesis and tumorigenesis.261 Similarly, the in vivo deletion of CXCR2 resulted in decreased chronic inflammation and development of colitis-associated tumorigenesis.285 Collectively, these experimental results suggest that CXCL8 may play an important role in recruiting MDSCs to the tumor microenvironment. Additional research is required to elucidate the exact mechanisms involved in these early findings.
Resistance to chemotherapy
Resistance to chemotherapeutics is an ongoing concern, and research has suggested that autocrine or paracrine expression of CXCL8 may contribute to resistance to chemotherapeutic agents in a variety of GI cancers. Currently, CRC chemotherapy involves the use of 5-fluorouracil and oxaliplatin.286 Indeed, the efficacy of oxaliplatin treatment has been found to be dependent on genetic variations within the CXCL8 and/or CXCR2 genes,8,287 and CXCL8 serum levels have been reported to be elevated in CRC patients who fail to respond to chemotherapy.288 Moreover, induced expression of CXCL8 in in vitro colonic adenocarcinoma cell lines resulted in decreased sensitivity to oxaliplatin treatment, while silencing of CXCL8 production produced the opposite effect.268 The former was associated with increased activation of the NF-κB pathway,268 which has been shown to induce transcription of genes that antagonize apoptosis.289 Similar results were observed using in vitro gastric adenocarcinoma cell lines.275 Additionally, blockade of CXCR2 signaling in in vitro colonic adenocarcinoma cell lines increased sensitivity to oxaliplatin and induced expression of apoptotic proteins.277 In addition, CXCL8-induced resistance to oxaliplatin therapy may occur following activation of upstream signaling cascades. Exposure of in vitro adenocarcinoma cells to oxaliplatin has been shown to induce interleukin-22 expression that subsequently promotes CXCL8 expression and resistance to 5-fluorouracil and oxaliplatin therapy.274 Gemcitabine is a commonly used chemotherapeutic agent used for treatment of pancreatic ductal adenocarcinomas.290 However, gemcitabine resistance is often reported,291 and experiments performed in vivo and in vitro have demonstrated that gemcitabine treatment induces CXCL8 expression in pancreatic cancer cells that subsequently results in neovascularization.292 Collectively, these results demonstrate that CXCL8 is involved in resistance to chemotherapeutic drugs in GI cancers, and targeting CXCL8 or its upstream inducers enhance the efficacy of chemotherapeutic agents. See Table 4 for a review of processes by which CXCL8 can impact neoplasia.
Specific targeting of CXCL8–CXCR1/2 signaling in cancer therapy
As noted above, CXCL8 and CXCR1/2 are upregulated in several cancers, and it is felt that cancer-related inflammation results in increased migration, invasiveness, cancer cell resistance to apoptosis, and increased cancer cell proliferation.262,268,293 Not only can chronic inflammation increase the risk of cancer (up to 20% of all cancers may result from chronic inflammation) but also cancer cells and cancer growth often promote inflammation.262,294 Numerous signaling pathways can be activated downstream of CXCL8–CXCR1/2, including MAPK, PI3K/Akt, PKC, FAK, and Src, all of which have been implicated in neoplasia and inflammatory processes. CXCL8–CXCR1/2 signaling can also activate numerous transcription factors, including NFκB, AP-1, β-catenin/Tcf, STAT3, and HIF, which can lead to upregulation of other receptor-signaling pathways, such as epidermal growth factor receptor and steroid receptors.262,295,296 CXCL8 signaling can be induced by numerous factors associated with both neoplasia and inflammation, including TNFα, IL-1β, ROS, death receptors (eg, Fas ligand), some steroid hormones (eg, androgens), hypoxia, acidosis, altered glucose levels, chemotherapeutic agents, and radiation therapy.262,297 Furthermore, the CXCL8 promoter contains consensus sites for transcription factors that are altered in inflammation and neoplasia, including NFκB, AP-1, β-catenin/Tcf, and HIF transcriptional factors.262,296 Oncogenic mutations and/or loss of tumor suppressor genes can alter CXCL8-CXCR1/2 signaling resulting in inflammation and cancer progression.262,296,298,299 See Table 4 for further details.
In view of the link between inflammation and cancer, several approaches have targeted inflammatory pathways in the fight against cancer, including the NFκB pathways and, more specifically, CXCL8–CXCR1/2 signaling. Numerous agents have been developed to block CXCL8–CXCR1/2 signaling directly or indirectly, including: 1) blocking signal transduction (inhibitors of MAPK pathways; ERK, JNK, p38, PI3K/Akt) that leads to CXCL8–CXCR1/2 expression; 2) NFκB inhibitors (downregulation of proinflammatory genes mediating, recruitment/invasion, apoptosis, proliferation, angiogenesis, and more specifically, CXCL8- CXCR1/2 expression); 3) nonsteroidal anti-inflammatory drugs (reduce inflammation and can decrease CXCL8- CXCR1/2 expression); and 4) direct targeting of CXCL8–CXCR1/2 signaling.262 The main approaches to directly target CXCL8–CXCR1/2 signaling include: 1) CXCL8 neutralizing antibodies; 2) CXCR1/2 neutralizing antibodies; and 3) small molecule CXCR1/2 antagonists and siRNA strategies. In summary, many studies have shown that CXCL8 and CXCR1/2 neutralizing antibodies, small molecule inhibitors, and siRNA can reduce inflammation and tumor growth in vitro, and some in preclinical models.262 More specifically, these agents can reduce inflammation in animal models of arthritis models, IBD (DSS300 and 2,4,6-trinitrobenzenesulfonic acid301 colitis), asthma, and tumor burden and metastasis in CRC models and other cancer cell types.262,277,302,303 Early clinical trials with primarily small molecule CXCR1/2 inhibitors are under way for IBD (UC), asthma, COPD, psoriasis, rheumatoid arthritis, solid organ transplant rejection, melanoma, CRC, breast cancer, and other malignancies262 (see Table 5).
 | Table 5 CXCR1/2 small molecule antagonists in development for inflammation and neoplasia |
Conclusion
Since its initial discovery, our understanding of how CXCL8 contributes to various GI inflammatory and malignant states has vastly improved. Clinical research has demonstrated that CXCL8 is elevated in a variety of disease states, including GI infection, IBD exacerbations, and GI malignancy, and CXCL8 levels have been shown to correlate with predicted clinical outcomes. In addition, in vitro and in vivo experiments have suggested potential mechanisms via which these processes occur. Much research has focused on CXCL8’s ability to induce various signaling events within PMNs, and indeed, experimental research has demonstrated that this factor contributes to a variety of proinflammatory PMN events, including its contribution to the oxidative burst, chemotaxis, and degranulation. However, research has also demonstrated that CXCL8 contributes to increased angiogenesis and EMT during malignancy via its actions on endothelial and epithelial cells, respectively. In addition, CXCL8 promotes the accumulation of MDSCs in GI tissues during malignancy. As our understanding of CXCL8 has improved, it has become apparent that certain CXCL8 polymorphisms are associated with altered susceptibility to a variety of disease states, including GI infection, IBD, and gastric cancer. Despite these findings, our understanding of the role that CXCL8 plays in the pathophysiology of various GI inflammatory and malignant disorders remains incompletely understood. Further understanding of the role of CXCL8–CXCL1/2 signaling in such events may help to increase our understanding of the pathophysiology of various disease states and, potentially, contribute to the further development of novel therapeutics.
Acknowledgments
James A Cotton is funded by Alberta Innovates: Health Solutions MD/PhD studentship. Jaye M Platnich is funded by Beverly Philips Rising Star Award from the Snyder Institute for Chronic Diseases. Daniel A Muruve and Paul L Beck are funded by Alberta Innovates: Health Solutions and Canadian Institutes of Health Research. Andre G Buret is funded by research grants from the Natural Sciences and Engineering Research Council of Canada, and Crohn’s and Colitis Foundation of Canada.
Disclosure
The authors report no conflicts of interest in this work.
References
O’Connor PM, Lapointe TK, Beck PL, Buret AG. Mechanisms by which inflammation may increase intestinal cancer risk in inflammatory bowel disease. Inflamm Bowel Dis. 2010;16(8):1411–1420. | |
Beaugerie L, Itzkowitz SH. Cancers complicating inflammatory bowel disease. N Engl J Med. 2015;372(15):1441–1452. | |
Kappelman MD, Rifas-Shiman SL, Porter CQ, et al. Direct health care costs of Crohn’s disease and ulcerative colitis in US children and adults. Gastroenterology. 2008;135(6):1907–1913. | |
Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142(1):46–54. e42. | |
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. | |
Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. | |
Peyrin-Biroulet L, Deltenre P, de Suray N, Branche J, Sandborn WJ, Colombel JF. Efficacy and safety of tumor necrosis factor antagonists in Crohn’s disease: meta-analysis of placebo-controlled trials. Clin Gastroenterol Hepatol. 2008;6(6):644–653. | |
Lurje G, Zhang W, Schultheis AM, et al. Polymorphisms in VEGF and IL-8 predict tumor recurrence in stage III colon cancer. Ann Oncol. 2008;19(10):1734–1741. | |
Yoshimura T, Matsushima K, Tanaka S, et al. Purification of a human monocyte-derived neutrophil chemotactic factor that has peptide sequence similarity to other host defense cytokines. Proc Natl Acad Sci U S A. 1987;84(24):9233–9237. | |
Schroder JM, Mrowietz U, Morita E, Christophers E. Purification and partial biochemical characterization of a human monocyte-derived, neutrophil-activating peptide that lacks interleukin 1 activity. J Immunol. 1987;139(10):3474–3483. | |
Walz A, Peveri P, Aschauer H, Baggiolini M. Purification and amino acid sequencing of NAF, a novel neutrophil-activating factor produced by monocytes. Biochem Biophys Res Commun. 1987;149(2):755–761. | |
Baggiolini M, Clark-Lewis I. Interleukin-8, a chemotactic and inflammatory cytokine. FEBS Lett. 1992;307(1):97–101. | |
Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12(2):121–127. | |
Rajarathnam K, Sykes BD, Dewald B, Baggiolini M, Clark-Lewis I. Disulfide bridges in interleukin-8 probed using non-natural disulfide analogues: dissociation of roles in structure from function. Biochemistry. 1999;38(24):7653–7658. | |
Oppenheim JJ, Zachariae CO, Mukaida N, Matsushima K. Properties of the novel proinflammatory supergene “intercrine” cytokine family. Annu Rev Immunol. 1991;9:617–648. | |
Hebert CA, Vitangcol RV, Baker JB. Scanning mutagenesis of interleukin-8 identifies a cluster of residues required for receptor binding. J Biol Chem. 1991;266(28):18989–18994. | |
Clark-Lewis I, Dewald B, Geiser T, Moser B, Baggiolini M. Platelet factor 4 binds to interleukin 8 receptors and activates neutrophils when its N terminus is modified with Glu-Leu-Arg. Proc Natl Acad Sci U S A. 1993;90(8):3574–3577. | |
Strieter RM, Polverini PJ, Arenberg DA, Kunkel SL. The role of CXC chemokines as regulators of angiogenesis. Shock. 1995;4(3):155–160. | |
Strieter RM, Polverini PJ, Kunkel SL, et al. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem. 1995;270(45):27348–27357. | |
Moser B, Dewald B, Barella L, Schumacher C, Baggiolini M, Clark-Lewis I. Interleukin-8 antagonists generated by N-terminal modification. J Biol Chem. 1993;268(10):7125–7128. | |
Modi WS, Dean M, Seuanez HN, Mukaida N, Matsushima K, O’Brien SJ. Monocyte-derived neutrophil chemotactic factor (MDNCF/IL-8) resides in a gene cluster along with several other members of the platelet factor 4 gene superfamily. Hum Genet. 1990;84(2):185–187. | |
Wolpe SD, Cerami A. Macrophage inflammatory proteins 1 and 2: members of a novel superfamily of cytokines. FASEB J. 1989;3(14):2565–2573. | |
Mukaida N, Shiroo M, Matsushima K. Genomic structure of the human monocyte-derived neutrophil chemotactic factor IL-8. J Immunol. 1989;143(4):1366–1371. | |
Murphy PM, Tiffany HL. Cloning of complementary DNA encoding a functional human interleukin-8 receptor. Science. 1991;253(5025):1280–1283. | |
Holmes WE, Lee J, Kuang WJ, Rice GC, Wood WI. Structure and functional expression of a human interleukin-8 receptor. Science. 1991;253(5025):1278–1280. | |
Sturm A, Baumgart DC, d’Heureuse JH, Hotz A, Wiedenmann B, Dignass AU. CXCL8 modulates human intestinal epithelial cells through a CXCR1 dependent pathway. Cytokine. 2005;29(1):42–48. | |
Schonbeck U, Brandt E, Petersen F, Flad HD, Loppnow H. IL-8 specifically binds to endothelial but not to smooth muscle cells. J Immunol. 1995;154(5):2375–2383. | |
Middleton J, Patterson AM, Gardner L, Schmutz C, Ashton BA. Leukocyte extravasation: chemokine transport and presentation by the endothelium. Blood. 2002;100(12):3853–3860. | |
Wuyts A, Van Osselaer N, Haelens A, et al. Characterization of synthetic human granulocyte chemotactic protein 2: usage of chemokine receptors CXCR1 and CXCR2 and in vivo inflammatory properties. Biochemistry. 1997;36(9):2716–2723. | |
Stillie R, Farooq SM, Gordon JR, Stadnyk AW. The functional significance behind expressing two IL-8 receptor types on PMN. J Leukoc Biol. 2009;86(3):529–543. | |
Jobin C, Panja A, Hellerbrand C, et al. Inhibition of proinflammatory molecule production by adenovirus-mediated expression of a nuclear factor kappaB super-repressor in human intestinal epithelial cells. J Immunol. 1998;160(1):410–418. | |
Mukaida N, Okamoto S, Ishikawa Y, Matsushima K. Molecular mechanism of interleukin-8 gene expression. J Leukoc Biol. 1994;56(5):554–558. | |
Yasumoto K, Okamoto S, Mukaida N, Murakami S, Mai M, Matsushima K. Tumor necrosis factor alpha and interferon gamma synergistically induce interleukin 8 production in a human gastric cancer cell line through acting concurrently on AP-1 and NF-kB-like binding sites of the interleukin 8 gene. J Biol Chem. 1992;267(31):22506–22511. | |
Stein B, Baldwin AS Jr. Distinct mechanisms for regulation of the interleukin-8 gene involve synergism and cooperativity between C/EBP and NF-kappa B. Mol Cell Biol. 1993;13(11):7191–7198. | |
Matsusaka T, Fujikawa K, Nishio Y, et al. Transcription factors NF-IL6 and NF-kappa B synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. Proc Natl Acad Sci U S A. 1993;90(21):10193–10197. | |
Mukaida N, Mahe Y, Matsushima K. Cooperative interaction of nuclear factor-kappa B- and cis-regulatory enhancer binding protein-like factor binding elements in activating the interleukin-8 gene by pro-inflammatory cytokines. J Biol Chem. 1990;265(34):21128–21133. | |
Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1(4):a000034. | |
Hayden MS, Ghosh S. NF-kappaB in immunobiology. Cell Res. 2011; 21(2):223–244. | |
Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol. 2010;11(5):373–384. | |
Girardin SE, Boneca IG, Carneiro LA, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science. 2003;300(5625):1584–1587. | |
Flannery S, Bowie AG. The interleukin-1 receptor-associated kinases: critical regulators of innate immune signalling. Biochem Pharmacol. 2010;80(12):1981–1991. | |
Vavricka SR, Musch MW, Chang JE, et al. hPepT1 transports muramyl dipeptide, activating NF-kappaB and stimulating IL-8 secretion in human colonic Caco2/bbe cells. Gastroenterology. 2004;127(5):1401–1409. | |
Jobin C, Holt L, Bradham CA, Streetz K, Brenner DA, Sartor RB. TNF receptor-associated factor-2 is involved in both IL-1 beta and TNF-alpha signaling cascades leading to NF-kappa B activation and IL-8 expression in human intestinal epithelial cells. J Immunol. 1999;162(8):4447–4454. | |
Labbé C, Boucher G, Foisy S, et al. Genome-wide expression profiling implicates a MAST3-regulated gene set in colonic mucosal inflammation of ulcerative colitis patients. Inflamm Bowel Dis. 2012;18(6):1072–1080. | |
Xie K. Interleukin-8 and human cancer biology. Cytokine Growth Factor Rev. 2001;12(4):375–391. | |
Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. 2010;2(3):a000158. | |
Delhase M, Hayakawa M, Chen Y, Karin M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science. 1999;284(5412):309–313. | |
Mercurio F, Zhu H, Murray BW, et al. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science. 1997;278(5339):860–866. | |
Kunsch C, Rosen CA. NF-kappa B subunit-specific regulation of the interleukin-8 promoter. Mol Cell Biol. 1993;13(10):6137–6146. | |
Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. | |
Maeda S, Akanuma M, Mitsuno Y, et al. Distinct mechanism of Helicobacter pylori-mediated NF-kappa B activation between gastric cancer cells and monocytic cells. J Biol Chem. 2001;276(48):44856–44864. | |
O’Connor PM, Lapointe TK, Jackson S, Beck PL, Jones NL, Buret AG. Helicobacter pylori activates calpain via toll-like receptor 2 to disrupt adherens junctions in human gastric epithelial cells. Infect Immun. 2011;79(10):3887–3894. | |
Eckmann L, Kagnoff MF, Fierer J. Epithelial cells secrete the chemokine interleukin-8 in response to bacterial entry. Infect Immun. 1993;61(11):4569–4574. | |
Zheng J, Meng J, Zhao S, Singh R, Song W. Campylobacter-induced interleukin-8 secretion in polarized human intestinal epithelial cells requires Campylobacter-secreted cytolethal distending toxin- and toll-like receptor-mediated activation of NF-kappaB. Infect Immun. 2008; 76(10):4498–4508. | |
Hansen A, Alston L, Tulk SE, et al. The P2Y6 receptor mediates Clostridium difficile toxin-induced CXCL8/IL-8 production and intestinal epithelial barrier dysfunction. PLoS One. 2013;8(11):e81491. | |
Maeda S, Yoshida H, Ogura K, et al. H. pylori activates NF-kappaB through a signaling pathway involving IkappaB kinases, NF-kappaB-inducing kinase, TRAF2, and TRAF6 in gastric cancer cells. Gastroenterology. 2000;119(1):97–108. | |
Sharma SA, Tummuru MK, Blaser MJ, Kerr LD. Activation of IL-8 gene expression by Helicobacter pylori is regulated by transcription factor nuclear factor-kappa B in gastric epithelial cells. J Immunol. 1998;160(5):2401–2407. | |
Dahan S, Busuttil V, Imbert V, Peyron JF, Rampal P, Czerucka D. Enterohemorrhagic Escherichia coli infection induces interleukin-8 production via activation of mitogen-activated protein kinases and the transcription factors NF-kappaB and AP-1 in T84 cells. Infect Immun. 2002;70(5):2304–2310. | |
Chandrakesan P, Ahmed I, Chinthalapally A, et al. Distinct compartmentalization of NF-kappaB activity in crypt and crypt-denuded lamina propria precedes and accompanies hyperplasia and/or colitis following bacterial infection. Infect Immun. 2012;80(2):753–767. | |
Neurath MF, Pettersson S, Meyer zum Buschenfelde KH, Strober W. Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-kappa B abrogates established experimental colitis in mice. Nat Med. 1996;2(9):998–1004. | |
Crabtree JE, Wyatt JI, Trejdosiewicz LK, et al. Interleukin-8 expression in Helicobacter pylori infected, normal, and neoplastic gastroduodenal mucosa. J Clin Pathol. 1994;47(1):61–66. | |
Rieder G, Einsiedl W, Hatz RA, Stolte M, Enders GA, Walz A. Comparison of CXC chemokines ENA-78 and interleukin-8 expression in Helicobacter pylori-associated gastritis. Infect Immun. 2001;69(1):81–88. | |
Shimoyama T, Everett SM, Dixon MF, Axon AT, Crabtree JE. Chemokine mRNA expression in gastric mucosa is associated with Helicobacter pylori cagA positivity and severity of gastritis. J Clin Pathol. 1998;51(10):765–770. | |
Ando T, Kusugami K, Ohsuga M, et al. Interleukin-8 activity correlates with histological severity in Helicobacter pylori-associated antral gastritis. Am J Gastroenterol. 1996;91(6):1150–1156. | |
Uemura N, Oomoto Y, Mukai T, et al. Gastric corpus IL-8 concentration and neutrophil infiltration in duodenal ulcer patients. Aliment Pharmacol Ther. 1997;11(4):793–800. | |
Yamaoka Y, Kodama T, Kita M, Imanishi J, Kashima K, Graham DY. Relation between clinical presentation, Helicobacter pylori density, interleukin 1beta and 8 production, and cagA status. Gut. 1999;45(6):804–811. | |
Rogler G, Brand K, Vogl D, et al. Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology. 1998;115(2):357–369. | |
Banks C, Bateman A, Payne R, Johnson P, Sheron N. Chemokine expression in IBD. Mucosal chemokine expression is unselectively increased in both ulcerative colitis and Crohn’s disease. J Pathol. 2003; 199(1):28–35. | |
McCormack G, Moriarty D, O’Donoghue DP, McCormick PA, Sheahan K, Baird AW. Tissue cytokine and chemokine expression in inflammatory bowel disease. Inflamm Res. 2001;50(10):491–495. | |
Nielsen OH, Rudiger N, Gaustadnes M, Horn T. Intestinal interleukin-8 concentration and gene expression in inflammatory bowel disease. Scand J Gastroenterol. 1997;32(10):1028–1034. | |
Wang W, Luo HS, Yu BP. Expression of NF-kappaB and human telomerase reverse transcriptase in gastric cancer and precancerous lesions. World J Gastroenterol. 2004;10(2):177–181. | |
Maihöfner C, Charalambous MP, Bhambra U, et al; Colorectal Cancer Group. Expression of cyclooxygenase-2 parallels expression of interleukin-1beta, interleukin-6 and NF-kappaB in human colorectal cancer. Carcinogenesis. 2003;24(4):665–671. | |
Balcerczak M, Balcerczak E, Pasz-Walczak G, Kordek R, Mirowski M. Expression of the p65 gene in patients with colorectal cancer: comparison with some histological typing, grading and clinical staging. Eur J Surg Oncol. 2004;30(3):266–270. | |
Neish AS. Microbes in gastrointestinal health and disease. Gastroenterology. 2009;136(1):65–80. | |
Gill SR, Pop M, Deboy RT, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312(5778):1355–1359. | |
Xu J, Mahowald MA, Ley RE, et al. Evolution of symbiotic bacteria in the distal human intestine. PLoS Biol. 2007;5(7):e156. | |
Littman DR, Pamer EG. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe. 2011;10(4):311–323. | |
Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. 2013;13(5):321–335. | |
Abreu MT, Vora P, Faure E, Thomas LS, Arnold ET, Arditi M. Decreased expression of toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J Immunol. 2001;167(3):1609–1616. | |
Melmed G, Thomas LS, Lee N, et al. Human intestinal epithelial cells are broadly unresponsive to toll-like receptor 2-dependent bacterial ligands: implications for host-microbial interactions in the gut. J Immunol. 2003;170(3):1406–1415. | |
Abreu MT, Arnold ET, Thomas LS, et al. TLR4 and MD-2 expression is regulated by immune-mediated signals in human intestinal epithelial cells. J Biol Chem. 2002;277(23):20431–20437. | |
Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. 2000;68(12):7010–7017. | |
Szebeni B, Veres G, Dezsõfi A, et al. Increased expression of toll-like receptor (TLR) 2 and TLR4 in the colonic mucosa of children with inflammatory bowel disease. Clin Exp Immunol. 2008;151(1):34–41. | |
Frolova L, Drastich P, Rossmann P, Klimesova K, Tlaskalova-Hogenova H. Expression of toll-like receptor 2 (TLR2), TLR4, and CD14 in biopsy samples of patients with inflammatory bowel diseases: upregulated expression of TLR2 in terminal ileum of patients with ulcerative colitis. J Histochem Cytochem. 2008;56(3):267–274. | |
Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol. 2001; 167(4):1882–1885. | |
Reed KA, Hobert ME, Kolenda CE, et al. The Salmonella typhimurium flagellar basal body protein FliE is required for flagellin production and to induce a proinflammatory response in epithelial cells. J Biol Chem. 2002;277(15):13346–13353. | |
Lee J, Mo JH, Katakura K, et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat Cell Biol. 2006;8(12):1327–1336. | |
O’Hara JR, Feener TD, Fischer CD, Buret AG. Campylobacter jejuni disrupts protective toll-like receptor 9 signaling in colonic epithelial cells and increases the severity of dextran sulfate sodium-induced colitis in mice. Infect Immun. 2012;80(4):1563–1571. | |
Zhang G, Ghosh S. Negative regulation of toll-like receptor-mediated signaling by Tollip. J Biol Chem. 2002;277(9):7059–7065. | |
Khan MA, Steiner TS, Sham HP, et al. The single IgG IL-1-related receptor controls TLR responses in differentiated human intestinal epithelial cells. J Immunol. 2010;184(5):2305–2313. | |
Otte JM, Cario E, Podolsky DK. Mechanisms of cross hyporesponsiveness to toll-like receptor bacterial ligands in intestinal epithelial cells. Gastroenterology. 2004;126(4):1054–1070. | |
Kadota C, Ishihara S, Aziz MM, et al. Down-regulation of single immunoglobulin interleukin-1R-related molecule (SIGIRR)/TIR8 expression in intestinal epithelial cells during inflammation. Clin Exp Immunol. 2010;162(2):348–361. | |
Nanthakumar N, Meng D, Goldstein AM, et al. The mechanism of excessive intestinal inflammation in necrotizing enterocolitis: an immature innate immune response. PLoS One. 2011;6(3):e17776. | |
Pimentel-Nunes P, Gonçalves N, Boal-Carvalho I, et al. Helicobacter pylori induces increased expression of toll-like receptors and decreased toll-interacting protein in gastric mucosa that persists throughout gastric carcinogenesis. Helicobacter. 2013;18(1):22–32. | |
Pimentel-Nunes P, Gonçalves N, Boal-Carvalho I, et al. Decreased toll-interacting protein and peroxisome proliferator-activated receptor gamma are associated with increased expression of toll-like receptors in colon carcinogenesis. J Clin Pathol. 2012;65(4):302–308. | |
Eftang LL, Esbensen Y, Tannaes TM, Bukholm IR, Bukholm G. Interleukin-8 is the single most up-regulated gene in whole genome profiling of H. pylori exposed gastric epithelial cells. BMC Microbiol. 2012;12:9. | |
Xiao H, Gulen MF, Qin J, et al. The toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity. 2007;26(4):461–475. | |
Maillard MH, Bega H, Uhlig HH, et al. Toll-interacting protein modulates colitis susceptibility in mice. Inflamm Bowel Dis. 2014;20(4):660–670. | |
Romero-Valdovinos M, Gudiño-Ramírez A, Reyes-Gordillo J, et al. Interleukin-8 and -10 gene polymorphisms in irritable bowel syndrome. Mol Biol Rep. 2012;39(9):8837–8843. | |
Ye BD, Kim SG, Park JH, Kim JS, Jung HC, Song IS. The interleukin-8-251 A allele is associated with increased risk of noncardia gastric adenocarcinoma in Helicobacter pylori-infected Koreans. J Clin Gastroenterol. 2009;43(3):233–239. | |
Vinagre RM, Corvelo TC, Arnaud VC, Leite AC, Barile KA, Martins LC. Determination of strains of Helicobacter pylori and of polymorphism in the interleukin-8 gene in patients with stomach cancer. Arq Gastroenterol. 2011;48(1):46–51. | |
Gyulai Z, Klausz G, Tiszai A, et al. Genetic polymorphism of interleukin-8 (IL-8) is associated with Helicobacter pylori-induced duodenal ulcer. Eur Cytokine Netw. 2004;15(4):353–358. | |
Yin YW, Hu AM, Sun QQ, et al. Association between interleukin-8 gene -251 T/A polymorphism and the risk of peptic ulcer disease: a meta-analysis. Hum Immunol. 2013;74(1):125–130. | |
Caleman Neto A, Rasmussen LT, de Labio RW, et al. Gene polymorphism of interleukin 1 and 8 in chronic gastritis patients infected with Helicobacter pylori. J Venom Anim Toxins Incl Trop Dis. 2014;20:17. | |
Taguchi A, Ohmiya N, Shirai K, et al. Interleukin-8 promoter polymorphism increases the risk of atrophic gastritis and gastric cancer in Japan. Cancer Epidemiol Biomarkers Prev. 2005;14(11 pt 1):2487–2493. | |
Jiang ZD, Okhuysen PC, Guo DC, et al. Genetic susceptibility to enteroaggregative Escherichia coli diarrhea: polymorphism in the interleukin-8 promotor region. J Infect Dis. 2003;188(4):506–511. | |
Jiang ZD, DuPont HL, Garey K, et al. A common polymorphism in the interleukin 8 gene promoter is associated with Clostridium difficile diarrhea. Am J Gastroenterol. 2006;101(5):1112–1116. | |
Jiang ZD, Garey KW, Price M, et al. Association of interleukin-8 polymorphism and immunoglobulin G anti-toxin A in patients with Clostridium difficile-associated diarrhea. Clin Gastroenterol Hepatol. 2007;5(8):964–968. | |
Garey KW, Jiang ZD, Ghantoji S, Tam VH, Arora V, Dupont HL. A common polymorphism in the interleukin-8 gene promoter is associated with an increased risk for recurrent Clostridium difficile infection. Clin Infect Dis. 2010;51(12):1406–1410. | |
Wang J, Pan HF, Hu YT, Zhu Y, He Q. Polymorphism of IL-8 in 251 allele and gastric cancer susceptibility: a meta-analysis. Dig Dis Sci. 2010;55(7):1818–1823. | |
Xue H, Liu J, Lin B, Wang Z, Sun J, Huang G. A meta-analysis of interleukin-8 -251 promoter polymorphism associated with gastric cancer risk. PLoS One. 2012;7(1):e28083. | |
Song JH, Kim SG, Jung SA, Lee MK, Jung HC, Song IS. The interleukin-8-251 AA genotype is associated with angiogenesis in gastric carcinogenesis in Helicobacter pylori-infected Koreans. Cytokine. 2010;51(2):158–165. | |
Ohyauchi M, Imatani A, Yonechi M, et al. The polymorphism interleukin 8 -251 A/T influences the susceptibility of Helicobacter pylori related gastric diseases in the Japanese population. Gut. 2005; 54(3):330–335. | |
Walczak A, Przybylowska K, Dziki L, et al. The lL-8 and IL-13 gene polymorphisms in inflammatory bowel disease and colorectal cancer. DNA Cell Biol. 2012;31(8):1431–1438. | |
Qadri Q, Rasool R, Afroze D, et al. Study of TLR4 and IL-8 gene polymorphisms in H. pylori-induced inflammation in gastric cancer in an ethnic Kashmiri population. Immunol Invest. 2014;43(4):324–336. | |
He Y, Liang X, Wu X, et al. Association between interleukin 8 -251 A/T and +781 C/T polymorphisms and osteoarthritis risk. Immunol Lett. 2014;162(1 pt A):207–211. | |
Koensgen D, Bruennert D, Ungureanu S, et al. Polymorphism of the IL-8 gene and the risk of ovarian cancer. Cytokine. 2015;71(2):334–338. | |
Rovin BH, Lu L, Zhang X. A novel interleukin-8 polymorphism is associated with severe systemic lupus erythematosus nephritis. Kidney Int. 2002;62(1):261–265. | |
Hull J, Thomson A, Kwiatkowski D. Association of respiratory syncytial virus bronchiolitis with the interleukin 8 gene region in UK families. Thorax. 2000;55(12):1023–1027. | |
Gimbrone MA Jr, Obin MS, Brock AF, et al. Endothelial interleukin-8: a novel inhibitor of leukocyte-endothelial interactions. Science. 1989;246(4937):1601–1603. | |
Schroder JM, Sticherling M, Henneicke HH, Preissner WC, Christophers E. IL-1 alpha or tumor necrosis factor-alpha stimulate release of three NAP-1/IL-8-related neutrophil chemotactic proteins in human dermal fibroblasts. J Immunol. 1990;144(6):2223–2232. | |
Piccardoni P, Evangelista V, Piccoli A, de Gaetano G, Walz A, Cerletti C. Thrombin-activated human platelets release two NAP-2 variants that stimulate polymorphonuclear leukocytes. Thromb Haemost. 1996;76(5):780–785. | |
Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood. 2000;96(8):2673–2681. | |
Padrines M, Wolf M, Walz A, Baggiolini M. Interleukin-8 processing by neutrophil elastase, cathepsin G and proteinase-3. FEBS Lett. 1994;352(2):231–235. | |
Mortier A, Berghmans N, Ronsse I, et al. Biological activity of CXCL8 forms generated by alternative cleavage of the signal peptide or by aminopeptidase-mediated truncation. PLoS One. 2011;6(8):e23913. | |
Loos T, Opdenakker G, Van Damme J, Proost P. Citrullination of CXCL8 increases this chemokine’s ability to mobilize neutrophils into the blood circulation. Haematologica. 2009;94(10):1346–1353. | |
Castaneda FE, Walia B, Vijay-Kumar M, et al. Targeted deletion of metalloproteinase 9 attenuates experimental colitis in mice: central role of epithelial-derived MMP. Gastroenterology. 2005;129(6):1991–2008. | |
Farkas K, Saródi Z, Bálint A, et al. The diagnostic value of a new fecal marker, matrix metalloprotease-9, in different types of inflammatory bowel diseases. J Crohns Colitis. 2015;9(3):231–237. | |
Kofla-Dlubacz A, Matusiewicz M, Krzesiek E, Noga L, Iwanczak B. Metalloproteinase-3 and -9 as novel markers in the evaluation of ulcerative colitis activity in children. Adv Clin Exp Med. 2014;23(1):103–110. | |
Makitalo L, Rintamaki H, Tervahartiala T, Sorsa T, Kolho KL. Serum MMPs 7-9 and their inhibitors during glucocorticoid and anti-TNF-alpha therapy in pediatric inflammatory bowel disease. Scand J Gastroenterol. 2012;47(7):785–794. | |
Kolho KL, Sipponen T, Valtonen E, Savilahti E. Fecal calprotectin, MMP-9, and human beta-defensin-2 levels in pediatric inflammatory bowel disease. Int J Colorectal Dis. 2014;29(1):43–50. | |
Swallow CJ, Murray MP, Guillem JG. Metastatic colorectal cancer cells induce matrix metalloproteinase release by human monocytes. Clin Exp Metastasis. 1996;14(1):3–11. | |
Zeng ZS, Huang Y, Cohen AM, Guillem JG. Prediction of colorectal cancer relapse and survival via tissue RNA levels of matrix metalloproteinase-9. J Clin Oncol. 1996;14(12):3133–3140. | |
Islekel H, Oktay G, Terzi C, Canda AE, Fuzun M, Kupelioglu A. Matrix metalloproteinase-9, -3 and tissue inhibitor of matrix metalloproteinase-1 in colorectal cancer: relationship to clinicopathological variables. Cell Biochem Funct. 2007;25(4):433–441. | |
Bendardaf R, Buhmeida A, Hilska M, et al. MMP-9 (gelatinase B) expression is associated with disease-free survival and disease-specific survival in colorectal cancer patients. Cancer Invest. 2010;28(1):38–43. | |
Chen SZ, Yao HQ, Zhu SZ, Li QY, Guo GH, Yu J. Expression levels of matrix metalloproteinase-9 in human gastric carcinoma. Oncol Lett. 2015;9(2):915–919. | |
Bailey CJ, Hembry RM, Alexander A, Irving MH, Grant ME, Shuttleworth CA. Distribution of the matrix metalloproteinases stromelysin, gelatinases A and B, and collagenase in Crohn’s disease and normal intestine. J Clin Pathol. 1994;47(2):113–116. | |
Tarlton JF, Whiting CV, Tunmore D, et al. The role of up-regulated serine proteases and matrix metalloproteinases in the pathogenesis of a murine model of colitis. Am J Pathol. 2000;157(6):1927–1935. | |
Mori N, Sato H, Hayashibara T, et al. Helicobacter pylori induces matrix metalloproteinase-9 through activation of nuclear factor kappaB. Gastroenterology. 2003;124(4):983–992. | |
Kitadai Y, Sasaki A, Ito M, et al. Helicobacter pylori infection influences expression of genes related to angiogenesis and invasion in human gastric carcinoma cells. Biochem Biophys Res Commun. 2003; 311(4):809–814. | |
Bergin PJ, Anders E, Sicheng W, et al. Increased production of matrix metalloproteinases in Helicobacter pylori-associated human gastritis. Helicobacter. 2004;9(3):201–210. | |
Kubben FJ, Sier CF, Schram MT, et al. Eradication of Helicobacter pylori infection favourably affects altered gastric mucosal MMP-9 levels. Helicobacter. 2007;12(5):498–504. | |
Rodrigues DM, Sousa AJ, Hawley SP, et al. Matrix metalloproteinase 9 contributes to gut microbe homeostasis in a model of infectious colitis. BMC Microbiol. 2012;12:105. | |
Shang K, Bai YP, Wang C, et al. Crucial involvement of tumor-associated neutrophils in the regulation of chronic colitis-associated carcinogenesis in mice. PLoS One. 2012;7(12):e51848. | |
Biasi F, Guina T, Maina M, et al. Progressive increase of matrix metalloprotease-9 and interleukin-8 serum levels during carcinogenic process in human colorectal tract. PLoS One. 2012;7(7):e41839. | |
Li Z, Wang Y, Dong S, et al. Association of CXCR1 and 2 expressions with gastric cancer metastasis in ex vivo and tumor cell invasion in vitro. Cytokine. 2014;69(1):6–13. | |
Munakata S, Tashiro Y, Nishida C, et al. Inhibition of plasmin protects against colitis in mice by suppressing matrix metalloproteinase 9-mediated cytokine release from myeloid cells. Gastroenterology. 2015;148(3):565–578. e564. | |
Liu H, Patel NR, Walter L, Ingersoll S, Sitaraman SV, Garg P. Constitutive expression of MMP9 in intestinal epithelium worsens murine acute colitis and is associated with increased levels of proinflammatory cytokine Kc. Am J Physiol Gastrointest Liver Physiol. 2013;304(9):G793–G803. | |
Garg P, Vijay-Kumar M, Wang L, Gewirtz AT, Merlin D, Sitaraman SV. Matrix metalloproteinase-9-mediated tissue injury overrides the protective effect of matrix metalloproteinase-2 during colitis. Am J Physiol Gastrointest Liver Physiol. 2009;296(2):G175–G184. | |
Semerad CL, Liu F, Gregory AD, Stumpf K, Link DC. G-CSF is an essential regulator of neutrophil trafficking from the bone marrow to the blood. Immunity. 2002;17(4):413–423. | |
Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. 2010;120(7):2423–2431. | |
Suratt BT, Petty JM, Young SK, et al. Role of the CXCR4/SDF-1 chemokine axis in circulating neutrophil homeostasis. Blood. 2004; 104(2):565–571. | |
Petty JM, Lenox CC, Weiss DJ, Poynter ME, Suratt BT. Crosstalk between CXCR4/stromal derived factor-1 and VLA-4/VCAM-1 pathways regulates neutrophil retention in the bone marrow. J Immunol. 2009;182(1):604–612. | |
Wengner AM, Pitchford SC, Furze RC, Rankin SM. The coordinated action of G-CSF and ELR + CXC chemokines in neutrophil mobilization during acute inflammation. Blood. 2008;111(1):42–49. | |
Laterveer L, Lindley IJ, Hamilton MS, Willemze R, Fibbe WE. Interleukin-8 induces rapid mobilization of hematopoietic stem cells with radioprotective capacity and long-term myelolymphoid repopulating ability. Blood. 1995;85(8):2269–2275. | |
Laterveer L, Lindley IJ, Heemskerk DP, et al. Rapid mobilization of hematopoietic progenitor cells in rhesus monkeys by a single intravenous injection of interleukin-8. Blood. 1996;87(2):781–788. | |
Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–175. | |
Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. 2011;17(11):1381–1390. | |
Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7(9):678–689. | |
Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11(8):519–531. | |
Massena S, Christoffersson G, Hjertström E, et al. A chemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood. 2010;116(11):1924–1931. | |
Rot A. Endothelial cell binding of NAP-1/IL-8: role in neutrophil emigration. Immunol Today. 1992;13(8):291–294. | |
Tanaka Y, Adams DH, Shaw S. Proteoglycans on endothelial cells present adhesion-inducing cytokines to leukocytes. Immunol Today. 1993;14(3):111–115. | |
Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med. 2006;203(12):2569–2575. | |
Jones DH, Anderson DC, Burr BL, et al. Quantitation of intracellular Mac-1 (CD11b/CD18) pools in human neutrophils. J Leukoc Biol. 1988;44(6):535–544. | |
Burns AR, Bowden RA, MacDonell SD, et al. Analysis of tight junctions during neutrophil transendothelial migration. J Cell Sci. 2000;113(pt 1):45–57. | |
Woodfin A, Voisin MB, Beyrau M, et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol. 2011;12(8):761–769. | |
Foxman EF, Campbell JJ, Butcher EC. Multistep navigation and the combinatorial control of leukocyte chemotaxis. J Cell Biol. 1997; 139(5):1349–1360. | |
Heit B, Tavener S, Raharjo E, Kubes P. An intracellular signaling hierarchy determines direction of migration in opposing chemotactic gradients. J Cell Biol. 2002;159(1):91–102. | |
McCormick BA, Colgan SP, Delp-Archer C, Miller SI, Madara JL. Salmonella typhimurium attachment to human intestinal epithelial monolayers: transcellular signalling to subepithelial neutrophils. J Cell Biol. 1993;123(4):895–907. | |
McCormick BA, Hofman PM, Kim J, Carnes DK, Miller SI, Madara JL. Surface attachment of Salmonella typhimurium to intestinal epithelia imprints the subepithelial matrix with gradients chemotactic for neutrophils. J Cell Biol. 1995;131(6 pt 1):1599–1608. | |
Mrsny RJ, Gewirtz AT, Siccardi D, et al. Identification of hepoxilin A3 in inflammatory events: a required role in neutrophil migration across intestinal epithelia. Proc Natl Acad Sci U S A. 2004;101(19):7421–7426. | |
Winkelstein JA, Marino MC, Johnston RB Jr, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine. 2000;79(3):155–169. | |
Segal AW, Harper AM, Garcia RC, Merzbach D. The action of cells from patients with chronic granulomatous disease on Staphylococcus aureus. J Med Microbiol. 1982;15(4):441–449. | |
Ellson CD, Davidson K, Ferguson GJ, O’Connor R, Stephens LR, Hawkins PT. Neutrophils from p40phox-/- mice exhibit severe defects in NADPH oxidase regulation and oxidant-dependent bacterial killing. J Exp Med. 2006;203(8):1927–1937. | |
Jackson SH, Gallin JI, Holland SM. The p47phox mouse knock-out model of chronic granulomatous disease. J Exp Med. 1995;182(3):751–758. | |
Morgenstern DE, Gifford MA, Li LL, Doerschuk CM, Dinauer MC. Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. J Exp Med. 1997;185(2):207–218. | |
Roberts AW, Kim C, Zhen L, et al. Deficiency of the hematopoietic cell-specific Rho family GTPase Rac2 is characterized by abnormalities in neutrophil function and host defense. Immunity. 1999;10(2):183–196. | |
Oshitani N, Kitano A, Okabe H, Nakamura S, Matsumoto T, Kobayashi K. Location of superoxide anion generation in human colonic mucosa obtained by biopsy. Gut. 1993;34(7):936–938. | |
Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. 2012;30:459–489. | |
El-Benna J, Dang PM, Gougerot-Pocidalo MA. Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin Immunopathol. 2008;30(3):279–289. | |
Groemping Y, Rittinger K. Activation and assembly of the NADPH oxidase: a structural perspective. Biochem J. 2005;386(pt 3):401–416. | |
Leto TL, Geiszt M. Role of Nox family NADPH oxidases in host defense. Antioxid Redox Signal. 2006;8(9–10):1549–1561. | |
Segal AW. How neutrophils kill microbes. Annu Rev Immunol. 2005;23:197–223. | |
Kucharzik T, Hudson JT 3rd, Lügering A, et al. Acute induction of human IL-8 production by intestinal epithelium triggers neutrophil infiltration without mucosal injury. Gut. 2005;54(11):1565–1572. | |
Daniels RH, Finnen MJ, Hill ME, Lackie JM. Recombinant human monocyte IL-8 primes NADPH-oxidase and phospholipase A2 activation in human neutrophils. Immunology. 1992;75(1):157–163. | |
Wozniak A, Betts WH, Murphy GA, Rokicinski M. Interleukin-8 primes human neutrophils for enhanced superoxide anion production. Immunology. 1993;79(4):608–615. | |
Jones SA, Wolf M, Qin S, Mackay CR, Baggiolini M. Different functions for the interleukin 8 receptors (IL-8R) of human neutrophil leukocytes: NADPH oxidase and phospholipase D are activated through IL-8R1 but not IL-8R2. Proc Natl Acad Sci U S A. 1996; 93(13):6682–6686. | |
Green SP, Chuntharapai A, Curnutte JT. Interleukin-8 (IL-8), melanoma growth-stimulatory activity, and neutrophil-activating peptide selectively mediate priming of the neutrophil NADPH oxidase through the type A or type B IL-8 receptor. J Biol Chem. 1996;271(41):25400–25405. | |
Nibbering PH, Pos O, Stevenhagen A, Van Furth R. Interleukin-8 enhances nonoxidative intracellular killing of Mycobacterium fortuitum by human granulocytes. Infect Immun. 1993;61(8):3111–3116. | |
Metzner B, Barbisch M, Parlow F, Kownatzki E, Schraufstatter I, Norgauer J. Interleukin-8 and GRO alpha prime human neutrophils for superoxide anion production and induce up-regulation of N-formyl peptide receptors. J Invest Dermatol. 1995;104(5):789–791. | |
Tithof PK, Peters-Golden M, Ganey PE. Distinct phospholipases A2 regulate the release of arachidonic acid for eicosanoid production and superoxide anion generation in neutrophils. J Immunol. 1998;160(2):953–960. | |
Gallois A, Bueb JL, Tschirhart E. Effect of SK&F 96365 on extracellular Ca2+ -dependent O2- production in neutrophil-like HL-60 cells. Eur J Pharmacol. 1998;361(2–3):293–298. | |
Valentin F, Bueb J, Capdeville-Atkinson C, Tschirhart E. Rac-1-mediated O2- secretion requires Ca2+ influx in neutrophil-like HL-60 cells. Cell Calcium. 2001;29(6):409–415. | |
Brechard S, Bueb JL, Tschirhart EJ. Interleukin-8 primes oxidative burst in neutrophil-like HL-60 through changes in cytosolic calcium. Cell Calcium. 2005;37(6):531–540. | |
Okamura N, Curnutte JT, Roberts RL, Babior BM. Relationship of protein phosphorylation to the activation of the respiratory burst in human neutrophils. Defects in the phosphorylation of a group of closely related 48-kDa proteins in two forms of chronic granulomatous disease. J Biol Chem. 1988;263(14):6777–6782. | |
Dang PM, Morel F, Gougerot-Pocidalo MA, El Benna J. Phosphorylation of the NADPH oxidase component p67(PHOX) by ERK2 and P38MAPK: selectivity of phosphorylated sites and existence of an intramolecular regulatory domain in the tetratricopeptide-rich region. Biochemistry. 2003;42(15):4520–4526. | |
Forbes LV, Moss SJ, Segal AW. Phosphorylation of p67phox in the neutrophil occurs in the cytosol and is independent of p47phox. FEBS Lett. 1999;449(2–3):225–229. | |
Guichard C, Pedruzzi E, Dewas C, et al. Interleukin-8-induced priming of neutrophil oxidative burst requires sequential recruitment of NADPH oxidase components into lipid rafts. J Biol Chem. 2005; 280(44):37021–37032. | |
Itzkowitz SH, Yio X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol. 2004;287(1):G7–G17. | |
Simmonds NJ, Allen RE, Stevens TR, Van Someren RN, Blake DR, Rampton DS. Chemiluminescence assay of mucosal reactive oxygen metabolites in inflammatory bowel disease. Gastroenterology. 1992; 103(1):186–196. | |
Davies GR, Simmonds NJ, Stevens TR, et al. Helicobacter pylori stimulates antral mucosal reactive oxygen metabolite production in vivo. Gut. 1994;35(2):179–185. | |
Suzuki H, Miura S, Imaeda H, et al. Enhanced levels of chemiluminescence and platelet activating factor in urease-positive gastric ulcers. Free Radic Biol Med. 1996;20(3):449–454. | |
McKenzie SJ, Baker MS, Buffinton GD, Doe WF. Evidence of oxidant-induced injury to epithelial cells during inflammatory bowel disease. J Clin Invest. 1996;98(1):136–141. | |
Baik SC, Youn HS, Chung MH, et al. Increased oxidative DNA damage in Helicobacter pylori-infected human gastric mucosa. Cancer Res. 1996;56(6):1279–1282. | |
Farinati F, Cardin R, Degan P, et al. Oxidative DNA damage accumulation in gastric carcinogenesis. Gut. 1998;42(3):351–356. | |
Jenks PJ, Jeremy AH, Robinson PA, Walker MM, Crabtree JE. Long-term infection with Helicobacter felis and inactivation of the tumour suppressor gene p53 cumulatively enhance the gastric mutation frequency in big blue transgenic mice. J Pathol. 2003;201(4):596–602. | |
Touati E, Michel V, Thiberge JM, Wuscher N, Huerre M, Labigne A. Chronic Helicobacter pylori infections induce gastric mutations in mice. Gastroenterology. 2003;124(5):1408–1419. | |
Chang CL, Marra G, Chauhan DP, et al. Oxidative stress inactivates the human DNA mismatch repair system. Am J Physiol Cell Physiol. 2002;283(1):C148–C154. | |
Hussain SP, Amstad P, Raja K, et al. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: a cancer-prone chronic inflammatory disease. Cancer Res. 2000;60(13):3333–3337. | |
Nusse O, Lindau M. The dynamics of exocytosis in human neutrophils. J Cell Biol. 1988;107(6 pt 1):2117–2123. | |
Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003;5(14):1317–1327. | |
Borregaard N. Neutrophils, from marrow to microbes. Immunity. 2010; 33(5):657–670. | |
Delclaux C, Delacourt C, D’Ortho MP, Boyer V, Lafuma C, Harf A. Role of gelatinase B and elastase in human polymorphonuclear neutrophil migration across basement membrane. Am J Respir Cell Mol Biol. 1996;14(3):288–295. | |
Singer II, Scott S, Kawka DW, Kazazis DM. Adhesomes: specific granules containing receptors for laminin, C3bi/fibrinogen, fibronectin, and vitronectin in human polymorphonuclear leukocytes and monocytes. J Cell Biol. 1989;109(6 pt 1):3169–3182. | |
Peveri P, Walz A, Dewald B, Baggiolini M. A novel neutrophil-activating factor produced by human mononuclear phagocytes. J Exp Med. 1988;167(5):1547–1559. | |
Thelen M, Peveri P, Kernen P, von Tscharner V, Walz A, Baggiolini M. Mechanism of neutrophil activation by NAF, a novel monocyte-derived peptide agonist. FASEB J. 1988;2(11):2702–2706. | |
Rainger GE, Rowley AF, Nash GB. Adhesion-dependent release of elastase from human neutrophils in a novel, flow-based model: specificity of different chemotactic agents. Blood. 1998;92(12):4819–4827. | |
Smith RJ, Sam LM, Leach KL, Justen JM. Postreceptor events associated with human neutrophil activation by interleukin-8. J Leukoc Biol. 1992;52(1):17–26. | |
Willems J, Joniau M, Cinque S, van Damme J. Human granulocyte chemotactic peptide (IL-8) as a specific neutrophil degranulator: comparison with other monokines. Immunology. 1989;67(4):540–542. | |
Chakrabarti S, Patel KD. Regulation of matrix metalloproteinase-9 release from IL-8-stimulated human neutrophils. J Leukoc Biol. 2005;78(1):279–288. | |
Chertov O, Michiel DF, Xu L, et al. Identification of defensin-1, defensin-2, and CAP37/azurocidin as T-cell chemoattractant proteins released from interleukin-8-stimulated neutrophils. J Biol Chem. 1996; 271(6):2935–2940. | |
Taub DD, Anver M, Oppenheim JJ, Longo DL, Murphy WJ. T lymphocyte recruitment by interleukin-8 (IL-8). IL-8-induced degranulation of neutrophils releases potent chemoattractants for human T lymphocytes both in vitro and in vivo. J Clin Invest. 1996;97(8):1931–1941. | |
Langhorst J, Elsenbruch S, Koelzer J, Rueffer A, Michalsen A, Dobos GJ. Noninvasive markers in the assessment of intestinal inflammation in inflammatory bowel diseases: performance of fecal lactoferrin, calprotectin, and PMN-elastase, CRP, and clinical indices. Am J Gastroenterol. 2008;103(1):162–169. | |
Schmid M, Fellermann K, Fritz P, Wiedow O, Stange EF, Wehkamp J. Attenuated induction of epithelial and leukocyte serine antiproteases elafin and secretory leukocyte protease inhibitor in Crohn’s disease. J Leukoc Biol. 2007;81(4):907–915. | |
Motta JP, Magne L, Descamps D, et al. Modifying the protease, antiprotease pattern by elafin overexpression protects mice from colitis. Gastroenterology. 2011;140(4):1272–1282. | |
Motta JP, Bermúdez-Humarán LG, Deraison C, et al. Food-grade bacteria expressing elafin protect against inflammation and restore colon homeostasis. Sci Transl Med. 2012;4(158):158ra144. | |
Ho AS, Chen CH, Cheng CC, et al. Neutrophil elastase as a diagnostic marker and therapeutic target in colorectal cancers. Oncotarget. 2014;5(2):473–480. | |
Wex T, Treiber G, Venerito M, et al. Helicobacter pylori-induced downregulation of the secretory leukocyte protease inhibitor (SLPI) in gastric epithelial cell lines and its functional relevance for H. pylori-mediated diseases. Biol Chem. 2006;387(7):893–901. | |
Pertuz Belloso S, Ostoa Saloma P, Benitez I, Soldevila G, Olivos A, Garcia-Zepeda E. Entamoeba histolytica cysteine protease 2 (EhCP2) modulates leucocyte migration by proteolytic cleavage of chemokines. Parasite Immunol. 2004;26(5):237–241. | |
Cotton JA, Bhargava A, Ferraz JG, Yates RM, Beck PL, Buret AG. Giardia duodenalis cathepsin B proteases degrade intestinal epithelial interleukin-8 and attenuate interleukin-8-induced neutrophil chemotaxis. Infect Immun. 2014;82(7):2772–2787. | |
Cotton JA, Motta JP, Schenck LP, Hirota SA, Beck PL, Buret AG. Giardia duodenalis infection reduces granulocyte infiltration in an in vivo model of bacterial toxin-induced colitis and attenuates inflammation in human intestinal tissue. PLoS One. 2014;9(10):e109087. | |
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | |
Albini A, Tosetti F, Li VW, Noonan DM, Li WW. Cancer prevention by targeting angiogenesis. Nat Rev Clin Oncol. 2012;9(9):498–509. | |
Fidler IJ, Ellis LM. Chemotherapeutic drugs – more really is not better. Nat Med. 2000;6(5):500–502. | |
Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1(1):27–31. | |
Raica M, Cimpean AM, Ribatti D. Angiogenesis in pre-malignant conditions. Eur J Cancer. 2009;45(11):1924–1934. | |
Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86(3):353–364. | |
Baluk P, Hashizume H, McDonald DM. Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev. 2005;15(1):102–111. | |
Nagy JA, Chang SH, Shih SC, Dvorak AM, Dvorak HF. Heterogeneity of the tumor vasculature. Semin Thromb Hemost. 2010;36(3):321–331. | |
Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med. 1997;3(2):177–182. | |
Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8(8):618–631. | |
Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7(3):211–217. | |
Sepulveda AR. Helicobacter, inflammation, and gastric cancer. Curr Pathobiol Rep. 2013;1(1):9–18. | |
Koch AE, Polverini PJ, Kunkel SL, et al. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science. 1992;258(5089):1798–1801. | |
Keeley EC, Mehrad B, Strieter RM. Chemokines as mediators of tumor angiogenesis and neovascularization. Exp Cell Res. 2011;317(5):685–690. | |
Yatsunami J, Tsuruta N, Ogata K, et al. Interleukin-8 participates in angiogenesis in non-small cell, but not small cell carcinoma of the lung. Cancer Lett. 1997;120(1):101–108. | |
Strieter RM, Kunkel SL, Elner VM, et al. Interleukin-8. A corneal factor that induces neovascularization. Am J Pathol. 1992;141(6):1279–1284. | |
Li A, Dubey S, Varney ML, Dave BJ, Singh RK. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol. 2003; 170(6):3369–3376. | |
Hu DE, Hori Y, Fan TP. Interleukin-8 stimulates angiogenesis in rats. Inflammation. 1993;17(2):135–143. | |
Addison CL, Daniel TO, Burdick MD, et al. The CXC chemokine receptor 2, CXCR2, is the putative receptor for ELR+ CXC chemokine-induced angiogenic activity. J Immunol. 2000;165(9):5269–5277. | |
Murdoch C, Monk PN, Finn A. Cxc chemokine receptor expression on human endothelial cells. Cytokine. 1999;11(9):704–712. | |
Heidemann J, Ogawa H, Dwinell MB, et al. Angiogenic effects of interleukin 8 (CXCL8) in human intestinal microvascular endothelial cells are mediated by CXCR2. J Biol Chem. 2003;278(10):8508–8515. | |
Kitadai Y, Haruma K, Sumii K, et al. Expression of interleukin-8 correlates with vascularity in human gastric carcinomas. Am J Pathol. 1998;152(1):93–100. | |
Kido S, Kitadai Y, Hattori N, et al. Interleukin 8 and vascular endothelial growth factor – prognostic factors in human gastric carcinomas? Eur J Cancer. 2001;37(12):1482–1487. | |
Vegran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011;71(7):2550–2560. | |
Lee YS, Choi I, Ning Y, et al. Interleukin-8 and its receptor CXCR2 in the tumour microenvironment promote colon cancer growth, progression and metastasis. Br J Cancer. 2012;106(11):1833–1841. | |
Kim SJ, Uehara H, Karashima T, McCarty M, Shih N, Fidler IJ. Expression of interleukin-8 correlates with angiogenesis, tumorigenicity, and metastasis of human prostate cancer cells implanted orthotopically in nude mice. Neoplasia. 2001;3(1):33–42. | |
Langeggen H, Johnson E, Hetland G. Effects of C5a and FMLP on interleukin-8 production and proliferation of human umbilical vein endothelial cells. Inflammation. 2001;25(2):83–89. | |
Li A, Varney ML, Valasek J, Godfrey M, Dave BJ, Singh RK. Autocrine role of interleukin-8 in induction of endothelial cell proliferation, survival, migration and MMP-2 production and angiogenesis. Angiogenesis. 2005;8(1):63–71. | |
Asfaha S, Dubeykovskiy AN, Tomita H, et al. Mice that express human interleukin-8 have increased mobilization of immature myeloid cells, which exacerbates inflammation and accelerates colon carcinogenesis. Gastroenterology. 2013;144(1):155–166. | |
Campbell LM, Maxwell PJ, Waugh DJ. Rationale and means to target pro-inflammatory interleukin-8 (CXCL8) signaling in cancer. Pharmaceuticals (Basel). 2013;6(8):929–959. | |
Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2(6):442–454. | |
Kitadai Y, Haruma K, Mukaida N, et al. Regulation of disease-progression genes in human gastric carcinoma cells by interleukin 8. Clin Cancer Res. 2000;6(7):2735–2740. | |
Terada H, Urano T, Konno H. Association of interleukin-8 and plasminogen activator system in the progression of colorectal cancer. Eur Surg Res. 2005;37(3):166–172. | |
Ueda T, Shimada E, Urakawa T. Serum levels of cytokines in patients with colorectal cancer: possible involvement of interleukin-6 and interleukin-8 in hematogenous metastasis. J Gastroenterol. 1994; 29(4):423–429. | |
Nastase A, Paslaru L, Herlea V, et al. Expression of interleukine-8 as an independent prognostic factor for sporadic colon cancer dissemination. J Med Life. 2014;7(2):215–219. | |
Ning Y, Manegold PC, Hong YK, et al. Interleukin-8 is associated with proliferation, migration, angiogenesis and chemosensitivity in vitro and in vivo in colon cancer cell line models. Int J Cancer. 2011;128(9):2038–2049. | |
Lee LF, Louie MC, Desai SJ, et al. Interleukin-8 confers androgen-independent growth and migration of LNCaP: differential effects of tyrosine kinases Src and FAK. Oncogene. 2004;23(12):2197–2205. | |
Li A, Varney ML, Singh RK. Expression of interleukin 8 and its receptors in human colon carcinoma cells with different metastatic potentials. Clin Cancer Res. 2001;7(10):3298–3304. | |
Hwang WL, Yang MH, Tsai ML, et al. SNAIL regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology. 2011;141(1):e271–e275. | |
Fernando RI, Castillo MD, Litzinger M, Hamilton DH, Palena C. IL-8 signaling plays a critical role in the epithelial-mesenchymal transition of human carcinoma cells. Cancer Res. 2011;71(15):5296–5306. | |
Brew R, Erikson JS, West DC, Kinsella AR, Slavin J, Christmas SE. Interleukin-8 as an autocrine growth factor for human colon carcinoma cells in vitro. Cytokine. 2000;12(1):78–85. | |
Wu T, Wang Z, Liu Y, et al. Interleukin 22 protects colorectal cancer cells from chemotherapy by activating the STAT3 pathway and inducing autocrine expression of interleukin 8. Clin Immunol. 2014;154(2):116–126. | |
Kuai WX, Wang Q, Yang XZ, Zhao Y, Yu R, Tang XJ. Interleukin-8 associates with adhesion, migration, invasion and chemosensitivity of human gastric cancer cells. World J Gastroenterol. 2012;18(9):979–985. | |
Xiao YC, Yang ZB, Cheng XS, et al. CXCL8, overexpressed in colorectal cancer, enhances the resistance of colorectal cancer cells to anoikis. Cancer Lett. 2015;361(1):22–32. | |
Ning Y, Labonte MJ, Zhang W, et al. The CXCR2 antagonist, SCH-527123, shows antitumor activity and sensitizes cells to oxaliplatin in preclinical colon cancer models. Mol Cancer Ther. 2012;11(6):1353–1364. | |
Ning Y, Lenz HJ. Targeting IL-8 in colorectal cancer. Expert Opin Ther Targets. 2012;16(5):491–497. | |
Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008; 181(8):5791–5802. | |
Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182(8):4499–4506. | |
Zhang B, Wang Z, Wu L, et al. Circulating and tumor-infiltrating myeloid-derived suppressor cells in patients with colorectal carcinoma. PLoS One. 2013;8(2):e57114. | |
Sun HL, Zhou X, Xue YF, et al. Increased frequency and clinical significance of myeloid-derived suppressor cells in human colorectal carcinoma. World J Gastroenterol. 2012;18(25):3303–3309. | |
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174. | |
Ugel S, Delpozzo F, Desantis G, et al. Therapeutic targeting of myeloid-derived suppressor cells. Curr Opin Pharmacol. 2009;9(4):470–481. | |
Katoh H, Wang D, Daikoku T, Sun H, Dey SK, Dubois RN. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell. 2013;24(5):631–644. | |
André T, Boni C, Mounedji-Boudiaf L, et al; Multicenter International Study of Oxaliplatin/5-Fluorouracil/Leucovorin in the Adjuvant Treatment of Colon Cancer (MOSAIC) Investigators. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med. 2004;350(23):2343–2351. | |
Zhang W, Stoehlmacher J, Park DJ, et al. Gene polymorphisms of epidermal growth factor receptor and its downstream effector, interleukin-8, predict oxaliplatin efficacy in patients with advanced colorectal cancer. Clin Colorectal Cancer. 2005;5(2):124–131. | |
Hayashi H, Arao T, Matsumoto K, et al. Biomarkers of reactive resistance and early disease progression during chemotherapy plus bevacizumab treatment for colorectal carcinoma. Oncotarget. 2014;5(9):2588–2595. | |
Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3(3):221–227. | |
Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15(6):2403–2413. | |
Paulson AS, Tran Cao HS, Tempero MA, Lowy AM. Therapeutic advances in pancreatic cancer. Gastroenterology. 2013;144(6):1316–1326. | |
Song Y, Baba T, Li YY, et al. Gemcitabine-induced CXCL8 expression counteracts its actions by inducing tumor neovascularization. Biochem Biophys Res Commun. 2015;458(2):341–346. | |
Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30(7):1073–1081. | |
De Marzo AM, DeWeese TL, Platz EA, et al. Pathological and molecular mechanisms of prostate carcinogenesis: implications for diagnosis, detection, prevention, and treatment. J Cell Biochem. 2004;91(3):459–477. | |
Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14(21):6735–6741. | |
Masckauchan TN, Shawber CJ, Funahashi Y, Li CM, Kitajewski J. Wnt/beta-catenin signaling induces proliferation, survival and interleukin-8 in human endothelial cells. Angiogenesis. 2005;8(1):43–51. | |
Imamura R, Konaka K, Matsumoto N, et al. Fas ligand induces cell-autonomous NF-kappaB activation and interleukin-8 production by a mechanism distinct from that of tumor necrosis factor-alpha. J Biol Chem. 2004;279(45):46415–46423. | |
Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6(5):447–458. | |
Maxwell PJ, Coulter J, Walker SM, et al. Potentiation of inflammatory CXCL8 signalling sustains cell survival in PTEN-deficient prostate carcinoma. Eur Urol. 2013;64(2):177–188. | |
Farooq SM, Stillie R, Svensson M, Svanborg C, Strieter RM, Stadnyk AW. Therapeutic effect of blocking CXCR2 on neutrophil recruitment and dextran sodium sulfate-induced colitis. J Pharmacol Exp Ther. 2009;329(1):123–129. | |
Bento AF, Leite DF, Claudino RF, Hara DB, Leal PC, Calixto JB. The selective nonpeptide CXCR2 antagonist SB225002 ameliorates acute experimental colitis in mice. J Leukoc Biol. 2008;84(4):1213–1221. | |
Jamieson T, Clarke M, Steele CW, et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J Clin Invest. 2012;122(9):3127–3144. | |
Varney ML, Singh S, Li A, Mayer-Ezell R, Bond R, Singh RK. Small molecule antagonists for CXCR2 and CXCR1 inhibit human colon cancer liver metastases. Cancer Lett. 2011;300(2):180–188. | |
Zlotnik A, Yoshie O, Nomiyama H. The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol. 2006;7(12):243. | |
Modi WS, Yoshimura T. Isolation of novel GRO genes and a phylogenetic analysis of the CXC chemokine subfamily in mammals. Mol Biol Evol. 1999;16(2):180–193. | |
Wuyts A, Haelens A, Proost P, et al. Identification of mouse granulocyte chemotactic protein-2 from fibroblasts and epithelial cells. Functional comparison with natural KC and macrophage inflammatory protein-2. J Immunol. 1996;157(4):1736–1743. | |
Fan X, Patera AC, Pong-Kennedy A, et al. Murine CXCR1 is a functional receptor for GCP-2/CXCL6 and interleukin-8/CXCL8. J Biol Chem. 2007;282(16):11658–11666. | |
Bozic CR, Gerard NP, von Uexkull-Guldenband C. The murine interleukin 8 type B receptor homologue and its ligands expression and biological characterization. J Biol Chem. 1994;269(47):29355–29358. | |
Cerretti DP, Nelson N, Kozlosky CJ, et al. The murine homologue of the human interleukin-8 receptor type B maps near the Ity-Lsh-Bcg disease resistance locus. Genomics. 1993;18(2):410–413. | |
Cacalano G, Lee J, Kikly K, et al. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science. 1994; 265(5172):682–684. | |
Lee J, Cacalano G, Camerato T, Toy K, Moore MW, Wood WI. Chemokine binding and activities mediated by the mouse IL-8 receptor. J Immunol. 1995;155(4):2158–2164. | |
Fu W, Zhang Y, Zhang J, Chen WF. Cloning and characterization of mouse homolog of the CXC chemokine receptor CXCR1. Cytokine. 2005;31(1):9–17. | |
Moepps B, Nuesseler E, Braun M, Gierschik P. A homolog of the human chemokine receptor CXCR1 is expressed in the mouse. Mol Immunol. 2006;43(7):897–914. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.