Back to Journals » Journal of Experimental Pharmacology » Volume 18
Integrative Computational and Transcriptional Analysis of NF-κB and HIF-1α Modulation Following Doxorubicin Treatment in Triple-Negative Breast Cancer Cells
Authors Utami N ![]() , Setiawansyah A, Bashari MH
, Setiawansyah A, Bashari MH ![]() , Usman HA
, Usman HA ![]() , Azhar RY
, Azhar RY ![]() , Khairani AF
, Khairani AF ![]()
Received 27 October 2025
Accepted for publication 31 January 2026
Published 11 February 2026 Volume 2026:18 576572
DOI https://doi.org/10.2147/JEP.S576572
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Abdelwahab Omri
Nurul Utami,1,2 Arif Setiawansyah,3 Muhammad Hasan Bashari,4 Hermin Aminah Usman,5 Raden Yohana Azhar,6 Astrid Feinisa Khairani4
1Doctoral Program in Medical Science, Faculty of Medicine, Universitas Padjadjaran, Bandung, Indonesia; 2Department of Anatomy, Anatomical Pathology, and Histology, Faculty of Medicine, Universitas Lampung, Bandar Lampung, Indonesia; 3Pharmacy Diploma Program, Akademi Farmasi Cendikia Farma Husada, Bandar Lampung, Indonesia; 4Department of Biomedical Sciences, Faculty of Medicine, Universitas Padjadjaran, Bandung, Indonesia; 5Department of Anatomical Pathology, Faculty of Medicine, Universitas Padjadjaran/Dr Hasan Sadikin General Hospital, Bandung, Indonesia; 6Division of Oncology Surgery, Department of Surgery, Faculty of Medicine, Universitas Padjadjaran/Dr Hasan Sadikin General Hospital, Bandung, Indonesia
Correspondence: Nurul Utami, Doctoral Program in Medical Science, Faculty of Medicine, Universitas Padjadjaran, Jl. Eijkman No. 38, Bandung, 40161, Indonesia, Tel +62 852 6952 9979, Email [email protected]
Purpose: Triple-negative breast cancer (TNBC) is an aggressive breast cancer subtype with limited targeted treatment options. Doxorubicin remains a cornerstone of TNBC treatment; however, its molecular effects beyond canonical cytotoxic mechanisms are not fully characterized. This study aimed to explore NF-κB- and HIF-1α-related transcriptional responses associated with doxorubicin treatment in TNBC cells using an integrative computational and experimental approach, in line with global cancer research priorities supporting Sustainable Development Goal (SDG) 3: Good Health and Well-Being.
Methods: Network pharmacology analysis, molecular docking, and molecular dynamics simulations were employed to explore potential pathway-level associations of doxorubicin with NF-κB and HIF-1α–related signaling. In vitro validation was performed using MTT cytotoxicity assays in MDA-MB-231 cells cultured in DMEM and RPMI-1640 media. Half-maximal inhibitory concentrations (IC50) were determined using four-parameter logistic regression. Transcriptional responses of NF-κB and HIF-1α were evaluated by RT-qPCR under normoxic conditions.
Results: Computational analyses suggested potential associations between doxorubicin and components of NF-κB and HIF-1α–related signaling pathways. In vitro assays demonstrated concentration-dependent cytotoxicity, with IC50 values of 2.34 μM (95% CI: 2.11– 2.74) in DMEM and 1.07 μM (95% CI: 0.92– 1.32 μM) in RPMI-1640. RT-qPCR analysis revealed downregulation of NF-κB and HIF-1α mRNA expression following doxorubicin treatment. These findings indicate transcriptional modulation associated with doxorubicin exposure, without establishing functional pathway inhibition.
Conclusion: This study provides transcriptional-level evidence suggesting the involvement of NF-κB– and HIF-1α–related pathways in the cellular response of TNBC cells to doxorubicin treatment. By integrating computational predictions with early experimental validation, the findings generate biologically plausible hypotheses for further mechanistic and functional investigations, contributing to foundational cancer research efforts aligned with SDG 3 (Good Health and Well-Being).
Keywords: doxorubicin, HIF-1α inhibition, molecular docking, NF-κB inhibition, quantitative PCR, triple-negative breast cancer, SDG 3, good health and well-being
Introduction
Triple negative breast cancer (TNBC) represents approximately 15–20% of all breast cancers and is characterized by the absence of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) expression.1 This molecular profile limits treatment options, as these tumors do not respond to hormonal or HER2-targeted therapies, leaving chemotherapy as the primary systemic treatment option.2,3 Doxorubicin remains a cornerstone in TNBC treatment despite its dose-limiting cardiotoxicity and the development of resistance.4–6
Among the molecular pathways implicated in TNBC progression, nuclear factor kappa B (NF-κB) and hypoxia-inducible factor-1 alpha (HIF-1α) have been extensively studied.7,8 Constitutive activation of NF-κB has been associated with cell survival, inflammation, and metastatic behavior,8 while HIF-1α plays a central role in hypoxia adaptation, angiogenesis, and metabolic reprogramming.7 Importantly, accumulating evidence suggests functional cross-talk between these pathways, which may contribute to aggressive tumor phenotypes and therapy resistance.9,10 This cross-talk involves shared molecular regulators, including transcriptional co-activators such as p300/CBP and co-regulation of downstream targets related to inflammation and angiogenesis (eg, VEGF and IL-8), which have been implicated in TNBC progression and therapeutic resistance.11–13 While doxorubicin’s primary mechanism of action involves DNA intercalation and topoisomerase II inhibition, emerging studies indicate that its cellular effects may extend beyond these canonical mechanisms.14 Several studies have indicated that doxorubicin may modulate key signaling pathways beyond its canonical cytotoxic mechanisms.15–17 While doxorubicin has been reported to modulate NF-κB and HIF-1α activity in various cancer contexts, including paradoxical activation or suppression depending on experimental conditions, its coordinated involvement in NF-κB- and HIF-1α-related responses within a TNBC-specific framework has not been systematically explored.13,18,19 However, the broader pathway-level responses associated with doxorubicin treatment in TNBC remain incompletely characterized, limiting opportunities to optimize treatment strategies and better understand resistance mechanisms.
Network pharmacology, an integrative approach combining systems biology and computational drug discovery, has emerged as a powerful tool for exploring drug–target and drug–pathway relationships.20 This methodology enables the systematic integration of multi-source biological data to generate hypotheses regarding potential pathway-level drug effects, particularly in complex diseases such as cancer.21,22 In this study, we employed a targeted network pharmacology framework, complemented by molecular modeling and transcriptional analysis, to explore NF-κB- and HIF-1α-related responses associated with doxorubicin treatment in TNBC cells. Rather than establishing definitive mechanistic inhibition, this approach aims to link computational predictions with early experimental evidence, providing a foundation for future mechanistic and functional investigations.
Material and Methods
Materials
Human TNBC cell line MDA-MB-231 (DKFZ, Germany), RPMI-1640 medium (Sygma-Aldrich), DMEM High Glucose Medium (Sygma-Aldrich), FBS, Penicillin-streptomycin (Sygma-Aldrich), PBS, trypsin EDTA, Quick-RNA™ Miniprep Kit (Zymo Research), SensiFast SYBR no-ROX kit (Bioline), SensiFast cDNA synthesis kit (Bioline), absolute ethanol. In silico studies were performed on a dedicated workstation running Ubuntu 20.04 LTS with an Intel® Core™ i9-12900KF processor (24 cores at 3.6 GHz), 32 GB RAM, and an NVIDIA RTX 4060 GPU with 16 GB VRAM.
Methods
Network Pharmacology Study
Novel targets of doxorubicin for TNBC treatment were identified using a targeted network pharmacology approach as described by Sadaqa et al (2025).23 The methodology commenced with comprehensive gene mining for doxorubicin targets across three established databases: SuperPRED, SwissTargetPrediction, and Sea Target Prediction. TNBC-associated genes were simultaneously retrieved from the Human Gene Cards database. All identified genes underwent standardization through the UniProt database, followed by cross-matching analysis between doxorubicin and TNBC gene sets using the Venny 2.0 platform. Protein-protein interaction (PPI) networks were subsequently constructed using the STRING database (https://string-db.org), with network visualization and topological parameter analysis performed in Cytoscape 3.10.1. The core targets identified through this analysis were then subjected to comprehensive pathway enrichment and biological process analyses using ShinyGO 0.80 (http://bioinformatics.sdstate.edu/go/). All enrichment analyses maintained rigorous statistical standards with a false discovery rate (FDR) threshold of 0.05.
Molecular Docking Study
Initial validation of doxorubicin potential against network pharmacology analysis results was conducted through molecular docking using Autodock 4.2 integrated with the Autodock Tools interface.24,25 The methodology encompassed systematic preparation of both macromolecular structures and ligands, with crystal structures of NF-κB, HIF-1α, TNF-α, EGFR, ERBB2, HSP90, STAT3, and p53 obtained from the Protein Data Bank using PDB identifiers 4DN5, 3KCX, 7JRA, 6DUK, 7JXH, 5H22, 6NUQ, and 4IPF, respectively. The three-dimensional structure of doxorubicin was retrieved from the PubChem database (PubChem CID: 31703). Macromolecular crystal structures underwent comprehensive preparation using Biovia Discovery Studio Visualizer client 2022 to eliminate water molecules, ligands, and heteroatoms from the original crystallographic data. Concurrently, the doxorubicin structure was optimized through molecular mechanics calculations employing the MMFF94 force field and steepest descent algorithms to achieve energetically favorable conformations. Both macromolecules and doxorubicin were subsequently imported into Autodock Tools for advanced preparation procedures, including addition of polar hydrogen atoms to macromolecular structures and configuration of structural flexibility parameters for doxorubicin. The molecular docking site was precisely defined at the active sites of target macromolecules, following native ligand coordinates with careful gridbox adjustments to encompass the entire active site region. The docking simulation employed genetic algorithm methodology with one hundred independent runs and Lamarckian genetic algorithm parameters for output generation. Optimal conformations were selected based on binding affinity scores and subsequently analyzed for detailed molecular interactions using Biovia Discovery Studio Visualizer client 2022 to elucidate binding mechanisms and interaction patterns between doxorubicin and target proteins.
Molecular Dynamic Simulation
Molecular dynamics simulations were conducted using the GROMACS 2024.3 software package26 to investigate protein-ligand interactions under physiological conditions, with system parameterization employing the CHARMM36m force field for protein components and the CHARMM General Force Field (CGenFF) for ligand structures. System preparation was conducted using CHARMM-GUI input generator,27 involving solvation within an octahedral TIP3P water box with sodium and chloride ions added for electrical neutrality and physiological ionic condition replication, followed by systematic energy minimization to resolve steric clashes and equilibration phases under constant volume and constant pressure ensemble conditions to establish system stability. Temperature regulation was maintained at 310 K using the velocity rescaling thermostat to reflect human physiological conditions, while pressure control was achieved at 1 atm using the Parrinello-Rahman barostat, with long-range electrostatic interactions calculated through the Particle Mesh Ewald algorithm for computational accuracy. Production molecular dynamics simulations were executed for 50 nanoseconds to capture conformational dynamics and binding behaviors, followed by comprehensive trajectory analysis evaluating root mean square deviation (RMSD) for structural stability, root mean square fluctuation (RMSF) for flexible region identification, solvent-accessible surface area (SASA) for hydrophobic interaction examination, radius of gyration (Rg) for compactness monitoring using native GROMACS analysis tools and MM-PBSA/MM-GBSA for binding energy calculation, with structural visualization and trajectory inspection accomplished through ChimeraX 2024.10.29 and Biovia Discovery Studio software 2022 platforms.
Cell Culture
The human triple-negative breast cancer (TNBC) cell line MDA-MB-231 was cultured in either Dulbecco’s Modified Eagle Medium (DMEM) High Glucose (D6429 HG, Sigma-Aldrich) or Roswell Park Memorial Institute (RPMI)-1640 (R8758, Sigma-Aldrich) medium, each supplemented with 10% fetal bovine serum (FBS) (F7524, Sigma-Aldrich) and 1% penicillin–streptomycin (P433, Sigma-Aldrich). The use of both DMEM and RPMI-1640 media was intentionally designed as an exploratory comparison to reflect commonly used culture conditions for MDA-MB-231 cells and to account for potential medium-dependent variability in cellular responses to doxorubicin. Previous work has shown that different cell culture media can strongly influence gene expression profiles in MDA-MB-231 cells, indicating that medium composition itself can impact cellular phenotypes and experimental outcomes (eg, differential expression of thousands of genes across DMEM and RPMI-1640).28 The MDA-MB-231 cells were obtained from Dr Thordur Oskarsson (DKFZ, Germany) and approved for use by the Ethics Committee of Universitas Padjadjaran (No. 265/UN6.KEP/EC/2025). Cells were maintained at 37°C in a humidified incubator with 5% CO2. Upon reaching 80–90% confluency, cells were washed with phosphate-buffered saline (PBS) and detached using trypsin–EDTA (T3924, Sigma-Aldrich) for use in subsequent experiments.
In vitro Cytotoxic Assay
The cytotoxic effects of doxorubicin were evaluated using the MTT assay. MDA-MB-231 cells were seeded at a density of 1 × 104 cells per well in 96-well plates containing 200 µL of complete DMEM High Glucose or RPMI-1640 medium. Plates were incubated at 37°C in 5% CO2 for 24 h to allow for cell adherence. Cells were then treated with seven concentrations of doxorubicin (0.5–5 µM), while untreated cells served as negative controls, and incubated for an additional 72 h under the same conditions. Cell viability was determined by adding 100 µL of fresh DMEM High Glucose or RPMI-1640 medium containing 10 µL of MTT reagent (InvitrogenTM) to each well, followed by a 3 h incubation at 37°C. Absorbance was measured at 550 nm using a microplate reader connected to a computer for data acquisition. All experiments were performed in triplicate in at least three independent biological replicates.
RT-qPCR Analysis
NF-κB and HIF-1α mRNA expression levels were quantified by reverse transcription quantitative polymerase chain reaction (RT-qPCR) to assess transcriptional changes following treatment. MDA-MB-231 cells were treated with doxorubicin at concentrations of 0.125, 0.25, and 0.5 µM for 72 hours prior to RNA extraction, based on the cytotoxicity profiling results. Total RNA was extracted from cells using the Quick-RNA™ Miniprep Kit (R1057, Zymo Research) following the manufacturer’s protocol. Complementary DNA (cDNA) was synthesized using the SensiFast cDNA Synthesis Kit (BIO-65053, Bioline Reagents Ltd). Quantitative PCR amplification was performed with the SensiFast SYBR No-ROX detection system (BIO-98005, Bioline Reagents Ltd.) on a real-time thermal cycler. Each reaction was carried out in technical duplicates. The thermal cycling protocol included an initial polymerase activation at 95°C for 2 min, followed by 40 cycles of denaturation at 95°C for 5 s and annealing/extension at 55–65°C for 20 s, with annealing temperatures optimized for each primer pair. GAPDH was used as the internal reference gene to normalize RNA input and reverse transcription efficiency. Relative gene expression was calculated using the comparative Ct (2^−ΔΔCt) method. Technical duplicates were performed for each biological replicate. The complete sequences of genes are summarized in Table 1.
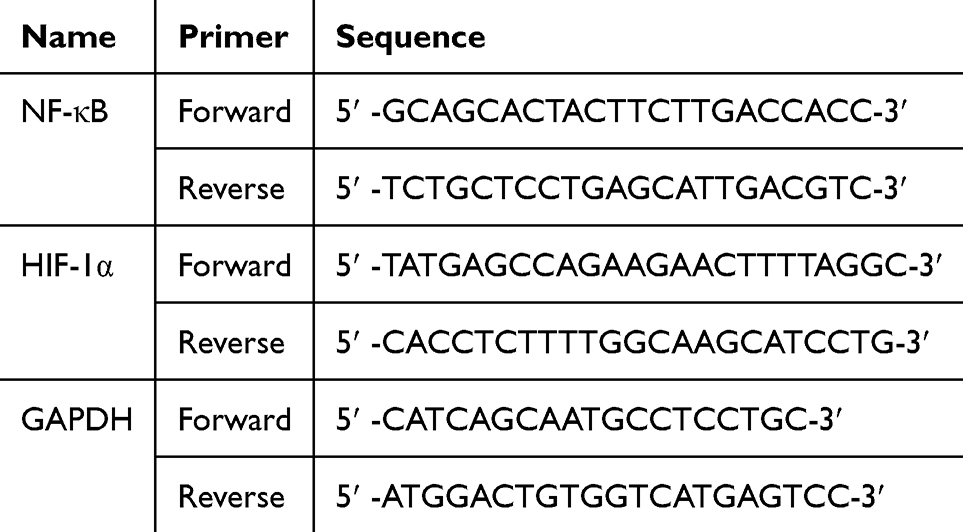 |
Table 1 Primer Sequences for RT-qPCR Analysis |
Data Analysis
Absorbance values from the MTT assay were normalized to untreated controls to calculate percentage cell viability. Dose–response curves were generated, and half-maximal inhibitory concentration (IC50) values were determined using GraphPad Prism version 10.3.1 (GraphPad Software, San Diego, CA, USA) by fitting the data to a four-parameter logistic (4PL) non-linear regression model (variable slope). IC50 values are reported together with their 95% confidence intervals (CI) derived from the model.29
For RT-qPCR analysis, quantitative data are presented as mean ± standard deviation (SD) calculated from three independent samples per group obtained within a single biological experiment. Technical duplicates were averaged prior to statistical analysis. IC50 values were derived from nonlinear regression analysis and are reported together with their 95% confidence intervals. Statistical comparisons between groups were performed using one-way ANOVA followed by Tukey’s post hoc test, with p < 0.05 considered statistically significant.
Results
Network Pharmacology
The research findings present a comprehensive molecular characterization of triple-negative breast cancer (TNBC) therapeutic targets through systematic bioinformatics analysis. The cross-matching procedure between doxorubicin-associated genes and TNBC-specific genes yielded 103 overlapping molecular targets. The subsequent application of topological network analysis algorithms identified ten core genes that demonstrate high connectivity and centrality within the molecular interaction network, as presented in Figure 1. These genes include established oncological markers such as TP53 (tumor suppressor p53), TNF (tumor necrosis factor), EGFR (epidermal growth factor receptor), and STAT3 (signal transducer and activator of transcription 3), alongside metabolic regulators including ALB (albumin) and molecular chaperones HSP90AA1 and HSP90AB1. The identification of these core genes highlights their potential roles in cellular homeostasis and dysregulated signaling processes associated with TNBC. The presence of both oncogenes and tumor suppressors within this core set reflects the complexity of regulatory networks involved in TNBC development and progression.
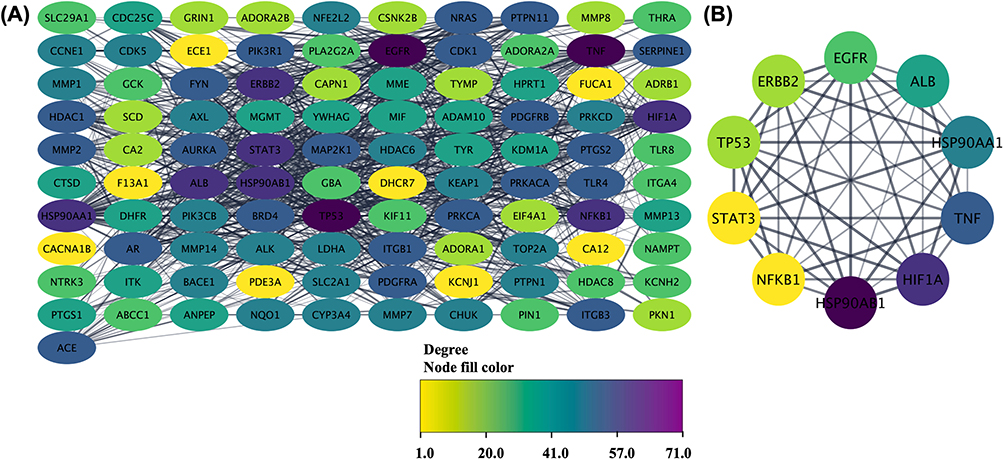 |
Figure 1 Protein-protein interaction network (A) and the core genes (B) related to TNBC. The color shift from yellow to purple indicating the shift of degree of connectivity. |
The KEGG pathway enrichment analysis revealed significant involvement of the core genes in cancer-related signaling pathways associated with TNBC-related biological processes (Figure 2A). Among these, the proteoglycan signaling pathway was identified, consistent with the known roles of proteoglycans as components of the tumor microenvironment that influence cell-matrix interactions and growth factor availability. Complementing this finding, the enrichment of HIF-1α pathway components indicates the importance of hypoxic responses in TNBC biology, which proves particularly relevant given the aggressive nature and poor vascularization often observed in these tumors. In addition, enrichment of the PI3K-Akt signaling pathway was detected, a pathway frequently reported to be dysregulated in various cancers. However, the analysis also revealed unexpected involvement of the core genes in non-oncological pathways, specifically Th17 cell differentiation and lipid metabolism pathways associated with atherosclerosis. This finding suggests broader biological roles for these molecular targets beyond cancer biology. The Th17 cell differentiation pathway involvement indicates potential immunological components in TNBC pathogenesis, which aligns with emerging understanding of immune system contributions to cancer development and progression.
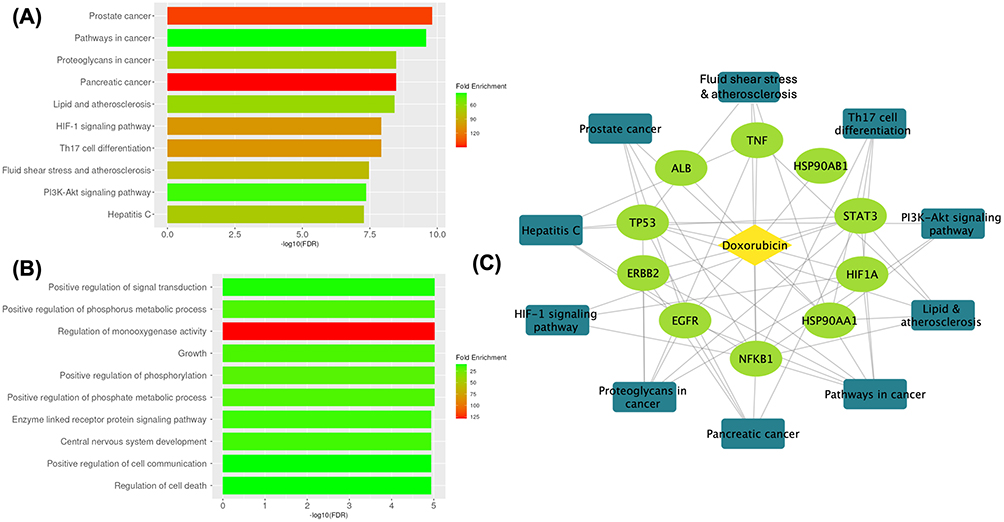 |
Figure 2 Enrichment analysis. (A) KEGG pathway, (B) gene ontology biological process, and (C) doxorubicin-genes-pathways network. |
The Gene Ontology biological process analysis (Figure 2B) identified enrichment of the core genes in several biological processes related to cellular regulation. Their involvement in signal transduction processes confirms their function as critical communication nodes within cellular networks, facilitating the transmission of molecular information essential for coordinated cellular responses. Building upon this foundational role, their participation in cellular development processes suggests their importance in maintaining proper cellular differentiation programs, the disruption of which represents a fundamental hallmark of cancer pathogenesis and contributes to the loss of normal cellular identity observed in malignant transformation. Most significantly, the involvement of these genes in apoptotic regulation indicates their crucial roles in controlling programmed cell death pathways, a process that maintains tissue homeostasis under normal physiological conditions. Since dysregulation of apoptosis represents a fundamental characteristic of cancer cells that enables their survival and proliferation despite cellular damage, the identification of multiple core genes involved in these processes suggests that apoptosis-related pathways may warrant further investigation in the context of TNBC. The complete network of doxorubicin-genes-pathway is presented in Figure 2C.
Molecular Docking
Initial validation of doxorubicin’s binding potential against the identified core targets was conducted through validated computational molecular docking analysis (Figure 3). This approach provided quantitative assessment of drug-target interactions by calculating binding free energy values, which serve as indicators of molecular affinity and binding stability. The comparative analysis presented in Table 2 demonstrates the binding free energy profiles of doxorubicin against each target protein, with corresponding native ligand values included as reference standards. The molecular docking results revealed heterogeneous binding affinities across the target protein panel, with doxorubicin exhibiting binding free energy values spanning from −5.53 kcal/mol to −12.06 kcal/mol. This range indicates variable degrees of molecular recognition and binding stability depending on the specific protein target. Notably, doxorubicin demonstrated favorable binding affinity for NF-κB and HIF-1α, achieving binding free energy values of −9.82 kcal/mol and −7.84 kcal/mol, respectively, both of which exceeded the binding energies of their corresponding native ligands.
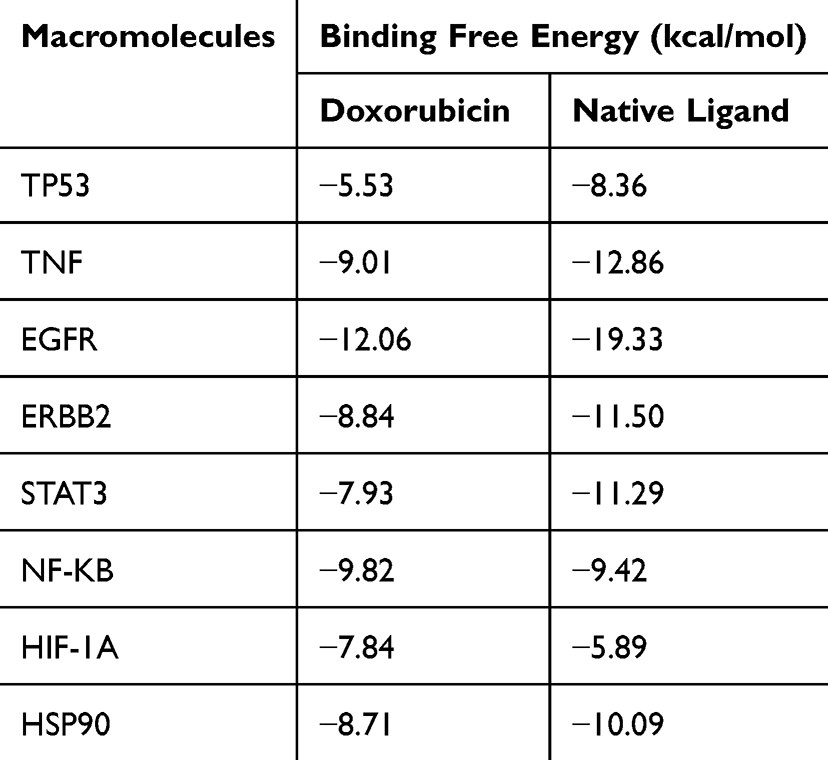 |
Table 2 Docking Score of Doxorubicin Against Core Targets |
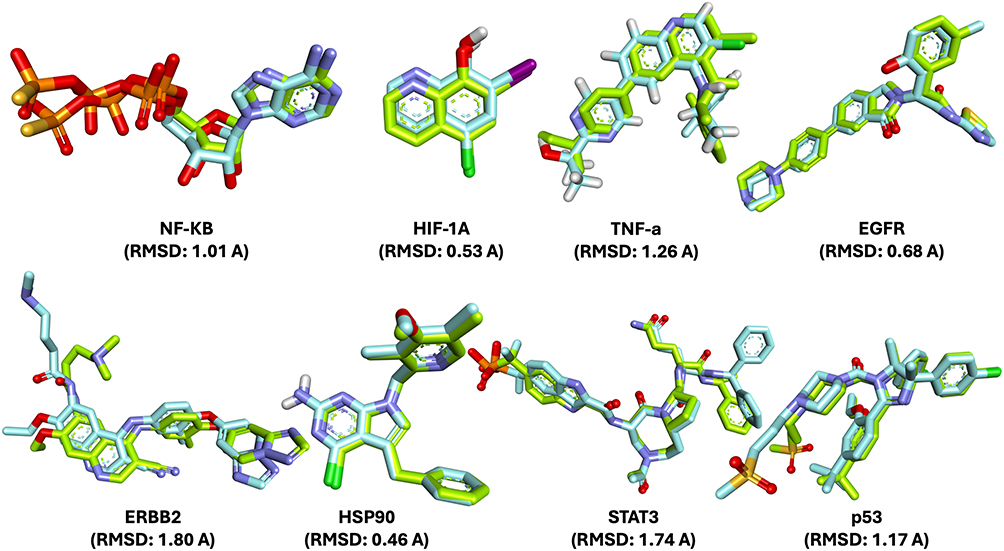 |
Figure 3 Validation of molecular docking protocol through RMSD analysis. Superimposition of the native (reference) ligand and the re-docked ligand poses for NF-κB, HIF-1A, TNF-α, EGFR, ERBB2, HSP90, STAT3, and p53 targets. The calculated RMSD values ranged from 0.46 to 1.80 Å, indicating good agreement between the docked and native conformations. RMSD values below 2.0 Å confirm the reliability and accuracy of the docking protocol applied in this study. |
The enhanced binding affinity of doxorubicin for NF-κB stems from distinctive intermolecular interaction patterns within the protein’s active site. Detailed interaction analysis, as presented in Figure 4, reveals that doxorubicin primarily engages the target protein through hydrogen bonding mechanisms, establishing seven discrete hydrogen bonds with critical amino acid residues including Asp 519, Leu 406, Lys 429, Ser 410, and Lys 517. This hydrogen bond-dominant interaction profile contrasts markedly with the native ligand’s more diverse binding mechanism. The native ligand establishes a complex interaction network encompassing multiple binding modalities: pi-alkyl interactions with Ala 427 and Leu 406, pi-sigma interactions with Leu 522 and Val 414, pi-sulfur interactions with Met 469 and His 537, and electrostatic attractive charge interactions with Lys 429, Asp 534, and Asp 515. Despite this interaction diversity, the native ligand demonstrates inferior overall binding affinity. Significantly, doxorubicin exhibits unique pi-anion interactions absent in the native ligand binding profile, contributing additional stabilization energy to the drug-protein complex and providing a structural basis for the observed binding affinity differences.
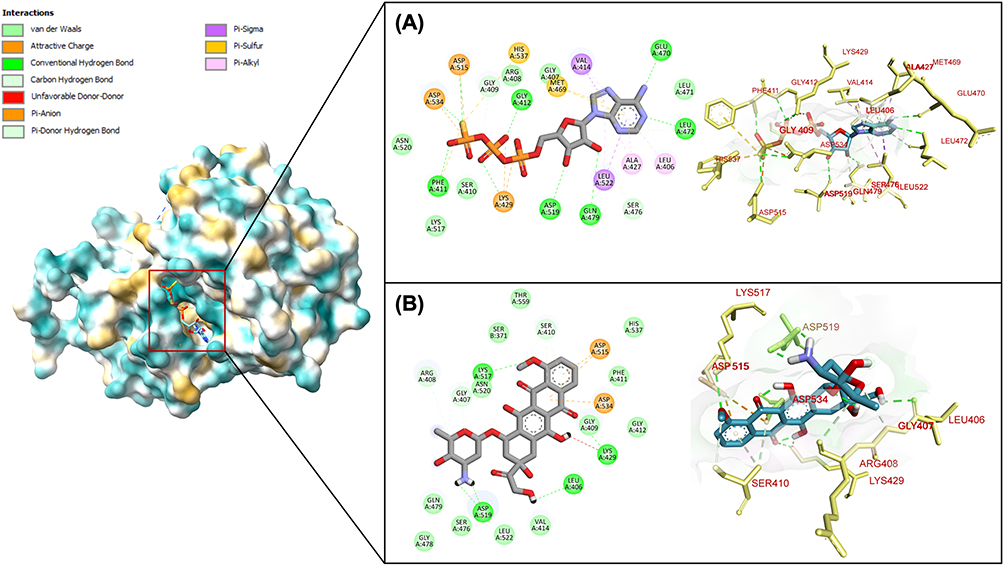 |
Figure 4 Molecular interaction of (A) native ligand and (B) doxorubicin with amino acid residues in NF-KB binding pocket. |
The molecular interaction profile between doxorubicin and HIF-1α demonstrates both quantitative and qualitative advantages compared to native ligand binding. Figure 5 illustrates that doxorubicin establishes six hydrogen bond interactions with amino acid residues Ser 184, His 279, Asp 201, Gln 239, Phe 100, and Gln 147. This extensive hydrogen bonding network represents a fundamental advantage, as the native ligand completely lacks hydrogen bond interactions within the HIF-1α active site. Beyond hydrogen bonding, doxorubicin forms three pi-sigma interactions specifically involving Trp 296 and Leu 186 residues, quantitatively surpassing the native ligand’s two pi-sigma interactions. While both compounds demonstrate equivalent pi-pi T-shaped interaction frequencies, their binding mechanisms diverge significantly in interaction type distribution. The native ligand predominantly relies on pi-alkyl interactions for protein binding stabilization, whereas doxorubicin achieves binding through the more energetically favorable combination of hydrogen bonds and pi-sigma interactions. This mechanistic distinction provides structural rationale for doxorubicin’s superior binding affinity toward HIF-1α. The predominance of hydrogen bonding and optimized pi-sigma interactions creates more stable drug-protein complexes with lower dissociation rates, which may contribute to increased complex stability and support further investigation of these interactions in biological contexts.
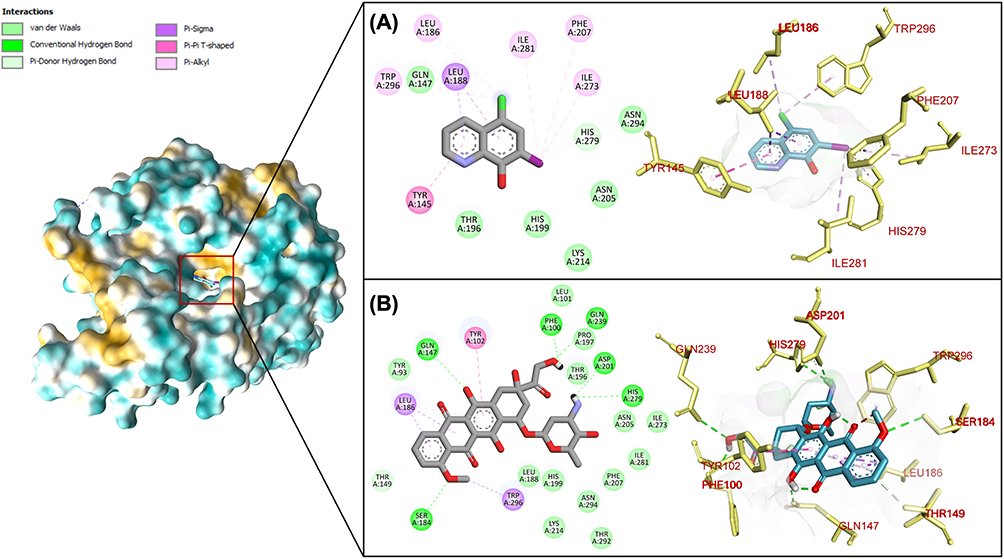 |
Figure 5 Molecular interaction of (A) native ligand and (B) doxorubicin with amino acid residues in HIF-1A binding pocket. |
Molecular Dynamic Simulation
Further evaluation of doxorubicin’s binding affinity toward NF-κB and HIF-1α was conducted through molecular dynamics (MD) simulations to comprehensively evaluate the structural characteristics, stability profiles, and conformational flexibility of the formed doxorubicin-protein complexes. The MD simulations provided dynamic insights into protein-drug interactions under physiological conditions, offering temporal resolution of binding behavior over extended time scales. Figure 5 presents the comprehensive dynamic behavior of doxorubicin-NF-κB and doxorubicin-HIF-1α complexes throughout a 50-nanosecond simulation period, revealing distinct stability patterns and structural characteristics for each complex.
Root Mean Square Deviation (RMSD) analysis demonstrated differential stability profiles between the two drug-target complexes, as illustrated in Figure 6A. The doxorubicin-NF-κB complex exhibited greater structural stability compared to the doxorubicin-HIF-1α complex throughout the entire simulation trajectory. Specifically, the doxorubicin-HIF-1α complex displayed considerable RMSD fluctuations during the 50-nanosecond simulation period, with pronounced structural deviations occurring particularly at the 20-nanosecond and 40-nanosecond time points, indicating significant conformational changes and reduced binding stability. In contrast, the doxorubicin-NF-κB complex maintained relatively minimal RMSD fluctuations throughout the simulation period, suggesting consistent structural integrity and stable drug-protein interactions.
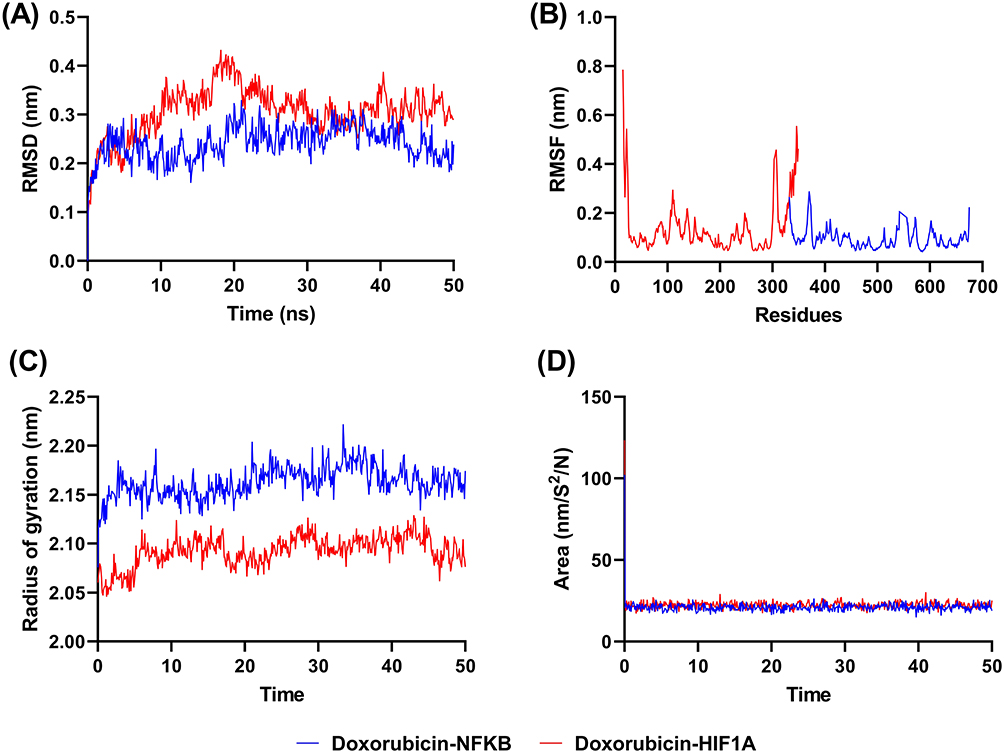 |
Figure 6 Molecular dynamic simulation of doxorubicin-NFKB/HIF-1A in 50 ns. (A) RMSD, (B) RMSF, (C) radius of gyration, and (D) SASA. |
The differential stability patterns observed in RMSD analysis were further corroborated by Root Mean Square Fluctuation (RMSF) measurements, as presented in Figure 6B, which quantify per-residue flexibility and local conformational changes during the simulation period. The RMSF data revealed that the doxorubicin-HIF-1α complex underwent extensive interaction rearrangements during the simulation, characterized by dynamic changes in amino acid residue interactions and binding site conformations. This high degree of conformational flexibility suggests increased dynamic rearrangement within the binding region. Conversely, the doxorubicin-NF-κB complex demonstrated minimal conformational changes, indicating consistent interaction patterns and stable binding site architecture throughout the simulation period.
Beyond stability and flexibility assessments, the ability of doxorubicin binding on the structural compactness of NF-κB and HIF-1α was evaluated through radius of gyration (Rg) measurements, which quantify the overall structural compactness and folding state of protein molecules. Lower Rg values indicate more compact or tightly folded protein structures, while higher values suggest structural expansion or partial unfolding. Figure 6C demonstrates that doxorubicin binding induces differential effects on target protein compactness, with the NF-κB complex exhibiting higher Rg values compared to the HIF-1α complex. This observation suggests that doxorubicin binding promotes greater structural expansion in NF-κB relative to HIF-1α, though the differences observed were not statistically significant across the simulation timeframe.
The Solvent Accessible Surface Area (SASA) parameter provides critical information regarding molecular surface exposure to aqueous environments, serving as an indicator of protein conformational changes and drug binding accessibility. SASA values reflect the extent to which protein surfaces remain available for solvent interaction, with changes in SASA indicating conformational alterations that may affect drug binding capacity and protein function. Figure 6D reveals that both doxorubicin-NF-κB and doxorubicin-HIF-1α complexes exhibit comparable SASA values throughout the simulation period. This similarity in solvent accessibility suggests that both complexes maintain equivalent degrees of conformational flexibility and demonstrate similar amenability to drug binding interactions, indicating that surface accessibility is not a limiting factor for doxorubicin association with either target protein under physiological conditions.
The binding free energies of doxorubicin toward HIF-1A and NF-κB were further evaluated using MM-PBSA and MM-GBSA approaches to validate the stability and strength of the ligand–protein interactions observed during molecular dynamics simulations (Table 3). Based on the MM-PBSA calculations, doxorubicin exhibited favorable binding affinities toward both targets, with total binding free energies (ΔG_bind) of −14.91 ± 5.27 kcal/mol for HIF-1A and −11.83 ± 6.82 kcal/mol for NF-κB. The binding was predominantly driven by van der Waals interactions, contributing −39.38 ± 5.41 kcal/mol and −41.64 ± 3.80 kcal/mol for HIF-1A and NF-κB, respectively. Electrostatic interactions (EEL) also contributed favorably, particularly for NF-κB (−27.64 ± 13.43 kcal/mol), indicating the importance of polar contacts in stabilizing the complex. However, these favorable gas-phase interactions were partially offset by unfavorable solvation free energies, consistent with typical MM-PBSA profiles for protein–ligand systems.
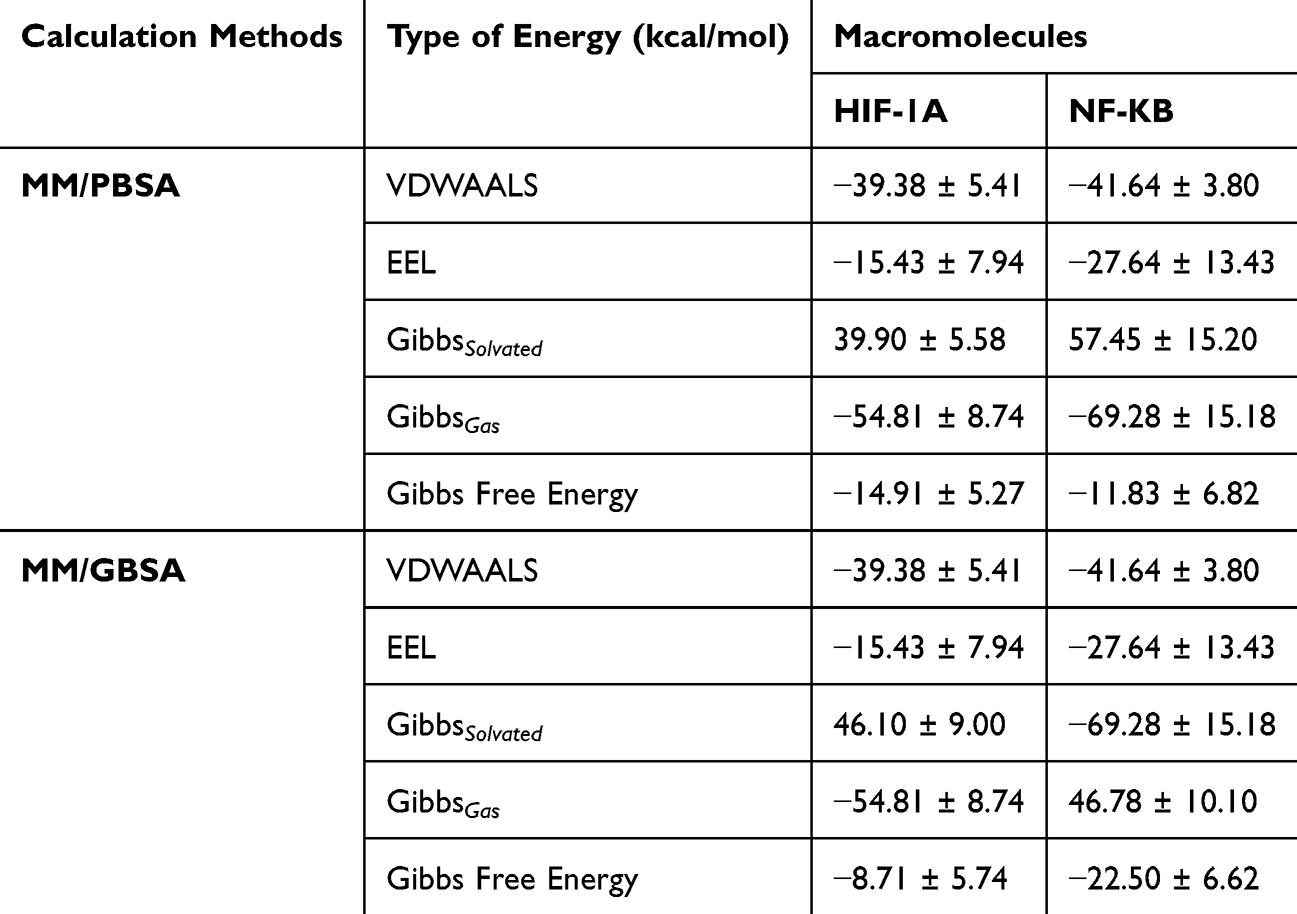 |
Table 3 Binding Free Energy Contributions Calculated Using MM-PBSA and MM-GBSA methods |
Similarly, MM-GBSA analysis supported the MM-PBSA findings and revealed stable binding of doxorubicin to both macromolecules. The calculated Gibbs free energies were −8.71 ± 5.74 kcal/mol for the HIF-1A complex and −22.50 ± 6.62 kcal/mol for the NF-κB complex, indicating a stronger binding preference toward NF-κB under the GBSA model. As observed in MM-PBSA results, van der Waals interactions were the dominant stabilizing force, while electrostatic contributions further enhanced binding affinity, especially in the NF-κB complex.
Overall, the consistent negative binding free energies obtained from both MM-PBSA and MM-GBSA calculations confirm the thermodynamic favorability of doxorubicin binding to HIF-1A and NF-κB. These results reinforce the reliability of the docking protocol and molecular dynamics simulations, providing additional quantitative support for the stability of the proposed ligand–target complexes.
In vitro Cytotoxic Activity Assay
The cytotoxic activity of doxorubicin against triple-negative breast cancer cells was evaluated through in vitro experimental studies following the in silico analyses. Cytotoxicity evaluation was conducted using MDA-MB-231 breast cancer cells, a well-established cellular model for TNBC research, with cells cultured in both RPMI-1640 and DMEM to assess media-dependent effects on drug efficacy. Figure 7 demonstrates a higher cytotoxic activity of doxorubicin’s anticancer activity when MDA-MB-231 cells were cultured in RPMI-1640 medium compared to DMEM, revealing significant media-dependent modulation of drug efficacy. In DMEM culture conditions, doxorubicin exhibited cytotoxic activity with an IC50 value of 2.34 µM (95% CI: 2.11–2.74). In contrast, culturing MDA-MB-231 cells in RPMI-1640 medium resulted in a lower IC50 value of 1.07 µM (95% CI: 0.92–1.32 µM), indicating increased cytotoxic sensitivity under these conditions.
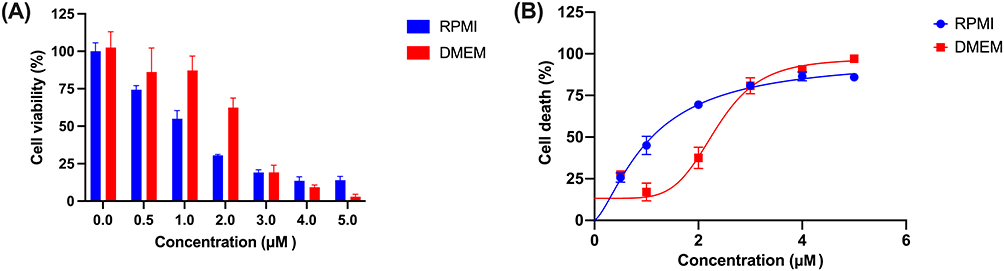 |
Figure 7 Cytotoxic assessment of doxorubicin against MDA-MB-231 breast cancer cells in RPMI and DMEM medium. (A) Histogram representation of cell viability (%) at varying concentrations after 72 hours evaluated using MTT assay; (B) Dose-response curves showing the concentration-dependent increase in cell death (%). |
This difference corresponds to an approximately 2.2-fold change in IC50 values between the two culture conditions, suggesting that the nutritional composition and ionic environment provided by RPMI-1640 medium may influence cellular conditions that affect doxorubicin response. The observed media-dependent differences highlight the influence of experimental conditions on cytotoxic responses and provide complementary in vitro support for the computational findings.
Gene Expression Analysis
Gene expression analysis of NF-κB and HIF-1α was conducted to further evaluate the in silico predictions regarding the effects of doxorubicin on the expression of these targets. The experimental validation employed MDA-MB-231 breast cancer cells treated with varying doxorubicin concentrations across two distinct culture media, providing comprehensive assessment of drug-target engagement under different nutritional and physiological conditions. Figure 8 presents a detailed analysis of gene expression patterns following doxorubicin supplementation, revealing concentration-dependent and medium-specific modulation of target gene levels.
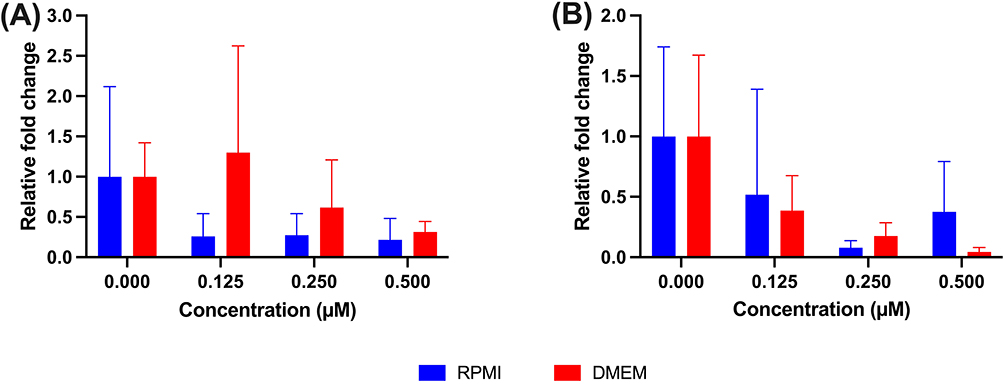 |
Figure 8 Relative fold change of gene expression upon the supplementation of doxorubicin evaluated from MDA-MB-231 breast cancer cells in RPMI and DMEM medium. (A) NF-κB and (B) HIF-1A. |
The analysis of NF-κB mRNA expression, as illustrated in Figure 8A, reveals markedly different dose-response profiles between RPMI-1640 and DMEM culture conditions, demonstrating the critical influence of cellular environment on drug-target interactions. In RPMI-1640 medium, NF-κB expression demonstrates a consistent dose-dependent reduction in expression, initiating from baseline expression levels (1.0 relative fold change in vehicle control) and progressively declining to approximately 0.25 relative fold change at both 0.125 μM and 0.250 μM doxorubicin concentrations. Notably, a slight recovery in expression to 0.30 relative fold change occurs at the highest tested concentration of 0.500 μM, suggesting potential adaptive cellular responses or saturation effects at elevated drug concentrations. In contrast, DMEM culture conditions reveal a biphasic response pattern characterized by initial expression maintenance followed by concentration-dependent modulation. The protein expression initially remains at baseline levels, subsequently exhibiting a paradoxical upregulation to approximately 1.25 relative fold change at 0.125 μM doxorubicin concentration. This transient increase is followed by progressive suppression, with expression levels declining to 0.60 and 0.30 relative fold changes at 0.250 μM and 0.500 μM concentrations, respectively. This biphasic pattern suggests complex regulatory mechanisms involving potential compensatory responses at lower drug concentrations before exhibiting greater expression reduction at higher concentrations.
HIF-1α gene expression analysis, presented in Figure 8B, demonstrates distinct concentration-dependent suppression patterns across both culture media, with notable differences in response magnitude and kinetics. In RPMI-1640 medium, HIF-1α expression maintains baseline levels initially, followed by substantial suppression to approximately 0.50 relative fold change at 0.125 μM doxorubicin concentration. The suppression continues progressively, reaching the lowest expression level of approximately 0.10 relative fold change at 0.250 μM, representing a 90% reduction in expression. Interestingly, partial recovery to 0.35 relative fold change occurs at the highest concentration of 0.500 μM, indicating potential cellular adaptation mechanisms or alternative regulatory pathways activation at elevated drug concentrations. DMEM culture conditions demonstrate comparable baseline expression levels but exhibit different dose-response kinetics and maximum suppression characteristics. The expression levels decline to 0.35 relative fold change at 0.125 μM, reaching the nadir of approximately 0.20 relative fold change at 0.250 μM concentration. At the highest tested concentration of 0.500 μM, HIF-1α expression remains minimally detectable at 0.05 relative fold change, representing the most profound suppression observed across all experimental conditions and suggesting sustained reduction in gene expression at higher concentrations.
The comprehensive gene expression analysis reveals significant medium-dependent differences in target suppression, with both culture environments demonstrating dose-dependent inhibition of HIF-1α expression, though RPMI-1640 medium exhibits greater response variability across the concentration range tested. The NF-κB expression patterns display more pronounced medium-specific differences, with DMEM culture conditions showing initial protein upregulation at lower concentrations before subsequent suppression, while RPMI-1640 demonstrates consistent downregulation across most tested concentrations. These findings provide experimental support for the computational predictions regarding doxorubicin-associated modulation of NF-κB and HIF-1α expression. The observed protein suppression patterns support the molecular docking and dynamics simulation results, supporting the observation that doxorubicin treatment is associated with reduced NF-κB and HIF-1α expression levels at the tested concentrations, and providing transcriptional-level evidence that complements the computational findings.
Discussion
The present study provides transcriptional-level evidence suggesting the concurrent involvement of NF-κB- and HIF-1α-related pathways in the cellular response of triple-negative breast cancer cells to doxorubicin treatment. This observation is relevant in the context of this aggressive cancer subtype, which accounts for approximately 15–20% of all breast cancers and demonstrates notably poor prognosis due to the absence of estrogen receptor, progesterone receptor, and HER2 expression.1–3 The concurrent involvement of NF-κB and HIF-1α may represent a biologically relevant concept for further investigation, given their interconnected roles in cancer-related signaling. NF-κB serves as a master regulator of inflammatory responses and cell survival pathways, promoting cancer cell proliferation, invasion, and resistance to apoptosis through the activation of numerous downstream target genes.30–32 Previous studies have demonstrated that constitutive NF-κB activation in breast cancer correlates with enhanced metastatic potential and chemotherapy resistance,33 with research by Shao et al34 showing that NF-κB inhibition sensitizes cancer cells to various chemotherapeutic agents. Similarly, HIF-1α functions as the key mediator of cellular responses to hypoxic conditions, facilitating angiogenesis, metabolic reprogramming, and epithelial-mesenchymal transition through the transcriptional activation of over 100 hypoxia-responsive genes.35–37 The work of Zheng et al38 and Nie et al39 demonstrated that HIF-1α overexpression in breast cancer correlates with increased mortality and resistance to therapy, supporting its relevance as a prognostic marker and potential therapeutic target.
The computational framework employed in this study provides evidence supporting favorable binding interactions between doxorubicin and both target proteins. The binding energy values of −9.82 kcal/mol for NF-κB and −7.84 kcal/mol for HIF-1α, which are lower than those of the corresponding native ligands, indicate thermodynamically favorable interactions within the context of molecular docking analyses.40,41 These findings align with previous computational studies demonstrating that binding energies lower than those of native ligands are generally associated with potential biological relevance of protein–drug interactions in the context of computational interaction prediction, as suggested by docking-based analyses.42,43 The molecular dynamics simulations further characterize these interactions by demonstrating complex stability over extended time periods, suggesting consistent protein-ligand association under the simulated conditions. The observed binding affinity and stability may be attributed to the formation of hydrogen and pi–sigma interactions between doxorubicin and amino acid residues within the binding sites of NF-κB and HIF-1α. Hydrogen bonds enhance binding strength by creating specific, directional electrostatic interactions that increase molecular stability and specificity while contributing to binding strength by enhancing molecular recognition and complex stability.44,45 Pi-sigma interactions, although individually weak, may contribute to overall binding affinity when combined with other non-covalent interactions such as hydrogen bonding, influencing overall binding affinity of doxorubicin to NF-κB and HIF-1α.
The in vitro studies provide experimental support complementary to the computational analyses through cytotoxicity assays that demonstrated IC50 values ranging from 1.07 to 2.40 μM. These concentrations fall within the range of plasma concentrations reported for doxorubicin following standard intravenous administration, as described by Huang et al46 (0.009–7.359 μM). It is important to note that previous studies have frequently reported activation of canonical NF-κB signaling by doxorubicin in breast cancer, including TNBC, primarily through protein-level mechanisms such as IκBα degradation and p65 nuclear translocation.31,47 The quantitative PCR results demonstrating 40–90% reduction in NF-κB and HIF-1α mRNA expression levels provide transcriptional-level experimental support consistent with the computational findings, with this substantial downregulation suggesting that doxorubicin treatment is associated with reduced expression of both transcription factors through mechanisms that may extend beyond simple competitive binding; however, these observations remain limited to transcriptional-level changes and do not establish functional inhibition or direct target engagement. The observation of concurrent transcriptional modulation of NF-κB and HIF-1α expression may offer hypotheses for future studies exploring mechanisms related to chemotherapy resistance in triple-negative breast cancer, a clinical challenge that has limited treatment options for patients with this aggressive disease subtype. NF-κB activation has been extensively linked to doxorubicin resistance through multiple mechanisms, including enhanced DNA repair capacity, increased anti-apoptotic protein expression, and activation of drug efflux pumps such as P-glycoprotein.48–50 Research has demonstrated that NF-κB inhibition significantly enhances doxorubicin sensitivity in resistant breast cancer cell lines,51,52 supporting the therapeutic potential of targeting this pathway in combination with conventional chemotherapy. Similarly, HIF-1α-mediated resistance mechanisms involve metabolic reprogramming toward glycolysis, enhanced angiogenesis, and activation of survival signaling pathways under hypoxic conditions commonly found in solid tumors.10,35,36
The proposed dual-pathway framework illustrated in Figure 9 represents a conceptual and hypothesis-generating model that differs from conventional single-target approaches in breast cancer research, which have often focused on single-target inhibition strategies that may exhibit limited efficacy due to compensatory pathway activation and resistance development. The work by Mahmoud et al53 and Xiong et al54 highlighted the challenges associated with conventional approaches in triple-negative breast cancer, emphasizing the need for alternative research strategies to better address resistance mechanisms present in this cancer subtype. Recent studies have explored combination-based approaches targeting multiple pathways simultaneously, with research by Brockmueller et al55 demonstrating that therapies targeting both NF-κB and HIF-1α pathways show enhanced efficacy in preclinical models, supporting the potential relevance of concurrently modulating NF-κB-and HIF-1α-related pathways, as suggested by the findings of this study.
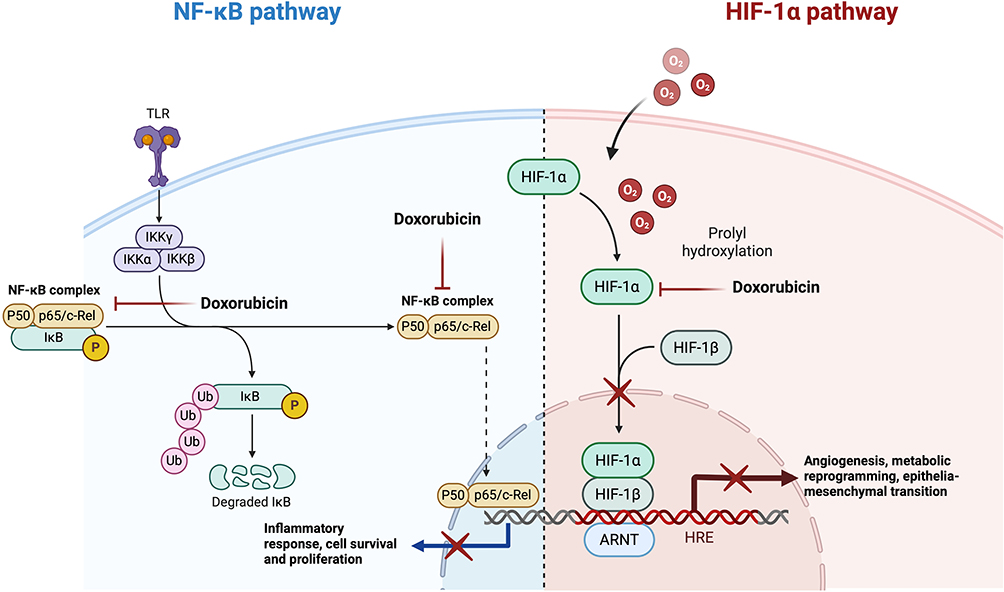 |
Figure 9 Proposed conceptual model illustrating NF-κB and HIF-1α-related signaling pathways potentially associated with doxorubicin treatment in triple-negative breast cancer (TNBC) cells. The model integrates computational predictions and transcriptional-level observations from the present study. Arrows and inhibitory symbols indicate hypothesized pathway interactions and do not represent experimentally validated functional inhibition. This figure is intended as a hypothesis-generating framework to guide future mechanistic and functional investigations. |
The potential relevance of these findings extends beyond mechanistic observations, as concurrent modulation of NF-κB- and HIF-1α-related pathways may offer conceptual advantages over single-pathway approaches in the context of resistance development. Studies by Roy et al56 and Giordano et al57 have shown that cancer cells with concurrent disruption of multiple survival pathways may exhibit reduced adaptive capacity compared to those with single-pathway perturbation. The substantial reduction in NF-κB and HIF-1α mRNA expression observed in this study suggests that concurrent transcriptional modulation of these pathways may be achievable within the concentration range tested.
Future research should aim to extend these findings across additional triple-negative breast cancer cell lines with diverse genetic backgrounds and resistance profiles to strengthen the generalizability of the observed transcriptional modulation. In vivo studies using appropriate animal models may provide additional evidence to support the translational relevance of these observations. Furthermore, future investigations incorporating protein-level validation and functional assays under hypoxic or hypoxia-mimicking conditions would help clarify the regulatory roles of NF-κB and HIF-1α beyond transcriptional changes observed under normoxic conditions. The development of biomarkers to monitor NF-κB– and HIF-1α–related pathway modulation could further facilitate hypothesis-driven translational studies. Overall, the identification of this proposed molecular framework opens new avenues for future research aimed at investigating coordinated pathway interactions in triple-negative breast cancer.
Conclusion
This study integrates network pharmacology, molecular docking, molecular dynamics simulations, and in vitro analyses to explore the molecular actions of doxorubicin in triple-negative breast cancer. The findings demonstrate that doxorubicin is associated with concurrent transcriptional modulation of NF-κB- and HIF-1α-related pathways at the transcriptional level, supported by favorable protein–ligand interactions predicted through computational approaches and consistent gene expression changes observed in vitro. Together, these results suggest that NF-κB and HIF-1α may represent interconnected components within the broader network of doxorubicin-responsive pathways in triple-negative breast cancer. While the present findings are limited to in vitro and transcriptional analyses under normoxic conditions, they provide a conceptual framework for future studies aimed at further elucidating coordinated pathway regulation and refining therapeutic strategies for this aggressive breast cancer subtype.
Acknowledgments
All in vitro experiments were conducted at the Biomedical Laboratory, Faculty of Medicine, Universitas Padjadjaran, Indonesia. We also acknowledge Widad Aghnia Shalannandia for technical assistance.
Funding
This work was supported by Internal Research Grant, Universitas Padjadjaran, Indonesia (Grant No. 904/UN6.3.1/PT.00/2025). The publication charge was supported by Universitas Padjadjaran (UNPAD) through the Indonesian Endowment Fund for Education (LPDP) under the Ministry of Higher Education, Science and Technology, as part of the EQUITY Program (Contract Nos. 4303/B3/DT.03.08/2025 and 3927/UN6.RKT/HK.07.00/2025).
Disclosure
The author(s) report no conflicts of interest in this work.
References
1. Zhang Z, Zhang R, Li D. Molecular Biology Mechanisms and Emerging Therapeutics of Triple-Negative Breast Cancer. Biologics. 2023;17:113–17. doi:10.2147/BTT.S426392
2. Dogra AK, Prakash A, Gupta S, Gupta M. Prognostic Significance and Molecular Classification of Triple Negative Breast Cancer: a Systematic Review. Eur J Breast Health. 2025;21(2):101–114. doi:10.4274/ejbh.galenos.2025.2024-10-2
3. Chen JQ, Russo J. ERα-negative and triple negative breast cancer: molecular features and potential therapeutic approaches. Biochim Biophys Acta Rev Cancer. 2009;1796(2):162–175. doi:10.1016/j.bbcan.2009.06.003
4. Bhutani V, Varzideh F, Wilson S, Kansakar U, Jankauskas SS, Santulli G. Doxorubicin-Induced Cardiotoxicity: a Comprehensive Update. J Cardiovasc Dev Dis. 2025;12(6). doi:10.3390/jcdd12060207
5. Sinha SJ, Kumar B, Prasad CP, Chauhan SS, Kumar M. Emerging Research and Future Directions on Doxorubicin: a Snapshot. Asian Pac J Cancer Prev. 2025;26(1):5–15. doi:10.31557/APJCP.2025.26.1.5
6. Wang C, Fan P, Wang Q. Evolving therapeutics and ensuing cardiotoxicities in triple-negative breast cancer. Cancer Treat Rev. 2024;130:102819. doi:10.1016/j.ctrv.2024.102819
7. Liu L, Bai J, Hu L, Jiang D. Hypoxia-mediated activation of hypoxia-inducible factor-1α in triple-negative breast cancer: a review. Medicine. 2023;102(43):E35493. doi:10.1097/MD.0000000000035493
8. Pavitra E, Kancharla J, Gupta VK, et al. The role of NF-κB in breast cancer initiation, growth, metastasis, and resistance to chemotherapy. Biomed. Pharmacother. 2023;163:114822. doi:10.1016/j.biopha.2023.114822
9. Jiang Y, Zhu Y, Wang X, et al. Temporal regulation of HIF-1 and NF-κB in hypoxic hepatocarcinoma cells. Oncotarget. 2015;6(11):9409–9419.
10. Kwon H-C, Kim SH, Oh SY, et al. Clinicopathological significance of nuclear factor-kappa B, HIF-1 alpha, and vascular endothelial growth factor expression in stage III colorectal cancer. Cancer Sci. 2010;101(6):1557–1561. doi:10.1111/j.1349-7006.2010.01553.x
11. Bandarra D, Biddlestone J, Mudie S, Muller HA, Rocha S. HIF-1α restricts NF-κB dependent gene expression to control innate immunity signals. Dis Model Mech. 2014;8(2):169–181. doi:10.1242/dmm.017285
12. Rius J, Guma M, Schachtrup C, et al. NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature. 2008;453(7196):7196):807–811. doi:10.1038/nature06905
13. Liu Q, Guan C, Liu C, Li H, Wu J, Sun C. Targeting hypoxia-inducible factor-1alpha: a new strategy for triple-negative breast cancer therapy. Biomed. Pharmacother. 2022;156:113861. doi:10.1016/j.biopha.2022.113861
14. Kciuk M, Gielecińska A, Mujwar S, et al. Doxorubicin—An Agent with Multiple Mechanisms of Anticancer Activity. Cells. 2023;12(4). doi:10.3390/cells12040659
15. Patricelli C, Lehmann P, Oxford JT, Pu X. Doxorubicin-induced modulation of TGF-β signaling cascade in mouse fibroblasts: insights into cardiotoxicity mechanisms. Sci Rep. 2023;13(1):18944. doi:10.1038/s41598-023-46216-7
16. Al-malky HS, Osman A-M-M, Damanhouri ZA, et al. Modulation of doxorubicin-induced expression of the multidrug resistance gene in breast cancer cells by diltiazem and protection against cardiotoxicity in experimental animals. Cancer Cell Int. 2019;19(1):191. doi:10.1186/s12935-019-0912-0
17. Taymaz-Nikerel H, Karabekmez ME, Eraslan S, Kırdar B. Doxorubicin induces an extensive transcriptional and metabolic rewiring in yeast cells. Sci Rep. 2018;8(1):13672. doi:10.1038/s41598-018-31939-9
18. Li Y, Zhao B, Peng J, et al. Inhibition of NF-κB signaling unveils novel strategies to overcome drug resistance in cancers. Drug Resist Updates. 2024;73:101042. doi:10.1016/j.drup.2023.101042
19. Lin Y, Bai L, Chen W, Xu S. The NF-κB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin Ther Targets. 2010;14(1):45–55. doi:10.1517/14728220903431069
20. Li L, Kar S. Leveraging network pharmacology for drug discovery: integrative approaches and emerging insights. Med Drug Discov. 2025;27:100220. doi:10.1016/j.medidd.2025.100220
21. Yabansabra YR, Bowaire AN, Setiawansyah A, Simaremare ES, Asmuruf FA. Integrating the Network Pharmacology and Molecular Dynamic Simulation to Reveal Pharmacological Mechanism of Arcangelisi flava (L.) Merr in Treating Inflammation. Biointerface Res Appl Chem. 2025;15(3):1–18. doi:10.33263/BRIAC153.044
22. Setiawansyah A. Network pharmacology and molecular docking simulation uncovered the potential of hexacyclinic acid as anti-osteoarthritis by regulating IL-17 signaling pathway. Acta Chimica Asiana. 2025;8(1):584–598. doi:10.29303/aca.v8i1.238
23. Sadaqa E, Setiawansyah A, Arsul MI, et al. pH-Sensitive Niosomal Nanoencapsulation of Beta-Caryophyllene and its Novel Pathway in Triple Negative Breast Cancer. Biointerface Res Appl Chem. 2025;15(4):1–18. doi:10.33263/BRIAC154.047
24. Morris GM. AutoDock Version 4.2 - User Guide. 2010:1–49.
25. Arsul MI, Setiawansyah A, Insanu M, Fidrianny I. Antihyperuricemia and chemical composition of Boehmeria virgata, in vitro and in silico approach with ADME prediction. Nat Prod Res. 2025. doi:10.1080/14786419.2025.2471848
26. Páll S, Zhmurov A, Bauer P, et al. Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J Chem Phys. 2020;153(13):134110. doi:10.1063/5.0018516
27. Lee J, Cheng X, Swails JM, et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J Chem Theory Comput. 2016;12(1):405–413. doi:10.1021/acs.jctc.5b00935
28. Kim SW, Kim S-J, Langley RR, Fidler IJ. Modulation of the cancer cell transcriptome by culture media formulations and cell density. Int J Oncol. 2015;46(5):2067–2075. doi:10.3892/ijo.2015.2930
29. Volpe DA, Hamed SS, Zhang LK. Use of Different Parameters and Equations for Calculation of IC50 Values in Efflux Assays: potential Sources of Variability in IC50 Determination. AAPS J. 2014;16(1):172–180. doi:10.1208/s12248-013-9554-7
30. Naugler WE, Karin M. NF-κB and cancer - identifying targets and mechanisms. Curr Opin Genet Dev. 2008;18(1):19–26. doi:10.1016/j.gde.2008.01.020
31. Guo Q, Jin Y, Chen X, et al. NF-κB in biology and targeted therapy: new insights and translational implications. Signal Transduct Target Ther. 2024;9(1):53. doi:10.1038/s41392-024-01757-9
32. Mao H, Zhao X, Sun S. NF-κB in inflammation and cancer. Cell Mol Immunol. 2025;22(8):811–839. doi:10.1038/s41423-025-01310-w
33. Devanaboyina M, Kaur J, Whiteley E, et al. NF-κB Signaling in Tumor Pathways Focusing on Breast and Ovarian Cancer. Oncol Rev. 2022;16. doi:10.3389/or.2022.10568.
34. Shao L, Wu L, Zhou D. Sensitization of tumor cells to cancer therapy by molecularly targeted inhibition of the inhibitor of nuclear factor κB kinase. Transl Cancer Res. 2012;1(2):100–108. doi:10.3978/j.issn.2218-676X.2012.05.04
35. Zhao Y, Xing C, Deng Y, Ye C, Peng H. HIF-1α signaling: essential roles in tumorigenesis and implications in targeted therapies. Genes Dis. 2024;11(1):234–251. doi:10.1016/j.gendis.2023.02.039
36. Yfantis A, Mylonis I, Chachami G, et al. Transcriptional Response to Hypoxia: the Role of HIF-1-Associated Co-Regulators. Cells. 2023;12(5). doi:10.3390/cells12050798
37. Malekan M, Ebrahimzadeh MA, Sheida F. The role of Hypoxia-Inducible Factor-1alpha and its signaling in melanoma. Biomed. Pharmacother. 2021;141:111873. doi:10.1016/j.biopha.2021.111873
38. Zheng X-D, Li H-Y, Gao S-Y, Wang Q, Liu J-B. High hypoxia inducible factor-1α expression is associated with reduced survival in patients with breast cancer: a meta-analysis. World J Clin Oncol. 2025;16(6). doi:10.5306/wjco.v16.i6.105691
39. Nie C, Lv H, Bie L, Hou H, Chen X. Hypoxia-inducible factor 1-alpha expression correlates with response to neoadjuvant chemotherapy in women with breast cancer. Medicine. 2018;97(51). doi:10.1097/MD.0000000000013551
40. Hu J, Hu S, Xia M, Zheng K, Zhang X. Drug-target binding affinity prediction based on power graph and word2vec. BMC Med Genomics. 2025;18(1):9. doi:10.1186/s12920-024-02073-5
41. Chen YZ, Ung CY. Prediction of potential toxicity and side effect protein targets of a small molecule by a ligand–protein inverse docking approach. J Mol Graph Model. 2001;20(3):199–218. doi:10.1016/S1093-3263(01)00109-7
42. Susanti G, Aldi Y, Handayani D, Ismed F, Setiawansyah A. Uncovering The Pharmacological Mechanism of Ficus elastica as Anti-hyperlipidemia Candidate: LC-HRMS, Network Pharmacology, In vitro and In vivo Studies. J Multidisciplinary Appl Natl Sci. 2025;5(1):332–351. doi:10.47352/jmans.2774-3047.249
43. Susanti G, Aldi Y, Handayani D, Ismed F, Setiawansyah A. Chemical and Pharmacological Potential of Ficus elastica Fractions for Anti-Hyperlipidemia: an Integrative Analysis from Molecular Docking, In Vitro, and In Vivo Studies. Chemical Methodologies. 2025;9(8):691–701. doi:10.48309/chemm.2025.517694.1944
44. Singh RK, Tiwari MK, Kim IW, Chen Z, Lee JK. Probing the role of sigma π interaction and energetics in the catalytic efficiency of endo-1,4-β-xylanase. Appl Environ Microbiol. 2012;78(24):8817–8821. doi:10.1128/AEM.02261-12
45. Chen D, Oezguen N, Urvil P, Ferguson C, Dann SM, Savidge TC. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci Adv. 2016;2(3):1 doi:10.1126/sciadv.1501240
46. Huang Y, Yang F, Guo L, et al. Plasma Pharmacokinetics and Tissue Distribution of Doxorubicin in Rats following Treatment with Astragali Radix. Pharmaceuticals. 2022;15(9). doi:10.3390/ph15091104
47. Dalmases A, González I, Menendez S, et al. Deficiency in p53 is required for doxorubicin induced transcriptional activation of NF-κB target genes in human breast cancer. Oncotarget. 2014;5(1):196–210. doi:10.18632/oncotarget.1556
48. Yi SY, Ahn JS, Uhm JE, et al. Favorable response to doxorubicin combination chemotherapy does not yield good clinical outcome in patients with metastatic breast cancer with triple-negative phenotype. BMC Cancer. 2010;10. doi:10.1186/1471-2407-10-527
49. Jeremias I, Kupatt C, Baumann B, Herr I, Wirth T, Debatin KM. Inhibition of Nuclear Factor κB Activation Attenuates Apoptosis Resistance in Lymphoid Cells. Blood. 1998;91(12):4624–4631. doi:10.1182/blood.V91.12.4624
50. Fang XJ, Jiang H, Zhu YQ, Zhang LY, Fan QH, Tian Y. Doxorubicin induces drug resistance and expression of the novel CD44st via NF-κB in human breast cancer MCF-7 cells. Oncol Rep. 2014;31(6):2735–2742. doi:10.3892/or.2014.3131
51. deGraffenried LA, Chandrasekar B, Friedrichs WE, et al. NF-κB inhibition markedly enhances sensitivity of resistant breast cancer tumor cells to tamoxifen. Ann Oncol. 2004;15(6):885–890. doi:10.1093/annonc/mdh232
52. Abdin SM, Tolba MF, Zaher DM, Omar HA. The impact of NF-κB inhibition on the sensitivity of breast cancer cells to chemotherapy-induced apoptosis. Cancer Res. 2020;80(16_Supplement):4107. doi:10.1158/1538-7445.AM2020-4107
53. Mahmoud R, Ordóñez-Morán P, Allegrucci C. Challenges for Triple Negative Breast Cancer Treatment: defeating Heterogeneity and Cancer Stemness. Cancers (Basel). 2022;14(17). doi:10.3390/cancers14174280
54. Xiong N, Wu H, Yu Z. Advancements and challenges in triple-negative breast cancer: a comprehensive review of therapeutic and diagnostic strategies. Front Oncol. 2024;14. doi:10.3389/fonc.2024.1405491.
55. Brockmueller A, Girisa S, Motallebi M, Kunnumakkara AB, Shakibaei M. Calebin A targets the HIF-1α/NF-κB pathway to suppress colorectal cancer cell migration. Front Pharmacol. 2023;14. doi:10.3389/fphar.2023.1203436.
56. Roy R, Ria T, RoyMahaPatra D, Sk UH. Single Inhibitors versus Dual Inhibitors: role of HDAC in Cancer. ACS Omega. 2023;8(19):16532–16544. doi:10.1021/acsomega.3c00222
57. Giordano S, Petrelli A. From Single- to Multi-Target Drugs in Cancer Therapy: When Aspecificity Becomes an Advantage. Curr Med Chem. 2008;15(5):422–432. doi:10.2174/092986708783503212
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.