Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Injectable long-acting systems for Radix Ophiopogonis polysaccharide based on mono-PEGylation and in situ formation of a PLGA depot
Authors Shi X, Lin X, Zheng X, Feng Y, Shen L
Received 26 July 2014
Accepted for publication 11 October 2014
Published 28 November 2014 Volume 2014:9(1) Pages 5555—5563
DOI https://doi.org/10.2147/IJN.S71819
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
XiaoLi Shi,1 Xiao Lin,1 XiangWei Zheng,2 Yi Feng,2 Lan Shen1,2
1College of Chinese Materia Medica, 2Engineering Research Center of Modern Preparation Technology of TCM of Ministry of Education, Shanghai University of Traditional Chinese Medicine, Shanghai, People’s Republic of China
Background: Radix Ophiopogonis polysaccharide (ROP), a highly hydrophilic macromolecule, has a unique anti-ischemic action in the myocardium. One of the main problems with its use is its relatively short half-life in vivo. To solve this problem, injectable long-acting drug delivery systems, which combine mono-PEGylation (PEG, polyethylene glycol) with the in situ formation of poly(D,L-lactide-co-glycolide) copolymer (PLGA) depots, were tested in this study.
Methods: Through a moderate coupling reaction between 20 kDa amino-terminated methoxy-PEG and excessive ROP with activated hydroxyls, a long-circulating and bioactive mono-PEGylated ROP was prepared and characterized. A reasonable and applicable range of PLGA formulations loaded with the mono-PEGylated ROP were prepared, characterized, and evaluated in vivo.
Results: Relative to ROP, the half-life of which was only 0.5 hours, the conjugate alone, following subcutaneous administration, showed markedly prolonged retention in the systemic circulation, with a mean residence time in vivo of approximately 2.76 days. In combination with in situ-forming PLGA depots, the residence time of the conjugate in vivo was prolonged further. In particular, a long-lasting and steady plasma exposure for nearly a month was achieved by the formulation comprising 40% 30 kDa PLGA in N-methyl-2-pyrrolidone.
Conclusion: Long-lasting and steady drug exposure could be achieved using mono-PEGylation in combination with in situ formation of PLGA depots. Such a combination with ROP would be promising for long-term prophylaxis and/or treatment of myocardial ischemia. For high-dose and highly hydrophilic macromolecular drugs like ROP, more than one preparation technology might be needed to achieve week-long or month-long delivery per dosing.
Keywords: Radix Ophiopogonis polysaccharide, polyethylene glycol, poly(D,L-lactide-co-glycolide) copolymer, conjugation, in situ-forming system
Introduction
An increasing number of new active pharmaceutical ingredients are macromolecules, eg, proteins, peptides, and polysaccharides, which not only suffer from low bioavailability after oral administration, but also have short half-lives after parenteral administration,1 thus requiring repeated administration. In this context, long-acting drug delivery systems are highly desirable to avoid continuous infusions or frequent injections. Currently, there are two main strategies by which hydrophilic macromolecular drugs can be made long-acting, ie, by extending drug release using sustained-release delivery systems or by improving the retention of the drug in plasma by modifying the drug chemically.2
For the first strategy, in situ-forming systems (ISFS), especially poly(D,L-lactide-co-glycolide) copolymer (PLGA) depots, which can be injected as a low viscous solution but transform in the body into a gel or solid depot, have received considerable attention. They are based on biodegradable polymers with excellent biocompatibility, are approved for parenteral administration, can be easily modulated, and cover a wide range of release periods.1–3 So far, there are two commercialized products (Eligard® and Atridox®), both of which use N-methyl-2-pyrrolidone (NMP) as the solvent and polylactic acid or PLGA as the biodegradable, biocompatible, and water-insoluble matrix. For the second strategy, PEGylation, the process of covalent attachment of one or more polyethylene glycol (PEG) polymer chains to a therapeutic molecule, is now established as the method of choice for improving the pharmacokinetics and pharmacodynamics of parenteral agents. It has been applied successfully to small-molecule drugs, oligonucleotides, polypeptides, proteins, and long-circulating colloidal drug delivery systems.4,5
Radix Ophiopogonis polysaccharide (ROP), a natural graminan-type fructan with an average molecular weight of 4.8 kDa,6 has a unique anti-ischemic action in the myocardium that protects myocardial cells from damage induced by ischemia and promotes formation of microvessels in ischemic zones.7–10 However, as a highly hydrophilic macromolecule, ROP is rarely absorbed after oral administration (absolute bioavailability ~2%) and rapidly excreted by the kidneys following intravenous administration (plasma half-life ~30 minutes).11,12 These undesirable pharmacokinetic properties limit its efficacy and clinical use to a large extent.
In our previous studies, in situ-forming PLGA depots and PEGylation were tested separately on ROP. It was found that sustained release of ROP for several days was achieved by PLGA formulations; however, only low plasma concentrations of ROP were observed after an approximately half-day burst release, mainly because of its high water solubility and rapid renal elimination.13 It was found that the effects of PEGylation on the bioactivity and pharmacokinetics of ROP depend mainly on the degree of grafting and the molecular weight of the conjugate, respectively.14 Mono-PEGylated ROPs were confirmed to be capable of reducing the frequency of ROP injections from twice a day to once every 1–3 days, without sacrificing therapeutic efficacy.15
In light of the above, a mono-PEGylated ROP was prepared using a moderate coupling reaction between 20 kDa amino-terminated methoxy-PEG and excessive ROP having activated hydroxyls. Next, PLGA formulations loaded with the conjugate were prepared, characterized, and evaluated in vivo to determine the feasibility of PEGylation in combination with in situ formation of PLGA depots to achieve a week-long or even month-long lasting and steady drug exposure for highly water-soluble polysaccharides.
Materials and methods
Materials and animals
ROP was prepared according to a previously reported method.6 Linear amino-terminated poly(ethylene glycol) methyl ether (mPEG-NH2) hydrochloride, with a molecular weight of 20 kDa, was purchased from Jenkem Technology Co Ltd (Beijing, People’s Republic of China). PLGA, with a molar lactide to glycolide ratio of 50/50, a molecular weight of ~10, ~30, or ~50 kDa, and free carboxylic acid at the end of the polymeric backbone chain was purchased from Jinan Daigang Biomaterial Co Ltd (Shandong, People’s Republic of China). NMP was purchased from Sinopharm Chemical Reagent Ltd (Shanghai, People’s Republic of China). All other chemicals were of reagent grade and purchased from commercial sources.
Male Sprague-Dawley rats weighing 230–260 g were supplied by the Lab Animal Center of Shanghai University of Traditional Chinese Medicine. The animal ethical experimentation committee of Shanghai University of Traditional Chinese Medicine approved all the procedures for the animal experiments, which were carried out according to the requirements of the National Act on the Use of Experimental Animals (People’s Republic of China).
Preparation and characterization of 20 kDa PEG mono-PEGylated ROP
First, 20 kDa PEG mono-PEGylated ROP (MP20k-R) was synthesized through a moderate coupling reaction between hydroxyl-activated ROP and amino-terminated mPEG according to a previous report.15 The synthesis process mainly comprised two steps.
In step 1 (hydroxyl-activation of ROP), ROP 1 g, p-nitrophenyl chloroformate 1 g, and 4-dimethylaminopyridine 100 mg were dissolved in 40 mL of a mixture of dimethyl sulfoxide, CH2Cl2, and pyridine (2/1/1 in volume ratio) and kept under stirring at 2°C for 2 hours. The product was precipitated in 360 mL of a cold diethyl ether/ethanol (1/1, v/v) mixture and filtered. The final product was washed at least nine times with the precipitation reagent and dried in vacuo for 24 hours. The yield was approximately 90%.
In step 2 (conjugate of activated ROP with mPEG-NH2), the activated ROP and mPEG20k-NH2 in a molar ratio of 4:1 were dissolved in a mixture of dimethyl sulfoxide/pyridine (1:1, v/v). The reaction mixture was stirred in the dark for 4 days at room temperature, followed by dialysis with water to remove organic solvents and then hydrolysis with 0.1 M NaOH to recover unreacted activated hydroxyls. After further dialysis with water to a neutral pH, the solution was finally lyophilized.
Characterization of the conjugate was carried out by high-performance gel permeation chromatography (HPGPC) in conjunction with anthrone-sulfuric acid colorimetry, according to a previous report.14 The high-performance liquid chromatography system consisted of a Waters liquid chromatograph and a Waters 2414 refractive index detector (Milford, MA, USA). A 300×8.0 mm SB-803 HQ gel-filtration column (Shodex OHpak, Tokyo, Japan) was used with 0.1 M phosphate buffer (pH 7.4) as mobile phase delivered at a flow rate of 0.5 mL per minute.
Preparation of FITC-labeled MP20k-R
Labeling of MP20k-R with fluorescein isothiocyanate (FITC; FMP20k-R) was carried out according to a previous report.14 MP20k-R (1 g) was dissolved in 10 mL of dimethyl sulfoxide. FITC 0.1 g was added, followed by 0.2 mL of a solution of dibutyltin dilaurate in dimethyl sulfoxide (1/4, v/v) and 0.5 mL of pyridine. The mixture was well vortexed and then heated for 30 minutes at 95°C. After the reaction, the product was precipitated by nine volumes of the cold diethyl ether/ethanol (3/1, v/v) mixture. The resulting precipitates were recovered by centrifugation, washed ten times with diethyl ether/ethanol mixture to fully remove free FITC and other excess reagents, and then dried in a vacuum.
Preparation and characterization of in situ-forming PLGA depots
PLGA solutions were prepared by dissolving PLGA in certain amounts of organic solvent (NMP) in a tube with intermittent shaking at room temperature. PLGA concentrations in sols were expressed as the weight percentage (wt%). The drug-containing PLGA formulations for further studies were prepared by dissolving a designated portion of MP20k-R in a certain volume of PLGA sol in a water bath (42°C). mPEG20k-NH2 and FMP20k-R were used to replace MP20k-R in different PLGA formulations for in vitro characterization and in vivo studies, respectively.
To determine the viscosity of each blank or mPEG-NH2-loaded formulation, 1 mL of each sol was added to a cone/plate viscometer (Physica MCR-101, Anton Paar, Graz, Austria) and continuously measured at a constant shear rate (75 per second) while varying the temperature from 42°C to 37°C (decreasing by 1°C per minute).
In vivo studies
The rats were dosed according to a predetermined dosage regimen (Table 1). All formulations were prepared in a water bath (42°C) and injected subcutaneously at the same position on the back of each rat using 12-gauge needles when the formulations were cooled to approximately 37°C. After administration, the pinpoint in the skin was sealed with 20 μL of medical glue in case the formulation leaked out. Blood samples (~0.3 mL) were collected into heparinized centrifuge tubes by retro-orbital puncture at predetermined time points. The samples were immediately centrifuged at 3,000 rpm for 10 minutes and plasma samples were collected and stored at −20°C for subsequent analysis. Next, 40 μL of 1 M perchloric acid was added to precipitate plasma proteins to a 100 μL portion of each plasma sample. After centrifugation at 10,000 rpm for 2 minutes, the supernatant of the sample was transferred to another clean tube and neutralized by 30 μL of 1 M NaOH. After another centrifugation, 100 μL of the supernatant was taken out and diluted with phosphate-buffered saline (pH 7.4) 40 times before being determined spectrofluorimetrically at λex 495 nm and λem 515 nm. Pharmacokinetic software (DAS 2.0, Chinese Pharmacology Society) was used to calculate major pharmacokinetic parameters by noncompartmental analysis. Values are reported as the mean ± standard deviation. One-way analysis of variance was used for comparisons among groups. A P-value <0.05 was considered to be statistically significant.
 | Table 1 In situ-forming PLGA formulations and the dosage regimen for MP20k-R (n=3) |
Results and discussion
Preparation and characterization of MP20k-R
MP20k-R was synthesized successfully through a coupling reaction between the hydroxyl-activated ROP and the amino-terminated mPEG20k. MP20k-R was characterized by HPGPC coupled with the carbohydrate-specific anthrone-sulfuric acid colorimetry. The calculated apparent molecular masses and PEG-grafting number for the conjugate are listed in Table 2. Compared with the mPEG20k-NH2 reagent, the polydispersity index and HPGPC peak of the conjugate were increased and slightly left-shifted, respectively (Table 2 and Figure 1). More importantly, the chromogenic reaction between the anthrone-sulfuric acid reagent and the eluate corresponding to the HPGPC peak for the conjugate was positive.
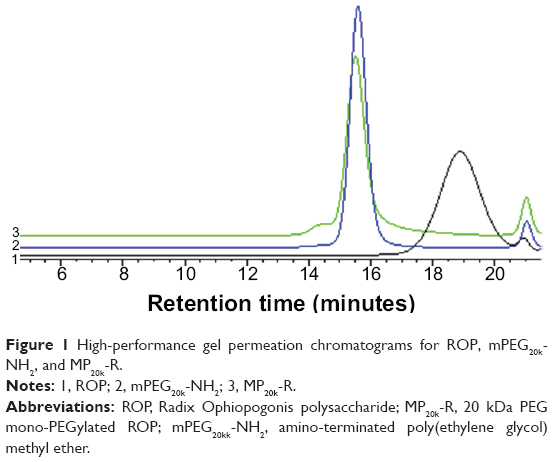 | Figure 1 High-performance gel permeation chromatograms for ROP, mPEG20k-NH2, and MP20k-R. |
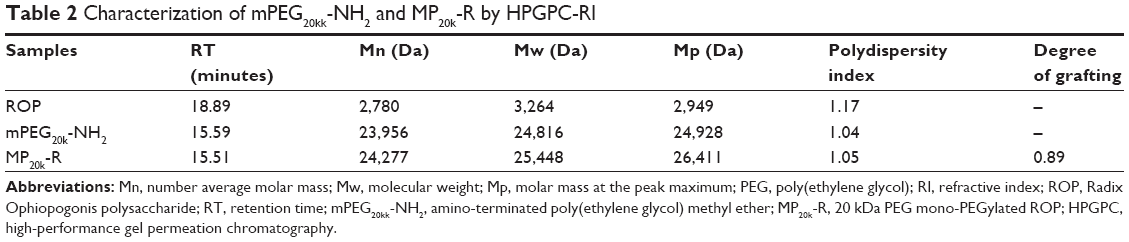 | Table 2 Characterization of mPEG20kk-NH2 and MP20k-R by HPGPC-RI |
Preparation and characterization of in situ-forming PLGA depots
Use of water-miscible solvents for PLGA like NMP and dimethyl sulfoxide and water-immiscible solvents like benzyl benzoate, ethyl acetate, benzyl alcohol, ethyl benzoate, and triacetin have been reported in the literature.1 Among these solvents, NMP is preferred because of its excellent solubility and more acceptable safety. It was reported that no safety concern about the use of NMP as the solvent for PLGA depots appeared. When researchers injected animals both subcutaneously and intramuscularly with in situ forming formulations based on NMP, animals maintained normal behavior during the study and histological analysis after 1 month showed tissue reactions similar to those reported for the usual preformed biodegradable implants.1 With regard to drug solubility in polymer solution and drug concentration, a drug can be either dissolved or dispersed in the ISFS. ROP is poorly soluble in NMP, while MP20k-R can be dissolved in NMP because its mass, and thus properties, mainly depend on the PEG moiety. Enhancing the viscosity of the PLGA formulation will result in decreased fluidity and further affect the formation of depots, while a reasonably low vehicle viscosity favors syringeability. Therefore, the effects of the concentration and molecular weight of PLGA and addition of the drug on the viscosity of PLGA sols were investigated.
As shown in Figure 2, the viscosities of PLGA30k sols at 37°C increased nonlinearly with increasing concentrations of PLGA in NMP and an abrupt change occurred at the concentration of 30% (w/w). The addition of mPEG20k-NH2 led to increased viscosity. The effect of the molecular weight of PLGA on viscosity was similar to that of concentration. Namely, viscosity increased nonlinearly with increasing molecular weight of PLGA and an abrupt change happened at 30 kDa, while addition of mPEG20k-NH2 also increased the viscosity (Figure 3). Decreasing the temperature from 42°C to 37°C and increasing mPEG20k-NH2 loading in the 40% PLGA30k/NMP formulation from 12.5% (125 mg/mL) to 20% (200 mg/mL) significantly enhanced the viscosity of the formulation (Figure 4). Thus, the 20% PLGA30k/NMP, 30% PLGA30k/NMP, 40% PLGA30k/NMP, and 30% PLGA50k/NMP formulations were chosen for in vivo studies to determine the applicability of in situ-forming PLGA depots for PEGylated polysaccharides.
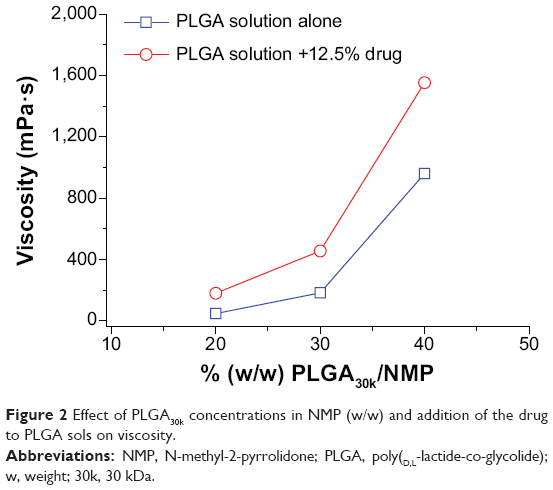 | Figure 2 Effect of PLGA30k concentrations in NMP (w/w) and addition of the drug to PLGA sols on viscosity. |
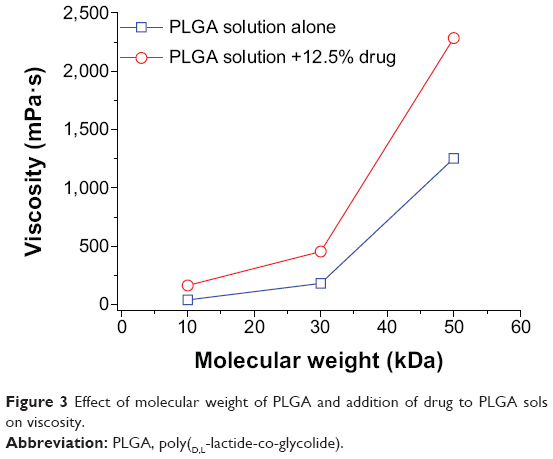 | Figure 3 Effect of molecular weight of PLGA and addition of drug to PLGA sols on viscosity. |
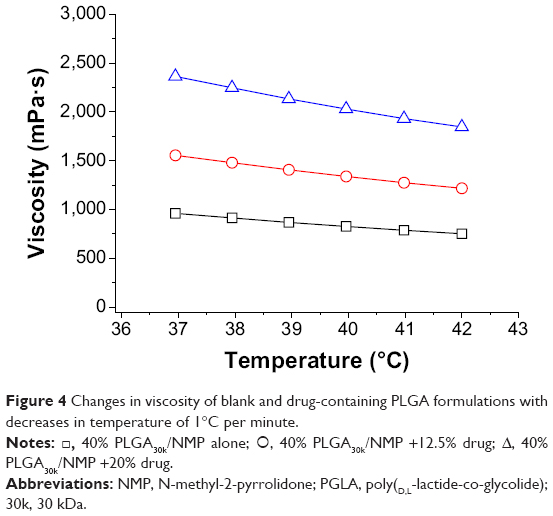 | Figure 4 Changes in viscosity of blank and drug-containing PLGA formulations with decreases in temperature of 1°C per minute. |
In vivo studies
Various types of drug delivery systems such as microspheres, ISFS, oily suspensions, and liposomes have been tested on drugs, and sustained release from several days to weeks, and even months, could be achieved.2 However, some time-drug plasma concentration profiles in published articles indicate an obvious burst release in the initial stages, while sustained but low steady plasma levels of drug have been observed at later stages.16–18 In some cases, a small burst release is appropriate for the effect of the drug by rapidly reaching the effective drug concentration; however, an obvious burst release might trigger drug toxicity. Many approaches have been adopted to modulate drug release kinetics, among which a combination of different delivery systems is a simple one. For example, to decrease burst release, microspheres in combination with in situ gels have been reported in some studies;19–21 however, few, if any, studies have reported on PEGylation in combination with ISFS. PEG is the most successful candidate for polymeric conjugation because of its unique advantages:22 little antigenicity, immunogenicity, and toxicity; high solubility in many solvents; highly hydrated and flexible backbone; and approval by the US Food and Drug Administration for human use in injectables. PEG and its conjugates are cleared from the body mainly by the kidney, and the excretion rate has a sigmoidal relationship with the molecular weight of the polymer, showing a greatly reduced rate over approximately 40 kDa.2 PEGs with a high molecular weight are eliminated slowly via the liver or become permanently entrapped into tissues or cells. Our previous studies indicated that, with proper design, PEGylated ROP that is both bioactive and long circulating in the blood could be obtained.14,15,22 For example, one with a PEG (20 kDa) grafting degree of approximately 1.0 showed a 47-fold increase in elimination half-life while preserving approximately 74% of ROP bioactivity.14 Further, when compared with PEGs of higher molecular weight, eg, 30 kDa or 40 kDa PEG, 20 kDa PEG is much less likely to cause accumulated toxicity. Therefore, MP20k-R might be the conjugate of choice for further development.
Polylactic acid, PLGA, and polycaprolactone are preferentially used for ISFS because of their approval by the US Food and Drug Administration and their long history of clinical use.3 Polymer grades with lactide/glycolide molar ratios from 100:0 to 0:100 and molecular weights from less than 10 kDa up to 200 kDa are available. Further, PLGA could be provided with a free carboxyl at the end of the polymeric backbone chain or end-capped with hydroxyl, which would influence the degradation of the polymer. As a result, PLGA copolymers with a wide range of physicochemical and degradation characteristics are available for controlled drug delivery applications.1 In this study, taking into consideration the solubility of PLGA in NMP and the pharmacokinetics of MP20k-R, PLGAs with a lactide/glycolide molar ratio of 50/50, molecular weights of ~10, ~30, and ~50 kDa, and a free carboxyl at the end of the polymeric backbone chain were selected as the depot matrix. Two key factors were investigated, ie, the PLGA concentration in NMP and the PLGA molecular weight. A drug dose (10 μmol/kg) that was 2.5 times higher than that in aqueous solution was used for all PLGA formulations to evaluate their sustained-release effects. The achieved plasma level versus time profiles are shown in Figure 5, and the main pharmacokinetic parameters determined by noncompartmental analysis are listed in Table 3. In general, the different PLGA formulations showed significant differences with regard to MP20k-R in vivo release kinetics.
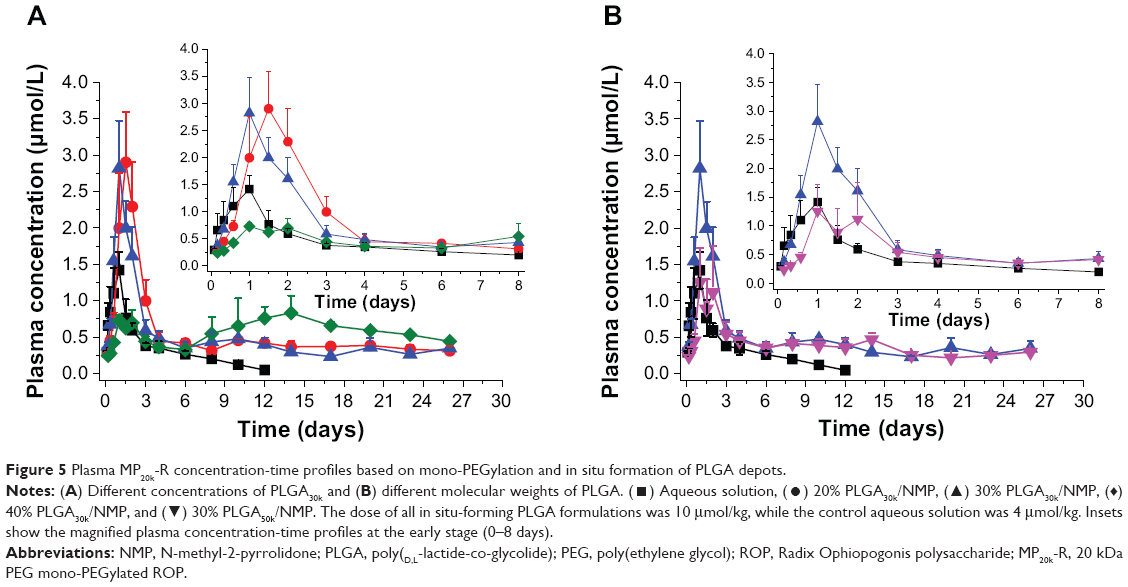 | Figure 5 Plasma MP20k-R concentration-time profiles based on mono-PEGylation and in situ formation of PLGA depots. |
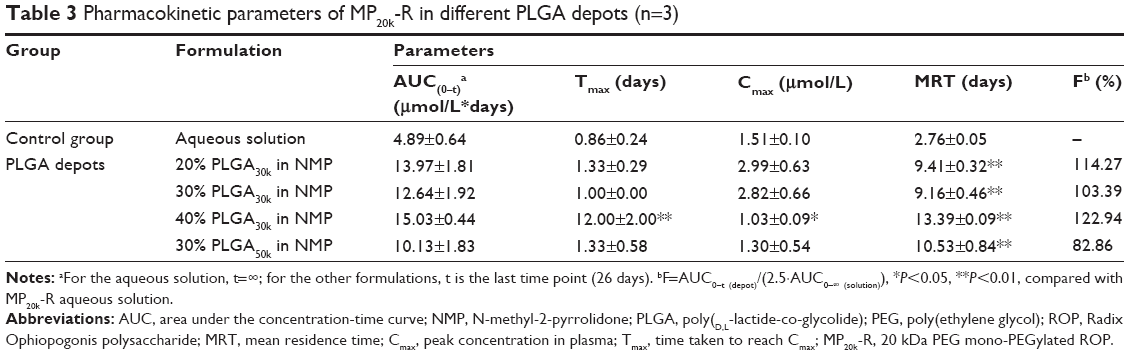 | Table 3 Pharmacokinetic parameters of MP20k-R in different PLGA depots (n=3) |
To determine the effects of PLGA concentration on release of MP20k-R, three different concentrations (20%, 30%, and 40%) of PLGA30k in NMP were investigated. The results showed that the effects were significant (Figure 5A and Table 3). Consistent with the changing trend in formulation viscosity, an abrupt change in pharmacokinetics occurred at a concentration of 30%. The maximum concentration (Cmax) of MP20k-R in plasma decreased sharply to become significantly (P<0.05) lower than that for the aqueous solution control when the concentration of PLGA30k was increased to 40%. Further, the almost flat profiles caused by the 40% PLGA30k-based formulation were observed not only at the early release stage, but also in the later period, indicating that long-lasting and stable plasma exposure of MP20k-R had been achieved. This, together with the observed gradual decrease in size of the depot formed in situ 1 month post dose, suggests that the in vivo degradation profile of the PLGA depot was related to the in vivo release profile of the drug. In addition, in terms of bioavailability, almost all of the MP20k-R should be released in a month, with gradual degradation of the depot formed in situ.
According to the relationship between formulation viscosity and the molecular weight of PLGA (Figure 3), with the same concentration (30%, w/w) in NMP, PLGAs with two different molecular weights (~30 kDa and ~50 kDa) were selected to investigate the effect of the molecular weight of PLGA on release of MP20k-R from the PLGA depots. The results showed that MP20k-R release was slowed down somewhat by increasing molecular weight of PLGA (Figure 5B and Table 3). The Cmax was decreased 2.17 times and the mean retention time was increased 1.15 times when the molecular weight of PLGA increased from ~30 kDa to ~50 kDa. A lower than control Cmax was obtained by the 30% PLGA50k/NMP formulation, but the difference is nonsignificant (P>0.05). In terms of the F value (82.86%), most of the MP20k-R in 30% PLGA50k/NMP was released in approximately 1 month.
Drug release from PLGA depots generally involves three stages: an initial burst during solidification of the matrix; drug diffusion; and drug release accompanied by degradation and erosion of the polymeric carrier.23 The above processes could be modulated by varying several parameters such as solvent, polymer, drug, additives, or injection site, thus obtaining ideal release kinetics. In this study, NMP was selected as the solvent for its water miscibility. Rapid diffusion of NMP toward body fluid occurred during solidification of the matrix, resulting in an obvious burst release of MP20k-R. However, this phenomenon was decreased by a high PLGA concentration in NMP (40% PLGA30k) and high molecular weight PLGA (30% PLGA50k) formulations. Increasing the PLGA concentration in NMP and increasing the molecular weight of PLGA would decrease the affinity of PLGA sols for water, which might result in rapid NMP diffusion but would form thicker solidified depot skins; the former showed an initial burst release and the latter could slow the solvent/water exchange and generate a less porous structure.1 Both processes would influence the release kinetics. These observations might explain why the 30% PLGA30k/NMP formulation showed lower plasma concentrations (1–4 days) than the 20% PLGA30k/NMP formulation, with a little shorter time taken to reach Cmax (Figure 5). However, for the 40% PLGA30k/NMP and 30% PLGA50k/NMP formulations, the solidified depot skins were thicker, and once formed, diffusion was slower. At the same drug loading (12.5%, w/v), the 40% PLGA30k/NMP formulation showed better controlled release for MP20k-R than did the 30% PLGA50k/NMP formulation at the early release stage (0–8 days), reflected in the lower observed Cmax. In addition, the sustained level of MP20k-R achieved by 40% PLGA30k was relatively higher than that achieved by 30% PLGA50k in the late period (8–26 days), which might be explained by the fact that higher molecular weight PLGA50k degraded more slowly than PLGA30k. Overall, for an MP20k-R loading of 12.5% (w/v), the 40% PLGA30k/NMP formulation was superior to the 30% PLGA50k/NMP formulation.
Generally speaking, a balance of the absorption rate and the elimination rate would produce steady drug levels. Adopting commonly used PEGs with molecular weights ranging from 5 kDa to 40 kDa as modification agents, the molecular weight of the drug increases accordingly by PEGylation, resulting in decreased absorption and elimination rates after subcutaneous administration.22 Meanwhile, the controlled-release effect of ISFS decreases the absorption rate. Among the various ISFS, thermally-induced hydrogel systems and solvent exchange-induced hydrophobic systems are particularly attractive, and these have been investigated extensively for drug delivery. For the former systems, hydrophilic copolymers of poly(ethylene oxide) and poly(propylene oxide) (PEO-PPO-PEO, known as Poloxamer) are the most widely studied synthetic polymers due to their proper reverse thermosensitivity. However, with their weak mechanical strength and high permeability, Poloxamer-based gels are rapidly eroded by dissolution from the surface, resulting in a delivery period that rarely exceeds a few days,3,24,25 which thus limits their use to short-term therapies, like management of pain,26 treatment of infection,27,28 and fertility control.29 For the solvent exchange-induced hydrophobic systems, hydrophobic PLGA copolymers have been used extensively for their significant controlled-release effect and their wide range of physicochemical and degradation characteristics. For example, the marketed drug Eligard®, containing leuprolide acetate (a luteinizing hormone-releasing hormone agonist), can be injected subcutaneously at 1–6-month intervals to treat advanced prostate cancer.30 In our previous study,31 the applicability of Poloxamer-based hydrogels was tested on PEGs with molecular weights of 5, 20, and 40 kDa. The results showed that at the same molar dose, sustained in vivo behavior was achieved by the 20% P407/10% P188, and 24% P407/10% P188 formulations for all the three PEGs; however, more remarkable improvements in pharmacokinetics were achieved by the gels incorporating lower molecular weight PEG. In the current study, the feasibility of mono-PEGylation in combination with in situ formation of PLGA depots was tested on ROP, and sustained release and steady plasma levels of MP20k-R were observed, especially when MP20k-R was loaded in the 40% PLGA30k/NMP formulation.
The above results indicate that mono-PEGylation in combination with ISFS is feasible to achieve long-lasting and steady drug levels in plasma, leading to long-acting drug formulations. However, it should be mentioned that ideal release kinetics would be obtained only when a PEGylated conjugate with appropriate molecular weight is loaded in a suitable ISFS. Specifically, the thermally-induced hydrogel will be more suitable for lower molecular weight PEGylated conjugates in terms of its rapid erosion, while the solvent exchange-induced hydrophobic ISFS will be more suitable for higher molecular weight PEGylated conjugates in terms of its slow degradation.
Conclusion
PEGylation and in situ formation of PLGA depots, which are examples of drug modification and sustained-release delivery technology, respectively, are two promising strategies that can be used to achieve long-acting drug formulations. By mono-PEGylation with 20 kDa PEG, the in vivo mean retention time of ROP was prolonged from approximately 1.0 hour to 2.76 days after subcutaneous administration; however, the “peak and valley” phenomenon of the plasma concentration versus time profile was still remarkable. When loaded into PLGA ISFS, the mean retention time of the conjugate was prolonged significantly further to 9.2–13.4 days. Both the concentration and the molecular weight of PLGA significantly influenced the in vivo release of the conjugate from the depot formed in situ, and with the correct choice, for example, the 40% 30 kDa PLGA formulation, month-long and steady plasma exposure of the conjugate could be achieved. Therefore, mono-PEGylation in combination with in situ formation of PLGA depots would be promising for long-term prophylaxis or/and treatment of myocardial ischemia with ROP. For macromolecular drugs that are highly hydrophilic and need be used at a large dose per administration, such as ROP, more than one preparation technology might be needed to achieve week-long or month-long delivery per dose.
Acknowledgments
This work was supported by the Program for New Century Excellent Talents in University (NCET-13-0906), the National Natural Science Foundation of China (81073065), the Key Discipline Project of Shanghai Education Committee (J50302), and the “085” Project (085ZY1219) of Shanghai University of Traditional Chinese Medicine.
Disclosure
The authors report no conflicts of interest in this work.
References
Parent M, Nouvel C, Koerber M, Sapin A, Maincent P, Boudiera A. PLGA in situ implants formed by phase inversion: critical physicochemical parameters to modulate drug release. J Control Release. 2013;172(1):292–304. | ||
Burgess DJ, Wright JC, editors. Long Acting Injections and Implants, Advances in Delivery Science and Technology. Berlin, Germany: Springer; 2012. | ||
Kempe S, Mäder K. In situ forming implants-an attractive formulation principle for parenteral depot formulations. J Control Release. 2012;161(2):668–679. | ||
Kang JS, DeLuca PP, Lee KC. Emerging PEGylated drugs. Expert Opin Emerg Drugs. 2009;14(2):363–380. | ||
Harris JM, Chess RB. Effect of PEGylation on pharmaceuticals. Nat Rev Drug Discov. 2003;2(3):214–221. | ||
Xu DS, Feng Y, Lin X, Deng HL, Fang JN, Dong Q. [Isolation, purification and structural analysis of a polysaccharide MDG-1 from Ophiopogon japonicas]. Yao Xue Xue Bao. 2005;40(7):636–639. Chinese. | ||
Zheng Q, Feng Y, Xu DS, Lin X, Chen YZ. Influence of sulfation on anti-myocardial ischemic activity of Ophiopogon japonicus polysaccharide. J Asian Nat Prod Res. 2009;11(4):306–321. | ||
Zheng Q, Feng Y, Xu DS. [Protective effect of Ophiopogonis polysaccharide MDG-1 on experimental myocardial ischemic rat]. Zhongguo Zhong Xi Yi Jie He Za Zhi. 2007;27(12):1116–1120. Chinese. | ||
Wang S, Zhang Z, Lin X, Xu DS, Feng Y, Ding K. A polysaccharide, MDG-1, induces S1P and bFGF expression and augments survival and angiogenesis in the ischemic heart. Glycobiology. 2010;20(4):473–484. | ||
Wang S, Lin X, Wang LY, Ruan KF, Feng Y, Li XY. A polysaccharide MDG-1 augments survival in the ischemic heart by inducing S1P release and S1P1 expression. Int J Biol Macromol. 2012;50(3):734–740. | ||
Lin X, Xu DS, Feng Y, Shen L. Determination of Ophiopogon japonicus polysaccharide in plasma by HPLC with modified postcolumn fluorescence derivatization. Anal Biochem. 2005;342(2):179–185. | ||
Lin X, Xu DS, Feng Y, Li SM, Lu ZL, Shen L. Release-controlling absorption enhancement of enterally administered Ophiopogon japonicus polysaccharide by sodium caprate in rats. J Pharm Sci. 2006;95(11):2534–2542. | ||
Shi XL, Lin X, Yao CX, Shen L, Feng Y. Injectable long-acting in situ forming systems for Radix Ophiopogonis polysaccharide. Int J Biol Macromol. 2015;72:553–559. | ||
Lin X, Wang S, Jiang Y, et al. Poly(ethylene glycol)-Radix Ophiopogonis polysaccharide conjugates: preparation, characterization, pharmacokinetics and in vitro bioactivity. Eur J Pharm Biopharm. 2010;76(2):230–237. | ||
Sun GL, Lin X, Shen L, et al. Mono-PEGylated radix ophiopogonis polysaccharide for the treatment of myocardial ischemia. Eur J Pharm Sci. 2013;49(4):629–636. | ||
Li ZP, Li L, Liu Y, et al. Development of interferon alpha-2b microspheres with constant release. Int J Pharm. 2011;410(1–2):48–53. | ||
Wang LX, Wang AP, Zhao XL, et al. Design of a long-term antipsychotic in situ forming implant and its release control method and mechanism. Int J Pharm. 2012;427(2):284–292. | ||
Lin X, Yang SS, Gou JX, et al. A novel risperidone-loaded SAIB-PLGA mixture matrix depot with a reduced burst release: effects of solvents and PLGA on drug release behaviors in vitro/in vivo. J Mater Sci Mater Med. 2012;23(2):443–455. | ||
Lee J, Tan CY, Lee SK, Kim YH, Lee KY. Controlled delivery of heat shock protein using an injectable microsphere/hydrogel combination system for the treatment of myocardial infarction. J Control Release. 2009;137(3):196–202. | ||
Machado HA, Abercrombie JJ, You T, Deluca PP, Leung KP. Release of a wound-healing agent from PLGA microspheres in a thermosensitive gel. Bio Med Res Int. 2013;2013:387863. | ||
Hiwale P, Lampis S, Conti G, et al. In vitro release of lysozyme from gelatin microspheres: effect of cross-linking agents and thermoreversible gel as suspending medium. Biomacromolecules. 2011;12(9):3186–3193. | ||
Lin X, Wang ZJ, Huang F, et al. Long-circulating delivery of bioactive polysaccharide from radix ophiopogonis by PEGylation. Int J Nanomedicine. 2011;6:2865–2872. | ||
Brodbeck KJ, DesNoyer JR, McHugh AJ. Phase inversion dynamics of PLGA solutions related to drug delivery. Part II. The role of solution thermodynamics and bath-side mass transfer. J Control Release. 1999;62(3):333–344. | ||
Ruel-Gariepy E, Leroux JC. In situ-forming hydrogels-review of temperature-sensitive systems. Eur J Pharm Biopharm. 2004;58(2):409–426. | ||
Jeong B, Gutowska A. Lessons from nature: stimuli-responsive polymers and their biomedical applications. Trends Biotechnol. 2002;20(7):305–311. | ||
Paavola A, Kilpelainen I, Yliruusi J, Rosenberg P. Controlled release injectable liposomal gel of ibuprofen for epidural analgesia. Int J Pharm. 2000;199(1):85–93. | ||
Veyries ML, Couarraze G, Geiger S, et al. Controlled release of vancomycin from poloxamer gels. Int J Pharm. 1999;192(2):183–193. | ||
Zhang L, Parsons DL, Navarre C, Kompella UB. Development and in-vitro evaluation of sustained release poloxamer 407 (P407) gel formulations of ceftiofur. J Control Release. 2002;85(1–3):73–81. | ||
Wenzel JG, Balaji KS, Koushik K, et al. Pluronic F127 gel formulations of deslorelin and GnRH reduce drug degradation and sustain drug release and effect in cattle. J Control Release. 2002;85(1–3):51–59. | ||
Sartor O. Eligard: leuprolide acetate in a novel sustained-release delivery system. Urology. 2003;61(2 Suppl 1):25–31. | ||
Zhang K, Shi XL, Lin X, Yao CX, Shen L, Feng Y. Poloxamer-based in situ hydrogels for controlled delivery of hydrophilic macromolecules after intramuscular injection in rats. Drug Deliv. March 7, 2014. [Epub ahead of print]. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.