Back to Journals » OncoTargets and Therapy » Volume 12
Inhibition of KPNB1 Inhibits Proliferation and Promotes Apoptosis of Chronic Myeloid Leukemia Cells Through Regulation of E2F1
Authors Wang T, Huang Z, Huang N, Peng Y, Gao M, Wang X, Feng W
Received 28 March 2019
Accepted for publication 14 November 2019
Published 2 December 2019 Volume 2019:12 Pages 10455—10467
DOI https://doi.org/10.2147/OTT.S210048
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Carlos E Vigil
Teng Wang,1 Zhenglan Huang,1 Ningshu Huang,2 Yuhang Peng,1 Miao Gao,3 Xin Wang,4 Wenli Feng1
1Department of Clinical Hematology, Key Laboratory of Laboratory Medical Diagnostics Designated by the Ministry of Education, Chongqing Medical University, Chongqing 400016, People’s Republic of China; 2Department of Clinical Laboratory, The Children’s Hospital of Chongqing Medical University, Chongqing 400016, People’s Republic of China; 3Department of Laboratory Medicine, The First Affiliated Hospital, Chongqing Medical University, Chongqing 400016, People’s Republic of China; 4Department of Hematology, The First Affiliated Hospital of Chongqing Medical University, Chongqing 400016, People’s Republic of China
Correspondence: Wenli Feng
Department of Clinical Hematology, Key Laboratory of Laboratory Medical Diagnostics Designated by the Ministry of Education, Chongqing Medical University, Chongqing 400016, People’s Republic of China
Tel +86-13983802837
Fax +86-23-68485239
Email [email protected]
Background: Karyopherin-β1 (KPNB1) belongs to the karyopherin superfamily, which functions as shuttling proteins from the cytoplasm to nuclear. A high level of KPNB1 has been reported in various cancers which promotes cell proliferation and inhibits apoptosis. However, the role of KPNB1 in chronic myeloid leukemia (CML) remains uncertain.
Methods: Expression level of KPNB1 in CML patient samples and cell lines was analyzed by Western blotting. The proliferation assays and colony formation assay were used to study the CML cell proliferation when KPNB1 knockdown in vitro. Next, Western blotting was used to evaluate the effects of KPNB1 on E2F1 and other cell cycle regulators. Then, the location of E2F1 was detected by immunofluorescence. Finally, flow cytometry was used to detect the effect of KPNB1 inhibitor importazole (IPZ) on CML cells.
Results: In this study, we firstly showed that KPNB1 is over-expressed in CML cells. Targeting KPNB1 with small interfering RNA (siRNA) and IPZ reduced proliferation and induced apoptosis of CML cells. The underlying mechanisms were also investigated that E2F1 nuclear transport was blocked after inhibiting KPNB1 with siRNA, suggesting KPNB1 over-expression mediates the excessive nuclear transport of E2F1 in CML cells. Moreover, the expression of the E2F1 targeted molecule such as c-Myc and KPNA2 was markedly reduced. The IPZ arrested CML cells at G2/M phase and induced cell apoptosis.
Conclusion: In summary, our results clearly showed that KPNB1 is over-expressed in CML cells and mediates the translocation of E2F1 into the nucleus of CML cells, thereby inhibition of KPNB1 reduced proliferation and induced apoptosis of CML cells which provides new insights for targeted CML therapies.
Keywords: CML, KPNB1, E2F1, c-Myc, IPZ, subcellular location
Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative malignant disease characterized by the presence of Philadelphia chromosome t(9:22). The translocation product is the pathognomonic fusion gene of BCR-ABL which encodes oncoprotein Bcr-Abl. The chimeric Bcr-Abl protein with constitutive kinase activity activates multiple downstream signaling pathways resulting in the survival and proliferation of CML cells.1,2 Tyrosine kinase inhibitors (TKIs) imatinib (IM) have been the most effective targeted drugs for patients with CML. However, a portion of patients failed to respond to IM. Even though next-generation TKIs such as nilotinib, dasatinib are unable to cruel all of the CML patients.3 Besides, TKI withdrawal in patients who have achieved complete molecular remission leads to relapse in most of the patients. Thus, it is urgent to explore the molecular resistance mechanisms and search for novel therapeutic targets of treatment for CML resistance.
E2F is the first cellular protein found to bind to the tumor suppressor, pRB.4,5 When associated with pRB family members, the E2Fs function as transcriptional repressors, whereas the free E2Fs activate transcription. E2F1 is one of the E2Fs and is known to upregulate target genes in different signaling pathways such as cell cycle, cell self-renewal, differentiation and apoptosis.6 E2F1 is down-regulated in many cancers including HCC,7 glioblastoma,8 pancreatic,9 renal10 and breast cancers.11 Interestingly, E2F1 over-expression is frequently found in glioblastoma12 and lymph node metastases of melanoma.13 Following its tumor-promoting function, E2F1 expression is correlated to tumor cell proliferation and antiapoptosis.14 E2F1 is also found to counteract with c-Myc-driven apoptosis via activation of PIK3CA/Akt/mTOR and c-Myc/COX-2 pathways.
Karyopherins are nuclear transport receptors that function as transporting cargo proteins into and out of the cell nucleus via the nuclear pore complex (NPC). The nucleocytoplasmic shuttling of large molecules is a highly regulated process controlled by specific nuclear importers and exporters. Karyopherin β1 (KPNB1), also known as importin β1, is a major nuclear importer belonging to the karyopherin family that transports proteins containing a nuclear localization signal (NLS) through the nuclear pore complex (NPC) into the nucleus. The classical nuclear import pathway is characterized by the recognition of the NLS on the cargo protein by the KPNB1 adaptor protein, Karyopherin α1 (KPNA2). After cargo recognition, KPNA2 binds KPNB1, and the trimeric complex translocates into the nucleus, via KPNB1-interactions with the nucleoporins (Nups) that comprise the NPC. The tight balance of KPNB1 is necessary for correct cell functioning. Recent studies have shown that KPNB1 expression is upregulated in various cancers such as cervical cancer,15 gastric cancer,16 breast cancer,17 hepatocellular cancer,18 diffuse large B-cell lymphoma19 and multiple myeloma.20 Many KPNB1 cargos are vital for tumorigenesis, including signaling transducers (STAT3, NF-κB, β-catenin21), growth factor receptors (ErbB-2, EGFR, c-Met), death receptors (DR5) and transcriptional factors (Snail). These lines of evidence suggest that the KPNB1 protein is associated with cellular transformation and cancer progression. These oncoproteins exhibit altered subcellular localization to sustain increased proliferation and decreased apoptosis in cancers. E2F1 is a transcription factor that plays an essential role in the development of tumors. However, the association between KPNB1 and E2F1 in CML has not been investigated. The expression of KPNB1 in CML and its function are worth exploring.
In this study, we firstly found that KPNB1 has a comparatively high expression level in CML patients' samples and CML cell lines. Further studies have found that interference with KPNB1 can significantly inhibit the proliferation of CML cells. By using KPNB1 selective inhibitor Importazole (IPZ), CML cells exhibited reduced cell proliferation and increased apoptosis. It has been shown that KPNB1 may involve in the CML progression though regulation of E2F1 entry into nuclear.
Materials and Methods
Clinical Samples
The CML patients we pick were newly diagnosed and at IM untreated step of the disease. We ensured that none of the patients has received preoperative IM treatment and obtained signed informed consents from all patients. This study was conducted in accordance with the Declaration of Helsinki and approved by the institutional ethics committee of Chongqing Medical University. Mononuclear cells were isolated using human bone marrow mononuclear cell isolation kit (Tbd science, Tianjin, China).
Cell Culture
The BP210 cell line was generated from Ba/F3 cell line by stably transformed by p210BCR-ABL. K562, K562R, KCL22, BP210 and HL60 cells were maintained in RPMI-1640 (Gibco, USA) supplemented with 10% fetal bovine serum (BI, Israel). For Ba/F3 cells, 1ng/mL of murine IL-3 (PeproTech, USA) was added to the medium. 293T cells were maintained in DMEM (Gibco, USA) supplemented with 10% fetal bovine serum (BI, Israel). All cells were cultured at 37°C in a humidified atmosphere with 5% CO2. All of the cells we used in this paper were purchased commercially from ATCC and were preserved in our laboratory.
Small Molecules, siRNA, Plasmid and Antibodies
The importinβ1-specific inhibitor importazole (IPZ) was purchased from MeChemExpress (MCE, USA). IM was obtained from Novartis (Basel, Switzerland). The siRNA targeting KPNB1 was purchased from Maobai Technology (Chongqing, China). The target sequences for KPNB1 siRNA are
① 5ʹ-GUGCAGAGAUCCCAGUAAAATTUUUACUGGGAUCUCUGCACTT-3ʹ
② 5ʹ-GGUGGUGAAUUCCUCAAGUTTACUUGAGGAAUUCACCACCTT-3ʹ,
③ 5ʹ-GCCCACCCUAAUAGAAUUATTUAAUUCUAUUAGGGGUGGGGCTT-3ʹ.
The plasmid of pAdTrack-Bcr-Abl was maintained in our Laboratory. The following antibodies were used in this study: anti-Abl, anti-KPNB1, anti-KPNA2, anti-P73, anti-P21, anti-P27 (Cell Signaling Technology, USA); anti-E2F1, anti-Myc, anti-NF-κB (Abcam, USA); anti-β-actin (Zhong Shan Jin Qiao, China).
Small Molecule Treatment and RNA Interference
1×106 cells were plated in 24-well plates and transfected with 30 μmol of siRNA using a nucleofector kit (Lonza, Basel, Switzerland), according to the manufacturer’s protocol. After transfection for 48 hrs, cells were harvested for viability, immunofluorescence analysis, clonal formation and Western blot analysis. Small molecule inhibitors IM and IPZ were dissolved in RPMI-1640. Cells treated with these inhibitors were collected at the indicated time for further analysis.
Cell Viability Assay
The viability of CML cells was evaluated by using the proliferation assay in liquid medium (CCK-8). Cells were seeded onto a 96-well plate at the concentration of 5×103 cells per well and then treated with the indicated agents for the indicated time. 10μL CCK-8 solution was added into each well at 37°C under dark condition for 2 hrs. After shaking for 5 mins, the absorbance at 492 nm was measured by a microplate reader (Eon, BioTeck, USA).
Apoptotic and Cell Cycle Analysis
Both cell apoptosis and cell cycle analysis were assessed by flow cytometry (FCM). Cells were collected after been treated with IM and IPZ at indicated concentration. Apoptosis was assessed using an Apoptosis Detection Kit (Becton-Dickinson) according to the manufacturer’s instruction. Moreover, nuclear morphology was examined by DAPI staining, and the results were observed with the fluorescence microscope. The cell cycle was analyzed by PI staining and quantified using FCM. The percentage of cells in different phases of the cell cycle was determined and quantitated.
Western Blotting
Cells were collected after been treated with IPZ and lysed by RIPA lysis buffer supplemented with proteinase and phosphatase inhibitors according to the manufacturer’s instruction. Protein concentration was determined by the BCA method (Beyotime Biotechnology, China). Then equal amounts of extracts (60 μg) were separated by 8–10% SDS-PAGE and transferred onto the PVDF membranes (Millipore, Boston, MA, USA), and was blocked in 5% skimmed milk powder/TBST, incubated with indicated antibodies overnight at 4°C, followed by incubation with HRP-conjugated secondary antibody for 1 hr at 37°C. Detection was performed using the enhanced chemiluminescence substrate (ECL) (Biogroud, China). Signals were visualized and analyzed by the Bio-Rad Gel Imaging System.
Immunofluorescence Analysis
CML cells were cultured in 12-well plates with a total of 5×104/well for 12 hrs and then were transfected with siRNA or treated with IPZ (5μM). After been cultured 24 hrs, cells were fixed by 4% paraformaldehyde (RT) for 1 hr. Cells were permeabilized for 15 mins with 1% Triton X-100 (Sigma), then blocked at 4°C for 60 mins with 10% normal goat serum (Boster, Wuhan), and incubated at 4°C with indicated antibody overnight. Cells were incubated at RT for 60 mins with Goat anti-Mouse IgG (H+L) Secondary Antibody CY3. After DNA staining with DAPI (Beyotime, China), coverslips were mounted onto glass slides with gel mount. Epifluorescence microscopy was performed with a Nikon microscope.
Colony Formation Assays
An amount of 500 K562 and K562R cells were seeded in 6-well plates. After been treated with KPNB1 siRNAs or inhibitor IPZ for 14 days later, cells were counted and analyzed.
Statistical Methods
The statistical analyses were performed by one-way ANOVA analysis to compare the mean of each group with that of the control group in GraphPad prism 5. Statistical significance is indicated as p<0.05.
Results
KPNB1 is Up-Regulated in Bone Marrow Cells from CML Patients' Samples and CML Cell Lines
KPNB1 has been shown highly expressed in many tumor cells, but its expression level in chronic myeloid leukemia is unclear. We then examined the KPNB1 and its adaptor KPNA2 protein expression levels in chronic myeloid leukemia (CML) patients' bone marrow mononuclear cells (BMMNCs) and CML cell lines. Our Western blotting results showed that KPNB1 and KPNA2 are highly expressed in bone marrow cells of CML patients compared to typical sample (Figure 1A and B). We further examined the expression levels of KPNB1 and KPNA2 in chronic myeloid leukemia cell lines (K562, K562R, Kcl22), mouse lymphocytes (BaF3, BP210) and acute myeloid leukemia cell line HL-60 (Bcr-Abl−). As a result (Figure 1C and D), we found that KPNB1 was much higher in Bcr-Abl+ BP210 cells than that in normal lymphocyte BaF3 cells. However, there is no difference in the expression of KPNB1 and KPNA2 between hematological tumor cell groups. For verifying whether there is a relationship between Bcr-Abl tyrosine kinase and the high expression of KPNB1, K562 and K562R cells were treated with tyrosine kinase inhibitor IM. Figure 1E shows that the treatment of IM (10μM) does not affect the expression of KPNB1 and KPNA2 in K562 and K562R cells. We then transfected pAdTrack-Bcr-Abl plasmid into 293T cells, and detected the expression of KPNB1 and KPNA2 in 293T with treatment of IM. The results showed that no difference had been found between groups. The above results indicate that KPNB1 is highly expressed in chronic myeloid leukemia cells. KPNB1 protein expression is not directly regulated by Bcr-Abl tyrosine kinase activity and KPNB1 may play a vital role in the malignant progression of CML.
Silencing of KPNB1 Inhibits the Proliferation of CML Cells
To investigate the effects of KPNB1 on CML cellular function, siRNA technology was utilized to inhibit KPNB1 expression. As shown in Figure 2A–D, we found that small interfering RNA No. 3 significantly inhibited the expression of KPNB1 in K562 and K562R cells. Surprisingly, the protein level of KPNA2 was also inhibited. We further detected the effect of small interfering RNA No. 3 on the proliferation of CML cells in vitro (Figure 2E and F), and we found that it significantly inhibits the clonal formation of K562 and K562R cells. The proliferation assay results (Figure 2G and H) also confirmed that interference with KPNB1 significantly reduced the growth and proliferation of K562 and K562R cells. Furthermore, our Western blotting data showed that the expression levels of c-Myc, E2F1 and NF-κB decreased after been silenced of KPNB1 (Figure 2I and J). More interestingly, the protein expression of Bcr-Abl was also inhibited. These results indicate that interference with KPNB1 can effectively inhibit the proliferation of CML cells. The mechanism may be associated with c-Myc, E2F1 and NF-κB.
Knockdown of KPNB1 Down-Regulated E2F1 Nuclear Localization
KPNB1 can mediate many transcription factors into the nucleus and play a vital role in promoting tumorigenesis. Francesca Pellicano’s study showed that hsa-mir183/EGR-mediated regulation of E2F1 is required for CML stem/progenitor cell survival. Thus, we consider that E2F1 may be one of the most important regulators in CML.22 To further explore the mechanism of KPNB1 in CML tumorigenesis, we used immunofluorescence to observe the transcription factor E2F1 subcellular localization changes after the interference of KPNB1. As we found (Figure 3), E2F1 localized in both cytoplasm and nucleus in CML cells K562 and K562R. However, after the interference of KPNB1, E2F1 failed to localize in the nucleus and is found only in the cytoplasm. c-Myc protein also exhibits the same subcellular localization after inhibition of KPNB1 (Figure S1). Previous studies have reported that E2F1 can regulate the expression of c-Myc and NF-κB. Proteasome inhibition by MG132 combined with IPZ was used to detect the protein expression of E2F1 and other regulators. Our results showed that MG132 plus IPZ prevent the decrease in E2F1 (Figure S3). However, the protein expression of KPNA2 and c-Myc still decreased after the treatment of IPZ and MG132, which means KPNB1 control E2F1 at the translation level. The decreased c-Myc protein expression and altered subcellular location is regulated by E2F1. These results indicate that interference of KPNB1 can promote CML apoptosis and inhibit proliferation, and its underlying mechanism may be related to the inability of E2F1 to enter the nucleus, and therefore fail to play a carcinogenic function.
KPNB1 Inhibitor IPZ Reduces the Proliferation of CML Cells and Enhances the Effect of IM
IPZ is a selective inhibitor of KPNB1 that inhibits the KPNB1 function of mediating protein entry into the nucleus. The results of the proliferation assay showed that 2.5 μM of IPZ could inhibit the proliferation of CML cells at 48 hrs, while 10μM of IPZ could exert significant inhibition after 6 hrs of treatment. The result of colony formation was as same as the proliferation assay. After 14 days of incubation with IPZ, the clonality of the cells was significantly decreased (Figure 4A–D). Compared with the group of IPZ treatment for 24 hrs, 2.5 μM of IPZ treatment for 48 hrs significantly reduced the proliferation of K562 and K562R cells (Figure 4E and F). We then detected the effect of IPZ on normal bone marrow mononuclear cells (BMMNCs) by CCK8. Our results showed that IPZ almost can not influence the proliferation of normal BMMNCs (Figure S4). Next, we examined the effect of IPZ, IPZ combined with IM on the proliferation of CML cells. It was found that a low concentration of IM affects the proliferation of K562 cells at 48 hrs other than K562R cells. IPZ (1.25μM) combined with 1μM of IM significantly reduced the number of K562 and K562R cells compared to a low concentration of IM (1μM) at 48 hrs (Figure 4G and H). The above results suggest that IPZ, an inhibitor of KPNB1, can reduce the proliferative capacity of CML cells and enhance the anticancer effect of IM. It can be used as a drug target for treatment.
IPZ Promotes Cell Cycle Arrest in the G2/M Phase
Flow cytometry analysis showed the cell cycle distribution of CML cells after been treated with IPZ. As in Figure 5A–H, compared with the untreated group, the number of cells in the G2/M phase of the IPZ group was significantly increased. E2F1, c-Myc, NFκB were associated with CML progression, and all of these molecules function as carcinogenic ability in the nucleus. Erk1/2 plays carcinogenic roles in the cytoplasm and nucleus. We then checked the expression levels of the above molecule and cell cycle regulators and found that KPNB1 inhibition decreased E2F1, c-Myc protein level and increased P21 and P27. The expression of P73 was slightly increased in K562R cells. Taken together, these results suggest that KPNB1 inhibition exerts its antitumor effect, in part, by inducing cell cycle arrest in the G2/M phase.
IPZ Promotes Apoptosis in CML Cells
As shown in the Figure 6A, K562 and K562R cells were treated with IPZ alone or in combination with IM; the nucleus of IPZ alone treatment group (5μM) and IPZ (5μM) combined with IM (1μM) treatment group was fragmented, indicating the effect of IPZ on CML apoptosis. Based on the previous results, we measured cell apoptosis using annexin V staining after treatment with 5μM IPZ alone or 5μM IPZ combined with 1μM IM. Flow cytometry analysis showed that IPZ combined with IM could significantly promote the apoptosis of both K562 and K562R cells (Figure 6B and C). However, K562R cells show higher sensitivity to IPZ than K562 cells, and the treatment of IPZ enhances the sensitivity of K562R to IM. In contrast, no meaningful effect on apoptosis was observed in the group of untreated and 1μM IM. These results indicate that inhibition of KPNB1 can enhance the sensitivity of K562R cells to IM, and KPNB1 may be a novel drug target for the treatment of CML.
Discussion
The primary treatment for chronic myeloid leukemia is the tyrosine kinase inhibitors (TKIs), which target the Bcr-Abl high tyrosine kinase. However, the presence of Bcr-Abl point mutation and leukemia stem cells, often leading to TKIs resistance. Furthermore, discontinuous use of TKIs leads to transition from chronic phase to blast crisis. Therefore, it is urgent to find novel drug targets for the treatment of chronic myeloid leukemia.
Trafficking of proteins across the nuclear envelope is essential to signal transduction and physiological regulation. Proper temporal-spatial localization of protein requires strict regulation by the karyopherin family. Many oncoproteins, such as transcription factors, accumulate in the nucleus to induce abnormal cell cycle in CML. KPNB1 is an importin protein that plays a crucial role in the nuclear import process, and KPNA2 is known as an essential adaptor between cargo and KPNB1. In the classical nuclear import pathway, the nuclear localization sequence (NLS) present in the cargo is recognized and bound by the adaptor protein KPNA2, which forms a trimer complex with KPNB1. Then, the complex is transported across the nuclear pore complex into the nucleus.
Many studies have shown that nuclear transport protein KPNB1 is highly expressed in a series of solid tumors and mediates several transcription factors into the nucleus. In our study, we firstly demonstrated that KPNB1 is highly expressed in chronic myeloid leukemia patients BMMNCs and CML cell lines. Treatment of IM (10μM) does not affect the expression of KPNB1 and KPNA2 in K562 and K562R cells. These results indicate that KPNB1 is highly expressed in chronic myeloid leukemia cells, and its expression may not be regulated directly by Bcr-Abl tyrosine kinase activity. Interferes with the KPNB1 significantly inhibit the proliferation of CML cell lines, suggesting that KPNB1 may play a vital role in the malignant proliferation of CML cells. Interestingly, KPNA2 expression was also decreased on KPNB1 knockdown. The possible reason for this is that KPNB1 may regulate the entry of the upstream transcription factors of KPNA2. However, it is unclear what role KPNB1 plays in the pathogenesis of chronic myeloid leukemia.
It has been reported that E2F1 plays a vital role in cancer promotion. Previous studies have shown that E2F1 is regulated by hsa-mir183/EGR to sustain CML stem cells’ survival and sternness. E2F1 also has been reported that it regulates the transcription of c-Myc,23 NF-κB,24 KPNB1 and KPNA2.25 Our Western blotting results displayed that the expression of E2F1, c-Myc and NF-κB was significantly decreased after interference with KPNB1. Since KPNB1 mainly plays a role in mediating cargo proteins' nuclear translocation, we then examined the change of E2F1, c-Myc and NF-κB subcellular localization in CML cells. According to our immunofluorescence results, E2F1 and c-Myc exhibit substantial nucleus accumulation in K562 and K562R cells. Based on the above results, we then propose that in chronic myeloid leukemia, E2F1 enters the nucleus via KPNB1-mediated nuclear entry, and promotes the expression of c-Myc, KPNA2 and KPNB1 through its transcription ability. Up-regulation of KPNB1 and KPNA2 mediates more E2F1 protein entry into the nucleus. The inhibition of KPNB1 disrupts the E2F1-KPNA2-KPNB1 complex and then leads to blockage of E2F1 in the cytoplasm. Decreased distribution of E2F1 in the nucleus down-regulates the expression of c-Myc and KPNA2, which explain the reason that inhibition of KPNB1 also inhibits the expression of KPNA2 (Figure 7). However, the above immunofluorescence results cannot fully explain why the protein expression of E2F1 decreased. Zhu ZC has demonstrated KPNB1 inhibition disrupts proteostasis in glioblastoma cells.26 However, it is not confirmed in CML. Besides, the regulation of KPNB1 in CML is still unclear, but there is evidence that it may be related to the EZH2-miR-30d signaling pathway.27 We analyze the expression of EZH2 at Broad Institute Cancer Cell Line Encyclopedia (CCLE), and we find that EZH2 exhibits a high mRNA level in CML cells (Figure S2). Therefore, we consider that over-expression of KPNB1 in CML cells would be controlled by EZH2. Further studies will be conducted to elucidate mechanisms of EZH2 regulation on miR-30d transcription and KPNB1 expression in CML cells.
As mentioned above, IPZ is a KPNB1 inhibitor that interferes with the interaction between Ran GTP and KPNB1. Given the fact that interference with KPNB1 can significantly reduce K562 and K562R cell number and clonal ability, we used IPZ at indicative concentration to investigate the effect of it on the proliferation and apoptosis in CML cells. Our results showed that targeting KPNB1 is a feasible cancer treatment option for CML. Based on the cell apoptosis experiment using annexin V staining, we found that IPZ can promote apoptosis of K562 and K562R cells. Besides, K562R exhibited more sensitive to IPZ than K562 cells, and the treatment of IPZ can enhance the sensitivity of K562R to IM. The underlying mechanism may be the accelerated nucleoporins-mediated protein import. In Veronica Rodriguez-Bravo’s work, they found that nucleoporins (Nups) promote lethal prostate cancer by increasing POM121-KPNB1-driven E2F1, c-Myc and AR nuclear import.28 Their studies suggest that specific Nups and KPNB1 may enhance the signaling activity of oncogenic and transcription factors. NPC composition and function may be of relevance for cancer progression and resistance. Thus, we consider that IPZ appears to be more efficient on K562R than on K562 cells, as part of the reason, due to the more active POM121-KPNB1-mediated nuclear import in K562R than in K562 cells.
Conclusion
In summary, our results demonstrated for the first time that KPNB1 is involved in the pathogenesis of CML. CML patients’ cells and cell lines exhibit KPNB1 over-expression. The silence of KPNB1 reduces the malignant proliferative capacity of K562 and K562R cells. The underlying mechanism may be involved in blocking E2F1 nuclear transport. IPZ, a selective inhibitor, promotes CML cell apoptosis and enhances the sensitivity of IM. All of these results further indicate that inhibiting the KPNB1 function may be a new strategy for CML treatment.
 |
Figure 1 Expressions of KPNB1 and KPNA2 in BMMNCs from CML patients and different leukemic cell lines. (A and B) The protein expression of KPNB1 in CML patients’ BMMNCs and normal BMMNCs. (C and D) The protein expression of KPNB1 in CML cell lines. (E) After been treated with IM (10μM) for 48 hrs, the expressions of KPNB1 and KPNA2 in K562 and K562R cells. (F) After been treated with IM (10μM) for 48 hrs, the expressions of KPNB1 and KPNA2 in 293T cells which transfected with pAdTrack-Bcr-Abl. Each error bar represents the SEM, **P <0.01, ***P<0.001. |
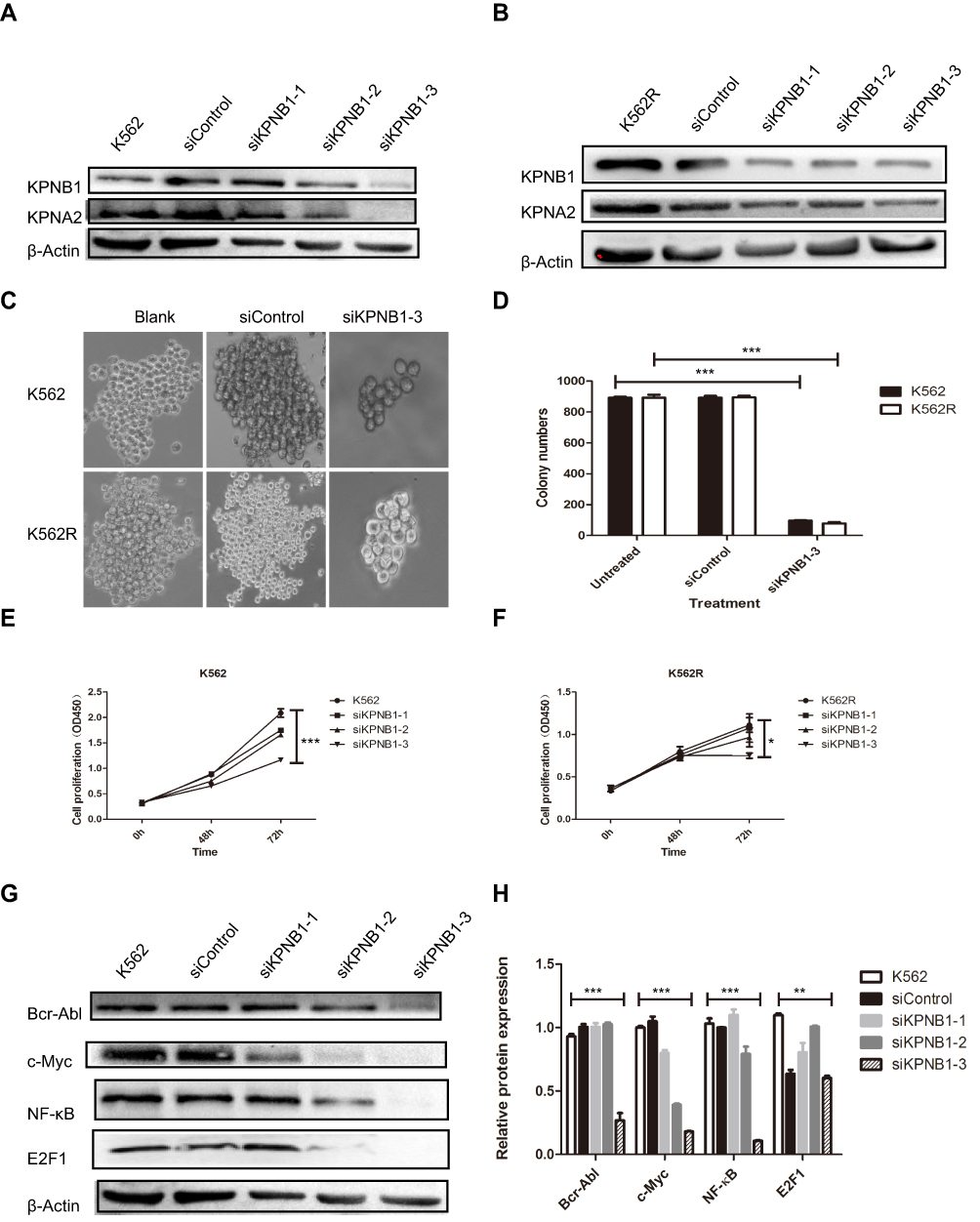 |
Figure 2 Silencing of KPNB1 inhibits the proliferation of CML cells. (A and B) si-KPNB1-3# is effective to inhibit the expression of KPNB1 in K562 and K562R cells. (C and D) si-KPNB1-3# significantly reduced the number of colony formation of K562 and K562R cells. (E and F) CCK-8 results show that si-KPNB1-3# inhibits the proliferation of K562 and K562R cells. (G and H) si-KPNB1-3# reduces the expression of c-Myc, E2F1, and NF-κB. *P<0.05, **P <0.01, ***P<0.001. |
 |
Figure 3 Silence of KPNB1 down-regulated the nuclear localization of E2F1. K562 and K562R cells were nuclear transfected with siKPNB1-3#, and then maintained for 48 hrs. Fixed cells were stained for E2F1 (red) and with DAPI (blue). |
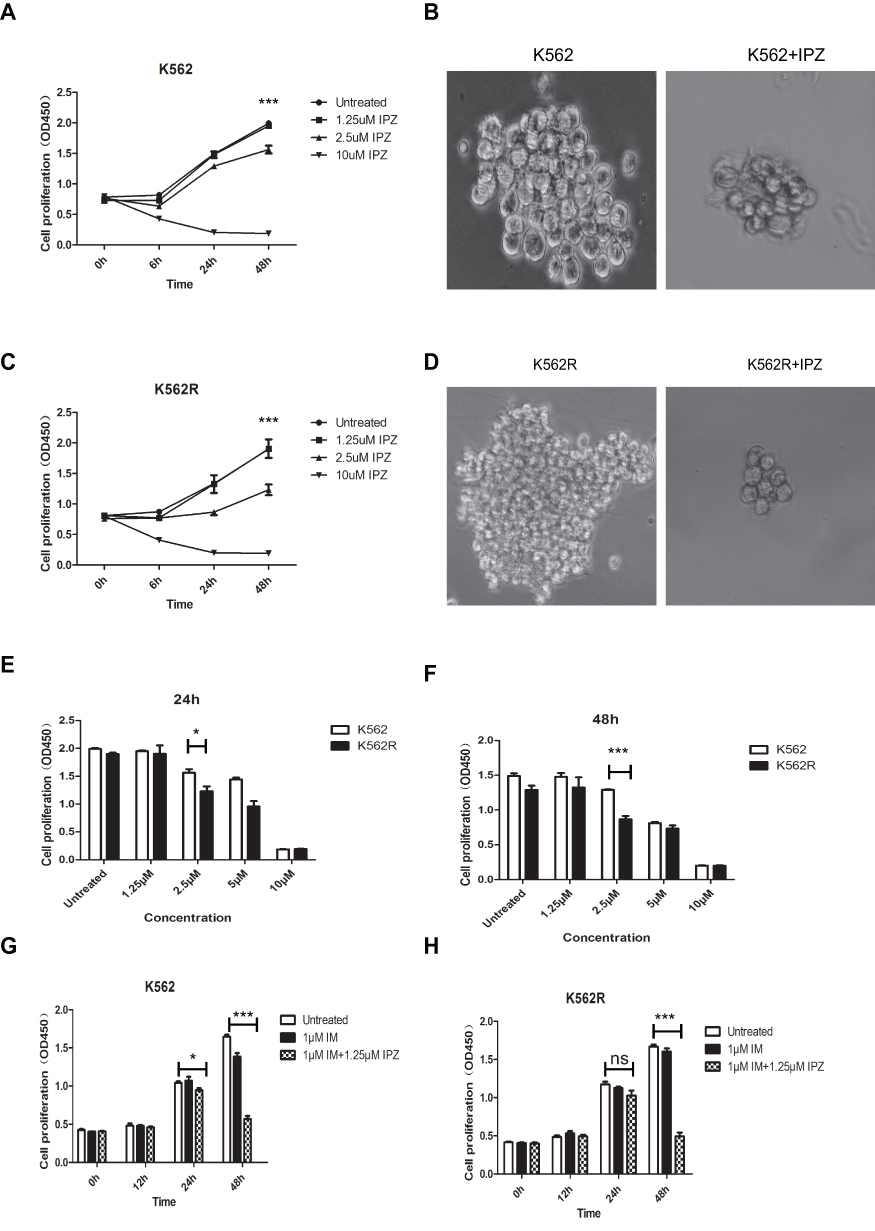 |
Figure 4 Importazole, an inhibitor of KPNB1, reduces the proliferation of CML cells and enhances the effect of IM. (A and B) Cell proliferation of K562 is inhibited after been treated with IPZ. (C and D) Cell proliferation of K562R is inhibited after been treated with IPZ. (E and F) 1.25μM IPZ significantly reduced the viability of K562 and K562R cells at 24 hrs and 48 hrs. (G and H) 1.25μM IPZ combined with 1μM IM significantly enhanced the effect on K562 and K562R cells. *P<0.05, ***P<0.001. |
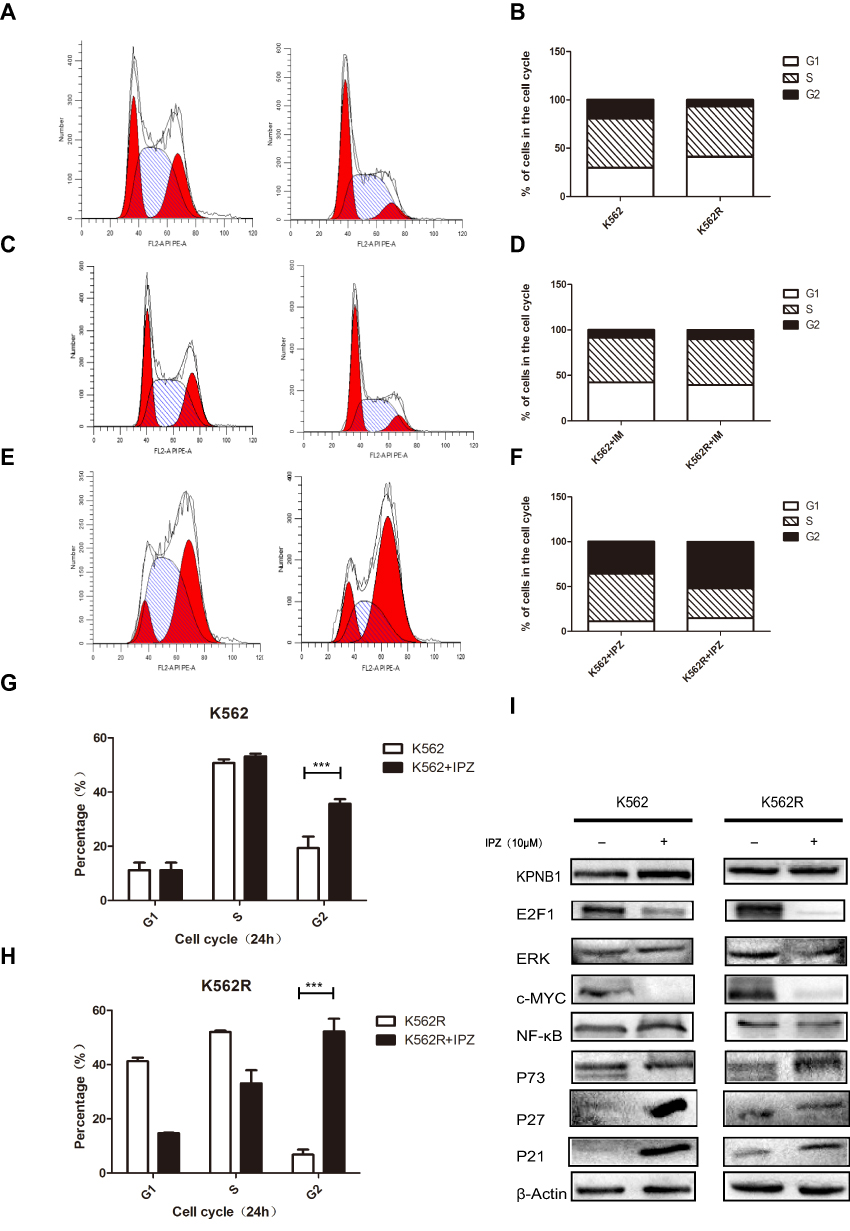 |
Figure 5 Importazole affects the biological function of CML cells, and preferentially changes the distribution of CML cell cell cycle. (A and B) Flow cytometry analysis of K562 and K562R. (C and D) Flow cytometry analysis of K562 and K562R treated with 1μM IM for 48 hrs. (E and F) Flow cytometry analysis of K562 and K562R treated with 1.25μM IPZ for 48 hrs. (G and H) IPZ treatment arrests CML cell cycle at the G2/M phase. (I) IPZ treatment reduces the protein expression of E2F1 and c-Myc, upregulates the expression of P21 and P27. ***P<0.001. |
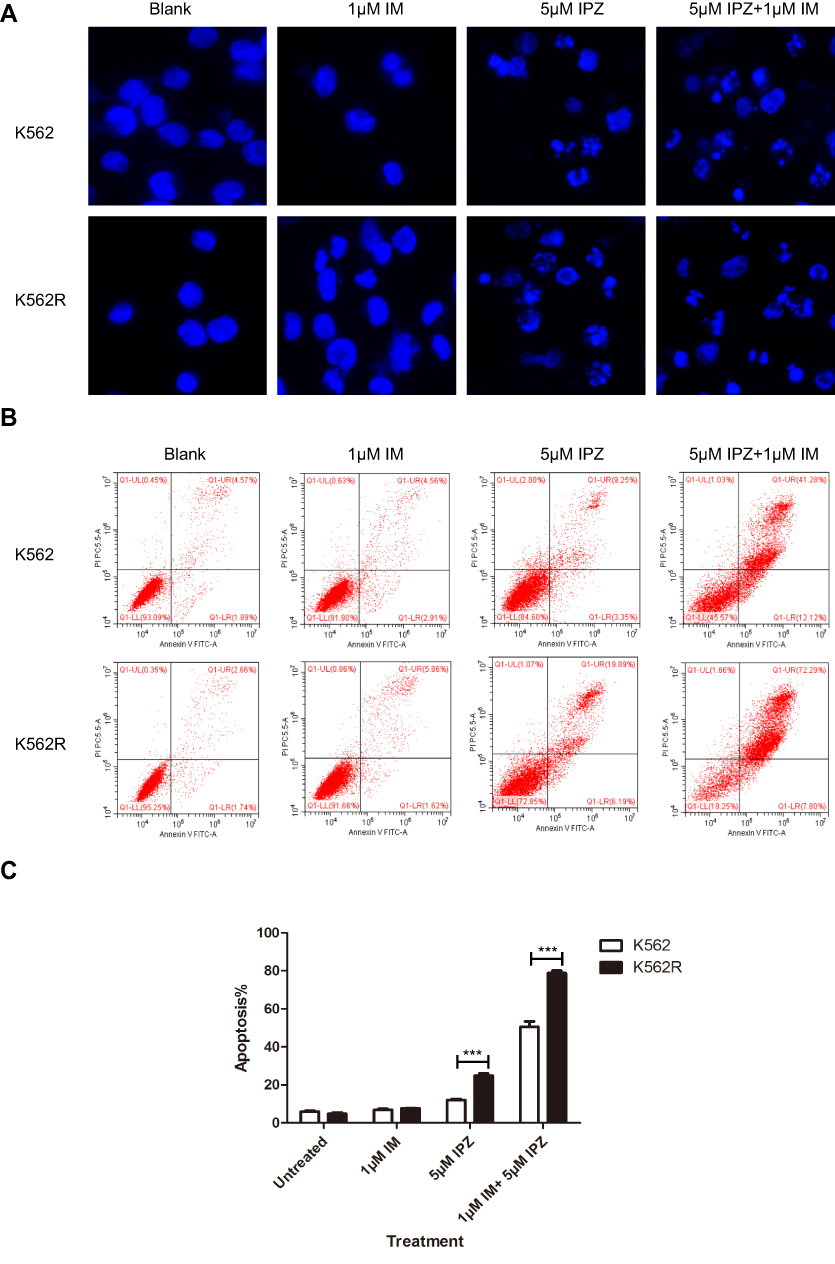 |
Figure 6 Importazole induces the death of CML cells. (A) The treatment of IPZ significantly broke the nuclear of K562 and K562R. (B and C) Apoptosis was analyzed by annexin V staining after the treatments of 5μM IPZ for 24 hrs in K562 and K562R cells. ***P<0.001. |
 |
Figure 7 Signal pathway of KPNB1-mediated E2F1 entry into the nucleus. KPNA2 recognizes the nuclear localization signal on E2F1 and binds to KPNB1 forming a nuclear translocation complex. KPNB1 is responsible for recognizing nuclear membrane proteins (Nups) and mediating E2F1 entry into the nucleus. When E2F1 was translocated into the nucleus, it functions as a transcription factor to regulate various gene expressions such as KPNB1, KPNA2, and c-Myc. The knockdown of KPNB1 decreased expression of KPNB1 and disrupted the formation of the E2F1-KPNA2-KPNB1 translocation complex. The obstruction of E2F1 in cytoplasm leads to inhibition of c-Myc and KPNA2 and then impairs the proliferation of CML progression. |
Abbreviations
CML, chronic myeloid leukemia; TKIs, tyrosine kinase; KPNB1, importin β1; IPZ, importazole; KPNA2, importin α1; BMMNCs, bone marrow mononuclear cells; IM, imatinib.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Quintás-Cardama A, Cortes J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood. 2009;113(8):1619–1630. doi:10.1182/blood-2008-03-144790
2. Hazlehurst LA, Bewry NN, Nair RR, et al. Signaling networks associated with BCR-ABL-dependent transformation. Cancer Control. 2009;16(2):100–107. doi:10.1177/107327480901600202
3. Swords R, Mahalingam D, Padmanabhan S, Carew J, Giles F. Nilotinib: optimal therapy for patients with chronic myeloid leukemia and resistance or intolerance to imatinib. Drug Des Devel Ther. 2009;3:89–101. doi:10.2147/dddt.s3069
4. Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009;9(11):785–797. doi:10.1038/nrc2696
5. Du W, Searle JS. The rb pathway and cancer therapeutics. Curr Drug Targets. 2009;10(7):581–589. doi:10.2174/138945009788680392
6. Müller H, Bracken AP, Vernell R, et al. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev. 2001;15(3):267–285. doi:10.1101/gad.864201
7. Zhan L, Huang C, Meng XM, et al. Promising roles of mammalian E2Fs in hepatocellular carcinoma. Cell Signal. 2014;26(5):1075–1081. doi:10.1016/j.cellsig.2014.01.008
8. Teplyuk NM, Uhlmann EJ, Wong AH, et al. MicroRNA-10b inhibition reduces E2F1-mediated transcription and miR-15/16 activity in glioblastoma. Oncotarget. 2015;6(6):3770–3783. doi:10.18632/oncotarget.3009
9. Batchu RB, Gruzdyn OV, Bryant CS, et al. Ritonavir-mediated induction of apoptosis in pancreatic cancer occurs via the RB/E2F-1 and AKT pathways. Pharmaceuticals (Basel). 2014;7(1):46–57. doi:10.3390/ph7010046
10. Ma X, Gao Y, Fan Y, et al. Overexpression of E2F1 promotes tumor malignancy and correlates with TNM stages in clear cell renal cell carcinoma. PLoS One. 2013;8(9):e73436. doi:10.1371/journal.pone.0073436
11. Wang Y, Deng O, Feng Z, et al. RNF126 promotes homologous recombination via regulation of E2F1-mediated BRCA1 expression. Oncogene. 2015;35(11):1363–1372. doi:10.1038/onc.2015.198
12. Alonso MM, Fueyo J, Shay JW, et al. Expression of transcription factor E2F1 and telomerase in glioblastomas: mechanistic linkage and prognostic significance. J Natl Cancer Inst. 2005;97(21):1589–1600. doi:10.1093/jnci/dji340
13. Meng P, Ghosh R. Transcription addiction: can we garner the Yin and Yang functions of E2F1 for cancer therapy? Cell Death Dis. 2014;5(8):e1360.
14. Pützer BM. E2F1 death pathways as targets for cancer therapy. J Cell Mol Med. 2007;11(2):239–251. doi:10.1111/jcmm.2007.11.issue-2
15. Stelma T. Leaner VD: KPNB1-mediated nuclear import is required for motility and inflammatory transcription factor activity in cervical cancer cells. Oncotarget. 2017;8(20):32833–32847. doi:10.18632/oncotarget.15834
16. Zhu J, Wang Y, Huang H, et al. Upregulation of KPNβ1 in gastric cancer cell promotes tumor cell proliferation and predicts poor prognosis. Tumour Biol. 2016;37(1):661–672. doi:10.1007/s13277-015-3839-7
17. Nordgard SH, Johansen FE, Alnaes GI, Kristensen VN. Genome-wide analysis identifies 16q deletion associated with survival, molecular subtypes, mRNA expression, and germline haplotypes in breast cancer patients. Genes Chromosomes Cancer. 2008;47:680–696. doi:10.1002/gcc.20569
18. Yang L, Hu B, Zhang Y, et al. Suppression of the nuclear transporterKPNbeta1 expression inhibits tumor proliferation in hepatocellular carcinoma. Med Oncol. 2015;32:128. doi:10.1007/s12032-015-0559-1
19. He S, Miao X, Wu Y, et al. Upregulation of nuclear transporter, Kpnbeta1, contributes to accelerated cell proliferation- and cell adhesion-mediated drug resistance (CAM-DR) in diffuse large B-cell lymphoma. J Cancer Res Clin Oncol. 2016;142:561–572. doi:10.1007/s00432-015-2057-4
20. Yan W, Li R, He J, Du J, Hou J. Importin beta1 mediates nuclear factor-kappaB signal transduction into the nuclei of myeloma cells and affects their proliferation and apoptosis. Cell Signal. 2015;27:851–859. doi:10.1016/j.cellsig.2015.01.013
21. Lu T, Bao Z, Wang Y, et al. Karyopherinβ1 regulates proliferation of human glioma cells via Wnt/β-catenin pathway. Biochem Biophys Res Commun. 2016;478(3):1189–1197. doi:10.1016/j.bbrc.2016.08.093
22. Pellicano F, Park L, Hopcroft LEM, et al. hsa-mir183/EGR1-mediated regulation of E2F1 is required for CML stem/progenitor cell survival. Blood. 2018;131(14):1532–1544. doi:10.1182/blood-2017-05-783845
23. Zhang Y, Chen L, Yang S, Fang D. E2F1: a potential negative regulator of hTERT transcription in healthy cells upon activation of oncogenic c-Myc. Med Sci Monit. 2012;18(1):RA12–15. doi:10.12659/MSM.882192
24. Crusz SM, Balkwill FR. Inflammation and cancer: advances and new agents. Nat Rev Clin Oncol. 2015;12(10):584–596. doi:10.1038/nrclinonc.2015.105
25. Van der Watt PJ, Ngarande E, Leaner VD. Overexpression of Kpnβ1 and Kpnα2 importin proteins in cancer derives from deregulated E2F activity. PLoS One. 2011;6(11):e27723. doi:10.1371/journal.pone.0027723
26. Zhu ZC, Liu JW, Li K, Zheng J, Xiong ZQ. KPNB1 inhibition disrupts proteostasis and triggers unfolded protein response-mediated apoptosis in glioblastoma cells. Oncogene. 2018;37(22):2936–2952. doi:10.1038/s41388-018-0180-9
27. Zhang P, Garnett J, Creighton CJ, et al. EZH2-miR-30d-KPNB1 pathway regulates malignant peripheral nerve sheath tumour cell survival and tumourigenesis. J Pathol. 2014;232(3):308–318. doi:10.1002/path.2014.232.issue-3
28. Rodriguez-Bravo V, Pippa R, Song WM, et al. Nuclear pores promote lethal prostate cancer by increasing POM121-driven E2F1, MYC, and AR nuclear import. Cell. 2018;174(5):1200–1215.e20. doi:10.1016/j.cell.2018.07.015
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.