Back to Journals » International Journal of Nanomedicine » Volume 13
Inducing angiogenesis with the controlled release of nitric oxide from biodegradable and biocompatible copolymeric nanoparticles
Authors Yang C, Hwang HH, Jeong S, Seo D, Jeong Y, Lee DY ![]() , Lee K
, Lee K
Received 25 May 2018
Accepted for publication 9 August 2018
Published 16 October 2018 Volume 2018:13 Pages 6517—6530
DOI https://doi.org/10.2147/IJN.S174989
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Chungmo Yang,1,* Hae Hyun Hwang,2,* Soohyun Jeong,1 Deokwon Seo,1 Yoon Jeong,1 Dong Yun Lee,2,3 Kangwon Lee1,4
1Department of Transdisciplinary Studies, Graduate School of Convergence Science and Technology, Seoul National University, Seoul 08826, Republic of Korea; 2Department of Bioengineering, College of Engineering, and BK21 PLUS Future Biopharmaceutical Human Resources Training and Research Team, Hanyang University, Seoul 04763, Republic of Korea; 3Institute of Nano Science & Technology (INST), Hanyang University, Seoul 04763, Republic of Korea; 4Advanced Institutes of Convergence Technology, Gyeonggi-do 16229, Republic of Korea
*These authors contributed equally to this work
Purpose: Nitric oxide (NO) can be clinically applied at low concentrations to regulate angiogenesis. However, studies using small molecule NO donors (N-diazeniumdiolate, S-nitrosothiol, etc) have yet to meet clinical requirements due to the short half-life and initial burst-release profile of NO donors. In this study, we report the feasibility of methoxy poly(ethylene glycol)-b-poly(lactic-co-glycolic acid) (mPEG-PLGA) nanoparticles (NPs) as NO-releasing polymers (NO-NPs) for inducing angiogenesis.
Materials and methods: The mPEG–PLGA copolymers were synthesized by typical ring-opening polymerization of lactide, glycolide and mPEG as macroinitiators. Double emulsion methods were used to prepare mPEG–PLGA NPs incorporating hydrophilic NONOate (diethylenetriamine NONOate).
Results: This liposomal NP encapsulates hydrophilic diethylenetriamine NONOate (70%±4%) more effectively than other previously reported materials. The application of NO-NPs at different ratios resulted in varying NO-release profiles with no significant cytotoxicity in various cell types: normal cells (fibroblasts, human umbilical vein endothelial cells and epithelial cells) and cancer cells (C6, A549 and MCF-7). The angiogenic potential of NO-NPs was confirmed in vitro by tube formation and ex vivo through an aorta ring assay. Tubular formation increased 189.8% in NO-NP–treated groups compared with that in the control group. Rat aorta exhibited robust sprouting angiogenesis in response to NO-NPs, indicating that NO was produced by polymeric NPs in a sustained manner.
Conclusion: These findings provide initial results for an angiogenesis-related drug development platform by a straightforward method with biocompatible polymers.
Keywords: mPEG-PLGA nanoparticles, sprouting angiogenesis, low concentration of nitric oxide, liposomal nanoparticles, amphiphilic polymers
Introduction
Nitric oxide (NO) can induce multiple biological functions by stimulating cellular signaling pathways. For example, representative NO-driven functions include various human physiological processes, such as immune responses, inhibition of platelet aggregation, angiogenesis, apoptosis, neurotransmission and inflammation.1 After these multiple functions of NO were identified, NO-based therapies began to be developed for clinical application. Specifically, the angiogenic activity of NO has been applied in repairing or regenerating damaged tissue due to the degradation of the extracellular matrix.2 Use of NO donor molecules, such as N-diazeniumdiolate (NONOate), S-nitrosothiol (RSNO) and nitrate/nitrite/nitroso compounds, has been a way to circumvent the short half-life of NO. NONOate is one of the most studied NO donors because it stoichiometrically generates two NO products. NONOate donors that have been developed to date include 1-(hydroxy-NNO-azoxy)-L-proline (PROLI/NONOate), 3,3′-(hydroxynitrosohydrazino)bis-1-propanamine (DPTA/NONOate), 1-(ethenyloxy-NNO-azoxy)-pyrrolidine (PYRRO/NONOate) and N-[bis(2-aminoethyl)amino]-N-hydroxynitrous amide (DETA/NONOate).3 However, the short half-life and the initial burst-release profile remain the challenges.3
Alternatively, polymer-based release of NO represents the most commonly applied strategy to overcome these problems.1,4,5 NONOates are generated by reacting NO gas at 5 atm with secondary amines in polymer chains.6 This reaction leads to the development of NO-functionalized polymeric self-assembled micelles and star-shaped polymers.7–9 These materials can successfully release NO at physiological levels. However, since none of these materials is US Food and Drug Administration approved, clinical applications with human patients are limited. Poly(lactic-co-glycolic acid) (PLGA) is the most successful material for tissue engineering and drug delivery because its hydrolysis renders natural metabolite monomers, lactic acid and glycolic acid.10 By imparting PLGA the ability to release NO, PLGA can be fabricated for multifunctional applications. NO donors can be dispersed within the PLGA matrix, creating a completely biodegradable NO-releasing material. When coated, PLGA can both promote and control the release of NO. Hydrolysis of the PLGA coating and its intrinsic acid residues provides the protons required for NO release.11 Previously, PLGA-based NO delivery vehicles had been examined for the treatment of vaginal dysfunction and wound healing.12–14 Polyethylene glycol (PEG)–PLGA (di- or triblock) copolymers have been widely studied as drug delivery systems with curcumin,15 5-fluorouracil16 and rhodamine-labeled dextran15,17 due to their biocompatibility and biodegradability. Their amphiphilicity enables the simultaneous loading of hydrophobic and hydrophilic drugs. Amphiphilic methoxy poly(ethylene glycol) (mPEG)–PLGA copolymer nanoparticles (NPs) had been prepared successfully previously.18 For example, hydrophobic doxorubicin and hydrophilic paclitaxel self-assembled with each counterpart polymer. Although mPEG–PLGA copolymers have not been approved by the US Food and Drug Administration, toxicity is not an issue when they hydrolyze.19 A previous report described mPEG-protected NPs with (O2-{2,4-dinitro-5-[4-(Nmethylamino) benzoyloxy]phenyl} 1-(N,N-dimethylamino)diazen-1-ium-1,2-diolate) (PABA/NONOate) that release NO.20 However, this study employed PS-b-PEG and PLA-b-PEG, not PEG–PLGA copolymers.
Here, we demonstrate the feasibility of using mPEG–PLGA copolymers as polymeric vehicles for the sustained release of NO to induce angiogenesis (Scheme 1). The double emulsion method was used to prepare mPEG–PLGA-incorporated NONOate (DETA/NONOate). The hydrophilic core, PEG, successfully encapsulated DETA/NONOate. PLGA segments prevented the initial burst release of NO as protecting layer and it had the important role for NO-releasing behavior of NPs. NPs with different PEG:PLGA ratios released NO for varying lengths of time and showed no cytotoxicity in cell lines, including mouse fibroblasts (3T3 L1), human umbilical vein endothelial cells (HUVECs) and human epithelial (EP) cells. Tube formation assays determined the optimum concentration of NO-releasing NPs (NO-NPs) for robust bridge formation. The aorta ring assay demonstrated that these carriers could promote angiogenesis. We expect that the mPEG–PLGA copolymer nanomaterial is a highly suitable scaffold for NO release, representing a promising material for therapeutic applications such as wound healing, treating hind limb ischemia and other angiogenesis-related treatments.
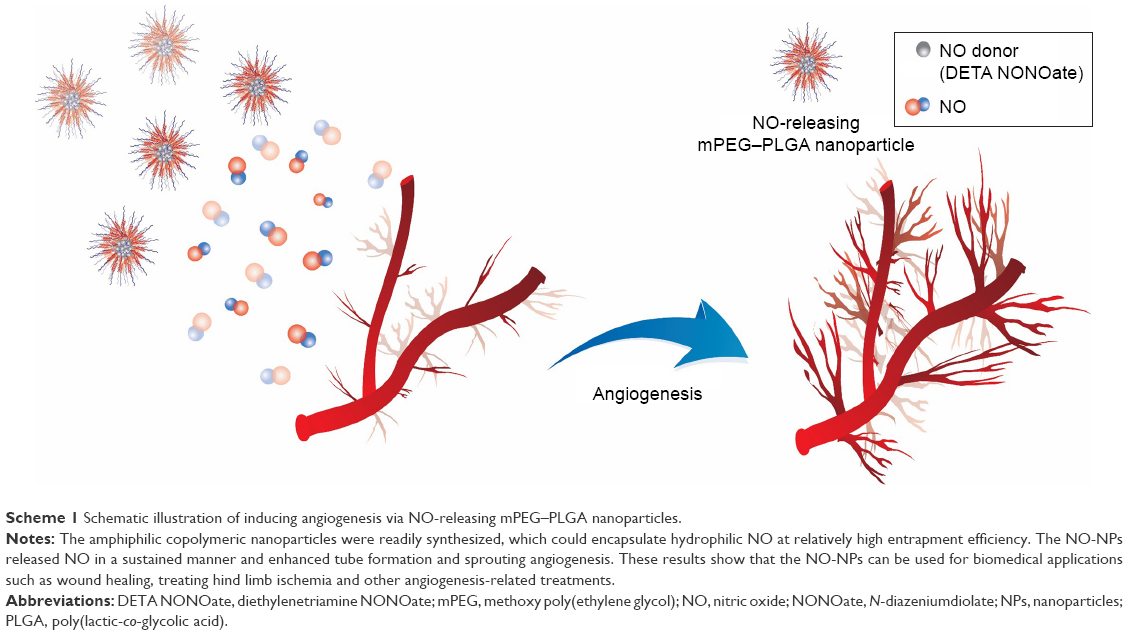 | Scheme 1 Schematic illustration of inducing angiogenesis via NO-releasing mPEG–PLGA nanoparticles. |
Materials and methods
Materials
Two types of mPEG (molecular weight [MW] 2,000 and 5,000 Da) were purchased from Acros Organics (Gael, Belgium) and Sigma Aldrich (St Louis, MO, USA), respectively. Lactide, glycolide and stannous octoate were purchased from Sigma Aldrich. Before use, mPEG was dried in vacuum to remove moisture, and D,L-lactide and glycolide were recrystallized from ethyl acetate and stored at −20°C. Diethylenetriamine NONOate (DETA NONOate) was obtained from Acros Organics. Dichloromethane (DCM), toluene, methanol and ethyl acetate were purchased from Samchun Chemical (Seoul, Korea). Poly(vinyl alcohol) was purchased from Junsei Chemical (Tokyo, Japan). HUVECs were purchased from Lonza (Basel, Switzerland). The 3T3 fibroblast, MCF-7 (breast adenocarcinoma), A549 (lung carcinoma) and C6 (mouse brain glial cell) cell lines were purchased from Korea Cell Line Bank (Seoul, Korea). Human EP cells (PCS-600-010) were purchased from ATCC (Manassas, VA, USA). The cell counting kit-8 (CCK-8) was purchased from Dojondo Molecular Technologies Inc. (Rockville, MD, USA). The Live/Dead Assay kit was purchased from Thermo Fisher Scientific (Waltham, MA, USA).
Synthesis of mPEG–PLGA block copolymers and NO-NPs
The typical ring-opening polymerization was conducted to synthesize mPEG–PLGA copolymers initiated by mPEG catalyzed by stannous octoate, as described previously (Figure 1).20 Briefly, pretreated monomers (mPEG, D,L-lactide and glycolide) were added to a dry round-bottom flask. Then, the flask was sealed and subsequently immersed in an oil bath. The catalyst, stannous octoate (0.05 wt % of the total monomer), was injected into the mixture. The temperature was increased to 130°C with gentle stirring, followed by injection of the catalyst. The polymerization proceeded for 8 hours in a nitrogen environment. After cooling down by quenching the reaction, the crude residue was dissolved in DCM and then precipitated in excess cold diethyl ether. The mixture was subsequently filtered and washed at least three times. Finally, hydroxyl-terminated mPEG–PLGA was recovered by drying at room temperature for 24 hours.
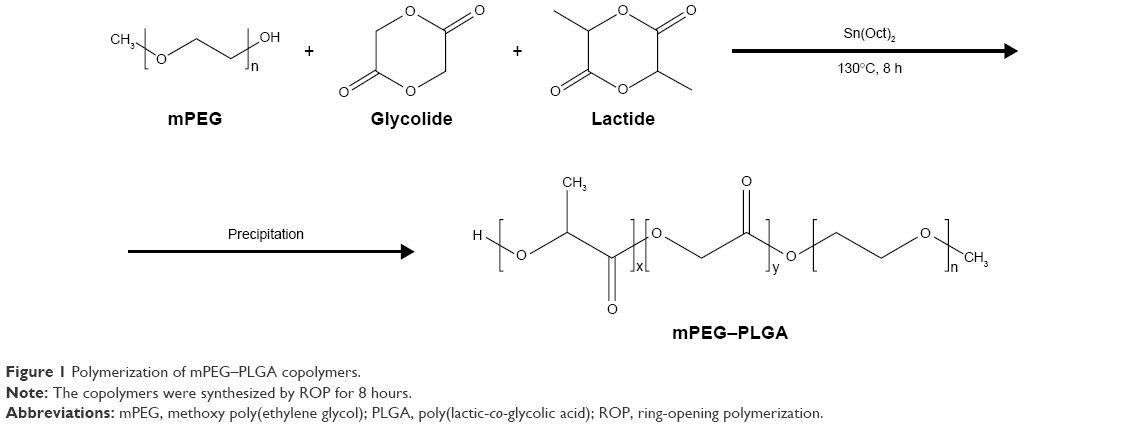 | Figure 1 Polymerization of mPEG–PLGA copolymers. |
mPEG–PLGA NPs containing DETA NONOate mPEG–PLGA and PLGA NPs encapsulating DETA NONOate were prepared using the modified double emulsion (water-in-oil-in-water) method, as previously reported (Figure 2).18 Briefly, mPEG–PLGA (20 mg) was dissolved in DCM (1 mL). Then, water or the DETA/NONOate solution (5wt%, 10 mM NaOH) was added and the mixture was emulsified using a probe sonicator in an ice bath for 3 minutes. Then, polyvinyl alcohol (2%) solution was added and emulsified for 5 minutes under the same conditions as the previous sonication. The emulsified mixture was poured into 0.6% polyvinyl alcohol solution (15 mL) and vigorously stirred for 30 minutes. After the DCM evaporated, the NPs were centrifuged and washed three times using distilled water. The NPs were then collected after freeze-drying for 3 days.
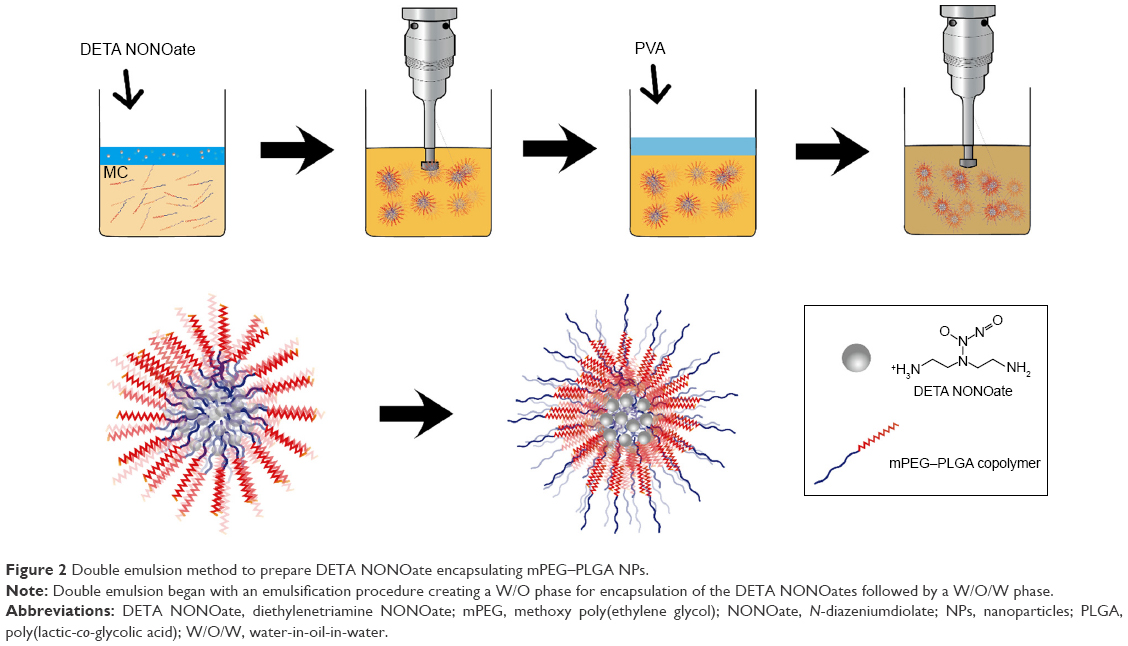 | Figure 2 Double emulsion method to prepare DETA NONOate encapsulating mPEG–PLGA NPs. |
Characterization of mPEG–PLGA copolymers
To confirm the existence of mPEG–PLGA copolymer structures, the prepared products were characterized using 1H nuclear magnetic resonance (1H NMR) spectrometer (ADVANCE II 400; Bruker Corporation, Billerica, MA, USA) with chloroform as a solvent. Fourier transform infrared spectrometer (FT-IR; SpectrumGX; PerkinElmer Inc., Waltham, MA, USA) was used to measure the copolymer structure. Gel permeation chromatography (GPC; Ultimate 3000; Thermo Fisher Scientific) confirmed the MW with tetrahydrofuran (THF) as a solvent.
Morphological characterization and size distribution
The morphological study was carried out using transmission electron microscopy (TEM; JEM-3100; JEOL, Tokyo, Japan). Negative staining with sodium phosphotungstate solution (1%) was conducted to pretreat the NPs. The size distribution and zeta potential of the NPs were measured using a Zetasizer (Malvern Instruments Ltd, Malvern, UK) with a fixed scattering angle of 173° at 25°C, and all samples were properly dispersed in deionized (DI) water.
Measurement of entrapment efficiency
The entrapment efficiency of DETA NONOate within NPs was confirmed using a previously reported method.21 The loading efficiency was measured by a ultraviolet–visible spectrometer (Lambda 35; PerkinElmer Inc.). The NPs were dispersed in 1 M sodium hydroxide and kept in a bath sonicator for 10 minutes. The mixture was continuously stirred until the NPs completely decomposed. After hydrolysis, the mixture was spectrophotometrically measured at a wavelength of 252 nm.22 The standard solution was prepared by combining blank NPs and known amounts of DETA NONOate in 1 M sodium hydroxide. The efficiency data were calculated using the following equation:
|

NO release measurement
NO released from the NO-NPs was detected by using the standard Griess assay with a Nitrite/Nitrate Assay Kit (Sigma Aldrich). The Griess assay is commonly used to determine nitrite and nitrate contents.9 Under normal physiological conditions, NONOate can easily produce NO. For the Griess assay, 10 mg of NO-NPs containing DETA NONOate was dissolved in PBS (2 mL). Then, the mixture was put into a dialysis bag (Cellu-Sep 1,000 molecular weight cut-off) to diffuse NO freely. The dialysis bag was immersed in PBS (6 mL) and maintained at 37°C. At each time point, a 100 μL sample was taken. For each sample, nitrate reductase and the enzyme cofactor, as indicated by the manufacturer’s instructions, were added to the solution, followed by incubation at room temperature for 30 minutes with gentle shaking. The nitrate reductase reduces the nitrate to nitrite. Then, Griess reagents were added to the sample, which was left to incubate for >30 minutes with gentle orbital shaking. After completely mixing, each sample was mixed with DI water. This procedure was repeated for each sample at each time point. The total concentration of nitrate and nitrite was determined by absorbance at 540 nm and calculated from a standard curve across different time points.
In vitro cytotoxicity assessment
Cytotoxicity was assessed in various cell types. HUVECs, human EP cells, mouse fibroblast, MCF-7 (breast adenocarcinoma), A549 (lung carcinoma) and C6 (mouse brain glial cell) (~5×103) were placed in media in 96-well plates at 37°C for each time point, that is, 12, 24 and 48 hours. For HUVECs, endothelial growth medium-2 was supplemented with fetal bovine serum (FBS) (10 mL), hydrocortisone (2 mL), ascorbic acid (0.5 mL), human epidermal growth factor (hEGF-2) (0.5 mL), heparin (0.5 mL), human fibroblast growth factor-b (hFGF-β) (2 mL), gentamicin sulfate-amphotericin (GA-1000) (0.5 mL), human recombinant insulin-like growth facto (VEGF) (0.5 mL) and R3-IGF (0.5 mL). The 3T3 L1 culture medium was DMEM supplemented with 10% bovine calf serum and 1% penicillin (PS). EP cells were cultured in DMEM with 10% FBS and 1% PS. The culture media were with 100 μL of 100 μg NP/mL or 50 μg NP/mL. At each time point, the viability of the cells was assessed using a CCK-8 assay kit. To further test the cytotoxicity of the NO-NPs, a Live/Dead assay was conducted. Likewise, the CCK-8 assay was used to assess normal and cancerous cell types (human EP cells, mouse fibroblast MCF-7, A549 and C6). The cell viability was assessed using the LIVE/DEAD™ Viability/Cytotoxicity Kit (Thermo Fisher Scientific). After the cells were cultured in a 96-well plate, the images were observed and captured randomly using a fluorescence microscope (Azio Observer Z1; Carl Zeiss, Oberkochen, Germany) at each time point (48 and 72 hours).
Tube formation assay
To confirm the angiogenic properties of NO-NPs, tube formation assays were conducted using HUVEC (passage 3) cells. Tube formation assay is one of the most established assays to investigate three-dimensional neovascularization mediated by endothelial cells. To construct a three-dimensional environment, 75 μL of Matrigel® Basement Membrane Matrix (growth factor reduced, #2354230; Corning Incorporated, Corning, NY, USA) was transferred to each well of a 96-well plate. The precoated plate was incubated at room temperature for 10 minutes and sequentially incubated in a 37°C incubator with 5% CO2 for 30 minutes. The endothelial basal medium-2 (EBM-2, Lonza, catalog number: CC-3156; without VEGF) or endothelial growth medium-2 (EGM-2, CC-3162; with 50 ng/mL VEGF, wherein the concentration of VEGF was determined by adjustment of angiogenic environment with considering self-generating growth factors in endothelial cells) were added to each well. Ans the medium containing blank NPs (50 μg/mL), DETA NONOate (CAS: 146724-94-9, 4 ng/mL, wherein the concentration of DETA NONOate is the same amount of NO released by 50 ng/mL NO-NPs) or NO-NPs (5, 10, 50, 75, 100 and 200 μg/mL) were treated at 2× concentration. The well plate was allowed to equilibrate at 37°C, 5% CO2, 90% humidity for at least 30 minutes. HUVECs (2×104 cells/well) were seeded into prepared precoated well plates and incubated under the same conditions. After 16 hours, tubule formation was assessed by microscopy at 40× magnification and the number of microvascular bridges was quantified using ImageJ software.
Aortic ring assay
First, 48-well cell culture plates were precoated with 100 μL Matrigel and incubated at 37°C and 5% CO2 for 30 minutes. Rat aortas (7-week-old male Sprague Dawley rats; DBL Company, Eumseong, Korea) were isolated and cut into rings with a thickness of 1.5 mm, and a single ring was placed in the top center of each well, which was then incubated for 10 minutes at 37°C. Then 100 μL of the Matrigel Matrix was supplemented on the top of each ring and incubated for 30 minutes at 37°C. Next, 500 μL of EBM-2 medium, containing blank NP (50 μg/mL), DETA NONOate (4 μg/mL) or NPs (50 μg/mL) with or without VEGF (50 ng/mL), was added to each well and incubated at 37°C and 5% CO2. The medium with or without growth factors and NPs was changed every other day. After 2 weeks, sprouting microvessel images were obtained using microscopy at 40× magnification and quantified using ImageJ software.
Statistical analysis
Data are represented as the mean±standard error of the mean. Statistically significant differences were analyzed by Student’s t-test or one-way ANOVA test (Sigma Plot; Systat Software Inc., San Jose, CA, USA). A P-value <0.05 was considered statistically significant.
Results
Characterization of mPEG–PLGA copolymers
The mPEG–PLGA copolymers were characterized by 1H NMR and FT-IR spectra (Figure 3). The peak at ~δ 1.55 ppm in 1H NMR of the copolymer (1) was the methyl protons in the D- and L-lactic acid repeat units. The methylene protons of the methoxy PEG caused a large peak at ~δ 3.6 ppm (2). The characteristic peaks revealed the successful synthesis of mPEG–PLGA copolymers from randomly ordered sequences of various D- and L-lactic and glycolic acid in the backbone of copolymer. The peaks at δ 4.8 ppm (3) and δ 5.2 ppm (4) were attributed to the protons in the CH2 groups in the glycolic acid and CH in the lactic acid segments, respectively.
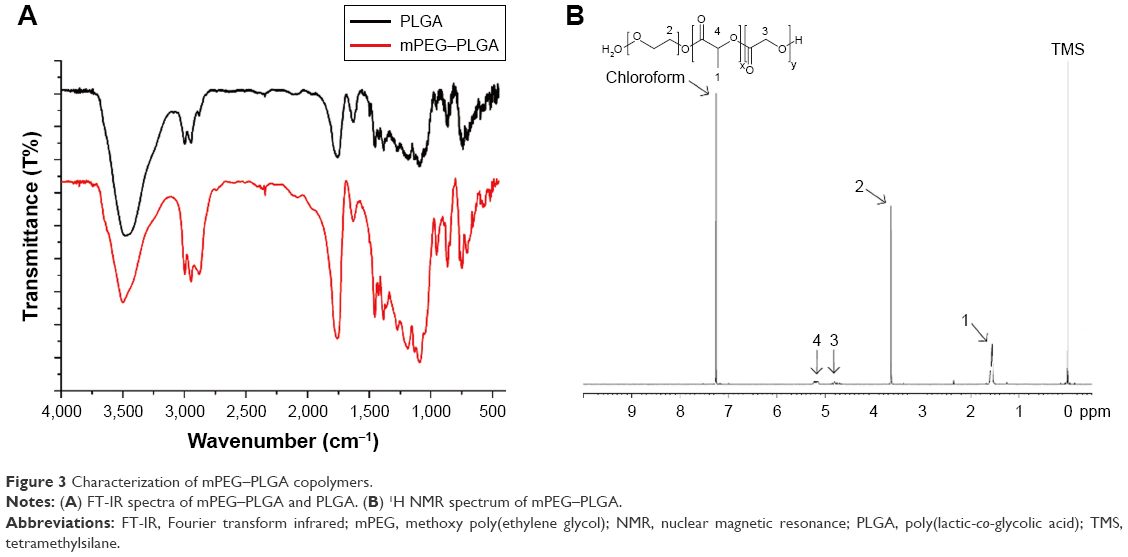 | Figure 3 Characterization of mPEG–PLGA copolymers. |
FT-IR spectra further confirmed the mPEG–PLGA copolymer structure. The C=O stretch attributed a strong band at 1,760 cm−1, and the bands at 1,090.92–1,188.61 cm−1 were assigned to the stretch of C−O bond. The bands at 2,995 and 2,943 cm−1 indicated C−H bond stretching in −CH2, and the peak at 2,875 cm−1 indicated C−H bonds. The strong band at 3,477 cm−1 was assigned to terminal hydroxyl group stretch in the copolymer. Since they possess the same functional groups, similar characteristic peaks were observed in the FT-IR spectra of the mPEG–PLGA and PLGA. The MW was confirmed by GPC with THF as a solvent. We measured four groups: PLGA and mPEG–PLGA synthesized by using mPEG MW 5,000 Da, and PEG MW 2,000 Da at different ratios (LE:GE), as shown in Table 1. The copolymers have a polydispersity index of 1.19–1.62.
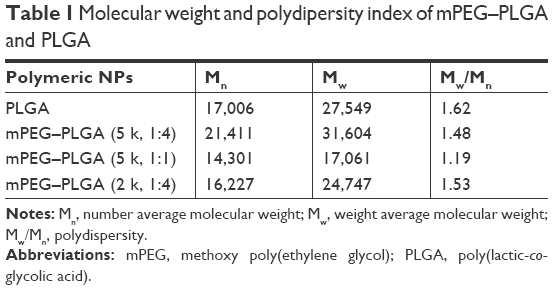 | Table 1 Molecular weight and polydipersity index of mPEG–PLGA and PLGA |
Characterization of mPEG–PLGA NPs: morphology, size, zeta potential, and entrapment efficiency
The mPEG–PLGA NP morphology was confirmed by TEM (Figure 4A). The NPs synthesized by the emulsification method using amphiphilic copolymers had a core–shell structure similar to those of other previously reported materials.23 The resulting NPs were characterized by dynamic light scattering (DLS), as shown in Figure 4B. mPEG–PLGA NPs showed an estimated size of 200 nm, which is small enough for pharmaceutical applications. The DLS results revealed that the NPs had similar size and a relatively narrow size distribution. The particle sizes of mPEG–PLGA 5 kDa and mPEG–PLGA 2 kDa differed. NPs having larger molecular weight than mPEG which were around 20 nm larger than another one. When the same emulsion methods were applied to prepare PLGA NPs, their particle size was ~350 nm with DETA/NONOates (data not shown). The zeta potential of NO-NPs displayed a slightly negative charge (avg. 1.59±0.254 mV; Figure 4C). These results are similar to other reports of PLGA-related NPs. The entrapment efficiency of DETA NONOate was calculated from hydrolyzed mPEG–PLGA NPs. The amount of NONOate was measured and calculated using a ultraviolet–visible spectrometer. The encapsulation efficiency was calculated to be 70%±4%. This result is a relatively higher value than that of previously reported hydrophobic micelle structures or NPs.12
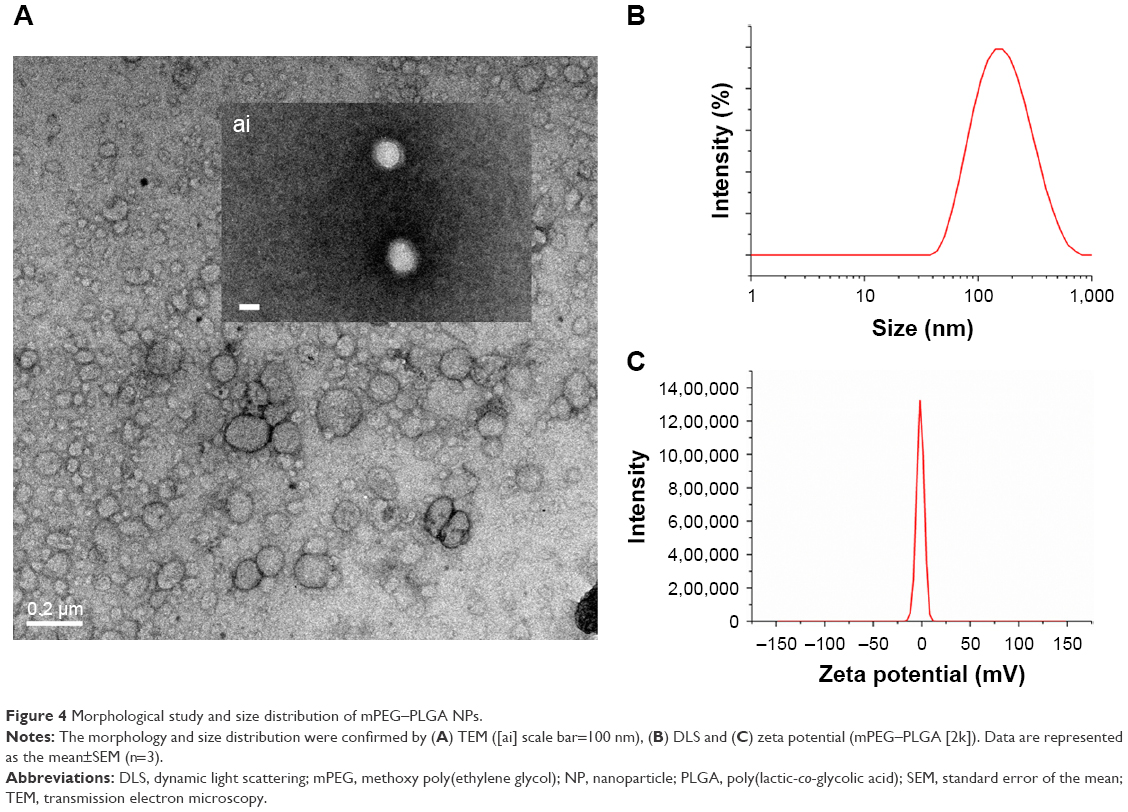 | Figure 4 Morphological study and size distribution of mPEG–PLGA NPs. |
NO release measurement
At physiological conditions, the free radical NO spontaneously oxidizes to nitrite or nitrate. The Griess assay is commonly used for NO detection by quantifying the amount of both nitrite and nitrate. Therefore, we conducted Griess assay to confirm that mPEG–PLGA efficiently releases NO in a controlled manner. We also tested NPs with PLGA segments of various lengths and found that as the PLGA portion shortens, more NO is released.
As free NO donors, DETA NONOates rapidly release NO under the same conditions, reaching ~80% in 24 hours. However, NO-NPs showed sustained-release behavior with a relatively lower flux of NO than that of free NO donors during the release period. mPEG–PLGA NPs (ratio 1:4) release NO that can promote angiogenesis, whereas mPEG–PLGA NPs (ratio 1:1) generate a relatively high concentration of NO, which can induce an antibacterial response or apoptosis. From this result, we conclude that the mPEG–PLGA ratio is a vital determinant for controlled NO release. To further investigate the role of PEG in the copolymer, we synthesized mPEG–PLGA with different MWs of mPEG (2 and 5 kDa). Our results showed that different polymer ratios affected NO release (Figure 5). The mPEG–PLGA NPs with high-MW mPEG released NO relatively rapidly, while NPs with higher proportions of PLGA or lower MW mPEG exhibited slower NO release.
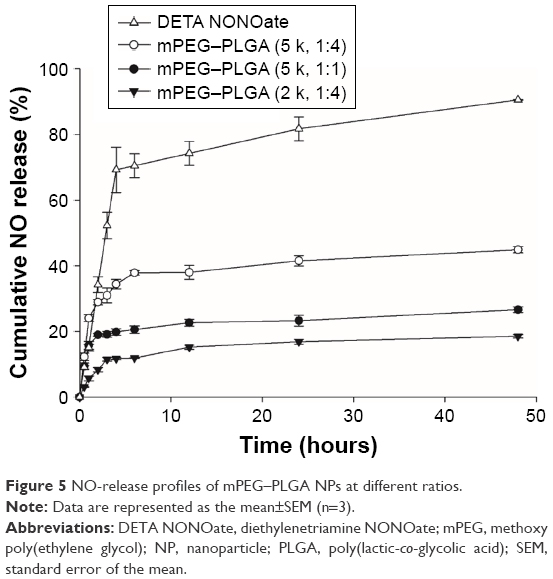 | Figure 5 NO-release profiles of mPEG–PLGA NPs at different ratios. |
In vitro cytotoxicity of NO-NPs
To apply these particles pharmaceutically, the particles must not be cytotoxic. Various cell lines were used to assess cytotoxicity, including mouse fibroblasts, HUVECs, human EP cells, C6, A549 and MCF-7. Cytotoxicity assays were carried out using different concentrations of NPs with NO over 48 hours. For all cell types tested, the results demonstrated that particles with NO were not cytotoxic at any of the experimental concentrations (Figure 6). The mouse fibroblast (3T3 L1) cell line showed a slightly higher cell density than the control group during early stages, but for cell viability, no significant difference was observed. To test our new materials in wound-healing applications, we tested the viability of the human EP cells. Similar to the other two cell types, mouse fibroblast and HUVEC, human EP cell viability did not decrease in response to NO-NPs. To observe cancer cell behavior when treated with NO-NPs compared to normal cells, further cytotoxicity tests were conducted. Like normal cells, the viability of the cancer cells showed no significant decrease in both the CCK-8 and Live/Dead assay (Figures 6 and 7). The Live/Dead assay showed result similar to that of cell viability in both normal cells and cancer cells. Low concentrations of NO can mediate angiogenesis in the early stages of new blood vessel formation, as NO participates in tip cell sprouting after extracellular matrix degradation.2 Additionally, HUVECs displayed no significant toxicity response to NO-releasing mPEG–PLGA NPs (Figure 6A).
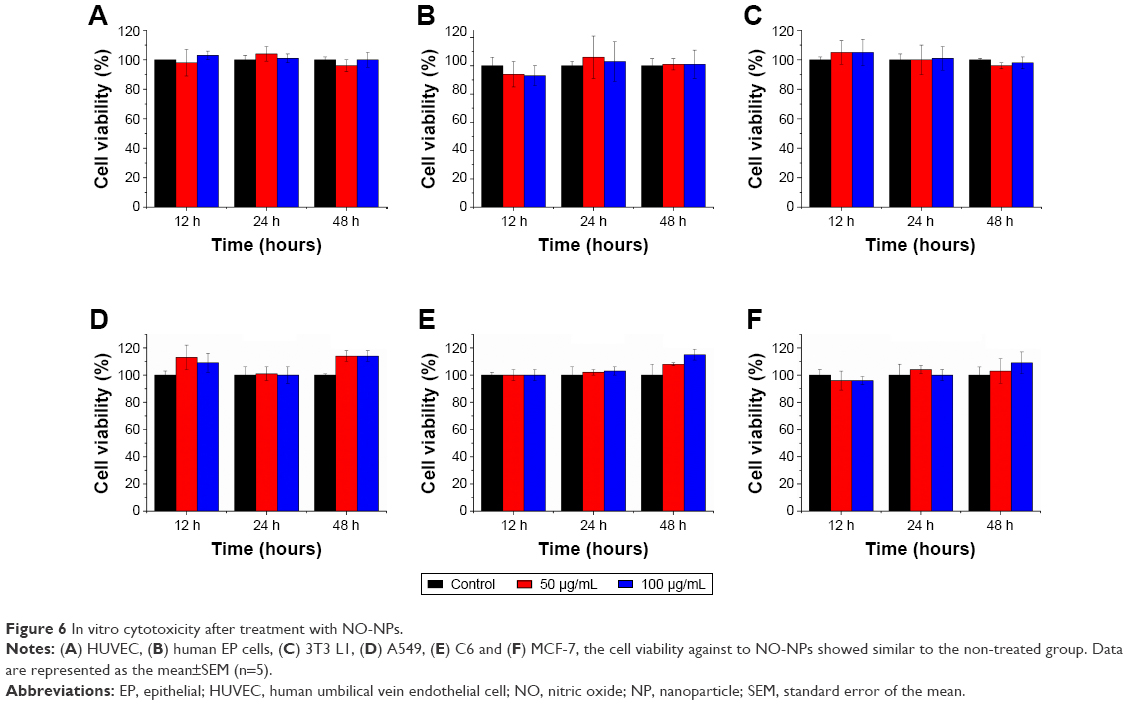 | Figure 6 In vitro cytotoxicity after treatment with NO-NPs. |
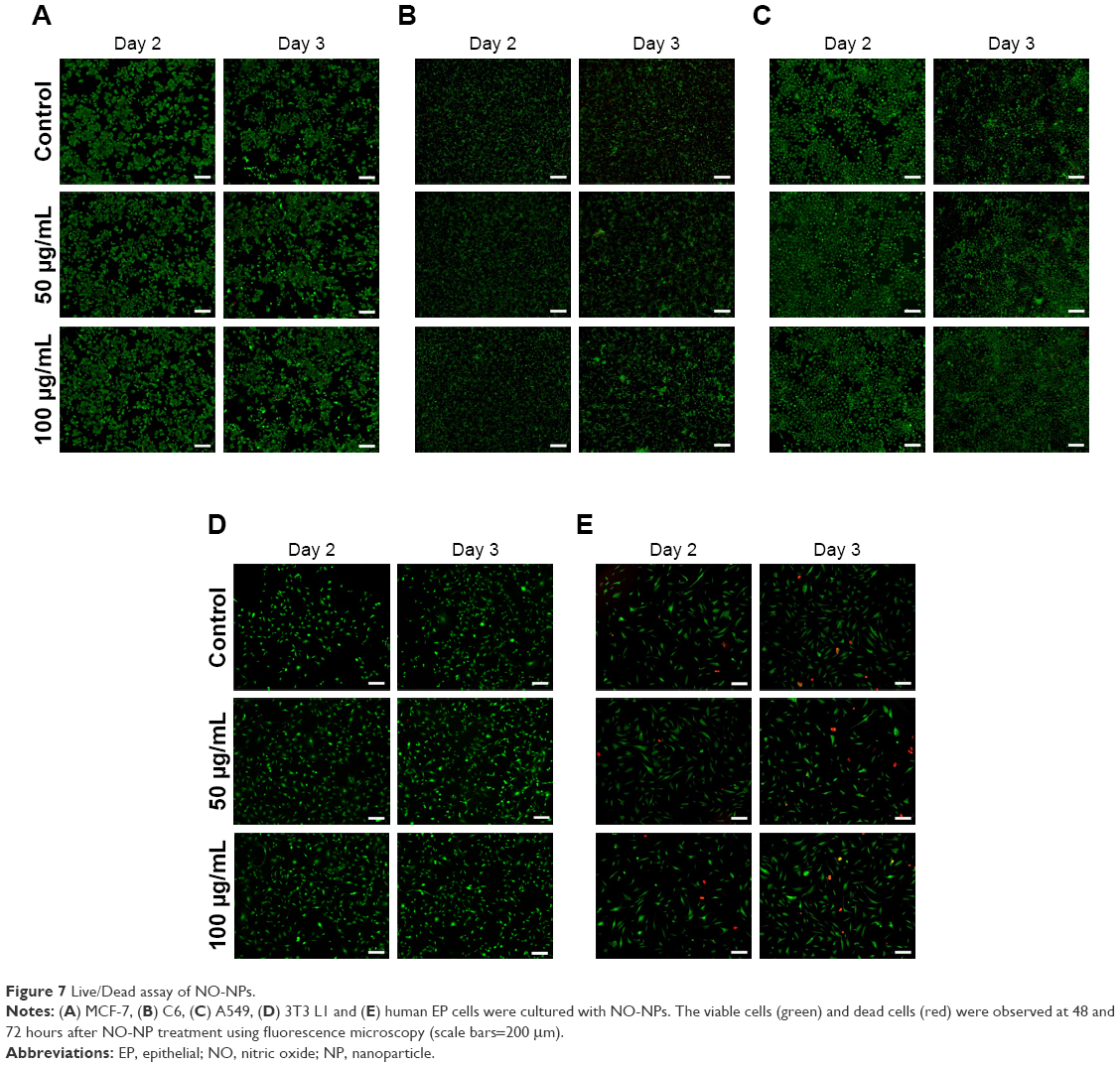 | Figure 7 Live/Dead assay of NO-NPs. |
Induction of angiogenesis by NO-NPs
To evaluate the induction of angiogenesis by NO-NPs, a tubular formation assay was performed in vitro using HUVECs. When NO-NPs were treated, HUVECs responded with more significant growth and formation of branched bridges than the negative control (without VEGF and NPs), as shown in Figure 8A. This induction of angiogenesis depended on the concentration when they were treated with up to 50 μg/mL of NO-NPs. In particular, at 50 μg/mL, more tubes were formed than those in the EBM-2+VEGF group (P<0.001), and the DETA NONOate group, which showed no significant differences but had high mean values, emitted the same amount of NO (Figure 8B). Interestingly, when treated with 75, 100 and 200 μg/mL of NO-NPs, the induction of angiogenesis was gradually inhibited.
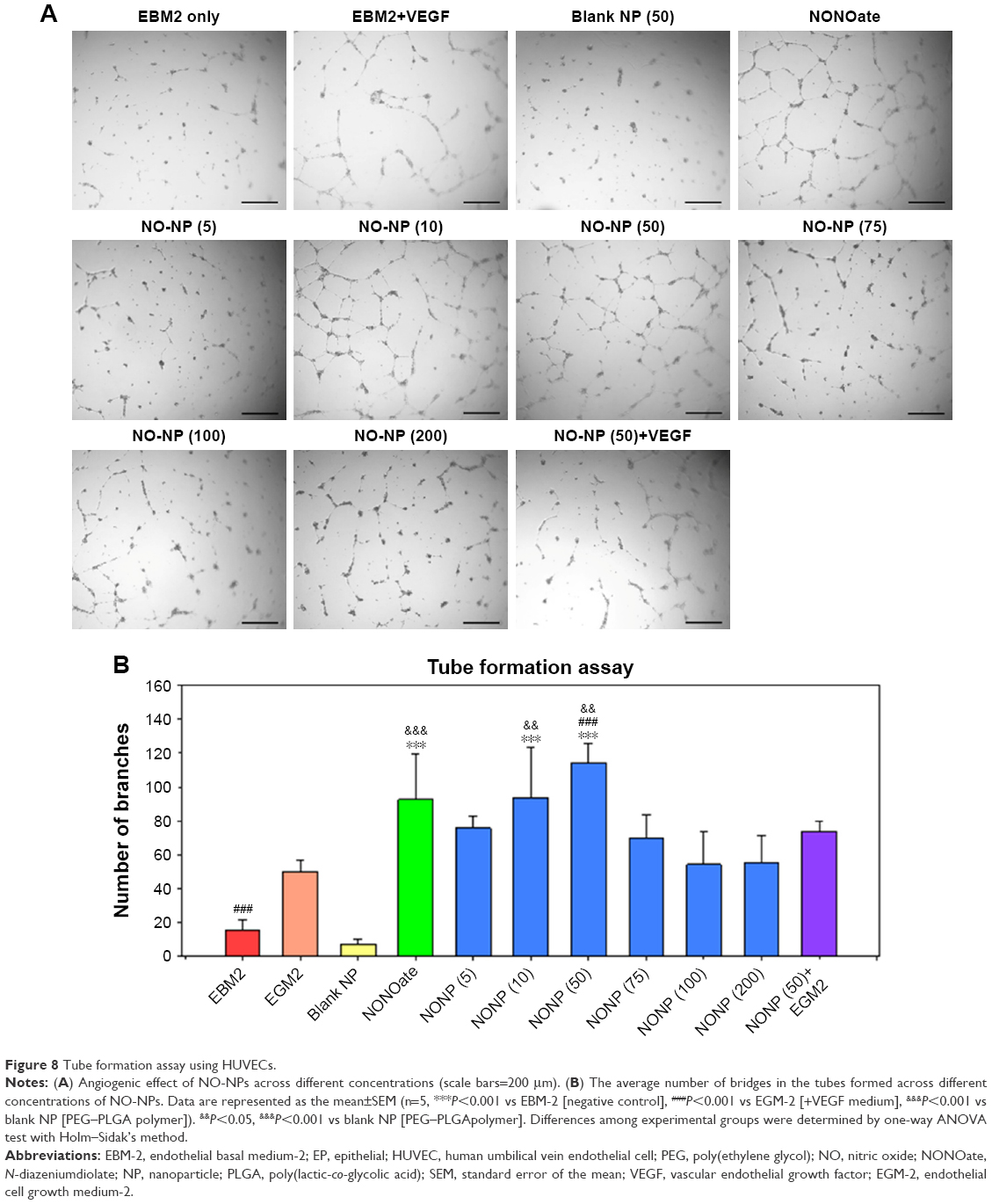 | Figure 8 Tube formation assay using HUVECs. |
In addition, we further evaluated tube formation with VEGF (50 ng/mL) addition to the NO-NPs group at 50 μg/mL concentration and found that tube formation was significantly decreased relative to that caused by NO-NPs alone (ie, no significant difference was observed). To assess the ex vivo induction of angiogenesis, the rat aorta ring assay was carried out according to relatively short- or long-term treatment with NO-NPs (50 μg/mL). This experimental system mimics in vivo neovascularization because the vessels grow out from the aorta ring and then engage with both smooth muscle cells and pericytes to establish endothelial cell tubes.24,25 When NO-NPs were added to rat aorta rings, new vessels were dramatically induced around the aorta ring (Figure 9A and B). No significant differences in the number of branches were observed between the controls, blank NPs and free NO donor groups. The NO-NP group exhibited significantly more branches, with ~586%, 344% and 424% relative to those of the control (P<0.001), blank NPs (P<0.05) and free NO donor (P<0.001) groups, respectively. Therefore, use of NO-NPs in vivo could potentially induce neovascularization in the body. In addition, we further evaluated whether NO-NPs could induce angiogenesis when accompanied by VEGF (50 ng/mL). Branches increased significantly in comparison to those in the control (517%; P<0.001), blank NPs (303%; P<0.05) and free NO donor (374%; P<0.001) groups in response to addition of VEGF. However, angiogenesis induction by NO-NPs was not significantly different in response to VEGF (Figure 9B).
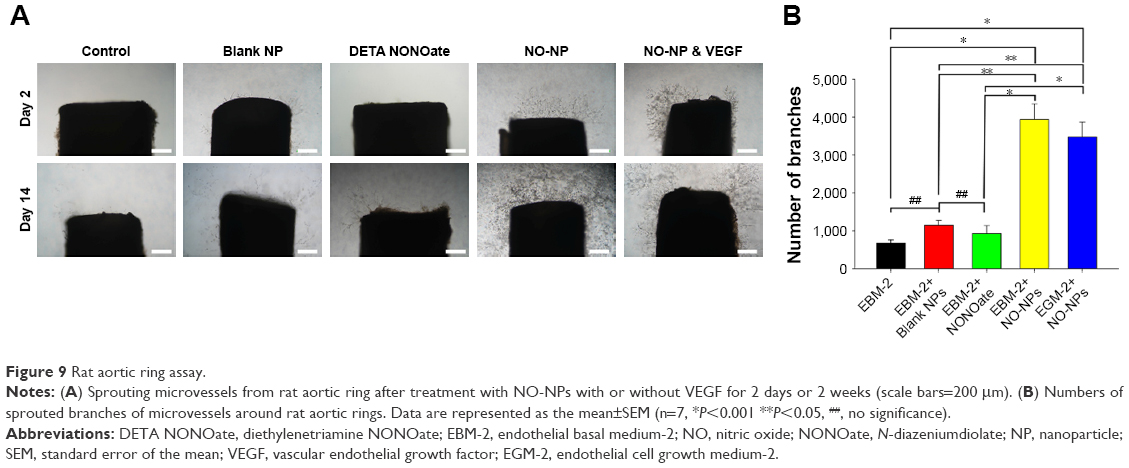 | Figure 9 Rat aortic ring assay. |
Discussion
NO was first discovered as a physiological modulator that is functionally identical to endothelium-derived relaxation factor.26 This finding gained notable scientific attention, and subsequently, biological processes that were mediated by NO, either directly or indirectly, were discovered. As the knowledge of physiological NO increases, studies on its clinical application have also been thriving. In particular, NO-related drugs such as L-arginine are used to treat cardiovascular disease and arteriosclerosis.27 As numerous studies have been conducted on drug delivery systems, attempts to exogenously deliver NO and other gaseous signaling molecules as therapeutic agents are increasing.
Several factors were considered when designing NO-releasing materials for clinical use. First, the concentration of NO is important because NO functions in a concentration-dependent manner. Taking this into account, a polymeric vehicle that could maintain appropriate drug concentrations could potentially be applied for NO release (Figure 1). Biocompatibility and biodegradability were the other factors considered for clinical NO delivery material designs. mPEG and PLGA are advantageous in that they are currently used in drug delivery systems and are both biocompatible and biodegradable. Thus, we designed mPEG–PLGA polymeric vehicles encapsulating NO donors to accomplish sustained NO release to promote angiogenesis. mPEG–PLGA copolymers were synthesized by common ring-opening polymerization and characterized by 1H NMR, FT-IR and GPC studies. Our 1H NMR and FT-IR results confirmed the copolymer structure, as shown in Figure 3. The MWs of various copolymers with different mPEG:PLGA ratios were assessed by GPC (Table 1). Our results illustrated that copolymers with increased mPEG ratios display lower MWs.
As mPEG–PLGA copolymers exhibit amphiphilic properties, they form NPs through emulsification (Figure 3). The NPs have an appropriate size to avoid destruction in the reticuloendothelial system and to allow for prolonged circulation in the human body.28 Accordingly, mPEG–PLGA forms various structures owing to their amphiphilicity.29–32 Linear amphiphilic copolymers with hydrophilic volume fraction factors of 25%–40% are known to produce NPs by self-assembly.33–35 With this knowledge, mPEG–PLGA block copolymers with appropriate MWs can be used to prepare NPs due to their high stability,36 potential various modifications15,37,38 and absence of significant toxicity in vivo.17,39–41 Biodegradable amphiphilic copolymers also facilitate vesicular formation, which facilitates delivery of both hydrophilic and hydrophobic compounds. Exemplary findings have demonstrated that mPEG–PLGA can load both hydrophilic doxorubicin and hydrophobic paclitaxel each to their affinitive site to enhance antitumor efficacy.18 To load either hydrophilic or hydrophobic drugs into polymeric NPs, the double emulsion method is most widely used.42,43 In this study, we used a two-step emulsification process of W/O emulsion followed by generating a water-in-oil-in-water emulsion to prepare the NPs (Figure 3).44
Because DETA NONOate is hydrophilic, encapsulating it in the micelle structure is difficult.45 As such, vesicular formation using linear amphiphilic copolymer nanocarriers has emerged as a method to enhance encapsulation efficiency46,47 and cellular uptake.48 Using vesicular formation with biodegradable amphiphilic copolymers improves the efficacy of nanocarrier preparation, encapsulation efficiency and usability. Therefore, we used hydrophilic-core NPs to encapsulate the hydrophilic NO donor and to leave room for the codelivery of other hydrophobic drugs. TEM and DLS results show the core–shell structure and size of mPEG–PLGA NPs (Figure 4A and B). As shown by the TEM and DLS results, mPEG–PLGA synthesized from various ratios resulted in various NP sizes: the longer the chain of mPEG, the more space that was occupied in the self-assembled structure (Figure 4A and B).
If NO released from NPs induces angiogenesis, then NO should be released at low concentrations. In this study, polymeric vehicles protected NO donors from stimuli sources such as protons, as specified in our report. We first hypothesized that the ratio of hydrophilic and hydrophobic chain lengths affects the NO release profile. As shown in Figure 5, the concentrations of released NO increase as the PLGA portion of the copolymer decreases. Likewise, the use of higher MW mPEG increases the particle size and slightly increases the NO release rate. Thus, we concluded that NO release can be controlled by varying the ratio of the copolymer components to determine the optimum ratio of mPEG–PLGA (2 kDa, 1:4). Subsequently, we used mPEG–PLGA (2 kDa, 1:4) for further investigation.
The NO-NPs exhibit no significant cytotoxicity against various cell lines, including 3T3 L1, HUVEC, human EP cells and other cancer cells, as shown by our results (Figures 6 and 7). The live cells overwhelmingly outnumbered the dead cells in both the normal and cancer cell types. In some cases, the cell population increased when treated with NO-NPs. This lack of cytotoxicity is evidence that NO-NPs can attain sustained release of NO while effectively protecting the contents from protons. In addition, our results suggest that the concentration of NO that is maintained is low even in the cancer environment.
To confirm NO as the sole contributor to angiogenesis, tube formation assay was conducted. This confirmed that NO promotes formation of bridge networks (tubule formation), as shown in Figure 8A and B. In addition, we conducted experiments with or without VEGF and blank NPs. Later, we confirmed that tubule formation was most robust at concentrations of 50 μg NP/mL and least induced at 200 μg NP/mL. This result is reasonable since gaseous NO promotes angiogenesis only in specific concentration ranges that exceed ~50 μg NP/mL. Altogether, our results indicate that angiogenesis is mainly stimulated by NO released from NPs rather than by the copolymer itself.
The aortic ring is a three-dimensional tool to evaluate the angiogenic potential of selected drugs or factors ex vivo.49 We used the aortic ring assay to confirm that NO induces angiogenesis without growth factors. Our results showed that NO released from NPs promotes angiogenesis (Figure 9A and B). These results provide further evidence that only NO can accelerate angiogenesis and that mPEG–PLGA NPs protect NO donors from protons and external stimuli to ensure a low concentration of NO is released. The extent of endothelial branching was significantly enhanced by treatment with NO (Figure 9B). On the other hand, rings treated with free NO donor groups showed limited branching compared to those in the NO-NP groups. Levels of proangiogenic microvessels branching out from the aortic ring were similar to those in the blank NP and free NO donor groups, with both exhibiting slightly more branches than the control. Furthermore, blank NPs induced more branches than did free NO donor groups, indicating that sprouting angiogenesis is dependent on a low concentration of NO. Interestingly, we observed slightly fewer branches in response to NO-NP with VEGF treatment in the aortic ring assay, as shown in the quantitative analysis. This finding could be due to the reciprocal regulation between NO and VEGF, where small amounts of NO upregulate VEGF expression through the hypoxia-inducible factor-1-mediated signal pathway, which enhances NO production from endothelial NOS by VEGF.50 Such reciprocal interaction could be responsible for the restrained angiogenesis observed in response to NO-NPs and VEGF treatment, which was expected to be robust. We believe NP concentration to be important in circumventing the diminishing effect of NO and VEGF interaction. Optimal concentrations of NPs for in vivo angiogenesis were not studied; however, our team plans to investigate this concentration in future studies. Additionally, our group also plans to investigate the applicability of NO-NPs to disease models that require neovascularization for successful recovery, such as in wound healing and lower limb ischemia disease models.
Conclusion
In this work, we prepared NO-releasing mPEG–PLGA NPs and confirmed that NO released from the NPs promotes angiogenesis in vitro and ex vivo. NO-NPs were prepared by mPEG–PLGA block copolymers, which encapsulated DETA NONOates as NO donors. We evaluated the NO release capacity, cytotoxicity and angiogenic potential of the NPs. The NPs displayed low cytotoxicity and released NO in a sustained manner. Furthermore, their angiogenic potential was confirmed by endothelial cell tubule formation and aortic ring assays. In the aortic ring assay, we identified that only NO released from the NPs promoted angiogenesis. To the best of our knowledge, this report is the first to confirm the promotion of angiogenesis using only NO-NPs without other biomolecules. These results suggest that liposomal mPEG–PLGA copolymer nanocarriers are a promising platform to promote angiogenesis, such as during wound healing, lower limb ischemia and cardiovascular disease.
Acknowledgments
This research was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Ministry of Science, ICT & Future Planning (grant numbers: NRF-2016M3A9B4919711, 2015R1A2A1A05001832, NRF-2015M3A9E2030125) and by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI14C2099).
Disclosure
The authors report no conflicts of interest in this work.
References
Seabra AB, Justo GZ, Haddad PS. State of the art, challenges and perspectives in the design of nitric oxide-releasing polymeric nanomaterials for biomedical applications. Biotechnol Adv. 2015;33(6 Pt 3):1370–1379. | ||
Clapp C, Thebault S, Jeziorski MC, Martínez de La Escalera G, de La Escalera GM. Peptide hormone regulation of angiogenesis. Physiol Rev. 2009;89(4):1177–1215. | ||
Keefer LK. Fifty years of diazeniumdiolate research. From laboratory curiosity to broad-spectrum biomedical advances. ACS Chem Biol. 2011;6(11):1147–1155. | ||
Jen MC, Serrano MC, van Lith R, Ameer GA. Polymer-based nitric oxide therapies: recent insights for biomedical applications. Adv Funct Mater. 2012;22(2):239–260. | ||
Quinn JF, Whittaker MR, Davis TP. Delivering nitric oxide with nanoparticles. J Control Release. 2015;205:190–205. | ||
Hrabie JA, Keefer LK. Chemistry of the nitric oxide-releasing diazeniumdiolate (“nitrosohydroxylamine”) functional group and its oxygen-substituted derivatives. Chem Rev. 2002;102(4):1135–1154. | ||
Riccio DA, Schoenfisch MH. Nitric oxide release: part I. Macromolecular scaffolds. Chem Soc Rev. 2012;41(10):3731–3741. | ||
Duong HT, Jung K, Kutty SK, et al. Nanoparticle (star polymer) delivery of nitric oxide effectively negates Pseudomonas aeruginosa biofilm formation. Biomacromolecules. 2014;15(7):2583–2589. | ||
Nguyen TK, Selvanayagam R, Ho KKK, et al. Co-delivery of nitric oxide and antibiotic using polymeric nanoparticles. Chem Sci. 2016;7(2):1016–1027. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Handa H, Brisbois EJ, Major TC, et al. In vitro and in vivo study of sustained nitric oxide release coating using diazeniumdiolate-oped poly(vinyl chloride) matrix with poly(lactide-co-glycolide) additive. J Mater Chem B. 2013;1(29):3578–3587. | ||
Yoo JW, Lee JS, Lee CH. Characterization of nitric oxide-releasing microparticles for the mucosal delivery. J Biomed Mater Res A. 2010;92(4):1233–1243. | ||
Kang Y, Kim J, Lee YM, Im S, Park H, Kim WJ. Nitric oxide-releasing polymer incorporated ointment for cutaneous wound healing. J Control Release. 2015;220(Pt B):624–630. | ||
Nurhasni H, Cao J, Choi M, et al. Nitric oxide-releasing poly(lactic-co-glycolic acid)-polyethylenimine nanoparticles for prolonged nitric oxide release, antibacterial efficacy, and in vivo wound healing activity. Int J Nanomedicine. 2015;10:3065–3080. | ||
Song Z, Feng R, Sun M, et al. Curcumin-loaded PLGA–PEG–PLGA triblock copolymeric micelles: preparation, pharmacokinetics and distribution in vivo. J Colloid Interface Sci. 2011;354(1):116–123. | ||
Qiao M, Chen D, Ma X, Liu Y. Injectable biodegradable temperature-responsive PLGA–PEG–PLGA copolymers: synthesis and effect of copolymer composition on the drug release from the copolymer-based hydrogels. Int J Pharm. 2005;294(1–2):103–112. | ||
Farokhzad OC, Cheng J, Teply BA, et al. Targeted nanoparticle–aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci U S A. 2006;103(16):6315–6320. | ||
Wang H, Zhao Y, Wu Y, et al. Enhanced anti-tumor efficacy by co-delivery of doxorubicin and paclitaxel with amphiphilic methoxy PEG–PLGA copolymer nanoparticles. Biomaterials. 2011;32(32):8281–8290. | ||
Zhang K, Tang X, Zhang J, et al. PEG–PLGA copolymers: their structure and structure-influenced drug delivery applications. J Control Release. 2014;183:77–86. | ||
Kumar V, Hong SY, Maciag AE, et al. Stabilization of the nitric oxide (NO) prodrugs and anticancer leads, PABA/NO and Double JS-K, through incorporation into PEG-protected nanoparticles. Mol Pharm. 2010;7(1):291–298. | ||
Bilati U, Allémann E, Doelker E, Poly DE. Poly(D,L-lactide-co-glycolide) protein-loaded nanoparticles prepared by the double emulsion method – processing and formulation issues for enhanced entrapment efficiency. J Microencapsul. 2005;22(2):205–214. | ||
Hrabie JA, Klose JR, Wink DA, Keefer LK. New nitric oxide-releasing zwitterions derived from polyamines. J Org Chem. 1993;58(6):1472–1476. | ||
Wang Y, Gao S, Ye WH, Yoon HS, Yang YY. Co-delivery of drugs and DNA from cationic core–shell nanoparticles self-assembled from a biodegradable copolymer. Nat Mater. 2006;5(10):791–796. | ||
Bellacen K, Lewis EC. Aortic ring assay. J Vis Exp. 2009;33:e1564. | ||
Aplin AC, Nicosia RF. The rat aortic ring model of angiogenesis. Methods Mol Biol. 2015;1214:255–264. | ||
Ignarro LJ, Byrns RE, Buga GM, Wood KS. Endothelium-derived relaxing factor from pulmonary artery and vein possesses pharmacologic and chemical properties identical to those of nitric oxide radical. Circ Res. 1987;61(6):866–879. | ||
Böger RH, Bode-Böger SM, Brandes RP, et al. Dietary L-arginine reduces the progression of atherosclerosis in cholesterol-fed rabbits: comparison with lovastatin. Circulation. 1997;96(4):1282–1290. | ||
Santander-Ortega MJ, Csaba N, González L, Bastos-González D, Ortega-Vinuesa JL, Alonso MJ. Protein-loaded PLGA–PEO blend nanoparticles: encapsulation, release and degradation characteristics. Colloid Polym Sci. 2010;288(2):141–150. | ||
Rösler A, Vandermeulen GWM, Klok H-A. Advanced drug delivery devices via self-assembly of amphiphilic block copolymers. Adv Drug Deliv Rev. 2012;64:270–279. | ||
Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv Drug Deliv Rev. 2001;47(1):113–131. | ||
Adams ML, Lavasanifar A, Kwon GS. Amphiphilic block copolymers for drug delivery. J Pharm Sci. 2003;92(7):1343–1355. | ||
Gaucher G, Dufresne MH, Sant VP, Kang N, Maysinger D, Leroux JC. Block copolymer micelles: preparation, characterization and application in drug delivery. J Control Release. 2005;109(1–3):169–188. | ||
Discher BM, Won YY, Ege DS, et al. Polymersomes: tough vesicles made from diblock copolymers. Science. 1999;284(5417):1143–1146. | ||
Bleul R, Thiermann R, Maskos M. Techniques to control polymersome size. Macromolecules. 2015;48(20):7396–7409. | ||
Lee JS, Feijen J. Polymersomes for drug delivery: design, formation and characterization. J Control Release. 2012;161(2):473–483. | ||
Discher DE, Eisenberg A, Vesicles P. Polymer vesicles. Science. 2002;297(5583):967–973. | ||
Chan JM, Zhang L, Yuet KP, et al. PLGA–lecithin–PEG core–shell nanoparticles for controlled drug delivery. Biomaterials. 2009;30(8):1627–1634. | ||
Gao N, Chen Z, Xiao X, et al. Surface modification of paclitaxel-loaded tri-block copolymer PLGA-b-PEG-b-PLGA nanoparticles with protamine for liver cancer therapy. J Nanopart Res. 2015;17(8):347. | ||
Alibolandi M, Sadeghi F, Abnous K, Atyabi F, Ramezani M, Hadizadeh F. The chemotherapeutic potential of doxorubicin-loaded PEG-b-PLGA nanopolymersomes in mouse breast cancer model. Eur J Pharm Biopharm. 2015;94:521–531. | ||
Zhao X, Li F, Li Y, et al. Co-delivery of HIF1α siRNA and gemcitabine via biocompatible lipid–polymer hybrid nanoparticles for effective treatment of pancreatic cancer. Biomaterials. 2015;46:13–25. | ||
Xue Y, Xu X, Zhang XQ, Farokhzad OC, Langer R. Preventing diet-induced obesity in mice by adipose tissue transformation and angiogenesis using targeted nanoparticles. Proc Natl Acad Sci U S A. 2016;113(20):5552–5557. | ||
Ganji F, Abdekhodaie MJ. Chitosan–g-PLGA copolymer as a thermosensitive membrane. Carbohydr Polym. 2010;80(3):740–746. | ||
Dong Y, Feng SS. Methoxy poly(ethylene glycol)-poly(lactide) (MPEG-PLA) nanoparticles for controlled delivery of anticancer drugs. Biomaterials. 2004;25(14):2843–2849. | ||
Hanson JA, Chang CB, Graves SM, Li Z, Mason TG, Deming TJ. Nanoscale double emulsions stabilized by single-component block copolypeptides. Nature. 2008;455(7209):85–88. | ||
Kim J, Saravanakumar G, Choi HW, Park D, Kim WJ. A platform for nitric oxide delivery. J Mater Chem B. 2014;2(4):341–356. | ||
Eloy JO, Claro de Souza M, Petrilli R, Barcellos JP, Lee RJ, Marchetti JM. Liposomes as carriers of hydrophilic small molecule drugs: strategies to enhance encapsulation and delivery. Colloids Surf B Biointerfaces. 2014;123:345–363. | ||
Suchyta DJ, Schoenfisch MH. Encapsulation of N-diazeniumdiolates within liposomes for enhanced nitric oxide donor stability and delivery. Mol Pharm. 2015;12(10):3569–3574. | ||
Straubinger RM, Hong K, Friend DS, Papahadjopoulos D. Endocytosis of liposomes and intracellular fate of encapsulated molecules: encounter with a low pH compartment after internalization in coated vesicles. Cell. 1983;32(4):1069–1079. | ||
Baker M, Robinson SD, Lechertier T, et al. Use of the mouse aortic ring assay to study angiogenesis. Nat Protoc. 2011;7(1):89–104. | ||
Kimura H, Esumi H. Reciprocal regulation between nitric oxide and vascular endothelial growth factor in angiogenesis. Acta Biochim Pol. 2003;50(1):49–59. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.