Back to Journals » International Journal of Nanomedicine » Volume 12
Improving intestinal absorption and oral bioavailability of curcumin via taurocholic acid-modified nanostructured lipid carriers
Authors Tian C, Asghar S, Wu Y, Chen Z, Jin X, Yin L ![]() , Huang L
, Huang L ![]() , Ping Q, Xiao Y
, Ping Q, Xiao Y
Received 10 July 2017
Accepted for publication 25 September 2017
Published 27 October 2017 Volume 2017:12 Pages 7897—7911
DOI https://doi.org/10.2147/IJN.S145988
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Cihui Tian,1 Sajid Asghar,2 Yifan Wu,1 Zhipeng Chen,3 Xin Jin,1 Lining Yin,1 Lin Huang,1 Qineng Ping,1 Yanyu Xiao1
1Department of Pharmaceutics, State Key Laboratory of Natural Medicines, China Pharmaceutical University, Nanjing, China; 2Faculty of Pharmaceutical Sciences, Government College University Faisalabad, Faisalabad, Pakistan; 3Department of Pharmacy, Nanjing University of Chinese Medicine, Nanjing, China
Abstract: The expression of multiple receptors on intestinal epithelial cells enables an actively targeted carrier to significantly enhance the oral delivery of payloads. Conjugating the receptors’ ligands on the surfaces of a particulate-delivery system allows site-specific targeting. Here, we used taurocholic acid (TCA) as a ligand for uptake of nanostructured lipid carriers (NLCs) mediated by a bile-acid transporter to improve oral bioavailability of curcumin (Cur). First, synthesis of TCA–polyethylene glycol 100–monostearate (S100-TCA) was carried out. Then, the physical and chemical properties of S100-TCA-modified Cur-loaded NLCs (Cur-TCA NLCs) with varying levels of S100-TCA modifications were investigated. Small particle size (<150 nm), high drug encapsulation (>90%), drug loading (about 3%), negative ζ-potential (–7 to –3 mV), and sustained release were obtained. In situ intestinal perfusion studies demonstrated improved absorption rate and permeability coefficient of Cur-TCA NLCs. Depending on the degree of modification, Cur-TCA NLCs displayed about a five- to 15-fold higher area under the curve in rats after oral administration than unmodified Cur NLCs, which established that the addition of S100-TCA to the NLCs boosted absorption of Cur. Further investigations of TCA NLCs might reveal a bright future for effective oral delivery of poorly bioavailable drugs.
Keywords: curcumin, nanostructured lipid carriers, taurocholic acid, oral administration, pharmacokinetics, bioavailability
Introduction
The patient compliance offered by oral administration, due to its ease of application, safety, and inexpensiveness, makes it a favored route for delivery of pharmaceutical actives. Despite the advantages, several main challenges for oral administration are the inactivation of drugs by gastric acid and digestive enzymes, poor permeation across epithelial membrane, and the insolubility of drugs at physiological pH.1,2
The expression of multiple receptors on intestinal epithelial cells enables noticeable transport of poorly absorbed drugs via specific ligand-modified drug-delivery systems. Vitamin B12-modified micelles prepared by Francis et al enhanced the transport of cyclosporine A in intestinal epithelial cells by binding to intrinsic factor receptors.3 Roger et al found increased oral absorption of folate-modified nanoparticles due to the expression of folate receptors on intestinal epithelial cells.4 Some reports have demonstrated that drug molecules connected to bile acids can interact with intestinal bile-acid transporters.5,6 Park et al combined the bile acid with heparin derivative to improve the drug uptake of intestinal epithelial cells.7 Khatun et al confirmed that using taurocholic acid (TCA), a bile salt, as a targeting moiety promoted the oral absorption of heparin–docetaxel nanoparticles via bile-acid transporters.8
Curcumin (Cur), a dietary polyphenolic compound, is the main active substance of the turmeric plant, which has extensive pharmacological activities (anti-inflammatory, antiatherosclerosis, antioxidant, hypolipidemic, and antimutagenic).9,10 Cur inhibits breast cancer-cell motility and invasiveness by modulating the adhesion molecules’ expression, and increases the sensitivity of chemotherapy drugs to cancer cells by regulating the expression of multidrug-resistance genes.11 Cur has been recognized as a third-generation chemopreventive agent for cancers by the US National Cancer Institute.12 However, a clinical trial showed that plasma concentration of Cur was only 22–41 ng/mL after oral ingestion of Cur at 8 g/day, indicating Cur was poorly absorbed after oral administration.13 The real therapeutic potential of Cur has not been fully utilized, due to lower absorption in the gut, rapid metabolism by the liver, and systemic elimination.14,15 Lately, numerous formulation platforms have been designed to address the poor oral bioavailability of drugs. Nanostructured lipid carriers (NLCs) stand tall over other strategies, due to their exceptional features: high bioavailability, reasonable capacity for loading of drugs with the ability to preserve their physical and chemical integrity for prolonged periods, controlled release of payload,16 and the absence of chemical cross-linking and toxic solvents during preparation are the main advantages offered by NLCs.17 They have low toxicity, due to their physiological lipid composition; therefore, NLCs are well tolerated in multiple-dosage therapy.18
In this study, TCA-functionalized NLCs were developed as novel bile-acid transporter-mediated nanocarriers to improve the oral delivery of Cur (Cur-TCA NLCs). Polyethylene glycol (PEG) 100–monostearate (S100), an amphiphilic polymer containing hydrophilic PEG attached to the hydrophobic stearate, can easily be incorporated into the lipid core of colloidal nanocarriers, with hydrophilic PEG chains extended toward their surface. PEG chains not only improve hydrophilicity and stability of the nanocarrier but also increase the residence time of the colloids in systemic circulation. Additionally, PEG chains can be easily utilized for ligand functionalization. As such, TCA was conjugated to S100 to form TCA-PEG100-monostearate (S100-TCA). Cur-loaded NLCs were modified with varying levels of S100-TCA and investigated for possible improvement in performance. Characteristic attributes (particle size, ζ-potential, morphology, drug loading [DL], encapsulation efficiency [EE], and in vitro release) of the NLCs were evaluated. Furthermore, intestinal absorption of Cur formulations in rats was investigated by intestinal circulation perfusion and in vivo pharmacokinetic studies.
Materials and methods
Materials
Cur (purity 98%), TCA sodium salt (purity >97%), cholesterol oleate (purity >85%), and S100 (polymerization degree of ethylene glycol was 100) were obtained from Aladdin Reagent (Shanghai, China), Yuanye Biotechnology (Shanghai, China), Tokyo Chemical Industry (Tokyo, Japan) and Chenrun Chemical (Nantong, China), respectively. Phosphatidylcholine (purity 90%) and glycerol trioleate (purity >98%) were gifted by Evonik Degussa China (Shanghai, China). 1-Ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDCI) was purchased from Energy Chemical (Shanghai, China). MTT was purchased from Sigma-Aldrich (St Louis, MO, USA).
Animals
The Experimental Animal Center of Nantong University (Nantong, China) provided the Sprague Dawley rats (180–220 g) for animal experimentation. All animal procedures were approved and conducted in strict accordance with approved standards for laboratory-animal care by the ethical committee of China Pharmaceutical University.
Synthesis of S100-TCA
A previous work with slight modifications was adopted for synthesis of S100-TCA.19 To obtain TCA, 2 mL distilled water containing 0.93 mmol TCA sodium salt was slowly dropped into hydrochloric acid solution (0.1 M, 3 mL) at 0°C and stirred at room temperature for 16 hours. Yellow crystalline mass was filtered using Whatman cellulose filters and vacuum-dried to obtain TCA.
To synthesize S100-TCA, EDCI (0.63 mmol) was added to 5 mL anhydrous N, N-dimethylformamide containing TCA (0.39 mmol) and stirred for 1 hour in an ice bath. Afterward, 25 mL of S100 (0.39 mmol) solution in CH2Cl2 was slowly dropped into the TCA solution and stirred for 1 hour in an ice bath, and subsequent stirring was continued for 24 hours at room temperature. Subsequently, vacuum was used to remove the organic solvent. Unreacted TCA and other minor molecular impurities were removed by dialysis of the resultant solution using a preswollen dialysis bag (5 kDa molecular weight cutoff [MWCO]; Sigma-Aldrich) at room temperature for 48 hours. After dialysis, S100-TCA was obtained by lyophilization. 1H nuclear magnetic resonance spectroscopy (1H-NMR; Avance AV 300; Bruker Optik GmbH, Ettlingen, Germany) was performed to characterize the structure of the conjugate using D2O as a solvent at 25°C. Furthermore, surface-tension measurements (Shibo Instruments, Shanghai, China) were also taken to measure critical micelle concentration.20
Preparation of Cur-TCA NLCs
Firstly, Cur NLCs were prepared according to our previous work.19 Briefly, 10 mL of ethanol–chloroform (1:1, v:v) was used to get a lipid–drug mixture of phosphatidylcholine (330 mg), cholesterol oleate (20 mg), glycerol trioleate (70 mg), and Cur (16 mg). A dried lipid film obtained through rotary evaporation at 37°C under vacuum was then hydrated with 10% (v:v) glycerol solution at 37°C for 30 minutes and subjected to ultrasonication for 5 minutes at 300 W in an ice bath. Unentrapped drug was removed from the final Cur-NLC suspension by extrusion using 0.45 μm cellulose nitrate membrane.
TCA-modified Cur NLCs (Cur-TCA NLCs) were prepared in a similar fashion to Cur NLCs, just with the addition of different quantities of S100-TCA into 10 mL of ethanol–chloroform mixture. The three levels (20, 50, and 100 mg) of Cur-TCA NLCs were named Cur-TCA20, Cur-TCA50, and Cur-TCA100 NLCs, respectively. Blank TCA NLCs were prepared the same way, only without Cur.
Determination of Cur content
Cur in NLCs was assayed by dissolving 100 μL Cur-TCA NLCs or Cur NLCs in methanol (10 mL) and subsequent centrifugation for 10 minutes at 12,000 rpm. Supernatant (20 μL) was analyzed by high-performance liquid chromatography (HPLC; LC-10AT; Shimadzu, Kyoto, Japan) at 420 nm. Acetonitrile:purified water:acetic acid at a ratio of 50:45:5 (v:v:v) was used as the mobile phase at a flow rate of 1 mL/min. A C18 column (150×4.6 mm, 5 μm; μBondapak) kept at 40°C was used as the stationary phase.21
Characterization of Cur-TCA NLCs
A Zetasizer 3000HS (Malvern Instruments, Malvern, UK) was employed to characterize Cur-TCA NLCs and Cur NLCs for particle size, polydispersity index (PDI), and ζ-potential. Morphology of the NLCs was observed under transmission electron microscopy (TEM; H-7000; Hitachi, Tokyo, Japan). A previously reported method was used to investigate the EE and DL of NLCs.22
Differential scanning calorimetry (DSC) was done with the aid of a TA 60WS with DSC-60 (Shimadzu) to analyze thermal properties of S100-TCA, Cur, blank TCA50 NLCs, and Cur-TCA50 NLCs. Each sample was sealed in an aluminum crucible and subjected to 50°C–300°C temperatures at 10 K/min in a nitrogen atmosphere. Ultraviolet-visible spectroscopy was utilized to probe the Cur orientation within the hydrophobic core of Cur NLCs or Cur-TCA50 NLCs.23,24 Spectra of aqueous suspension of NLCs and methanolic solution of Cur were taken from 200 to 800 nm at 1.5 μg/mL Cur concentration.
Surface PEG density and distance between adjacent PEG chains of S100-TCA NLCs
A colorimetric method with slight modifications was used for quantification of S100-TCA (W1).25 PEG can form colored complexes with iodine, with specific absorption at 500 nm and a linear relationship with PEG content. The unadhered S100-TCA was separated by ultrafiltration with a 100 kDa MWCO. Concentration of the unadhered S100-TCA was calculated from a standard curve in a range of 2.5–100 μg/mL S100-TCA. The total amount of S100-TCA (W2) was directly measured without ultrafiltration and deemed equal to the adhered and the unadhered S100-TCA on the surface of the modified NLCs. Therefore, the amount of S100-TCA adhered on the surface of modified NLCs (WPEG) was equal to (W2-W1). The surface density of PEG chains (SDPEG) was obtained thus:26
SDPEG = NPEG/SNP |
NPEG is the total number of PEG chains, and was calculated as:
NPEG = (WPEG/MPEG) × An |
where WPEG is equal to (W2 − W1), MPEG is S100-TCA MW, and An denotes Avogadro’s number. SNP is the surface area of the NLCs, and was calculated as:
SNP = 4πr2 × NNP |
where NNP and r denote the total number of NLCs and the approximate radii of NLCs, respectively. NNP was calculated as:
NNP = Mt/Ms |
where Mt is the total weight of lyophilized NLCs and Ms the mass of one particle. Resolving, Ms = ρ × V = ρ × 4πr3/3, where ρ is the density of NLC suspension and the value is approximately equal to water density, V is the volume of one particle, and r is the radii of NLCs. DPEG (distance between the adjacent PEG chains) on the TCA NLCs’ surface was calculated thus:
DPEG = (1/SDPEG)1/2 |
In vitro Cur release
Drug release from Cur NLCs and Cur-TCA NLCs was monitored in vitro in a way to simulate gastrointestinal conditions. In vitro release studies were carried out by placing 2 mL formulations (equivalent to 100 μg of Cur) in preswollen 8–10 kDa MWCO dialysis bags (Sigma-Aldrich) in 50 mL pH 1.2 0.1 M HCl as release media (simulating gastric pH) for 2 hours, then the release medium was replaced with the same volume of pH 6.8 PBS (simulating intestinal pH) at 37°C for the next 22 hours (n=3). Additionally, the release of Cur was also assessed in physiological saline to simulate plasma environment. All media contained 1% (w:v) Tween 80 to meet sink conditions. Cur solution (1.5 mg Cur dissolved in 10 mL PEG400, and then diluted to 50 μg/mL with deionized water) was regarded as control. The samples (1 mL) were withdrawn at predetermined times and replaced with the same amount of fresh medium maintained at 37°C. Centrifugation of the samples was carried out for 5 minutes at 12,000 rpm, and resultant supernatants were quantified for drug content by HPLC. Samples were protected from light during the experiments. DDSolver software was used to evaluate drug-release kinetics.27
Rat intestinal perfusion study
Krebs–Ringer (KR) buffer solution was obtained by dissolving 0.35 g KCl, 7.8 g NaCl, 0.02 g MgCl2, 1.37 g NaHCO3, 0.2 g CaCl2, 0.22 g NaH2PO4, and 1.48 g glucose in 1,000 mL deionized water. As needed, pH was adjusted to 6.8 with KOH or HCl solution.28 The Cur intestinal circulating perfusion solution (40 μg/mL Cur) was prepared by diluting Cur solution, Cur NLCs, and Cur-TCA NLCs with KR buffer solution.
Rat intestinal perfusion studies were done as per a previous report with slight modifications.29 Sprague Dawley rats were randomly grouped into five groups (three rats per group). After being fasted for 24 hours (water ad libitum), each rat was injected intraperitoneally with 10% chloral hydrate and fixed in a supine position. Intestinal segments (10 cm; duodenum, jejunum, and ileum) were exposed after laparotomy. Careful cannulation was done at both ends of the exposed organ. Afterward, intestines were washed with physiological saline (37°C and flow rate of 0.5 mL/min) with a peristaltic pump (HL-2; Huxi Instruments, Shanghai, China).30 Cur intestinal circulating perfusion solution (25 mL) was used for single-pass intestinal perfusion. First, 25 mL of the solution at 37°C was flowed back (flow rate 0.5 mL/min) for 30 minutes through the segment to obtain the steady state. Afterward, the solution was allowed to flow at a reduced flow rate of 0.2 mL/min for the following 2 hours. Samples (100 μL) were collected from distal portions of three intestinal segments at predetermined intervals and vortexed with 900 μL methanol for 30 seconds. The resulting mixtures were subjected to centrifugation at 12,000 rpm for 10 minutes. Supernatants (20 μL) were analyzed for Cur determination by HPLC.
Intestinal segments were removed from the killed animals for length (l) and radius (r) measurements. Furthermore, gravimetry was employed to find the overall flux of water (absorption and efflux) in the given segment. The following equation was used to correct the perfusate drug concentration:
Cout(corr) = Cout × (Qout/Qin) |
where Cout(corr) is corrected drug concentration in exiting fluid, Cout drug concentration in exiting fluid, Qin inlet perfusate flux (mL/min), and Qout outlet perfusate flux (mL/min). The following equations were used to calculate the effective permeability (Peff) coefficient and Ka (absorption rate) constant:
Peff = [− Q × ln(Cout(corr)/Cin)]/2πrl |
Ka = (1 − Cout(corr)/Cin) × Q/πr2l |
where Q is flow rate of fluid (0.2 mL/min), Cin drug concentration in the entering fluid, l length (cm) of the intestinal segment and r radius (cm) of the intestinal segment.
Pharmacokinetics
Five groups of male Sprague Dawley rats (n=6) were fasted overnight with free access to water. Cur equivalent of Cur solution (50 mg/kg dissolved in water–PEG400 [7:3, v:v]), Cur NLCs, Cur-TCA20 NLCs, Cur-TCA50 NLCs, and Cur-TCA100 NLCs were given orally to the rats. Blood samples (0.5 mL) collected from eyeground veins at predetermined intervals were centrifuged at 4,000 rpm for 10 minutes to obtain plasma and frozen at −20°C until analysis by HPLC.
Direct observation of the plasma-profile curve yielded maximum concentration (Cmax) and time to Cmax. DAS 2.0 software was used to get other useful pharmacokinetic parameters. The following equation was employed to determine relative bioavailability (F):
F (%) = AUCpreparation/AUCCur solution × 100 |
where AUCpreparation and AUCCur solution are area under the curve of Cur of different Cur preparations and Cur solution, respectively.
Statistical analysis
Results are given as means ± standard deviation. Statistical significance was tested by analysis of variance or two-tailed Student’s t-test with SPSS 19.0 software. Statistical significance was set at P<0.05.
Results and discussion
S100-TCA synthesis and characterization
The synthesis process of S100-TCA is shown in detail in Figure 1A. Lyophilized S100-TCA was white, odorless, and fibrous. In S100-TCA synthesis, the sulfonic acid of TCA was coupled to the hydroxyl of S100. The 1H-NMR spectrum (Figure 1B) shows hydrogen signals at 2.2–2.4 ppm depicting the existence of −COCH2− of TCA in S100-TCA, thus confirming that TCA had been successfully grafted onto the S100 structure to form the new polymer (S100-TCA). Critical micelle concentrations of S100 and S100-TCA were 62.85±0.11 and 15.86±0.53 μg/mL, respectively, which revealed that the conjugation of TCA did not affect the amphiphilicity of S100. As per calculation by TCA reference-standard curve, 11.79%±3.98% of TCA was coupled with the S100-TCA.
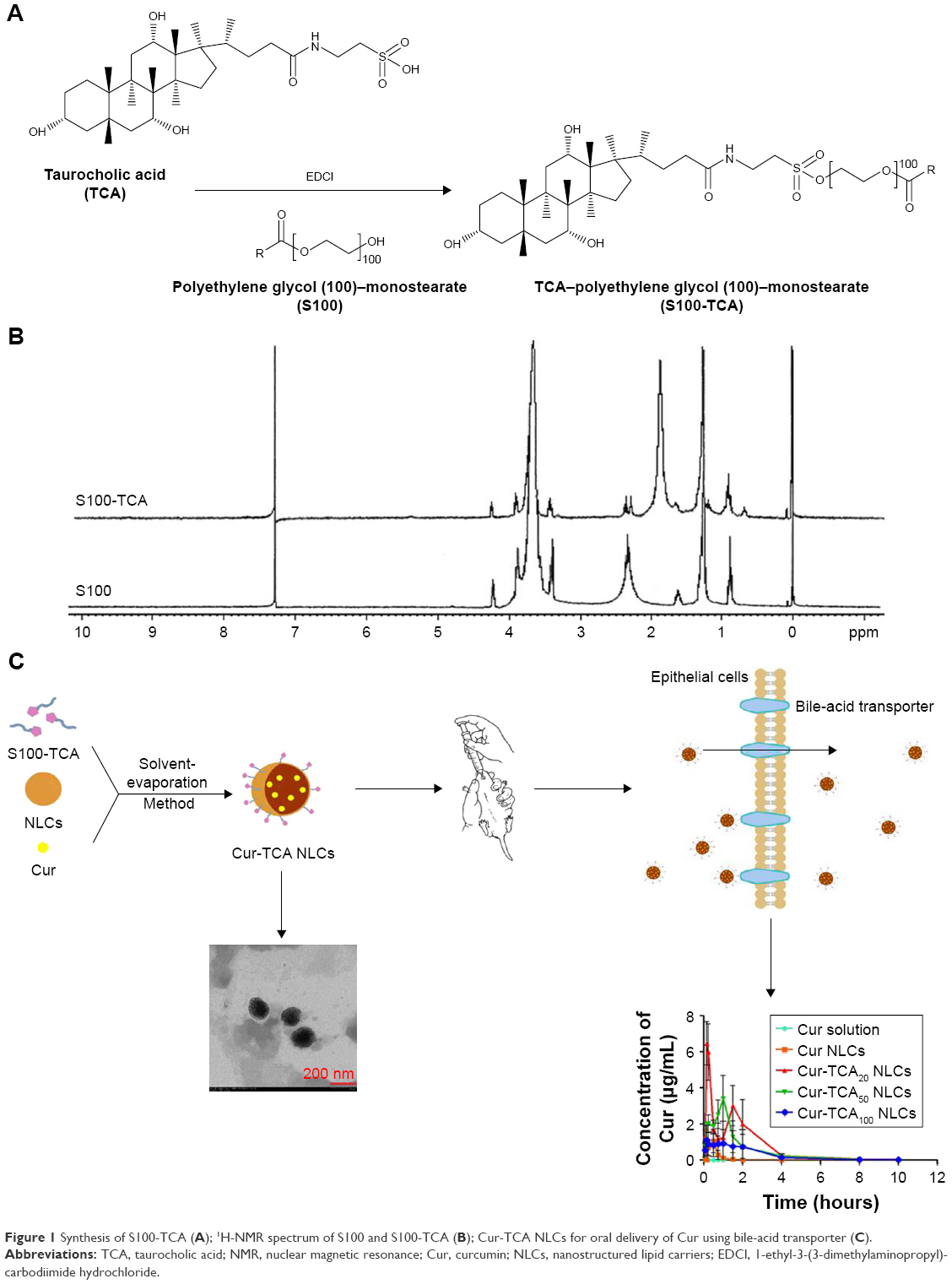 | Figure 1 Synthesis of S100-TCA (A); 1H-NMR spectrum of S100 and S100-TCA (B); Cur-TCA NLCs for oral delivery of Cur using bile-acid transporter (C). |
Preparation and characterization of Cur-TCA NLCs
Figure 1C shows Cur-TCA NLC construction with the hydrophobic monostearate of S100-TCA located inside and TCA presence on the surface of NLCs, along with receptor-mediated permeation of obtained NLCs. Modification of Cur-TCA NLCs with varying degrees of S100-TCA affected the characteristic features of the formulations, as shown in Table 1. Mean Cur NLC size was 141.2 nm; however, after modification with S100-TCA, NLC particle size decreased to 131.6 nm, 118.7 nm, and 99.6 nm for Cur-TCA20, Cur-TCA50, and Cur-TCA100 NLCs, respectively. Increase in S100-TCA in the NLCs inversely affected the particle size. The ability of the S100 to reduce the surface tension might explain the observed phenomenon.31 Moreover, the presence of PEG chains in the S100-TCA would also restrict the size of NLCs.19 All Cur-loaded NLCs showed good monodispersity, as a PDI value <0.3 has been associated with narrow particle-size distribution.32 The substantial increase in PDI value for a higher S100-TCA level (100 mg) was attributed to the excess S100-TCA that failed to get completely adsorbed on to the NLCs and could have assembled into the small sized, thus leading to the increased PDI value.33 The localization of PEG over the NLC surfaces could have resulted in increased ζ-potential of NLCs,34 as reduced surface negativity of the nanocarrier on modification with different amounts of S100-TCA is evident from Table 1. It has been reported that −20 to −11 mV surface charge might not provide sufficient stability to the dispersions.31 However, S100-TCA contains hydrophilic PEG, which might impart a supplementary steric shield to the nanoparticles.33 Consequently, electrostatic repulsion in tandem with steric hindrance would ensure stable dispersion.
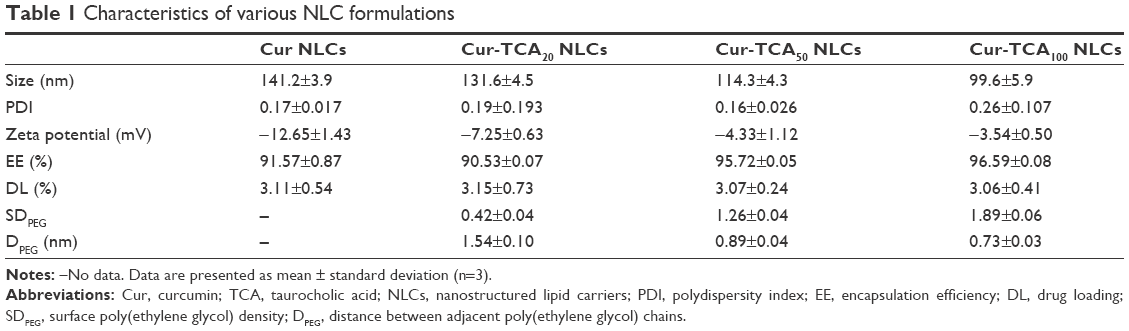 | Table 1 Characteristics of various NLC formulations |
More than 90% EE and about 3% DL were obtained for all the preparations. The quasispherical shapes of Cur NLCs and Cur-TCA50 NLCs were demonstrated by TEM (Figure 2A and B). The results showed that both NLCs were uniformly dispersed without any aggregation. In addition, Cur-TCA50 NLC types could have been dyed black by water-soluble phosphotungstic acid, on account of the increased hydrophilicity of NLCs after modification with S100-TCA.
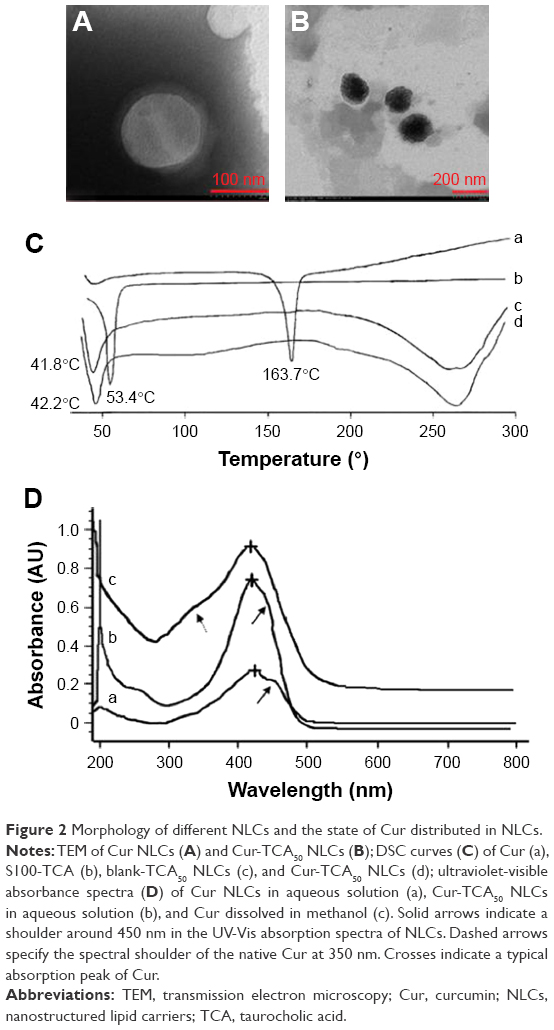 | Figure 2 Morphology of different NLCs and the state of Cur distributed in NLCs. |
Figure 2C displays the DSC thermograms of Cur, S100-TCA, blank-TCA50 NLCs, and Cur-TCA50 NLCs. As mentioned in the literature, a sharp endothermic peak is observed for pure Cur at 163.7°C.35 However, the Cur peak at 163.7°C was not visible in Cur-TCA50 NLCs, possibly due to the entrapment of drug molecules in TCA50 NLCs.22 In addition, the DSC thermograms show a melting peak for S100-TCA at 53.4°C, while melting peaks for blank-TCA50 NLCs and Cur-TCA50 NLCs were 42.2°C and 41.8°C, respectively. This indicates alteration in the crystallinity and melting behavior of the lipids, due to the presence of the amphiphilic molecule (S100-TCA) in the molten-lipid core of NLCs, as discussed in the literature.36
Figure 2D shows a typical absorption band for Cur at around 423–425 nm in the ultraviolet-visible spectrum. Furthermore, solid arrows indicate a shoulder around 450 nm in the ultraviolet-visible absorption spectra of NLCs. This depicts similar surroundings for Cur in both Cur NLCs and Cur-TCA50 NLCs, which could be described by Cur encapsulation in the apolar core.37 However, dashed arrows in Figure 2D specify the spectral shoulder of the native Cur at 350 nm, thus implying Cur presence in the polar environment, as supported by Leung and Kee.38
SDPEG and DPEG of modified NLCs
Hydrophilic molecules, such as PEG, not only stabilize colloidal dispersions but also improve their diffusivity across mucus.39 SDPEG values of Cur-TCA20, Cur-TCA50, and Cur-TCA100 NLCs were 0.42±0.043, 1.26±0.041, and 1.89±0.062, respectively (Table 1). The MW of PEG used, degree of modification, and size of the colloidal dispersions have been reported to affect SDPEG.40 An increase in S100-TCA led to higher SDPEG in the modified formulation. In addition, Cur-TCA20 NLCs and Cur-TCA50 NLCs displayed 2.11- and 1.22-times greater DPEG, respectively, than Cur-TCA100 NLCs. A decrease in DPEG was noted to raise the modification-material feed from 20 mg to 100 mg, which entailed a thicker PEG coat over NLC surfaces with reduced spacing among adjacent PEG chains. A DPEG <2-5 nm does not favor the adsorption of complementary proteins, and hence phagocytosis by the mononuclear phagocyte system can be hindered to achieve longer circulation in vivo.41 A DPEG <2 nm was obtained for all modifications, which indicated that this is enough to achieve prolonged in vivo circulation of preparations.
Cur-release study
Figure 3 shows the cumulative drug release from different Cur preparations in pH 1.2 HCl solution, pH 6.8 PBS solution, and physiological saline. Except for the Cur solution (60% in the first 2 hours), a sustained release of Cur was observed for all formulations. Modification of NLCs with TCA caused much slower release of drug than the simple NLCs. In the first 2 hours in acidic medium, only 5% of the loaded Cur was released from the TCA NLCs. In pH 6.8 PBS and physiological saline, the formulation released almost 70% of drug within 24 hours. It can be safely assumed from the release experiments that the formulations remain intact in the gastrointestinal tract to allow controlled release of the active moiety. This significantly increases the chances of the TCA-modified nanoparticles being absorbed via bile-acid transporter-mediated endocytosis and to maintain a good level of Cur in the bloodstream for a prolonged time.
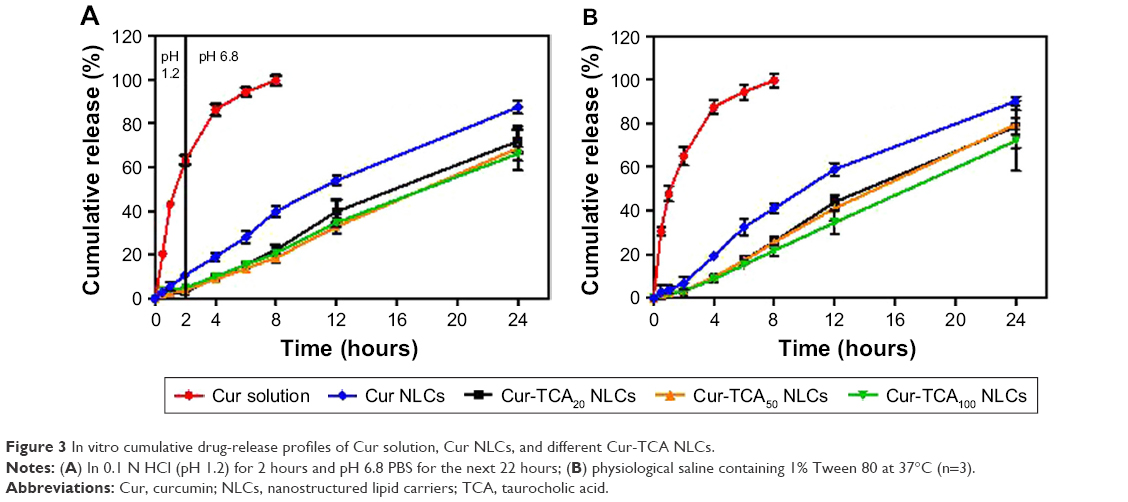 | Figure 3 In vitro cumulative drug-release profiles of Cur solution, Cur NLCs, and different Cur-TCA NLCs. |
The mechanism of drug release was found to be explained by the Weibull model for all the preparations (Table S1). As explained by Papadopoulou et al, when the exponent parameter β>1, drug transport follows a complex release mechanism.42 Here, β>1 for all TCA NLCs, so Cur release from TCA NLCs followed a nonlinear increase in the rate, which decreased afterward.
Rat intestinal perfusion study
To assess the possible causes of the removal of drug from intestinal lumen, an intestinal perfusion study was conducted. Cur stability in the KR solution was tested for 2 hours at 37°C, with evaluation of the possible complexation of Cur with components of the KR solution. Cur neither degraded in the KR solution nor had any aspecific complexation with components of the KR solution.
The time-dependent loss of drug (rate and extent) in the intestinal perfusion experiment is associated with the absorption of drug across the intestine.43 Strong correlation between in situ perfusion and cell-uptake experiments has been reported in an effort to evaluate the efficiency of a drug-delivery carrier in transport of a drug.44 Here, this method was used to observe the absorption performances of Cur solution and different Cur-loaded NLCs in different rat intestinal segments, because it allows experimentation in the actual intestinal environment.45
As shown in Figure 4A and B, Cur solution had maximal Ka and Peff values in the duodenum among the three intestinal segments. Intestinal absorption was obviously enhanced by loading Cur into NLCs, since Ka and Peff values were significantly elevated in Cur NLCs in comparison to Cur solution in every corresponding intestinal section. Unsurprisingly, Cur-TCA NLCs displayed better absorption from all intestinal segments compared to Cur NLCs. Increases of 1.09- to 1.53-fold in Cur-TCA NLC Ka values were observed in the small intestine. As shown in Figure 4B, Cur-TCA20 NLC Peff values in the duodenum, jejunum, and ileum increased by 1.43-, 1.71- and 2.44-fold, respectively, compared to Cur NLCs. These results show that specific binding of the ligand-coated formulation with the bile-acid transporter boosted intestinal absorption of Cur.
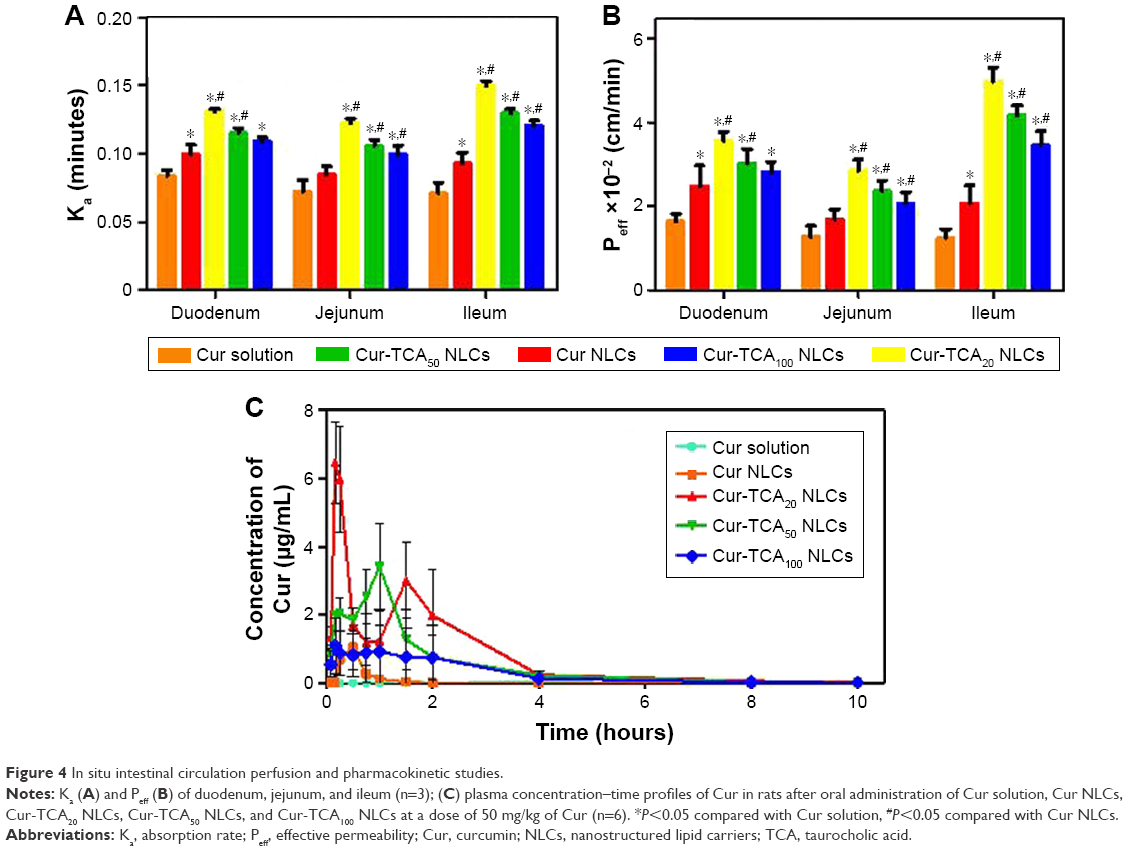 | Figure 4 In situ intestinal circulation perfusion and pharmacokinetic studies. |
However, crowding the carrier with the targeting molecule does not always lead to greater uptake, as evidenced by superior absorption of TCA20 NLCs than TCA50 NLCs and TCA100 NLCs. TCA100 NLCs possessed higher surface0ligand density (1.89) compared with TCA20 NLCs and TCA50 NLCs; however, lower absorption was found for TCA100 NLCs. When ligand density is too high, steric hindrance interferes with ligand and transporter binding, thus affecting the absorption of the preparation. Our results comply with a previous finding about iRGD-modified doxorubicin-loaded liposomes.46 In addition, the higher surface ligand-density of TCA100 NLCs might lead to intermolecular hydrogen bonds, which could have reduced the density of free OH– of TCA on NLC surfaces.47 Intestinal transport of bile acids involves transporters that follow the hard-and-fast rule of sodium dependence.48 Therefore, the absorption of Cur-TCA50 NLCs and Cur-TCA100 NLCs could have been reduced on account of reduction of free hydroxyl groups bound to Na+. As such, the degree of S100-TCA coating on NLC surfaces was found to determine the absorption of Cur.
TCA modification obviously improved both Ka and Peff in intestinal segments via bile-acid transporter-mediated intestinal absorption. Near-neutral NLC surface charge has been reported to enhance permeability by averting mucus entrapment of the carrier, owing to the mucus–carrier electrostatic interaction.49 The ζ-potential of TCA NLCs was about −7 to −3 mV, and might have assisted in the penetration of NLCs across the mucus layer. Cur-TCA20 NLCs were selected for further studies on identifying site-specific absorption through the small intestine. The results showed that Cur-TCA20 NLCs were taken up throughout the small intestine; however, the ileum showed maximum absorption (Ka ×10−2 and Peff ×10−2 of Cur-TCA20 NLCs were 6.82 minutes and 3.55 cm/min, 6.58 minutes and 2.84 cm/min, and 7.01 minutes and 4.98 cm/min in duodenum, jejunum, and ileum, respectively). The expression of apical sodium-dependent bile-acid transporters responsible for the absorption of bile acids through the small intestine is prevalent in the distal portion of the small intestine (ileum).48 As such, we observed maximum absorption of Cur-TCA20 NLCs through the ileum in comparison to other segments of the small intestine.
Pharmacokinetic studies
Figure 4C and Table 2 show the plasma profiles of drug and useful pharmacokinetic parameters obtained from the in vivo evaluation of various Cur preparations in rats after oral administration. Incorporation of hydrophobic drug in the NLCs markedly increased in Cmax and shortened peak plasma time. Cur NLCs made 4.27-fold more drug bioavailable than the Cur solution. Inadequate solubility of Cur in the gastric fluids and significantly higher drug biotransformation and clearance are the root causes of its subpar bioavailability. Wahlström and Blennow observed that most Cur was metabolized and actively excreted through the biliary pathway.15 Rapid biotransformation of Cur (90% within 30 minutes) to sulfate or glucuronide conjugates was attributed to liver microsomal enzymes.14 Therefore, higher plasma levels of drug are highly unlikely for longer periods after ingestion. Possible reasons for increased oral absorption of Cur NLCs are increased drug solubility in nanoparticle form, obviated degradation of drugs by gastric acid and digestive enzymes,17 and NLCs’ property of eluding drug efflux of through the Pgp pump.49
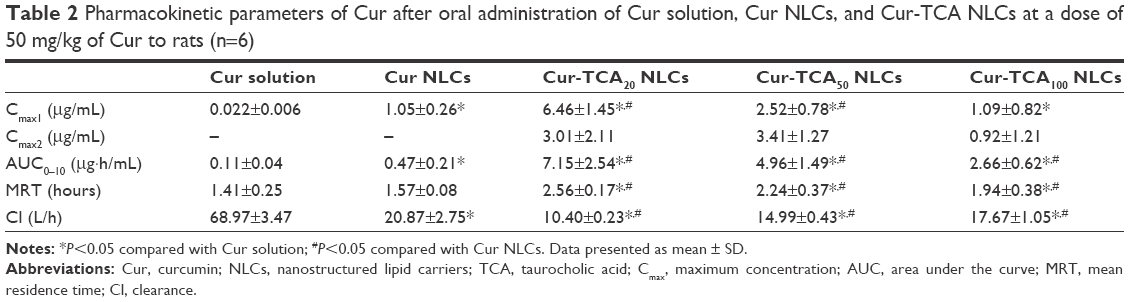 | Table 2 Pharmacokinetic parameters of Cur after oral administration of Cur solution, Cur NLCs, and Cur-TCA NLCs at a dose of 50 mg/kg of Cur to rats (n=6) |
As shown in Figure 4C, oral administration of Cur-TCA NLCs and Cur NLCs produced quite different plasma profiles of Cur. TCA-NLC formulations of Cur exhibited superior oral bioavailability. Cur-TCA NLCs gave rise to the double-peak phenomenon in their Cur plasma profile. After oral administration of Cur-TCA NLCs, some TCA-modified NLCs could have been excreted into the hepatobiliary system and secreted with bile into the small intestine, and this would have led to repeated absorption from the small intestine, thus showing a bimodal concentration–time curve.50,51 TCA-NLC formulations increased mean residence time and decreased clearance of Cur compared to Cur NLCs. PEG chains improve surface hydrophilicity and resist plasma protein adsorption, and thus TCA-NLC formulations avoided rapid removal from the body to prolong circulation, which might have contributed to the increased residence time of drug.52
The AUC0–10 after oral administration of Cur-TCA20, Cur-TCA50, and Cur-TCA100 NLCs in rats increased 15.21-, 10.55-, and 5.66-fold, respectively, compared with Cur NLCs. Khatun et al performed a competitive oral absorption study in mice to assess oral absorption due to bile-acid transporters.8 The results demonstrated that absorption of TCA-modified nanoparticles may occur via both specific binding with bile-acid transporters and aspecific binding in the entire small intestine. Therefore, TCA NLCs showed higher bioavailability compared to NLCs due to the interaction between TCA and intestinal bile-acid transporters. As a result, excellent bioavailability of TCA NLCs was observed in rats. PEG chains present in the S100-TCA might also have played a role in the distinctive oral absorption of TCA NLCs, on account of PEG ability to facilitate the movement of drug through the stagnant water layer adjacent to the intestinal membrane. With the increase in S100-TCA, the AUC of Cur-TCA NLCs decreased, confirming the results obtained from the intestinal circulation perfusion. Therefore, TCA NLCs eliciting a bile acid-mediated mechanism could prove a potent vehicle for improved intestinal absorption of poorly water-soluble drugs.
Conclusion
TCA NLCs were developed as a novel strategy to augment oral bioavailability of Cur. TCA NLCs showed sustained release in physiological media and also prolonged circulation in vivo. Our findings in animal studies suggested that TCA NLCs perked up the absorption of Cur, and TCA20 NLCs possessed an excellent oral absorption profile compared to the other preparations. In a nutshell, the encouraging in vitro and in vivo performance of TCA NLCs suggested that this could provide a flexible drug-delivery platform for hydrophobic molecules. However, the absorption mechanism of TCA NLCs at the cellular level needs further investigation and confirmation.
Acknowledgments
This work was supported by the Natural Science Foundation of Jiangsu Province (BK20130655), the National Natural Science Foundation of China (81503027), and College Students Innovation Project for the R&D of Novel Drugs (J1030830).
Disclosure
The authors report no conflicts of interest in this work.
References
Fletcher J, Wirz A, Young J, Vallance R, McColl KE. Unbuffered highly acidic gastric juice exists at the gastroesophageal junction after a meal. Gastroenterology. 2001;121(4):775–783. | ||
Mao HQ, Roy K, Troung-Le VL, et al. Chitosan-DNA nanoparticles as gene carriers: synthesis, characterization and transfection efficiency. J Control Release. 2001;70(3):399–421. | ||
Francis MF, Cristea M, Winnik FM. Exploiting the vitamin B12 pathway to enhance oral drug delivery via polymeric micelles. Biomacromolecules. 2005;6(5):2462–2467. | ||
Roger E, Kalscheuer S, Kirtane A, et al. Folic acid functionalized nanoparticles for enhanced oral drug delivery. Mol Pharm. 2012;9(7):2103–2110. | ||
Mirvish SS. Bile acids and other lipids in the gall-bladder biles of Africans with primary cancer of the liver. Br J Cancer. 1964;18(3):478–483. | ||
Park K, Lee GY, Kim YS, et al. Heparin-deoxycholic acid chemical conjugate as an anticancer drug carrier and its antitumor activity. J Control Release. 2006;114(3):300–306. | ||
Park KS, Lee SK, Son DH, et al. The attenuation of experimental lung metastasis by a bile acid acylated-heparin derivative. Biomaterials. 2007;28(16):2667–2676. | ||
Khatun Z, Nurunnabi M, Cho KJ, Byun Y, Bae YH, Lee YK. Oral absorption mechanism and anti-angiogenesis effect of taurocholic acid-linked heparin-docetaxel conjugates. J Control Release. 2014;177:64–73. | ||
Naksuriya O, Okonogi S, Schiffelers RM, Hennink WE. Curcumin nanoformulations: a review of pharmaceutical properties and preclinical studies and clinical data related to cancer treatment. Biomaterials. 2014;35(10):3365–3383. | ||
Nayak AP, Tiyaboonchai W, Patankar S, Madhusudhan B, Souto EB. Curcuminoids-loaded lipid nanoparticles: novel approach towards malaria treatment. Colloids Surf B Biointerfaces. 2010;81(1):263–273. | ||
Shim JS, Kim JH, Cho HY, et al. Irreversible inhibition of CD13/aminopeptidase N by the antiangiogenic agent curcumin. Chem Biol. 2003;10(8):695–704. | ||
Lin JK, Lin-Shiau SY. Mechanisms of cancer chemoprevention by curcumin. Proc Natl Sci Counc Repub China B. 2001;25(2):59–66. | ||
Dhillon N, Aggarwal BB, Newman RA, et al. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin Cancer Res. 2008;14(14):4491–4499. | ||
Vareed SK, Kakarala M, Ruffin MT, et al. Pharmacokinetics of curcumin conjugate metabolites in healthy human subjects. Cancer Epidem Biomarkers Prev. 2008;17(6):1411–1417. | ||
Wahlström B, Blennow G. A study on the fate of curcumin in the rat. Acta Pharmacol Toxicol (Copenh). 1978;43(2):86–92. | ||
Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery: a review of the state of the art. Eur J Pharm Biopharm. 2000;50(1):161–177. | ||
Müller RH, Mehnert W, Lucks JS, et al. Solid lipid nanoparticles (SLN): an alternative colloidal carrier system for controlled drug delivery. Eur J Pharm Biopharm. 1995;41(1):62–69. | ||
Attama AA, Schicke BC, Paepenmüller T, Müller-Goymann CC. Solid lipid nanodispersions containing mixed lipid core and a polar heterolipid: characterization. Eur J Pharm Biopharm. 2007;67(1):48–57. | ||
Meng F, Asghar S, Gao S, et al. A novel LDL-mimic nanocarrier for the targeted delivery of curcumin into the brain to treat Alzheimer’s disease. Colloids Surf B Biointerfaces. 2015;134:88–97. | ||
Ding F, Huang J, Lin J, et al. A study of the binding of CI mordant red 3 with bovine serum albumin using fluorescence spectroscopy. Dyes Pigm. 2009;82(1):65–70. | ||
Meng FF, Asghar S, Xu YR, et al. Design and evaluation of lipoprotein resembling curcumin-encapsulated protein-free nanostructured lipid carrier for brain targeting. Int J Pharm. 2016;506(1–2):46–56. | ||
Yang L, Gao S, Asghar S, et al. Hyaluronic acid/chitosan nanoparticles for delivery of curcuminoid and its in vitro evaluation in glioma cells. Int J Biol Macromol. 2015;72:1391–1401. | ||
Mohanty C, Sahoo SK. The in vitro stability and in vivo pharmacokinetics of curcumin prepared as an aqueous nanoparticulate formulation. Biomaterials. 2010;31(25):6597–6611. | ||
Puvvada N, Rajput S, Kumar BN, Mandal M, Pathak A. Exploring the fluorescence switching phenomenon of curcumin encapsulated niosomes: in vitro real time monitoring of curcumin release to cancer cells. RCS Adv. 2013;3(8):2553–2557. | ||
Peracchia MT, Vauthier C, Passirani C, Couvreur P, Labarre D. Complement consumption by poly(ethylene glycol) in different conformations chemically coupled to poly(isobutyl 2-cyanoacrylate) nanoparticles. Life Sci. 1997;61(7):749–761. | ||
Su Z, Niu J, Xiao Y, et al. Effect of octreotide-polyethylene glycol(100) monostearate modification on the pharmacokinetics and cellular uptake of nanostructured lipid carrier loaded with hydroxycamptothecine. Mol Pharm. 2011;8(5):1641–1651. | ||
Zhang Y, Huo MR, Zhou JP, et al. DDSolver: an add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010;12(3):263–271. | ||
Rathore R, Jain JP, Srivastava A, Jachak SM, Kumar N. Simultaneous determination of hydrazinocurcumin and phenol red in samples from rat intestinal permeability studies: HPLC method development and validation. J Pharm Biomed Anal. 2008;46(2):374–380. | ||
Dezani TM, Dezani AB, Junior JB, Serra CH. Single-pass intestinal perfusion (SPIP) and prediction of fraction absorbed and permeability in humans: a study with antiretroviral drugs. Eur J Pharm Biopharm. 2016;104:131–139. | ||
Jain R, Duvvuri S, Kansara V, Mandava NK, Mitra AK. Intestinal absorption of novel-dipeptide prodrugs of saquinavir in rats. Int J Pharm. 2007;336(2):233–240. | ||
Lim SJ, Kim CK. Formulation parameters determining the physicochemical characteristics of solid lipid nanoparticles loaded with all-trans retinoic acid. Int J Pharm. 2002;243(1):135–146. | ||
Luo Y, Zhang Y, Pan K, Critzer F, Davidson PM, Zhong Q. Self-emulsification of alkaline-dissolved clove bud oil by whey protein, gum arabic, lecithin, and their combinations. J Agric Food Chem. 2014;62(19):4417–4424. | ||
Su Z, Shi Y, Xiao Y, et al. Effect of octreotide surface density on receptor-mediated endocytosis in vitro and anticancer efficacy of modified nanocarrier in vivo after optimization. Int J Pharm. 2013;447(1):281–292. | ||
Luo Y, Teng Z, Li Y, Wang Q. Solid lipid nanoparticles for oral drug delivery: chitosan coating improves stability, controlled delivery, mucoadhesion and cellular uptake. Carbohydr Polym. 2015;122:221–229. | ||
Sanphui P, Goud NR, Khandavilli UB, Bhanoth S, Nangia A. New polymorphs of curcumin. Chem Commun. 2011;47(17):5013–5015. | ||
Fang JY, Fang CL, Liu CH, Su YH. Lipid nanoparticles as vehicles for topical psoralen delivery: solid lipid nanoparticles (SLN) versus nanostructured lipid carriers (NLC). Eur J Pharm Biopharm. 2008;70(2):633–640. | ||
Sahu A, Kasoju N, Bora U. Fluorescence study of the curcumin-casein micelle complexation and its application as a drug nanocarrier to cancer cells. Biomacromolecules. 2008;9(10):2905–2912. | ||
Leung MH, Kee TW. Effective stabilization of curcumin by association to plasma proteins: human serum albumin and fibrinogen. Langmuir. 2009;25(10):5773–5777. | ||
Rieux AD, Pourcelle V, Cani PD, Marchand-Brynaert J, Préat V. Targeted nanoparticles with novel non-peptidic ligands for oral delivery. Adv Drug Deliv Rev. 2013;65(6):833–844. | ||
Soong R, Macdonald PM. PEG molecular weight and lateral diffusion of PEG-ylated lipids in magnetically aligned bicelles. Biochim Biophys Acta. 2007;1768(7):1805–1814. | ||
Gref R, Domb A, Quellec P, et al. The controlled intravenous delivery of drugs using PEG-coated sterically stabilized nanospheres. Adv Drug Delivery Rev. 1995;16(2):215–233. | ||
Papadopoulou V, Kosmidis K, Vlachou M, Macheras P. On the use of the Weibull function for the discernment of drug release mechanisms. Int J Pharm. 2006;309(1):44–50. | ||
Dahan A, West BT, Amidon GL. Segmental-dependent membrane permeability along the intestine following oral drug administration: evaluation of a triple single-pass intestinal perfusion (TSPIP) approach in the rat. Eur J Pharm Sci. 2009;36(2):320–329. | ||
Chu XY, Sánchez-Castaño GP, Higaki K, Oh DM, Hsu CP, Amidon GL. Correlation between epithelial cell permeability of cephalexin and expression of intestinal oligopeptide transporter. J Pharmacol Exp Ther. 2001;299(2):575–582. | ||
Holmstock NF, Annaert PP, Augustijns P. Boosting of HIV protease inhibitors by ritonavir in the intestine: the relative role of Cyp and P-gp inhibition based on Caco-2 monolayers versus in situ intestinal perfusion in mice. Drug Metab Dispos. 2012;40(8):1473–1477. | ||
Zhao B, Fan YC, Wang XQ, Dai WB, Zhang Q, Wang XL. [Cellular toxicity and anti-tumor efficacy of iRGD modified doxorubicin loaded sterically stabilized liposomes]. Yao Xue Xue Bao. 2013;48(3):417–422. Chinese. | ||
Dietschy JM. Mechanisms for the intestinal absorption of bile acids. J Lipid Res. 1968;9(3):297–309. | ||
Dawson PA, Lan T, Rao A. Bile acid transporters. J Lipid Res. 2009;50(12):2340–2357. | ||
Beloqui A, Solinís MÁ, Rodríguez-Gascón A, Almeida AJ, Préat V. Nanostructured lipid carriers: promising drug delivery systems for future clinics. Nanomedicine. 2016;12(1):143–161. | ||
Xiao Y, Chen X, Yang L, et al. Preparation and oral bioavailability study of curcuminoid-loaded microemulsion. J Agric Food Chem. 2013;61(15):3654–3660. | ||
Ravindranath V, Chandrasekhara N. Absorption and tissue distribution of curcumin in rats. Toxicology. 1980;16(3):259–265. | ||
Shen J, Wang Y, Ping Q, Xiao Y, Huang X. Mucoadhesive effect of thiolated PEG stearate and its modified NLC for ocular drug delivery. J Control Release. 2009;137(3):217–223. |
Supplementary materials
Procedure
Determination of coupling ratio of TCA on S100-TCA
The amount of taurocholic acid (TCA) in S100-TCA conjugate was determined via ultraviolet-visible spectrophotometry according to a previous method.1 In brief, the amount of TCA was calculated from a reference-standard curve from the anhydrous ethanol TCA solution mixed with S100 as standard solution of 0.045–1.2 mg/mL. S100-TCA (10 mg) was dissolved in anhydrous ethanol (5 mL) and evaluated for TCA content at 268 nm. This procedure was performed in triplicate. The coupling ratio (CR) of reaction was calculated thus:
CR (%) = (C × V/W) × 100 |
where C is the concentration of TCA in S100-TCA solution, V the volume of S100-TCA solution, and W the amount of S100-TCA. Unreacted TCA was removed by dialysis of the resultant solution using pre-swelled dialysis bag (5 kDa molecular weight cutoff; Sigma-Aldrich, St Louis, MO, USA) at room temperature for 48 hours.
Stability of Cur-TCA NLCs in different media
The stability of Cur-TCA NLCs was examined by monitoring changes in particle size under various environmental conditions (simulated gastric fluid, pH 1.2), artificial intestinal fluid (pH 6.8), and simulated blood (pH 7.4). Briefly, an appropriate amount of Cur preparation was diluted in 5 mL different media (Cur concentration 0.4 mg/mL). This mixture was incubated for 2 hours for simulated gastric fluid (pH 1.2) and 8 hours for artificial intestinal fluid (pH 6.8) and simulated blood (pH 7.4). The tubes were placed in an orbital shaker and maintained at 37°C at 100 rpm. At different times, 0.1 mL suspension was taken out and diluted in 5 mL distilled water. Particle size was measured by the Zetasizer 3000 HS (Malvern Instruments, Malvern, UK).
Cytotoxicity studies
Caco2 cells (human colon cancer) were obtained from the cell bank of the Chinese Academy of Sciences (Shanghai, China) and used as an intestinal absorption model in vitro.2 The cells were routinely grown in DMEM/high (KeyGen Biotech, Nanjing, China) medium with 10% FBS, 1% nonessential amino acids, and 1% penicillin–streptomycin. Cells were maintained at 37°C in an incubator (Thermo Fisher Scientific, Waltham, MA, USA) with 5% CO2.
Cytotoxicity of blank TCA NLCs and Cur-TCA NLCs was evaluated in Caco2 cells with MTT method with slight modifications.3 Briefly, Caco2 cells were seeded in 96-well culture plates at a density of 5×103 cells/well for 3 weeks and then incubated with blank TCA NLCs and Cur formulations. Blank TCA NLC concentrations were 50–1,600 μg/mL. Cur concentrations were 0.5–16 μg/mL. After 24 hours’ incubation, 10 μL MTT (5 mg/mL) was added to each well for 4 hours, and then the medium was replaced with 150 μL dimethyl sulfoxide. Absorbance at a 570 nm (A570) was read using a microplate reader (Thermo Fisher Scientific). Ratios of A570 to preparations of untreated cells (percentages) were used to express cytotoxicity.
Cur stability in intestinal circulation-perfusion solution and physical absorption of rat intestinal segments
The Cur or Cur-loaded NLC intestinal circulation-perfusion solution (40 μg/mL) was prepared by dissolving Cur or Cur-loaded NLCs (equivalent to 4 mg Cur) in 100 mL Krebs–Ringer buffer solution (pH 6.8). Cur and Cur-loaded NLCs solutions were incubated at 37°C with the circulating perfusion tubes for 2 hours, and then samples were collected and analyzed by high-performance liquid chromatography.
Intestinal tracts was removed from rat abdomens and cut into three segments in vitro, which were 10 cm-long segments of duodenum (1 cm distal to pyloric sphincter), jejunum (15 cm to pyloric sphincter), and ileum (20 cm proximal to cecum). Cur solution or Cur-loaded NLCs with segments were incubated at 37°C for 2 hours. Sample solutions were collected at 0, 30, 60, 90, and 120 minutes and analyzed by high-performance liquid chromatography. Each experiment was performed in triplicated. The remaining ration (%) was calculated by: remaining ratio (%) = C2/C0 ×100, where C0 and C2 are concentrations of Cur intestinal circulation-perfusion solution at 0 and t, and t=30, 60, 90, and 120 minutes.
Results and discussion
Stability of Cur-TCA NLCs
To evaluate the stability of Cur-TCA NLCs, nanoparticles were incubated in different buffer solutions. A significant change was observed in Cur-TCA NLC sizes after incubation in simulated gastric fluid for 2 hours (Figure S1A; nanoparticles exhibited significant dimension increases to about 400 nm, almost three times larger than original size). These results were in accordance with previous research.4 The stability and integrity of an oral drug-delivery system in gastric conditions is of great significance in ensuring its in vivo efficacy. Though NLCs holds a promising potential for oral drug delivery, its poor stability in acidic conditions (such as the stomach) limits practical applications. Chitosan-coated NLCs5 can be potentially applied in oral drug-delivery systems in future research. As shown in Figure S1B and C, Cur-TCA NLCs showed good stability in simulated intestinal fluid and simulated blood, with no significant increase in particle size, suggesting that Cur-TCA NLCs remained intact in the intestinal tract and were then taken up by epithelial cells.
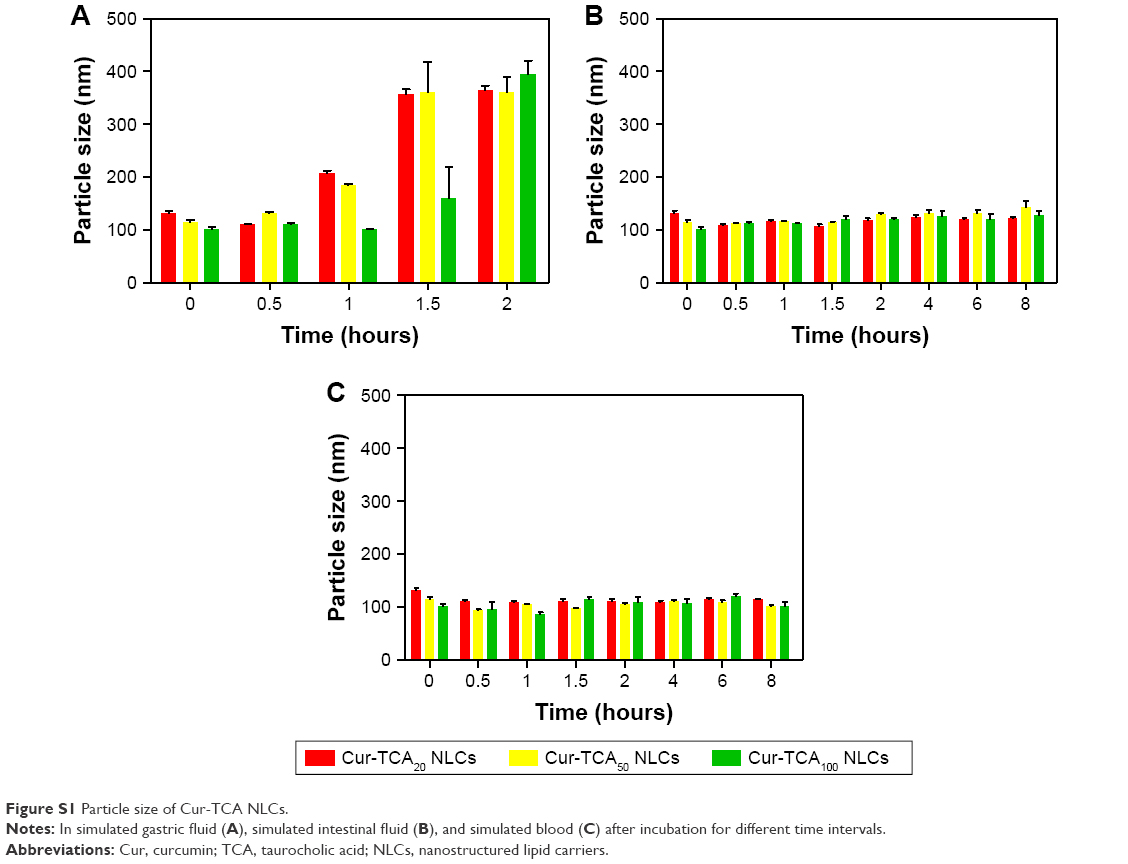 | Figure S1 Particle size of Cur-TCA NLCs. |
In vitro cytotoxicity assay against Caco2 cells
Figure S2A displays the percentage of cell viability as a function of the concentration of the blank NLCs. Results showed that blank NLCs in the studied concentrations had no effect on the cell viability of Caco2 cells, suggesting blank NLCs might be good biocompatible carriers. Figure S2B shows the percentage of cell viability as a function of Cur concentration. Results showed that for both Cur solution and Cur-loaded NLCs, cell viability decreased concentration-dependently. The increased cytotoxic effect of the Cur-TCA NLCs could be attributed to better penetration of the drug in Caco2 cells due to the carrier. The mechanism of drug release was utilized by zero-order, first-order, Higuchi, and Weibull models to simulate the release profiles of Cur from different NLCs (Table S1).
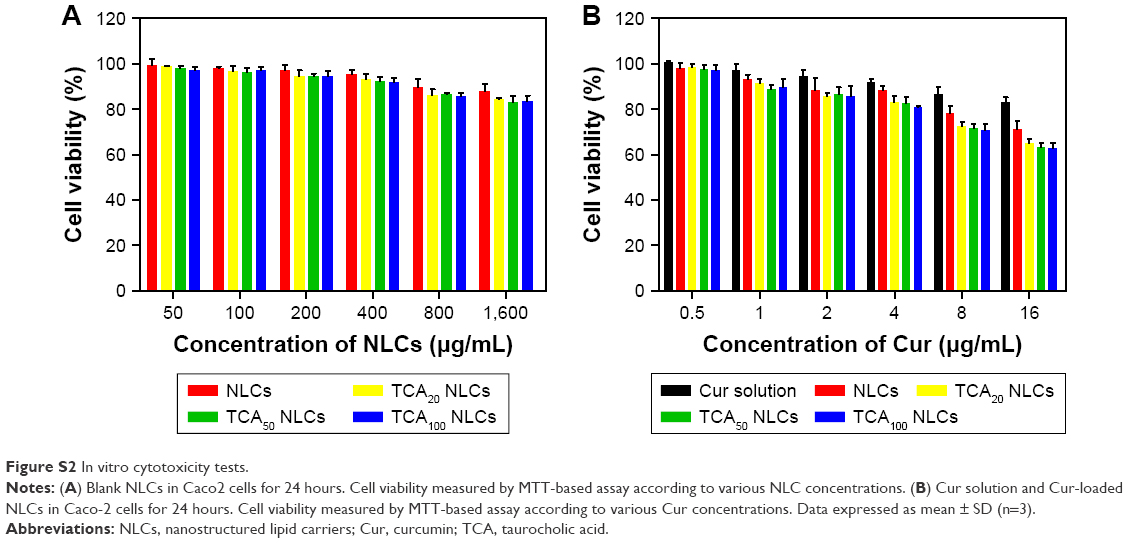 | Figure S2 In vitro cytotoxicity tests. |
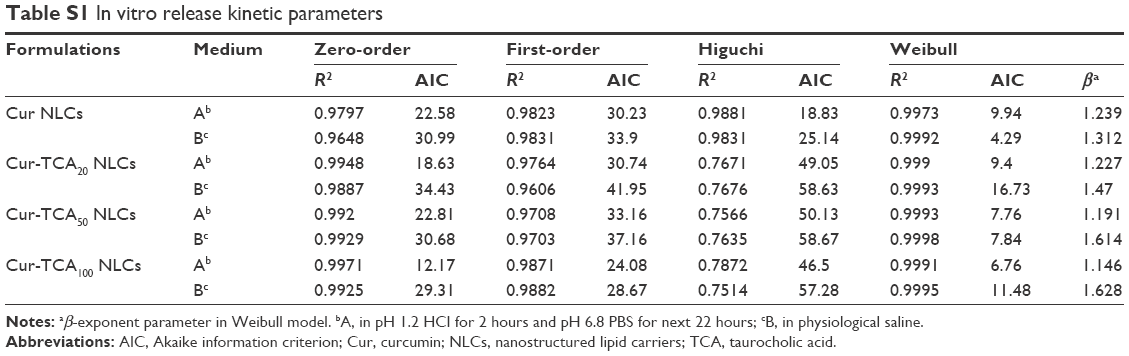 | Table S1 In vitro release kinetic parameters |
References
Su Z, Niu J, Xiao Y, et al. Effect of octreotide-polyethylene glycol(100) monostearate modification on the pharmacokinetics and cellular uptake of nanostructured lipid carrier loaded with hydroxycamptothecine. Mol Pharm. 2011;8(5):1641–1651. | ||
Kim SK, Lee DY, Lee E, et al. Absorption study of deoxycholic acid-heparin conjugate as a new form of oral anti-coagulant. J Control Release. 2007;120(1–2):4–10. | ||
Xiao J, Nian S, Huang QR. Assembly of kafirin/carboxymethyl chitosan nanoparticles to enhance the cellular uptake of curcumin. Food Hydrocoll. 2015;51:166–175. | ||
Paliwal R, Rai S, Vaidya B, et al. Effect of lipid core material on characteristics of solid lipid nanoparticles designed for oral lymphatic delivery. Nanomedicine. 2009;5(2):184–191. | ||
Luo Y, Teng Z, Li Y, Wang Q. Solid lipid nanoparticles for oral drug delivery: chitosan coating improves stability, controlled delivery, mucoadhesion and cellular uptake. Carbohydr Polym. 2015;122:221–229. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.