Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 13
Implementation of Pharmacogenetics to Individualize Treatment Regimens for Children with Acute Lymphoblastic Leukemia
Authors Maamari D ![]() , El-Khoury H, Saifi O, Muwakkit SA, Zgheib NK
, El-Khoury H, Saifi O, Muwakkit SA, Zgheib NK ![]()
Received 29 April 2020
Accepted for publication 20 July 2020
Published 12 August 2020 Volume 2020:13 Pages 295—317
DOI https://doi.org/10.2147/PGPM.S239602
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Dimitri Maamari1 ,* Habib El-Khoury1 ,* Omran Saifi,1 Samar A Muwakkit,2 Nathalie K Zgheib3
1Faculty of Medicine, American University of Beirut, Beirut, Lebanon; 2Department of Pediatrics and Adolescent Medicine, American University of Beirut Medical Center, Beirut, Lebanon; 3Department of Pharmacology and Toxicology, American University of Beirut, Faculty of Medicine, Beirut, Lebanon
*These authors contributed equally to this work
Correspondence: Samar A Muwakkit; Nathalie K Zgheib Email [email protected]; [email protected]
Abstract: Despite major advances in the management and high cure rates of childhood acute lymphoblastic leukemia (ALL), patients still suffer from many drug-induced toxicities, sometimes necessitating dose reduction, or halting of cytotoxic drugs with a secondary risk of disease relapse. In addition, investigators have noted significant inter-individual variability in drug toxicities and disease outcomes, hence the role of pharmacogenetics (PGx) in elucidating genetic polymorphisms in candidate genes for the optimization of disease management. In this review, we present the PGx data in association with main toxicities seen in children treated for ALL in addition to efficacy, with a focus on the most plausible germline PGx variants. We then follow with a summary of the highest evidence drug-gene annotations with suggestions to move forward in implementing preemptive PGx for the individualization of treatment regimens for children with ALL.
Keywords: pharmacogenetics, childhood ALL, implementation
Introduction
Acute lymphoblastic leukemia (ALL) is the most common cancer in children, accounting for about 30% of childhood cancers worldwide. Despite major advances in the management of the disease with cure rates reaching up to 94%, patients still suffer from many drug-induced toxicities, sometimes necessitating dose reduction, or halting of cytotoxic drugs with a secondary risk of disease relapse. In addition, investigators have noted significant inter-individual variability in drug toxicities and disease outcomes, hence the role of pharmacogenetics (PGx) in elucidating genetic polymorphisms in candidate genes for the optimization of disease management.1
There are currently various types and versions of childhood ALL treatment protocols, but they all share many commonalities and capture several toxicity and efficacy outcomes, hence making childhood ALL an ideal platform for PGx analyses. The typical treatment course of childhood ALL entails three major phases and lasts for 2–3 years depending on disease risk stratification. Treatment starts with the induction phase that mainly includes anthracyclines such as doxorubicin (DOX) or daunorubicin (DAU), L-Asparaginase (LASPA), glucocorticoids such as prednisone (PRED) or prednisolone (PRDL), and vinca-alkaloids such as vincristine (VINC) to eradicate leukemic cells. This is followed by a consolidation phase that is mainly comprised of high dose methotrexate (MTX) and 6-mercaptopurine (6-MP) to kill any residual leukemic cells. This phase is sometimes followed by re-induction cycles of dexamethasone (DEXA) and VINC depending on residual disease. Lastly, a prolonged maintenance or continuation phase follows intending to maintain remission. It mostly includes treatment with 6-MP and MTX with initial rotating cycles of DEXA, VINC or cyclophosphamide (CTX).2
Some relatively recently published reviews are available on the PGx of childhood leukemia most of which listed the evidence by individual drugs.3–7 Nevertheless, a major downside of PGx research in childhood ALL is that in each phase of the treatment protocol, patients receive a combination of different drugs with many times overlapping toxicities such as hepatotoxicity and myelosuppression. Besides, at times, drug-gene interactions are compounded by drug-drug interactions such as the case for 6-MP and MTX.8 Accordingly, it may be difficult to determine the drug to which the PGx toxicity or efficacy are related to adjust the dose accordingly.9–11 Therefore, in this review, we chose to present the PGx data in association with main toxicities seen in children treated for ALL in addition to efficacy, with a focus on the most plausible germline PGx variants. We summarize the cited gene variants with allele frequencies into a Table 1, and show the overlap between the different candidate genes and drug toxicity and outcome in a Venn diagram (Figure 1). We then follow with a summary of the highest evidence drug-gene annotations with suggestions to move forward in implementing PGx for the individualization of treatment regimens for children with ALL.
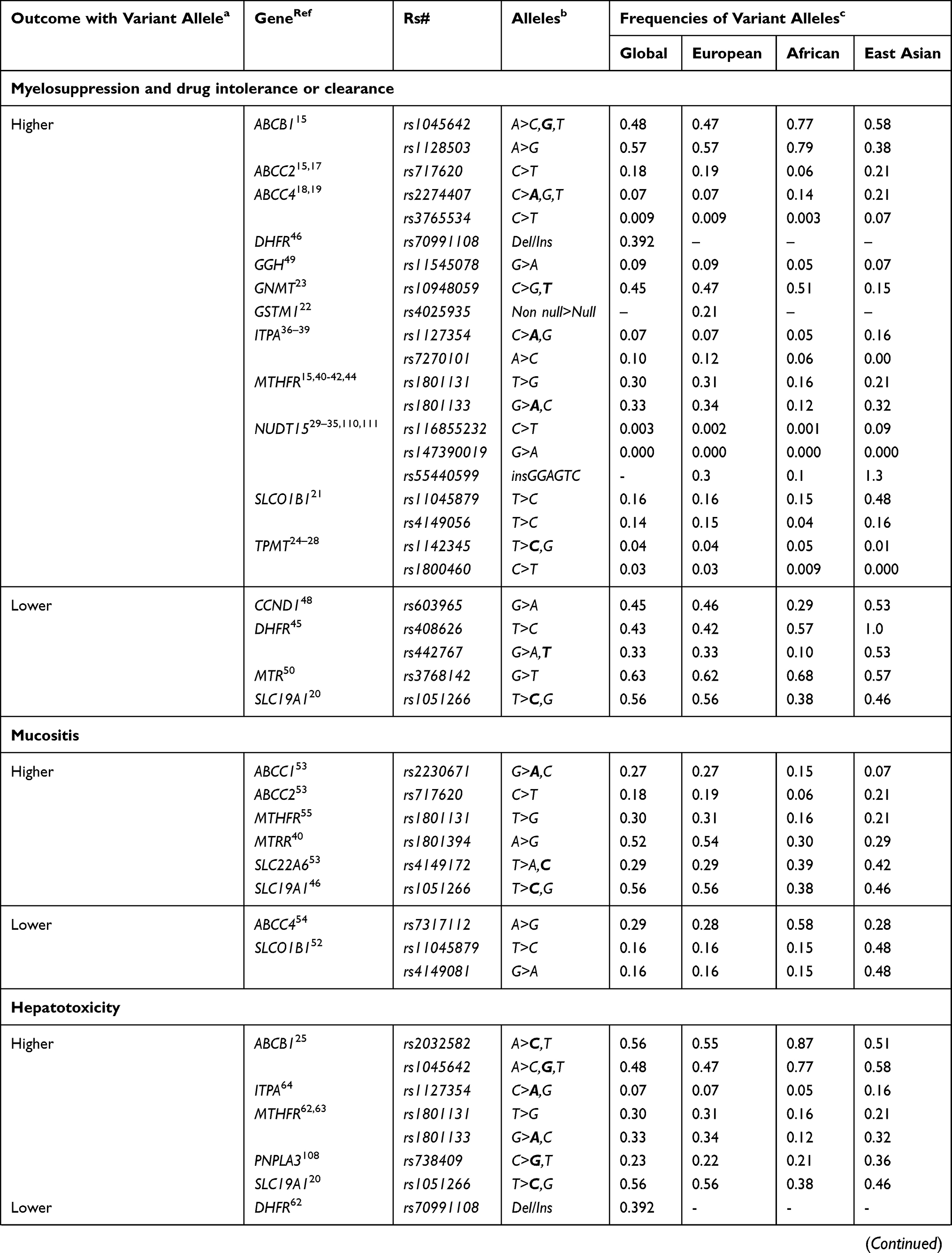 | 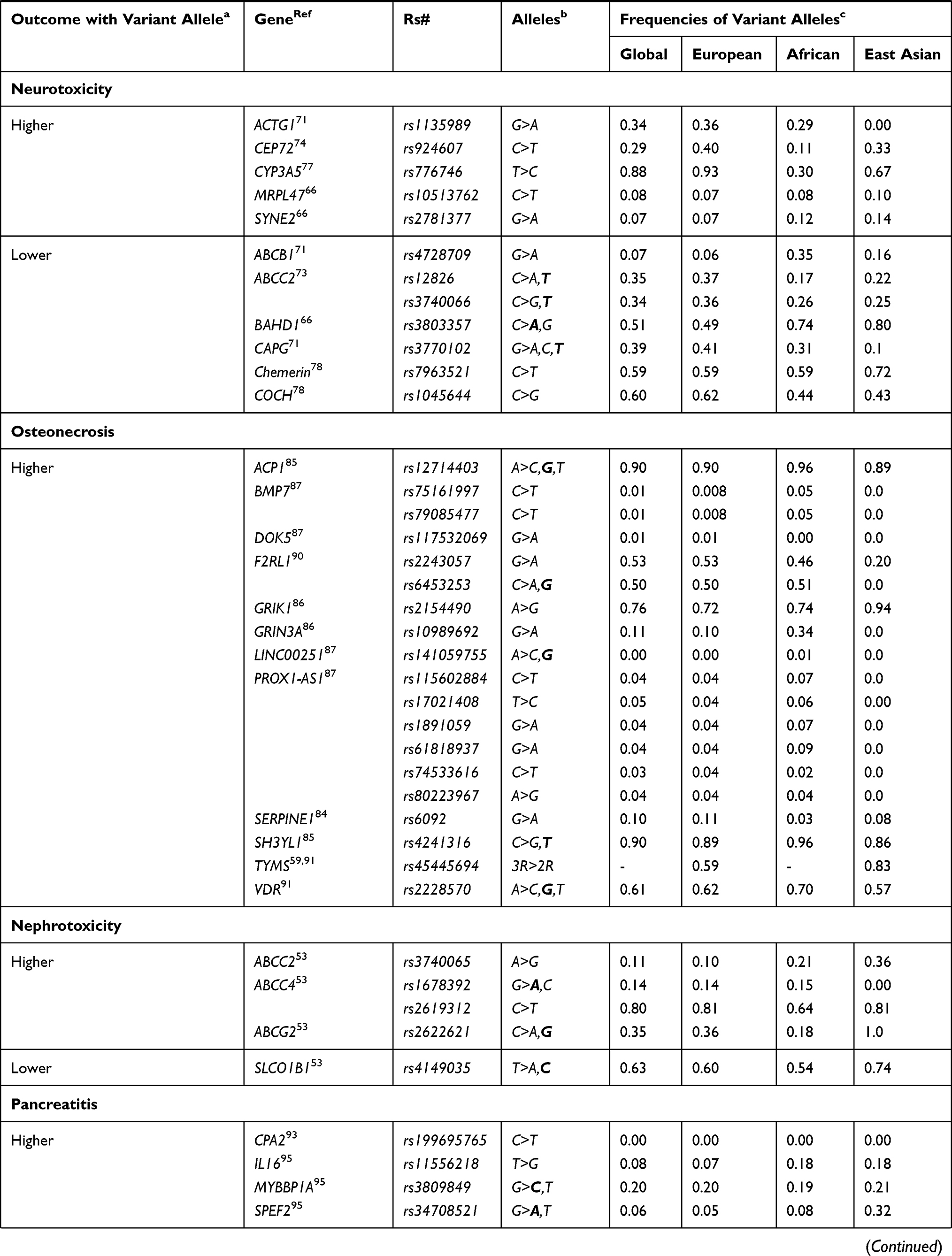 | 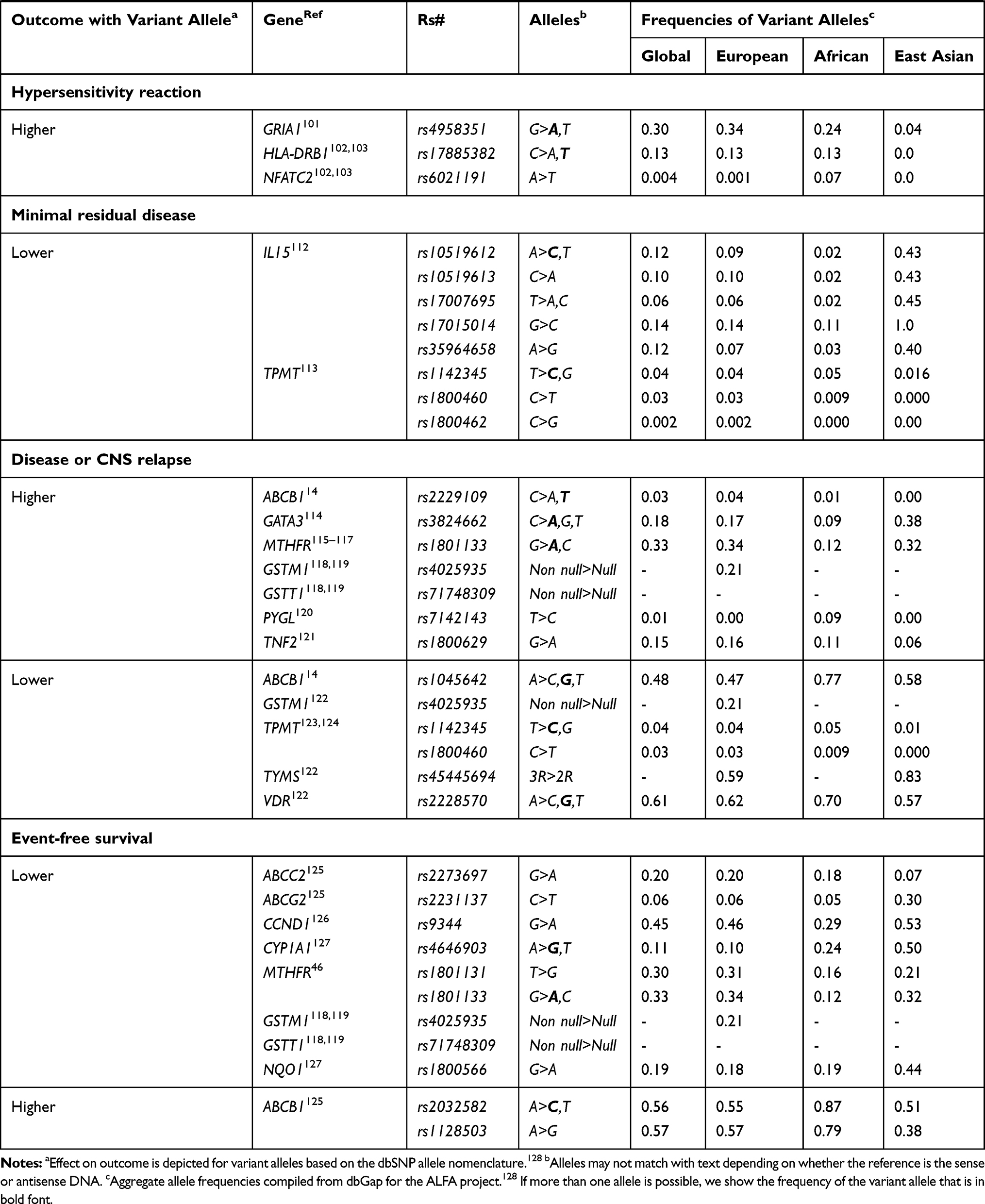 |
Table 1 List of Gene Variants Associated with Major Toxicities and Efficacies of Drugs Used for the Treatment of Childhood ALL |
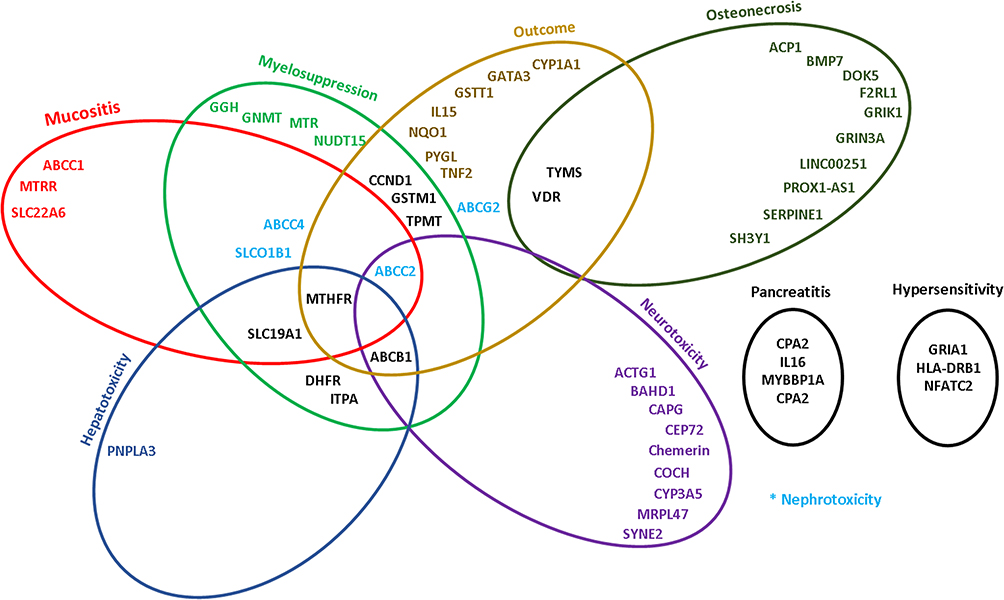 |
Figure 1 Ven diagrams showing the overlap between different genes and drug toxicity and outcome in the treatment of childhood ALL. |
Variants Associated with All Treatment Toxicities
Myelotoxicity and Drug Intolerance or Clearance
Cytotoxic chemotherapeutic drugs especially kill actively replicating cells such as in the bone marrow, hence leading to myelotoxicity and drug intolerance. In ALL therapy, almost all drugs may be associated with hematologic toxicity, yet most PGx evaluations were focused on 6-MP and MTX as both drugs are typically provided in combination during the consolidation and maintenance phases of all childhood ALL treatment protocols. 6-MP is a prodrug and its active metabolites, thioguanine and thiodeoxyguanine nucleotide triphosphate (TGTP and Td GTP), lead to cytotoxicity by inducing RNA and DNA damage respectively,12 while MTX is an antifolate with a complex pharmacodynamic (Pd) pathway that ultimately leads to inhibition of nucleotide synthesis.13 Candidate genes that were most extensively evaluated with myelotoxicity and drug intolerance or clearance included genes that code for transporters, detoxifying enzymes, and enzymes in the pathways of purines and antifolates.
Gene candidate studies have shown significant associations between polymorphisms in the ATP Binding Cassette Subfamily B Member 1 (ABCB1), which codes for a membrane transporter P-glycoprotein, and myelotoxicity during ALL treatment.14 For example, a significant association was found between neutropenia (absolute neutrophil count <500) and variant allele carriers of ABCB1 rs1045642 and rs1128503 in 127 Lebanese ALL patients,15 and one of these polymorphisms, rs1045642 in ABCB1, may even contribute to potentially life-threatening infections in ALL therapy as found in 70 Saudi patients.16 Similarly, ATP-binding cassette subfamily C member 2 (ABCC2) −24C>T polymorphism (rs717620) contributed to variability in MTX kinetics, increasing its plasma concentration, and causing a significantly higher risk of leukopenia, anemia, and thrombocytopenia in 117 Lebanese and 112 Chinese children.15,17 ATP-binding cassette subfamily C member 4 (ABCC4) polymorphisms in 70 Egyptian patients were associated with 6-MP induced myelotoxicity as well, whereby the rs2274407 variant allele was significantly associated with neutropenia, agranulocytosis, and leukopenia.18 Another variant in ABCC4 (rs3765534), studied in 95 Japanese patients, was also found to be associated with severe leukopenia.19
In addition to ATP-binding cassettes, genetic polymorphisms in reduced folate carriers or solute carriers (RFC or SLC) were also shown in gene candidate studies to play a role in ALL-related myelotoxicity. A study on 563 Danish patients showed that SLC19A1 is involved in the transport of MTX across the cell membrane, and a higher degree of bone marrow toxicity was observed in patients with the SLC19A1 wild type genotype (rs1051266).20 Moreover, variant alleles in SLCO1B1 (rs4149056 and rs1104587) among 48 Turkish patients were found to be associated with lower 6-MP and MTX tolerance.21 Furthermore, genetic variants in detoxifying enzymes were also identified. Glutathione S-transferase Mu 1 (GSTM1) encodes for an enzyme that functions in the detoxification of carcinogens and therapeutic drugs by conjugation with glutathione. The risk of severe infections was increased in 36 Italian subjects with the GSTM1 null genotype (rs4025935) compared to those with the GSTM1 non-null genotype who had significantly more moderate degree infections.22 Likewise, polymorphisms 1298CT or TT in Glycine N-methyltransferase (GNMT) (rs10948059), a gene that codes for a hepatic detoxifying enzyme, demonstrated a higher risk of hematologic toxicity in 308 Slovenian patients when compared to the GNMT CC wild type.23
Another subset of genes is involved in the pharmacokinetic (Pk) pathway of thiopurines, and genetic polymorphisms in these genes were found to affect 6-MP’s toxicity and tolerance. Thiopurine S-methyltransferase (TPMT) is an enzyme that converts thiopurine drugs into inactive metabolites. Defected alleles of this enzyme decrease the activity of the coding gene and increase the concentration of the thiopurine drugs, specifically 6-MP, causing higher toxicities.12 A large number of PGx studies were conducted with TPMT. For example, candidate gene studies on 68 Belgradian and 1135 British children with variant alleles of the single nucleotide polymorphisms (SNPs) TPMT*3B and *3C (rs1800460 and rs1142345 respectively) showed significantly higher frequency of cytopenias; therefore, requiring dose adjustments below target levels significantly more often than those with the TPMT wild-type variant.24,25 Such group of children have significantly lower leukocyte counts and percentage of target 6-MP dosage, and longer periods with more than grade 2 infections and chemotherapy interruptions during maintenance therapy as evident in the 164 Turkish and 203 Polish children studied.26–28
Another important PGx player with thiopurine-related myelotoxicity is nucleoside diphosphate-linked moiety X-type motif (NUDT15) that encodes for an enzyme that is a negative regulator of thiopurine activation. Polymorphisms in this gene can result in toxic accumulation of thiopurines metabolites. Variants of NUDT15 (rs116855232, rs55440599, and rs147390019) lead to a higher risk of leukopenia and 6-MP drug intolerance as seen in 404 Chinese, 124 Uruguayan, and 270 Asian children.29–31 GWAS study done on 1028 children from different races showed that patients with the TT genotype at rs116855232 were more sensitive to 6-MP when compared to those with TC and CC genotypes. These results were replicated and retained significance in 371 ALL patients.32 Similar results were reported by a candidate gene study done on 105 Chinese children.33 Moreover, Japanese carriers of the T allele of this polymorphism had more frequent leukopenia,34 and Thai patients with NUDT15 CT or TT of that same SNP had a significantly increased risk of neutropenia as early as two months after 6-MP administration.35
Finally, variants in Inosine triphosphatase (ITPA) which codes for ITPase, an enzyme that recycles purines trapped in the form of ITP, were also described. For example, 19 British ALL patients with ITPA IVS2+21A>C (rs7270101) variant had significantly higher concentrations of the active cytotoxic metabolite 6-thioguanine which was associated with thrombocytopenia.36 Moreover, the risk of prolonged neutropenia was higher in 63 Indian, 136 Lebanese, and 74 Kurdish children heterozygous for ITPA 94C>A (rs1127354) polymorphism37,38 and variant allele carriers of that same SNP.39
Polymorphisms in genes coding for enzymes in the folate metabolic pathway can alter tolerance to MTX. Methylenetetrahydrofolate reductase (MTHFR) encodes the rate-limiting enzyme in the methyl cycle. Most studies, including a candidate gene study done on 81 Dutch patients, described the MTHFR polymorphisms A1298C (rs1801131) and C677T (rs1801133) to be associated with MTX-related myelotoxicity.40 Carriers of at least one MTHFR 677T variant allele showed an increased risk of developing severe leukopenia and neutropenia in 286 Argentinian children,41 and anemia in 127 Lebanese children.15 Besides, 78 European Caucasian children homozygotes (TT genotype) for the same SNP had more pronounced myelosuppression.42 On the same line, 20 Japanese patients with an increasing number of T alleles at MTHFR C677T experienced more frequent interruptions in both 6-MP and MTX.43 However, one gene candidate study done on 27 Turkish children among few others suggested that the A1298C polymorphism, rather than C677T, was associated with MTX-related toxicity. It also showed contradicting results concerning the risk allele. For instance, subjects with the MTHFR C677T polymorphism (CT, TT) had significantly higher MTX levels at 24-hours but did not seem to suffer from toxicity. As for subjects with the MTHFR A1298C polymorphism (AC, CC), they had significantly higher MTX levels at 48-hours, more frequent anemia, thrombocytopenia, and febrile neutropenia.44
Furthermore, variants in Dihydrofolate reductase (DHFR), which codes for an enzyme that reduces dihydrofolate to tetrahydrofolate (THF), were also described. The wild types of two genetic polymorphisms, −317AG (rs408626) and −680CA (rs442767), were found to be associated with severe leukopenia in 70 Indian patients receiving MTX.45 In addition, 141 Spanish patients showed thrombocytopenia with the I allele of the 19pb D/I polymorphism in the DHFR gene (rs70991108), and severe neutropenia with the CC genotype in the −680CA SNP of DHFR.46 Cyclin D1 (CCND1) is a cell cycle regulator that is involved in gene expression such as the DHFR gene.47 Accordingly, homozygous individuals for the variant allele of CCND1 G870A allele (rs603965) had a significantly lower frequency of weeks with high-grade hematologic toxicity from MTX treatment as evident by the Canadian gene candidate study on 186 ALL patients.48 Additional candidate genes include the Lysosomal enzyme gamma glutamate hydrolase (GGH) that codes for the enzyme that metabolizes MTX polyglutamates (MTX-PG) back to MTX, hence resulting in diminished drug efficacy. As such, Garcia-Bournissen et al49 showed higher rates of grade 2 thrombocytopenia in 239 European descendant carriers for GGH polymorphism (rs11545078) A compared to non-carriers. Finally, Methionine synthase (MTR), another gene involved in the folate metabolic pathway, affects the Pk and toxicity of MTX, and a significant association between the MTR rs3768142 polymorphism and granulocytopenia was reported in 118 Hungarian (Caucasian) patients.50
Mucositis
Treatment-induced mucositis during ALL therapy has been well studied and mostly attributed to the use of MTX in different regimens.51 With limited treatment options available for mucositis, such a complication remains debilitating and can potentially lead to treatment interruption or even cessation. Some PGx studies were conducted with mucositis and ALL, and the most significant findings are with SNPs in drug transporters and enzymes involved in the transport and Pd pathway of MTX.
Concerning drug transporters, in a GWAS of 434 patients with newly diagnosed ALL enrolled and treated on St. Jude Children’s Research Hospital Total XIIIB protocol, Trevino et al52 surveyed 500,568 germline SNPs to identify new candidate genes whose polymorphisms can forecast which patients would benefit from a tailored dosage of MTX. Two SNPs in SLCO1B1, rs11045879 T, and rs4149081 G wild type alleles, were associated with mucositis during both consolidation and continuation phases. However, those same findings were not reproducible in patients receiving the Total XV protocol. This was attributed to the fact that MTX doses were adjusted to achieve a common steady-state plasma concentration for patients treated under the Total XV protocol. In support of the results of the above GWAS using a candidate gene approach, Salazar et al46 also showed that Spanish children who had the variant genotype of another SNP in the SLC19A1 gene (rs1051266) had higher rates of mucositis when treated for ALL. This relationship remained significant in multivariate analysis considering the clinical risk categories as a covariate. Furthermore, concerning additional candidate variants in drug transporters, Lopez-Lopez et al53 analyzed polymorphisms in up to 12 of the most important genes involved in MTX transport in 151 Spanish pediatric ALL patients diagnosed with B-ALL at four different hospitals. Among many SNPs linked to treatment toxicities, polymorphisms in SLC22A6, ABCC2, and ABCC1 (rs4149172, rs717620, and rs2230671, respectively) were significantly associated with mucositis. Finally, den Hoed et al,54 contrasted their findings to other studies while looking at candidate genes in ALL patients treated according to the Dutch Child Oncology Group ALL-10 protocol. Interestingly, they found no association between previously studied SNPs and mucositis such as those in SLCO1B1. However, their data showed that wild-type genotype rs7317112 in ABCC4 was a predictor of mucositis compared to the G variant allele. Following correction for age and gender, patients with the wild-type polymorphism were shown to be more prone to severe mucositis. All other SNPs studied were not associated with treatment-induced mucositis.54
Concerning enzymes involved in the MTX Pd pathway, MTHFR genotypes (C677T/rs1801133 and A1298T/rs1801131) were assessed by Moulik et al55 in children treated with a uniform Children’s Cancer Group (CCG)-1961 based chemotherapy protocol at a tertiary care academic hospital in North India. They only found that a higher proportion of children with rs1801131 variant allele carriers developed mucositis as compared to children with wild type 1298 genotype. This relationship was amplified in patients with folate deficiency.55 Finally in a retrospective study on 81 children with ALL treated according to the Dutch Childhood Oncology Group ALL-9 protocol, Huang et al40 showed that significantly more patients with a 5-methyltetrahydrofolate-homocysteine methyltransferase reductase (MTRR) (rs1801394) AG or GG genotype experienced mucositis as a treatment toxicity compared to wild-type genotype patients. The MTRR enzyme is responsible for maintaining levels of methylcobalamin, an activating cofactor for methionine synthase which catalyzes the remethylation of homocysteine to methionine.13
Hepatotoxicity
Hepatotoxicity is an important side effect of many of the cytotoxic drugs administered for ALL treatment, and worsening hepatic function often leads to treatment interruption with increased risk of disease relapse.56 Various genetic polymorphisms have been linked with hepatic toxicity. These can occur in genes not directly involved in any known drug interaction, such as in Patatin-like phospholipase domain-containing protein 3 (PNPLA3); while others can affect genes that play a role in the metabolism and action of chemotherapeutic agents such as ITPA, DHFR and MTHFR, as well cellular entry and interaction of drugs as seen with ABCB1 and SLC19A1.
In a cohort of 138 Spanish children of European descent with ALL receiving CTX, DAU, LASPA, PRED, and VINC, a genetic polymorphism in PNPLA3 (rs738409) was associated with a 2.6-fold risk of liver injury via a candidate gene approach. Around 31% of the children carrying the polymorphism suffered from grade 2 to 4 hepatotoxicity, described as an elevation of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) 2.6 times above the normal. Of these, 18% were categorized as high toxicity, with AST or ALT being 5-fold above the normal levels.57 PNPLA3 codes for adiponutrin, an enzyme found in hepatocytes and involved in triacylglycerol catabolism and remodeling.58 Similarly, a GWAS conducted on 715 children from St. Jude undergoing ALL treatment with 6-MP, CTX, DAU, LASPA, PRED and VINC showed that patients with that polymorphism were also at a significantly higher risk of developing an elevation in ALT following induction chemotherapy.59 This study was also replicated in 2285 children from the Children’s Oncology Group (COG). This genetic variant could explain the onset of steatohepatitis in children treated for ALL, especially that it has been linked to hepatotoxicity and possible development of non-alcoholic steatohepatitis in knock-in mice.60
Polymorphisms in other genes, such as those encoding for drug transporters, were also studied with hepatotoxicity in ALL, though will lesser evidence. For example, 68 children of Serbian origin were studied by Milosevic et al25 using a candidate gene approach, and the polymorphism rs2032582 in ABCB1 was shown to be associated with hepatotoxicity. Of note that the same polymorphism was studied by Gregers et al,14 in a candidate gene approach, and it was not associated with significant hepatotoxicity in a cohort of 522 Danish children. Nevertheless in this same study,14 the C variant allele of another SNP in ABCB1 (rs1045642) was associated with more liver toxicity upon treatment with high dose MTX.
In addition to ABCB1, a genetic variant in SLC19A1 has been associated with MTX-related hepatotoxicity. For instance, a candidate gene study done on a subset of 182 children of European Nordic descent undergoing treatment for ALL showed a significantly higher elevation in ALT levels in patients receiving high dose MTX with the SLC19A1 rs1051266 polymorphism.20 Although the patients in this study received different chemotherapy agents, the liver toxicity was linked to MTX because of the known function and importance of the RFC protein for the MTX therapeutic effect. Also, a meta-analysis in 2017 showed higher MTX toxicities, including hepatotoxicity, in patients with rheumatoid arthritis receiving MTX monotherapy and carrying the rs1051266 polymorphism.61
In addition to drug transporters, polymorphisms in other candidate genes were linked to MTX-related hepatotoxicity. For example, in a study on 122 Italian children with ALL, a deletion in DHFR (rs70991108) has been associated with a 4.57-fold increase in hepatotoxicity.62 Furthermore, in the same study, a commonly studied genetic polymorphism in MTHFR (rs1801133) has also been associated with a 5.23-fold increase in hepatic toxicity.62 In another candidate gene study conducted on 73 Japanese children who were receiving 6-MP and MTX during maintenance phase therapy, Tanaka et al63 used a candidate gene approach to describe additional genetic polymorphisms in MTHFR linked to hepatotoxicity. As such, children who had the combined MTHFR 677TT, 677CT and 1298AC polymorphisms had a significantly higher risk of severe and life-threatening liver injury, as well as a faster onset of hepatic injury requiring hospitalization. In addition, a candidate gene approach in a cohort of 63 Eastern Asian children receiving maintenance therapy for ALL, ITPA 94C>A (rs1127354) polymorphism was associated with a significantly higher risk of hepatotoxicity.64
Neurotoxicity
In patients receiving chemotherapy, neurotoxicity can present under various forms, including sensory, motor, and autonomic dysfunction.65 The peripheral nervous system is commonly affected as the central nervous system is usually protected from toxic agents by the blood-brain barrier.65 Various polymorphisms have been shown to either predispose or protect patients with regard to neurologic toxicities during ALL treatment with some variants being traced directly to VINC. This includes genetic polymorphisms in centrosomes and microtubules that are targeted by VINC, and in Cytochrome P450A5 (CYP3A5) that is responsible for its metabolism. Other studies have described polymorphisms thought to be linked to VINC-induced neurotoxicity although the mechanism describing VINC as the cause of nervous system dysfunction is not clearly highlighted.
In a cohort of 237 children with ALL, Abaji et al66 identified 2 genetic polymorphisms that put patients receiving chemotherapy at risk for peripheral neurotoxicity. The first polymorphism is rs2781377 in spectrin repeat-containing nuclear envelope protein 2 (SYNE2), which codes for nesprin, a nuclear envelope spectrin repeat protein that maintains cellular cytoskeleton.67 Interestingly, variants of the SYNE2 have been linked to neurological diseases in mice.68 In addition, the rs10513762 variant in mitochondrial ribosomal protein L47 (MRPL47) was shown by the same authors66 to increase the risk of neurotoxicity. MRPL47 has also been shown to play a role in neurologic disorders.69 Furthermore, the rs3803357 in the bromo adjacent homology domain containing 1 (BAHD1) played a protective role against peripheral neurotoxicity in those patients.66 Previous in vitro studies have demonstrated that BAHD1 acts as a regulatory factor in inflammation and contributes via epigenetic mechanisms to autonomic and sensory neuropathies.70 Those polymorphisms, which were studied by Abaji et al66 by a whole-genome sequencing approach, were validated by an independent replication cohort of 405 children. In addition to these studies, Ceppi et al71 evaluated a population of 339 children of European descent with ALL, with the majority being of French-Canadian origin. By using a candidate gene approach, and a validation cohort from the Dana-Farber Cancer Institute (DFCI), 3 polymorphisms that put patients at either increased or decreased risk of neurotoxicity were identified. A polymorphism in actin gamma 1 (ACTG1) (rs1135989), which codes for a protein that maintains cellular cytoskeleton, has been shown to increase the risk for toxicities of the nervous system.71 On the other hand, variants of capping actin protein, gelsolin like (CAPG) (rs3770102) lead to a lower risk of neurotoxicity.71 Products encoded by CAPG play an important role in the motility of non-muscle cells by interacting with the cytoskeleton.71 Interactions between these 2 proteins and VINC have been described by Verillis et al,72 and cell lines with resistance to VINC had decreased expression of both ACTG1 and CAPG protein products. Furthermore, a polymorphism in ABCB1 (rs4728709) has been shown to carry a protective effect against neurotoxicity.71 Also, a study conducted by Lopez-Lopez et al73 on 152 children of European descent with ALL identified, by using a candidate gene approach, 2 more polymorphisms (rs12826 and rs3740066) in ABCC2 to be linked with lower neurotoxicity.
Other studies have found that some polymorphisms predisposing to neurotoxicity can occur in genes that code for proteins involved in vinca alkaloids’ downstream targets. In 2015, 222 children of mixed ancestry, receiving chemotherapy for ALL were studied with a GWAS, and 56% of patients with the TT allele at rs924607 in CEP72 had neuropathies, as compared to 21.4% of patients carrying the wild type CC or heterozygous CT alleles.74 Children with the homozygous TT allele were also more likely to have more severe neurotoxicity.74 The polymorphism occurred in the promoter region of Centrosomal Protein 72 (CEP72), a gene responsible for a protein involved in centrosome formation.74 The authors have linked neurotoxicity in this cohort of children to VINC, as vinca alkaloids target microtubules and centrosomal proteins.75 In addition to CEP72, CYP3A5 was found to metabolize VINC in in-vitro studies,76 and Egbelakin et al77 demonstrated, by using a candidate gene approach on 107 children of mixed origins, that patients treated for ALL and carrying the CYP3A5*3A (rs776746) polymorphism had more severe neurotoxicity, a higher frequency of neuropathies, and a longer duration of the neurologic dysfunction. However, there was no significant difference in the occurrence of neurotoxicity between children with this polymorphism and those not having it.77 In this study, the authors attributed this toxicity to VINC because of previous in vitro studies and because neurotoxicity occurred mostly in early ALL treatment when the dosage of VINC is highest.77
More recently, Li et al78 incorporated 2 GWAS to try to identify polymorphisms linked to neurotoxicity and found that rs1045644, a polymorphism in a gene that contributes to cochlin formation, provided relative protection from neurotoxicity.78 Cochlin has been previously described to play a role in hearing loss and vestibular disorders of imbalance.79 Furthermore, studies have shown that cochlin can be induced by bone morphogenic protein,80 which was shown to play a role in neurogenesis, and neural stem cell maturation.81 Li et al78 studied variants in 1696 DNA samples from the Pediatric Oncology Group, and 99 samples from the ADVANCE trial of patients from mixed origins. The authors identified another polymorphism, rs7963521 in a gene coding for chemerin. This genetic variant decreased the risk of suffering from neurotoxicity in children with ALL, and chemerin has been shown to play a role in various pathways, including inflammation and adipogenesis.78
Osteonecrosis
Osteonecrosis is a pathologic process that affects osteocytes and causes cellular death. It is a progressive condition that destroys joints within months to a year, ultimately leading to mechanical failure of the affected bone.82 Osteonecrosis can be caused by traumatic or atraumatic conditions, with the most common cause of non-traumatic osteonecrosis being glucocorticoid administration.82 Many studies have tried to identify genetic polymorphisms that can predict a higher risk of osteonecrosis in children receiving ALL chemotherapy. Because, in murine models receiving only corticosteroids, disruption of bone vasculature appeared to be an inciting event that preceded steroid-induced osteonecrosis,83 DEXA is frequently perceived as the culprit of osteonecrosis in children with ALL; however, variants in enzymes involved in MTX’s mechanism of action have also been linked to this toxicity.
In 2008, a candidate SNP approach conducted on 291 children of mixed race showed the variant rs6092 in plasminogen activator inhibitor-1 (PAI1 or SERPINE1), a gene linked with inhibition of fibrinolysis and thrombus formation, to be associated with a significantly higher risk of osteonecrosis, whereby 27% of children with the GA/AA had osteonecrosis as compared to only 11.7% of children with the GG variant.84 Following this study, a GWAS was performed in 2011 on 364 children with ALL who received the St. Jude protocol Total XV and revealed a significantly increased risk for osteonecrosis with a variant in acid phosphatase 1 (ACP1) (rs12714403), a regulator for osteoblast differentiation, and another in SH3 domain-containing YSC84-like protein 1 (SH3YL1) (rs4241316).85 More recently in 2015, a much larger GWAS that entailed 2285 patients of mixed origins with two validation cohorts, one from the COG and another from St. Jude, revealed highly significant associations with two relatively common variants in glutamate NMDA receptor subunit 3A (GRIN3A) (rs10989692) and glutamate ionotropic receptor kainate type subunit 1 (GRIK1) (rs2154490).86 Notably, compelling evidence showed the GRIN3A variant to be involved in various vascular phenotypes, including cerebral ischemia, arterial embolism and thrombosis.86 In 2016, a GWAS was performed to assess the risk of osteonecrosis in children less than 10 years of age with ALL. The GWAS was conducted on a discovery cohort of 369 children and validated on 817 children below 10 years of age and of mixed ethnic backgrounds. The study identified 10 polymorphisms linked to osteonecrosis: rs79085477 and rs75161997 in bone morphogenic protein 7 (BMP7), rs1891059, rs115602884, rs74533616, rs80223967, rs17021408 and rs61818937 in prospero homeobox 1 antisense RNA (PROX1-AS1), rs141059755 in long intergenic non-protein coding RNA 251 (LINC00251), and rs117532069 in docking protein 5 (DOK5).87 BMP7 has been shown to decrease osteoclast formation and hence bone resorption.88 Furthermore, BMP7 has been demonstrated to induce apoptosis in vascular smooth muscle cells,89 which could explain the mechanism of vascular injury as a cause of osteonecrosis as described previously by Janke et al.83 Another GWAS performed on 391 children supported further the vascular injury mechanism of osteonecrosis by identifying 2 polymorphisms (rs2243057 and rs6453253) in coagulation factor II thrombin receptor-like trypsin receptor 1 (F2RL1).90 Studies have shown that the interaction between F2RL1 and its receptor F2R, play a role in clot formation, angiogenesis and arteriopathy.90
Interestingly, other studies have described an increased risk of osteonecrosis with polymorphisms affecting antimetabolite pathways. For instance, a candidate gene study on 64 children of mixed origins with ALL identified the rs45445694 2R/2R low enzyme activity polymorphism in thymidylate synthase (TYMS) to be linked with osteonecrosis.91 Previous studies have shown that this polymorphism is associated with lower levels of thymidylate synthase in the cell which could explain increased toxicity from antimetabolites such as MTX.91 In addition, the allele variant of a polymorphism in vitamin D receptor (VDR) was also identified (rs10735810 that is now merged with rs2228570); this gene was included in the candidate gene approach as previous studies have linked genetic variants in VDR to low bone mineral density.91
Nephrotoxicity
Worsening kidney function is an important complication of cancer treatment including in children with ALL,92 and patients carrying certain genetic polymorphisms in efflux pumps and transporters, of which MTX is a common substrate, were shown to be at an increased risk of chemotherapy-induced nephrotoxicity. As a matter of fact, a candidate gene approach by Lopez-Lopez et al53 described the presence of 5 genetic polymorphisms that predispose to nephrotoxicity in a cohort of 151 Spanish children receiving ALL treatment. As such, variant alleles of polymorphisms rs3740065 in ABCC2, rs2619312, and rs1678392 in ABCC4, rs2622621 in ABCG2, with the wild type allele of rs4149035 in SLCO1B1 were associated with nephrotoxicity and significantly increased creatinine levels.
Pancreatitis
To determine PGx risk factors for treatment-induced pancreatitis in pediatric ALL patients, a GWAS was performed in a cohort of more than 5000 children and young adults with ALL. A rare nonsense variant in Carboxypeptidase 2 (CPA2) (rs199695765) had the strongest association with the risk of treatment-induced pancreatitis, after adjusting for clinical features.93 The CPA2 gene encodes a pancreatic carboxypeptidase proenzyme,94 and the previously-described high-risk SNP (rs199695765) resulted in early termination in the pro-peptide region.93 Also, 15 other variants in CPA2 were associated with pancreatitis. Hence, this largest of its kind study regarding risk of pancreatitis in childhood ALL patients suggests that carriers of CPA2 variants might benefit from a modified treatment regimen that does not heavily rely on LASPA being the most commonly known drug responsible for pancreatitis during pediatric ALL therapy.93 More recently in their whole exome sequencing study, Abaji et al95 filtered through a multi-step selection process their top-ranking signals related to ASP toxicity in a discovery cohort of 302 children with ALL from Quebec. Three SNPs were shown to be associated with treatment-induced pancreatitis: rs72755233 in ADAMTS17, rs3809849 in MYBBP1A, and rs9908032 in SPECC1. Two other SNPs that predicted thrombosis in the same patient cohort were also found to correlate with the risk of pancreatitis: variant allele carriers of the rs11556218 in IL16 and variant allele carriers of the rs34708521 in SPEF2. Of those findings, the only variants that retained significance in a replication cohort of 282 children treated for ALL at the Dana-Farber Cancer Institute were rs3809849 in MYBBP1A, rs11556218 in IL16, and rs34708521 in SPEF2. The association of the latter two genes was seen to be more prominent when combining both discovery and replication cohorts.95 Interestingly, the rs3809849 in the MYBBP1A gene was not commonly studied before. This gene encodes for the MYB Binding Protein 1A which plays an important role in various cellular processes including mitosis and cell cycle control, and it was most recently discovered to act as a co-repressor of nuclear factor kappaB (NF-kB) transcription factor.96 Notably, NF-kB was shown lately to play a key role in the development of acute pancreatitis.97
Hypersensitivity Reactions
Although treatment regimens for childhood ALL include a wide variety of chemotherapeutic drugs, hypersensitivity reactions have mostly been linked to LASPA, and have been associated with the production of anti-asparaginase immunoglobulin G (IgG) rather than immunoglobulin E (IgE) antibodies.98 However, a small subtype of patients receiving PEGylated Escherichia coli asparaginase develop hypersensitivity reactions without evidence of detectable antibodies.99 This leaves the door open to multiple possible reasonings behind the hypersensitivity reactions during ALL treatment and the exact culprit agent or combination of agents.
Only a few studies have investigated germline genomics that predispose patients to hypersensitivity reactions during ALL therapy. In a study on 485 ALL pediatric patients treated on St. Jude Children Research Hospital’s Total XV protocol, and after testing on a validation cohort, a SNP in glutamate ionotropic receptor AMPA type subunit 1 (GRIA1) (rs4958351) was shown to be associated with the risk of hypersensitivity reactions. GRIA1 encodes a subunit of a ligand-gated ion channel that transmits glutaminergic signals in the brain.100 The overall cumulative incidence of hypersensitivity reactions for patients with the AA, AG, or GG genotypes was 74%, 44%, and 32% respectively. There was no association between the different genotypes and the severity of the reaction.101 More recently in a large GWAS on more than 3000 patients treated on different ALL protocols, SNPs in the nuclear factor of activated T Cells 2 (NFATC2) (rs6021191) and major histocompatibility complex, class II, DR beta 1 (HLA-DRB1) (rs17885382) were associated with hypersensitivity reactions, and results were similar taking into consideration the asparaginase preparation used.102 These findings were asserted by Kutszegi et al103 later in 2017, in addition to HLA-DRB1*07:01–HLA-DQA1*02:01–HLA-DQB1*02:02 haplotype reconstruction which also correlated positively and significantly with increased risk of hypersensitivity reactions.103 Finally, in the same whole-exome sequencing study mentioned above by Abaji et al,95 three additional variants were shown to be significantly associated with the risk of treatment-induced allergy: the variant alleles of rs9656982 in SLC7A13 and of rs3809849 in MYBBP1A in an additive manner, and rs75714066 variant allele in YTHDC2. These significant associations were however not validated in the replication cohort.
Role of Genetic Polymorphisms in miRNA
Although most of the literature focused on variants in exonic and sometimes intronic or promoter areas in candidate genes, there is current interest in epigenetic markers of toxicity, and more specifically on the role of SNPs in microRNA (miRNA), small non-coding RNAs that regulate gene expression to include those involved in drug disposition, in modulating drug toxicity in childhood ALL.104 A team of investigators from Spain took the lead in this area, and they were the first to show that the T variant allele (rs639174) in DROSHA, the gene encoding for the ribonuclease III enzyme, is associated with more vomiting in children treated for ALL.105 Additional variants in genes coding for miRNAs were then revealed in association with combination drug toxicities such as vomiting, diarrhea, mucositis and neurotoxicity106–108 as well as MTX clearance.109 For instance, Iparraguirre et al109 genotyped for a large number of miRNA SNPs in peripheral blood of 167 Spanish children treated for ALL and revealed three SNPs to be associated with MTX plasma levels: miR-5189 (rs56292801), miR-595 (rs4909237) and miR-6083 (rs78790512). These were predicted in-silico to affect the expression of some SLC transporters. Afterwards, the same team of investigators ascertained that a SNP in miR-1206 (rs2114358) is associated with MTX-related mucositis,106 followed by the identification of three SNPs to be linked with mucositis (miR-4268; rs4674470), diarrhea (miR-4751;rs8667) and vomiting (miR-3117;rs12402181).106 In addition, a study conducted on the same cohort of Spanish children with ALL (N=155), the rs12402181 variant of miR-3117-3p was associated with lower neurotoxicity.108 This specific miRNA has also been shown to target ABCC1 as well as RalA Binding Protein 1 (RALBP1), which could, therefore, affect the efflux of chemotherapeutic agents.108 In addition, another polymorphism, rs7896283 in miR-4481, was shown to cause a 2.62-fold increase in the rate and severity of neurotoxicity. The product of this gene affects genetic processes that help in the guidance of axons and regeneration of peripheral nerves.108
Variants Associated with ALL Treatment Efficacy
The use of treatment regimens based on progressive intensification and risk-directed chemotherapy has drastically improved survival and outcome in pediatric ALL.129 However, prognosis remains poor for a small subtype of patients. Outcomes in ALL are highly influenced by minute changes in drug doses or exposure, making inter-individual variability in drug Pk and Pd a big determinant of response to treatment.122 Inherited germline variations showed promising results in predicting both early response to treatment measured by minimal residual disease (MRD), the risk of relapse, and event-free survival (EFS) in ALL.130
Early Treatment Response and Minimal Residual Disease
Genetic polymorphisms in genes involved in purines’ pathways such as TPMT were shown to affect treatment response. As a matter of fact, in a study on 814 pediatric ALL patients treated on the BFM-ALL trial between 1999 and 2002, there was a 2.9-fold reduction in risk of having positive MRD in TPMT heterozygous patients (rs1800462, rs1800460, rs1142345) following treatment induction, indicating better therapy response.113 On the other hand, looking at genes encoding inherent immunologic markers, certain variants were shown to predict response to treatment. For example, in the first and oldest genome-wide interrogation for PGx published by Yang et al in 2009,112 the role of SNPs located in the Interleukin 15 (IL15) locus in predicting response to ALL therapy has been asserted. As such, IL15 was found to play a role in protecting malignant hematologic cells from glucocorticoid-induced apoptosis in vitro,131 and higher IL15 expression in leukemic blasts has also been linked to increased risk of leukemia CNS relapse.132 The SNP in the IL15 locus (rs17007695) showed the strongest association with lower MRD in both St. Jude and COG cohorts of the study. This SNP was also flanked by 4 other IL15 SNPs (rs17015014, rs10519612, rs10519613, and rs35964658), and they were also all associated with lower MRD in both cohorts.112
Disease Relapse
With the drastic improvement in overall survival and outcome in childhood ALL, disease relapse still carries a poor prognosis.2 Predicting disease relapse and identifying patient subgroups at high-risk of relapse is needed to improve disease outcomes. Recently, the role of germline genomics with ALL relapse has been receiving increasing attention.
In a “discovery versus replication” effort, Yang et al120 performed a genetic screen on a large group of children (N=2535) enrolled in trials in the COG and at St. Jude Children’s Research Hospital, looking for associations between germline SNP genotypes and risk of relapse.120 Accounting for the confounding of known prognostic factors, a total of 134 SNPs were successfully identified. The strongest association with relapse risk was observed with a SNP in glycogene phosphorylase (PYGL) (rs7142143) whereby the presence of one C allele copy at the PYGL locus showed a 3.6-fold increased risk of relapse. Being a target of adenosine monophosphate, PYGL plays an important role in response to chemotherapeutic agents such as 6-MP and MTX.133 Besides, in vitro evidence supported the overexpression of PYGL in multi-drug resistance cancer cell lines.134
Looking further into genes encoding targets of the chemotherapeutic agent MTX, in a study on 520 patients treated according to the CCG-1891, Aplenc et al115 established a significant association between the MTHFR C677T (rs1801133) polymorphism and the risk of relapse. This relationship was also supported by another study done on a smaller cohort, showing that MTHFR 677TT genotype carriers had a higher risk of relapse and lower 7-year overall survival.116 This same genotype was shown to exhibit a 50% rate of disease relapse in 2 years in Egyptian carrier patients.117 Also, two separate reports supported the association between TPMT (rs1800460, rs1142345) heterozygosity and decreased risk of relapse, in addition to its previously mentioned influence on MRD.123,124 After accounting for other confounding prognostic factors, Rocha et al122 showed the GSTM1 non-null (rs4025935) genotype alone, or the combined GSTM1 non-null and TYMS 3R/3R (rs45445694) genotypes, to be independent prognostic factors for more hematologic relapse in high-risk pediatric ALL patients. These genotypes were not predictive in the low-risk group. On the other hand, Takanashi et al118 also studied the GST gene patterns in 82 patients with childhood B-precursor ALL. Their analysis showed that, in contrast to Rocha et al,122 the GST double null genotype (simultaneous deletion of both the GSTM1 (rs4025935) and GSTT1 (rs71748309) genes) was a significant independent predictor of early relapse and lower EFS at 3 years. These findings were also supported by Leonardi et al119 in their retrospective study on 140 Argentinian patients with childhood ALL. In contrast to other studies done later on in Western pediatric patients, Kim et al135 reported no association between survival or relapse and any of the genotypes studied in a population of 100 Korean pediatric patients. This highlights the possible role of genetic ancestry and the need for studies looking at additional alleles that might be peculiar to certain regions of the world.
In a GWAS of 511 ALL cases from the COG, Perez-Andreu et al114 identified a single susceptibility locus in the GATA binding protein 3 GATA3 (rs3824662) as a somatic lesion that predisposed children to Philadelphia chromosome-like (Ph-like) ALL. The GATA3 SNP was also associated with a higher risk of ALL relapse; however, this association with prognosis was thought to be largely driven by the gene’s relationship with Ph-like ALL, which inherently predicts poor prognosis for ALL patients. GATA3 belongs to a group of transcription factors thought to play a critical role during T-cell development and differentiation from T-lineage progenitor cells.136 Interestingly, this study highlights the interaction between host germline and tumor somatic genomes and the possible implication of such a relationship in tumorigenesis and prognosis.114
Also, in a Danish population-based study, Gregers et al14 explored the impact of four ABCB1 polymorphisms (rs2229109, rs1128503, rs2032582 and rs1045642) on disease relapse. Risk of relapse was increased by 2.9-fold for patients with the rs2229109 heterozygous genotype versus wild type, and reduced by 61% and 40%, respectively, for patients with the rs1045642 heterozygous or homozygous variant versus wild type. These findings were also seen during subgroup analysis of the high-risk patients, though no statistical significance was shown in the low-risk group by either univariate or multivariate analysis. Finally, and knowing that plasma levels of tumor necrosis factor (TNF) and interleukin 10 (IL-10) have been associated with treatment outcome in hematologic malignancies, Lauten et al121 looked at germline polymorphisms within the TNF and IL-10 genes. The study done on 135 ALL pediatric patients treated according to BFM protocols did not show a statistically significant relationship between the expression of the TNF2 risk allele (rs1800629) and the risk of relapse in the total population. However, when looking at the expression of the SNP in patients with poor response to PRED the expression of the TNF2 risk allele (rs1800629) showed a higher risk of relapse. PRED response was assessed using the in vivo resistance test.121 This is of use to predict the risk of relapse in patients treated on regimens that depend highly on PRED for induction therapy.
CNS Relapse
A major improvement in ALL therapy was the elimination of prophylactic cranial irradiation from treatment regimens, without impacting the risk of CNS relapse.137 Currently, tailoring the therapy according to the risk of CNS relapse to reduce treatment-related sequelae by reducing toxicity, remains a challenge.138 Combining PGx to previously known risk factors for such outcomes might be of help to better stratify patients at risk.
Using a candidate gene approach to determine SNPs that might influence treatment outcome in 247 children with newly diagnosed ALL treated on the St. Jude Children’s Research Hospital Total XIIIB study, Rocha et al122 identified certain polymorphisms that predicted the risk of CNS relapse. In the high-risk population, the VDR Fokl T allele (rs2228570) was linked to a higher risk of CNS relapse. VDR is thought to play a role in regulating the expression of CYP3A4 and p-glycoprotein, however, the exact role of the polymorphism in the gene is not clear.139 In fact in this study, all CNS relapses occurred in patients with at least 1 VDR Fokl T allele, however, the frequency of the genotypes did not differ between low-risk and high-risk patients for CNS relapse at diagnosis. Also, among those with at least 1 VDR Fokl T allele, the VDR intron 8 GG genotype was associated with a greater risk of CNS relapse, and the combination of both VDR Fokl and VDR intron 8 genotypes were associated with CNS relapse in the multivariate analysis including other prognostic factors. Therefore, 2 polymorphisms in the VDR locus were associated with risk of CNS relapse and remained so after adjusting for other prognostic factors.122
Event-Free Survival
Salazar et al46 showed in their study of 141 patients from Spanish medical centers that MTHFR genotype subdivided their patient population into two groups with quite different EFS at 48 months, as patients with unfavorable MTHFR genotype (homozygous 677T - rs1801133, homozygous 1298C - rs1801131 and compound heterozygous) had significantly lower EFS. SNPs in the remaining genes analyzed, including the A80G polymorphism in SLCO1B1 (rs1051266), the A870G polymorphism in CCND1 (rs9344) and the A317G (rs408626) and C680A (rs442767) promoter changes in DHFR, did not show any significant association with EFS.46 Another study showed that children of French-Canadian origin with ALL and homozygous for the CCND1 A (rs9344) variant had poorer EFS when compared to carriers of the G variant. This result retained its significance in the presence of other prognostic variables and had a more important impact in individuals who were also homozygous for the TYMS 3R (rs45445694) polymorphism.126 In addition in a retrospective candidate gene approach study on 320 ALL children treated according to the LAL or DFCI protocols, Krajinovic et al127 showed that patients carrying variant alleles of the CYP1A1*2A (rs4646903) and NQO1*2 (rs1800566) genotypes had lower EFS. This significance was retained in a multivariate analysis model that included other known prognostic factors. Using another candidate gene approach, Zhai et al125 analyzed several SNPs in relation to EFS. The study recruited a total of 138 Han Chinese children with ALL. Analyses showed that patients carrying the wild type alleles of the rs2032582 or rs1128503 in ABCB1 or allele variants in ABCC2 (rs2273697) or ABCG2 (rs2231137) had lower EFS. Combining these SNPs also lead significant results. Finally, Kunkele et al140 retrospectively genotyped a series of 182 patients with childhood ALL diagnosed and treated at the Children’s University Hospital Essen, for a BCL2 polymorphism (rs2279115). The genotype was shown to be non-correlated with patient outcome such as EFS. Furthermore, during their multivariate regression analyses of the high-risk group, this SNP remained non-significant for EFS even after correction for other variables.
Evidence for the Individualization of Treatment Regimens
Despite the relatively large number of PGx studies in childhood ALL, and the significant results with many variants from candidate gene and whole-genome studies, so far not many variants have been supported by Level 1 or 2 evidence as per PharmGKB criteria (Table 2).141 Notably, 6-MP is the only drug whose approved updated labels entail either actionable PGx (European Medicines Agency, Health Canada Santé, and Swissmedic) or recommend testing (US Food and Drug Administration). In addition, both the Clinical Pharmacogenetics Implementation Consortium (CPIC) and the Dutch Pharmacogenetics Working Group include annotations in their clinical guidelines for 6-MP dose adjustment with selected TPMT and NUDT15 variants.142
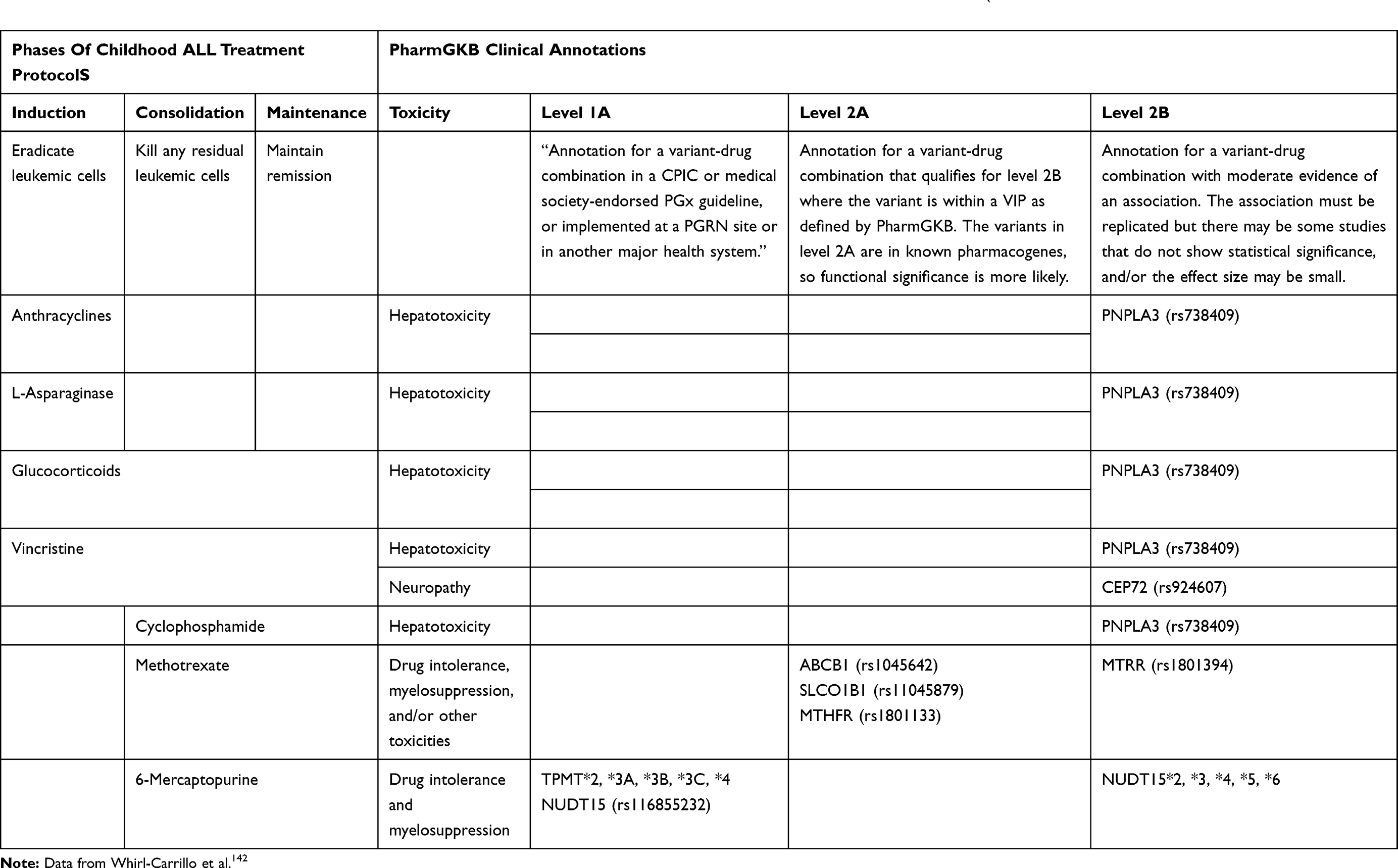 |
Table 2 Clinical Annotations with Levels 1 or 2 Based on the PharmGKB of Drugs Used for the Treatment of Childhood ALL` |
TPMT is the earliest and most extensively studied enzyme in the context of 6-MP tolerability and toxicity. For instance, back in 1980, Weinshilboum and Sladek reported a trimodal distribution of enzyme activity in Caucasians.143 It took more than a decade to ascertain that TPMT poor metabolizers are associated with increased severity of hematologic toxicity and necessitate major dose adjustment in childhood ALL.144–147 Fast forward, four TPMT SNPs (*2 rs1800584, *3B rs1800460, *3C rs1142345, and *3A that combines *3B and *3C) and potentially *4 rs1800584 have been extensively studied and replicated in multiple different populations of children treated for ALL with various protocols.148 Results showed marked decreased enzyme activity and myelotoxicity with these SNPs,39,149 and they were hence included in the CPIC guidelines.150 Although homozygous TPMT variants are highly predictive of 6-MP intolerance, they are quite uncommon (the variant alleles are no more than 5% in Whites, Black, and Asians)7,151,152 hence the need to elicit actionable PGx variants that are more prevalent. As a matter of fact, in 2014, a GWAS in Koreans with inflammatory bowel disease lead to the identification of the rs116855232 C variant allele in NUDT15 to be associated with significant thiopurine toxicity,153 an effect that was later validated in some studies in children receiving 6-MP for ALL.29,31,34,110 Accordingly, the updated CPIC guidelines in 2018 recommend reducing 6-MP dosing based on TPMT and NUDT15 genotype,154,155 and similar guidelines are also listed in the US, European and Swiss drug labels.142 Additional NUDT15 polymorphisms (rs554405994, rs186364861, and rs746071566) were also shown to affect 6-MP tolerance and myelotoxicity but they are not yet integrated into the guidelines.156–158 Notably, NUDT15 genetic polymorphisms are especially common in Asians only, hence the need for further studies for the incorporation of more common and impactful variants in other candidate genes such as the enzyme ITPA (rs1127354 and rs7270101).151–153
Concerning other drugs, probably the largest number of candidate genes were evaluated with MTX toxicity and efficacy. In addition, and since MTX levels are typically measured during the consolidation phase with secondary MTX dose adjustment, there have been a large number of PGx studies with MTX Pks.7,13,15 Nevertheless, and due to some inconsistencies in patient populations, phenotypes, and treatment protocols, none of the genetic markers has yet reached a level to be included in clinical practice. As seen in Table 2, only three variants in three genes (ABCB1 rs1045642, SLCO1B1 rs1104879, and MTHFR rs1801133) are listed as Level 2A for MTX by the PharmGKB. Nevertheless, and based on two meta-analyses, it appears that the T variant allele of the C677T MTHFR polymorphism (rs1801133) is significantly associated with increased risk of relapse159 and toxicities (such as hepatotoxicity, hematologic toxicity, and mucositis) in childhood malignancies.160 It is therefore plausible that sometime soon, physician prescribers would be able to adjust MTX dosing based on the MTHFR C677T genotype.
Few variants are currently annotated as Level 2B for several drugs including MTX and 6-MP with a variant in the PNPLA3 gene being associated with hepatotoxicity in DNR, L-ASPA, PRDL, VINC, and CTX combination therapy, and another in CEP72 (rs924607) with VINC-related peripheral neuropathy. The variant in the latter gene was initially reported in a candidate gene study and another large GWAS with VINC-related neuropathy in two independent cohorts of patients treated with the St. Jude and the Children’s Oncology Group (COG) protocols.74,161 Results were however not replicated in two other studies.162,163 More recently whole-exome sequencing of DNA from 240 European children with ALL revealed an association between four variants in four genes (BAHD1 rs3803357, MRPL47 rs10513762, SYNE2 rs2781377, and CDH2 rs1944294) with VINC-related neuropathy,66 however these results were not yet validated in other populations.
Finally, relatively few PGx studies are currently available for other drugs such as steroids and L-ASPA. Probably the most compelling evidence is on the association of HLA-DRB1 variants with hypersensitivity, an association that appeared in two large GWAS with mixed populations102,164 and in another more recent candidate SNP study with a smaller sample of European children.103 Of note that anthracyclines are commonly associated with acute and chronic cardiotoxicity, yet no variants were classified as level 1 or 2 by the PharmGKB although the Canadian Pharmacogenomics Network for Drug Safety (CPNDS) has annotated recommendations for genetic testing of RARG (rs2229774), SLC28A3 (rs7853758), and UGT1A6 (rs17863783) to reduce the incidence of DAU or DOX-induced cardiotoxicity in children with cancer.142
Clinical Implementation
Although both candidate gene and GWAS have elucidated many inherited genetic variants to be associated with inter-individual variability in ALL treatment toxicity and outcome, further investigations are needed to replicate some of the findings, and more concerted efforts are necessary to move these tests into the clinic. For instance, and despite all of the available clinical annotations and clinical practice guidelines, and despite some evidence for the clinical utility of PGx testing, at least for childhood ALL,1 it appears that the clinical adoption of PGx testing did not yet reach its potential with variable uptake worldwide. As a matter of fact in a recent global survey of the implementation of PGx in clinical practice, we have shown that although TPMT genotyping is the most commonly available clinical PGx worldwide, many institutions do not offer it yet, especially for those outside of Northern Europe and America. In addition to previously reported challenges such as cost, reimbursement, and unawareness or skepticism of physicians, new themes emerged concerning data management and applications of clinical decision supports into the electronic medical records.165 As depicted in Figure 1, drugs are affected by a multitude of genes and gene variants; it may hence be much more cost-effective and practical to preemptively genotype for an array of candidate genes, store data, and release them when needed for individualized therapy.166,167 Such approach is currently adopted by a number of academic hospitals in the USA166 including the St. Jude Children’s Research Hospital PG4KDS protocol,168 and is currently being collaboratively tested at a large scale in Europe.169 It is hoped that with the implementation of more NGS studies and collaborative efforts to include large and adequately powered multinational cohorts, the clinical implementation of PGx for ALL becomes a daily reality, with the potential for showing a benefit of the integration of preemptive PGx information on clinical outcome.166,170
Disclosure
The authors declare that there is no conflict of interest in this work.
References
1. Evans WE, Crews KR, Pui CH. A health-care system perspective on implementing genomic medicine: pediatric acute lymphoblastic leukemia as a paradigm. Clin Pharmacol Ther. 2013;94(2):224–229.
2. Stanulla M, Schrappe M, Yang S-K. Treatment of childhood acute lymphoblastic leukemia. Semin Hematol. 2009;46(1):52–63. doi:10.1053/j.seminhematol.2008.09.007
3. Lopez-Lopez E, Gutierrez-Camino A, Bilbao-Aldaiturriaga N, Pombar-Gomez M, Martin-Guerrero I, Garcia-Orad A. Pharmacogenetics of childhood acute lymphoblastic leukemia. Pharmacogenomics. 2014;15(10):1383–1398. doi:10.2217/pgs.14.106
4. Al-Mahayri ZN, Patrinos GP, Ali BR. Pharmacogenomics in pediatric acute lymphoblastic leukemia: promises and limitations. Pharmacogenomics. 2017;18(7):687–699. doi:10.2217/pgs-2017-0005
5. Lee SHR, Yang JJ. Pharmacogenomics in acute lymphoblastic leukemia. Best Pract Res Clin Haematol. 2017;30(3):229–236. doi:10.1016/j.beha.2017.07.007
6. Mei L, Ontiveros EP, Griffiths EA, Thompson JE, Wang ES, Wetzler M. Pharmacogenetics predictive of response and toxicity in acute lymphoblastic leukemia therapy. Blood Rev. 2015;29(4):243–249. doi:10.1016/j.blre.2015.01.001
7. Rudin S, Marable M, Huang RS. The Promise of Pharmacogenomics in Reducing Toxicity During Acute Lymphoblastic Leukemia Maintenance Treatment. Genomics Proteomics Bioinformatics. 2017;15(2):82–93. doi:10.1016/j.gpb.2016.11.003
8. Schmiegelow K, Nielsen SN, Frandsen TL, Nersting J. Mercaptopurine/Methotrexate maintenance therapy of childhood acute lymphoblastic leukemia: clinical facts and fiction. J Pediatr Hematol Oncol. 2014;36(7):503–517. doi:10.1097/MPH.0000000000000206
9. Davidsen ML, Dalhoff K, Schmiegelow K. Pharmacogenetics influence treatment efficacy in childhood acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2008;30(11):831–849. doi:10.1097/MPH.0b013e3181868570
10. Maxwell RR, Cole PD. Pharmacogenetic predictors of treatment-related toxicity among children with acute lymphoblastic leukemia. Curr Hematol Malig Rep. 2017;12(3):176–186. doi:10.1007/s11899-017-0376-z
11. Relling MV, Ramsey LB. Pharmacogenomics of acute lymphoid leukemia: new insights into treatment toxicity and efficacy. Hematology Am Soc Hematol Educ Program. 2013;2013(1):126–130. doi:10.1182/asheducation-2013.1.126
12. Zaza G, Cheok M, Krynetskaia N, et al. Thiopurine pathway. Pharmacogenet Genomics. 2010;20(9):573–574. doi:10.1097/FPC.0b013e328334338f
13. Mikkelsen TS, Thorn CF, Yang JJ, et al. PharmGKB summary: methotrexate pathway. Pharmacogenet Genomics. 2011;21(10):679–686. doi:10.1097/FPC.0b013e328343dd93
14. Gregers J, Green H, Christensen IJ, et al. Polymorphisms in the ABCB1 gene and effect on outcome and toxicity in childhood acute lymphoblastic leukemia. Pharmacogenomics J. 2015;15(4):372–379. doi:10.1038/tpj.2014.81
15. Zgheib NK, Akra-Ismail M, Aridi C, et al. Genetic polymorphisms in candidate genes predict increased toxicity with methotrexate therapy in Lebanese children with acute lymphoblastic leukemia. Pharmacogenet Genomics. 2014;24(8):387–396. doi:10.1097/FPC.0000000000000069
16. ElFayoumi RI, Hagras MM, Abozenadaha A, et al. The influence of polymorphisms in the drug transporter, ABCB1 on the toxicity of glucocorticoids in Saudi children with acute lymphoblastic leukaemia. Pharmacol Rep. 2019;71(1):90–95. doi:10.1016/j.pharep.2018.09.010
17. Liu Y, Yin Y, Sheng Q, et al. Association of ABCC2-24C>T polymorphism with high-dose methotrexate plasma concentrations and toxicities in childhood acute lymphoblastic leukemia. PLoS One. 2014;9(1):e82681. doi:10.1371/journal.pone.0082681
18. Hareedy MS, El Desoky ES, Woillard JB, et al. Genetic variants in 6-mercaptopurine pathway as potential factors of hematological toxicity in acute lymphoblastic leukemia patients. Pharmacogenomics. 2015;16(10):1119–1134. doi:10.2217/PGS.15.62
19. Tanaka Y, Nakadate H, Kondoh K, Nakamura K, Koh K, Manabe A. Interaction between NUDT15 and ABCC4 variants enhances intolerability of 6-mercaptopurine in Japanese patients with childhood acute lymphoblastic leukemia. Pharmacogenomics J. 2018;18(2):275–280. doi:10.1038/tpj.2017.12
20. Gregers J, Christensen IJ, Dalhoff K, et al. The association of reduced folate carrier 80G>A polymorphism to outcome in childhood acute lymphoblastic leukemia interacts with chromosome 21 copy number. Blood. 2010;115(23):4671–4677. doi:10.1182/blood-2010-01-256958
21. Eldem I, Yavuz D, Cumaogullari O, et al. SLCO1B1 Polymorphisms are Associated With Drug Intolerance in Childhood Leukemia Maintenance Therapy. J Pediatr Hematol Oncol. 2018;40(5):e289–e294. doi:10.1097/MPH.0000000000001153
22. Marino S, Verzegnassi F, Tamaro P, et al. Response to glucocorticoids and toxicity in childhood acute lymphoblastic leukemia: role of polymorphisms of genes involved in glucocorticoid response. Pediatr Blood Cancer. 2009;53(6):984–991. doi:10.1002/pbc.22163
23. Smid A, Karas-Kuzelicki N, Jazbec J, Mlinaric-Rascan I. PACSIN2 polymorphism is associated with thiopurine-induced hematological toxicity in children with acute lymphoblastic leukaemia undergoing maintenance therapy. Sci Rep. 2016;6(1):30244. doi:10.1038/srep30244
24. Lennard L, Cartwright CS, Wade R, Vora A. Thiopurine dose intensity and treatment outcome in childhood lymphoblastic leukaemia: the influence of thiopurine methyltransferase pharmacogenetics. Br J Haematol. 2015;169(2):228–240. doi:10.1111/bjh.13240
25. Milosevic G, Kotur N, Krstovski N, et al. Variants in TPMT, ITPA, ABCC4 and ABCB1 Genes As Predictors of 6-mercaptopurine Induced Toxicity in Children with Acute Lymphoblastic Leukemia. J Med Biochem. 2018;37(3):320–327. doi:10.1515/jomb-2017-0060
26. Albayrak M, Konyssova U, Kaya Z, et al. Thiopurine methyltransferase polymorphisms and mercaptopurine tolerance in Turkish children with acute lymphoblastic leukemia. Cancer Chemother Pharmacol. 2011;68(5):1155–1159. doi:10.1007/s00280-011-1599-7
27. Tumer TB, Ulusoy G, Adali O, Sahin G, Gozdasoglu S, Arinc E. The low frequency of defective TPMT alleles in Turkish population: a study on pediatric patients with acute lymphoblastic leukemia. Am J Hematol. 2007;82(10):906–910. doi:10.1002/ajh.20947
28. Peregud-Pogorzelski J, Tetera-Rudnicka E, Kurzawski M, et al. Thiopurine S-methyltransferase (TPMT) polymorphisms in children with acute lymphoblastic leukemia, and the need for reduction or cessation of 6-mercaptopurine doses during maintenance therapy: the Polish multicenter analysis. Pediatr Blood Cancer. 2011;57(4):578–582. doi:10.1002/pbc.23013
29. Liang DC, Yang CP, Liu HC, et al. NUDT15 gene polymorphism related to mercaptopurine intolerance in Taiwan Chinese children with acute lymphoblastic leukemia. Pharmacogenomics J. 2016;16(6):536–539. doi:10.1038/tpj.2015.75
30. Soler AM, Olano N, Mendez Y, et al. TPMT and NUDT15 genes are both related to mercaptopurine intolerance in acute lymphoblastic leukaemia patients from Uruguay. Br J Haematol. 2018;181(2):252–255.
31. Moriyama T, Nishii R, Perez-Andreu V, et al. NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet. 2016;48(4):367–373. doi:10.1038/ng.3508
32. Yang JJ, Landier W, Yang W, et al. Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J Clin Oncol. 2015;33(11):1235–1242. doi:10.1200/JCO.2014.59.4671
33. Zhou H, Li L, Yang P, et al. Optimal predictor for 6-mercaptopurine intolerance in Chinese children with acute lymphoblastic leukemia: NUDT15, TPMT, or ITPA genetic variants? BMC Cancer. 2018;18(1):516. doi:10.1186/s12885-018-4398-2
34. Tanaka Y, Kato M, Hasegawa D, et al. Susceptibility to 6-MP toxicity conferred by a NUDT15 variant in Japanese children with acute lymphoblastic leukaemia. Br J Haematol. 2015;171(1):109–115. doi:10.1111/bjh.13518
35. Chiengthong K, Ittiwut C, Muensri S, et al. NUDT15 c.415C>T increases risk of 6-mercaptopurine induced myelosuppression during maintenance therapy in children with acute lymphoblastic leukemia. Haematologica. 2016;101(1):e24–26. doi:10.3324/haematol.2015.134775
36. Hawwa AF, Millership JS, Collier PS, et al. Pharmacogenomic studies of the anticancer and immunosuppressive thiopurines mercaptopurine and azathioprine. Br J Clin Pharmacol. 2008;66(4):517–528. doi:10.1111/j.1365-2125.2008.03248.x
37. Khera S, Trehan A, Bhatia P, Singh M, Bansal D, Varma N. Prevalence of TPMT, ITPA and NUDT 15 genetic polymorphisms and their relation to 6MP toxicity in north Indian children with acute lymphoblastic leukemia. Cancer Chemother Pharmacol. 2019;83(2):341–348. doi:10.1007/s00280-018-3732-3
38. Moradveisi B, Muwakkit S, Zamani F, Ghaderi E, Mohammadi E, Zgheib NK. ITPA, TPMT, and NUDT15 Genetic Polymorphisms Predict 6-Mercaptopurine Toxicity in Middle Eastern Children With Acute Lymphoblastic Leukemia. Front Pharmacol. 2019;10:916. doi:10.3389/fphar.2019.00916
39. Stocco G, Cheok MH, Crews KR, et al. Genetic polymorphism of inosine triphosphate pyrophosphatase is a determinant of mercaptopurine metabolism and toxicity during treatment for acute lymphoblastic leukemia. Clin Pharmacol Ther. 2009;85(2):164–172. doi:10.1038/clpt.2008.154
40. Huang L, Tissing WJ, de Jonge R, van Zelst BD, Pieters R. Polymorphisms in folate-related genes: association with side effects of high-dose methotrexate in childhood acute lymphoblastic leukemia. Leukemia. 2008;22(9):1798–1800. doi:10.1038/leu.2008.66
41. Araoz HV, D’Aloi K, Foncuberta ME, et al. Pharmacogenetic studies in children with acute lymphoblastic leukemia in Argentina. Leuk Lymphoma. 2015;56(5):1370–1378. doi:10.3109/10428194.2014.951844
42. Chiusolo P, Reddiconto G, Casorelli I, et al. Preponderance of methylenetetrahydrofolate reductase C677T homozygosity among leukemia patients intolerant to methotrexate. Ann Oncol. 2002;13(12):1915–1918. doi:10.1093/annonc/mdf322
43. Shimasaki N, Mori T, Torii C, et al. Influence of MTHFR and RFC1 polymorphisms on toxicities during maintenance chemotherapy for childhood acute lymphoblastic leukemia or lymphoma. J Pediatr Hematol Oncol. 2008;30(5):347–352. doi:10.1097/MPH.0b013e318165b25d
44. Kantar M, Kosova B, Cetingul N, et al. Methylenetetrahydrofolate reductase C677T and A1298C gene polymorphisms and therapy-related toxicity in children treated for acute lymphoblastic leukemia and non-Hodgkin lymphoma. Leuk Lymphoma. 2009;50(6):912–917. doi:10.1080/10428190902893819
45. Kodidela S, Pradhan SC, Dubashi B, Basu D. Influence of dihydrofolate reductase gene polymorphisms rs408626 (−317A>G) and rs442767 (−680C>A) on the outcome of methotrexate-based maintenance therapy in South Indian patients with acute lymphoblastic leukemia. Eur J Clin Pharmacol. 2015;71(11):1349–1358. doi:10.1007/s00228-015-1930-z
46. Salazar J, Altes A, Del Rio E, et al. Methotrexate consolidation treatment according to pharmacogenetics of MTHFR ameliorates event-free survival in childhood acute lymphoblastic leukaemia. Pharmacogenomics J. 2012;12(5):379–385. doi:10.1038/tpj.2011.25
47. Wang Q, He G, Hou M, et al. Cell Cycle Regulation by Alternative Polyadenylation of CCND1. Sci Rep. 2018;8(1):6824. doi:10.1038/s41598-018-25141-0
48. Costea I, Moghrabi A, Laverdiere C, Graziani A, Krajinovic M. Folate cycle gene variants and chemotherapy toxicity in pediatric patients with acute lymphoblastic leukemia. Haematologica. 2006;91(8):1113–1116.
49. Garcia-Bournissen F, Moghrabi A, Krajinovic M. Therapeutic responses in childhood acute lymphoblastic leukemia (ALL) and haplotypes of gamma glutamyl hydrolase (GGH) gene. Leuk Res. 2007;31(7):1023–1025. doi:10.1016/j.leukres.2006.08.007
50. Csordas K, Lautner-Csorba O, Semsei AF, et al. Associations of novel genetic variations in the folate-related and ARID5B genes with the pharmacokinetics and toxicity of high-dose methotrexate in paediatric acute lymphoblastic leukaemia. Br J Haematol. 2014;166(3):410–420. doi:10.1111/bjh.12886
51. Oosterom N, Berrevoets M, den Hoed MAH, et al. The role of genetic polymorphisms in the thymidylate synthase (TYMS) gene in methotrexate-induced oral mucositis in children with acute lymphoblastic leukemia. Pharmacogenet Genomics. 2018;28(10):223–229. doi:10.1097/FPC.0000000000000352
52. Trevino LR, Shimasaki N, Yang W, et al. Germline genetic variation in an organic anion transporter polypeptide associated with methotrexate pharmacokinetics and clinical effects. J Clin Oncol. 2009;27(35):5972–5978. doi:10.1200/JCO.2008.20.4156
53. Lopez-Lopez E, Ballesteros J, Pinan MA, et al. Polymorphisms in the methotrexate transport pathway: a new tool for MTX plasma level prediction in pediatric acute lymphoblastic leukemia. Pharmacogenet Genomics. 2013;23(2):53–61. doi:10.1097/FPC.0b013e32835c3b24
54. den Hoed MA, Lopez-Lopez E, Te Winkel ML, et al. Genetic and metabolic determinants of methotrexate-induced mucositis in pediatric acute lymphoblastic leukemia. Pharmacogenomics J. 2015;15(3):248–254. doi:10.1038/tpj.2014.63
55. Roy Moulik N, Kumar A, Agrawal S, Awasthi S, Mahdi AA, Kumar A. Role of folate status and methylenetetrahydrofolate reductase genotype on the toxicity and outcome of induction chemotherapy in children with acute lymphoblastic leukemia. Leuk Lymphoma. 2015;56(5):1379–1384. doi:10.3109/10428194.2014.947608
56. Hunger SP, Loh ML, Whitlock JA, et al. Children’s Oncology Group’s 2013 blueprint for research: acute lymphoblastic leukemia. Pediatr Blood Cancer. 2013;60(6):957–963. doi:10.1002/pbc.24420
57. Gutierrez-Camino A, Martin-Guerrero I, Garcia-Orad A. PNPLA3 rs738409 and Hepatotoxicity in Children With B-cell Acute Lymphoblastic Leukemia: A Validation Study in a Spanish Cohort. Clin Pharmacol Ther. 2017;102(6):906. doi:10.1002/cpt.756
58. Kienesberger PC, Oberer M, Lass A, Zechner R. Mammalian patatin domain containing proteins: a family with diverse lipolytic activities involved in multiple biological functions. J Lipid Res. 2009;50(Suppl):S63–68. doi:10.1194/jlr.R800082-JLR200
59. Liu Y, Fernandez CA, Smith C, et al. Genome-Wide Study Links PNPLA3 Variant With Elevated Hepatic Transaminase After Acute Lymphoblastic Leukemia Therapy. Clin Pharmacol Ther. 2017;102(1):131–140. doi:10.1002/cpt.629
60. Smagris E, BasuRay S, Li J, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology. 2015;61(1):108–118. doi:10.1002/hep.27242
61. Qiu Q, Huang J, Lin Y, et al. Polymorphisms and pharmacogenomics for the toxicity of methotrexate monotherapy in patients with rheumatoid arthritis: A systematic review and meta-analysis. Medicine. 2017;96(11):e6337. doi:10.1097/MD.0000000000006337
62. Ongaro A, De Mattei M, Della Porta MG, et al. Gene polymorphisms in folate metabolizing enzymes in adult acute lymphoblastic leukemia: effects on methotrexate-related toxicity and survival. Haematologica. 2009;94(10):1391–1398. doi:10.3324/haematol.2009.008326
63. Tanaka Y, Manabe A, Nakadate H, et al. Methylenetetrahydrofolate reductase gene haplotypes affect toxicity during maintenance therapy for childhood acute lymphoblastic leukemia in Japanese patients. Leuk Lymphoma. 2014;55(5):1126–1131. doi:10.3109/10428194.2013.825902
64. Wan Rosalina WR, Teh LK, Mohamad N, et al. Polymorphism of ITPA 94C>A and risk of adverse effects among patients with acute lymphoblastic leukaemia treated with 6-mercaptopurine. J Clin Pharm Ther. 2012;37(2):237–241. doi:10.1111/j.1365-2710.2011.01272.x
65. Argyriou AA, Bruna J, Marmiroli P, Cavaletti G. Chemotherapy-induced peripheral neurotoxicity (CIPN): an update. Crit Rev Oncol Hematol. 2012;82(1):51–77. doi:10.1016/j.critrevonc.2011.04.012
66. Abaji R, Ceppi F, Patel S, et al. Genetic risk factors for VIPN in childhood acute lymphoblastic leukemia patients identified using whole-exome sequencing. Pharmacogenomics. 2018;19(15):1181–1193. doi:10.2217/pgs-2018-0093
67. Warren DT, Zhang Q, Weissberg PL, Shanahan CM. Nesprins: intracellular scaffolds that maintain cell architecture and coordinate cell function? Expert Rev Mol Med. 2005;7(11):1–15. doi:10.1017/S1462399405009294
68. Mross C, Marko M, Munck M, et al. Depletion of Nesprin-2 is associated with an embryonic lethal phenotype in mice. Nucleus. 2018;9(1):503–515. doi:10.1080/19491034.2018.1523664
69. Kenmochi N, Suzuki T, Uechi T, et al. The human mitochondrial ribosomal protein genes: mapping of 54 genes to the chromosomes and implications for human disorders. Genomics. 2001;77(1–2):65–70. doi:10.1006/geno.2001.6622
70. Pavlovic S, Kotur N, Stankovic B, Zukic B, Gasic V, Dokmanovic L. Pharmacogenomic and Pharmacotranscriptomic Profiling of Childhood Acute Lymphoblastic Leukemia: paving the Way to Personalized Treatment. Genes. 2019;10(3):3. doi:10.3390/genes10030191
71. Ceppi F, Langlois-Pelletier C, Gagne V, et al. Polymorphisms of the vincristine pathway and response to treatment in children with childhood acute lymphoblastic leukemia. Pharmacogenomics. 2014;15(8):1105–1116. doi:10.2217/pgs.14.68
72. Verrills NM, Liem NL, Liaw TY, Hood BD, Lock RB, Kavallaris M. Proteomic analysis reveals a novel role for the actin cytoskeleton in vincristine resistant childhood leukemia–an in vivo study. Proteomics. 2006;6(5):1681–1694. doi:10.1002/pmic.200500417
73. Lopez-Lopez E, Gutierrez-Camino A, Astigarraga I, et al. Vincristine pharmacokinetics pathway and neurotoxicity during early phases of treatment in pediatric acute lymphoblastic leukemia. Pharmacogenomics. 2016;17(7):731–741. doi:10.2217/pgs-2016-0001
74. Diouf B, Crews KR, Lew G, et al. Association of an inherited genetic variant with vincristine-related peripheral neuropathy in children with acute lymphoblastic leukemia. JAMA. 2015;313(8):815–823. doi:10.1001/jama.2015.0894
75. Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4(4):253–265. doi:10.1038/nrc1317
76. Dennison JB, Kulanthaivel P, Barbuch RJ, Renbarger JL, Ehlhardt WJ, Hall SD. Selective metabolism of vincristine in vitro by CYP3A5. Drug Metab Dispos. 2006;34(8):1317–1327. doi:10.1124/dmd.106.009902
77. Egbelakin A, Ferguson MJ, MacGill EA, et al. Increased risk of vincristine neurotoxicity associated with low CYP3A5 expression genotype in children with acute lymphoblastic leukemia. Pediatr Blood Cancer. 2011;56(3):361–367. doi:10.1002/pbc.22845
78. Li L, Sajdyk T, Smith EML, et al. Genetic Variants Associated With Vincristine-Induced Peripheral Neuropathy in Two Populations of Children With Acute Lymphoblastic Leukemia. Clin Pharmacol Ther. 2019;105(6):1421–1428. doi:10.1002/cpt.1324
79. Robertson NG, Cremers CW, Huygen PL, et al. Cochlin immunostaining of inner ear pathologic deposits and proteomic analysis in DFNA9 deafness and vestibular dysfunction. Hum Mol Genet. 2006;15(7):1071–1085. doi:10.1093/hmg/ddl022
80. Zhang J, Fei T, Li Z, et al. BMP induces cochlin expression to facilitate self-renewal and suppress neural differentiation of mouse embryonic stem cells. J Biol Chem. 2013;288(12):8053–8060. doi:10.1074/jbc.M112.433995
81. Bond AM, Bhalala OG, Kessler JA. The dynamic role of bone morphogenetic proteins in neural stem cell fate and maturation. Dev Neurobiol. 2012;72(7):1068–1084. doi:10.1002/dneu.22022
82. Mont MA, Hungerford DS. Non-traumatic avascular necrosis of the femoral head. J Bone Joint Surg Am. 1995;77(3):459–474. doi:10.2106/00004623-199503000-00018
83. Janke LJ, Liu C, Vogel P, et al. Primary epiphyseal arteriopathy in a mouse model of steroid-induced osteonecrosis. Am J Pathol. 2013;183(1):19–25. doi:10.1016/j.ajpath.2013.03.004
84. French D, Hamilton LH, Mattano LA, et al. A PAI-1 (SERPINE1) polymorphism predicts osteonecrosis in children with acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Blood. 2008;111(9):4496–4499. doi:10.1182/blood-2007-11-123885
85. Kawedia JD, Kaste SC, Pei D, et al. Pharmacokinetic, pharmacodynamic, and pharmacogenetic determinants of osteonecrosis in children with acute lymphoblastic leukemia. Blood. 2011;117(8):2340–2347. doi:10.1182/blood-2010-10-311969
86. Karol SE, Yang W, Van Driest SL, et al. Genetics of glucocorticoid-associated osteonecrosis in children with acute lymphoblastic leukemia. Blood. 2015;126(15):1770–1776. doi:10.1182/blood-2015-05-643601
87. Karol SE, Mattano LA, Yang W, et al. Genetic risk factors for the development of osteonecrosis in children under age 10 treated for acute lymphoblastic leukemia. Blood. 2016;127(5):558–564. doi:10.1182/blood-2015-10-673848
88. Maurer T, Zimmermann G, Maurer S, Stegmaier S, Wagner C, Hansch GM. Inhibition of osteoclast generation: a novel function of the bone morphogenetic protein 7/osteogenic protein 1. Mediators Inflamm. 2012;2012:171209. doi:10.1155/2012/171209
89. Zhang S, Fantozzi I, Tigno DD, et al. Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2003;285(3):L740–754. doi:10.1152/ajplung.00284.2002
90. Ramsey LB, Pounds S, Cheng C, et al. Genetics of pleiotropic effects of dexamethasone. Pharmacogenet Genomics. 2017;27(8):294–302. doi:10.1097/FPC.0000000000000293
91. Relling MV, Yang W, Das S, et al. Pharmacogenetic risk factors for osteonecrosis of the hip among children with leukemia. J Clin Oncol. 2004;22(19):3930–3936. doi:10.1200/JCO.2004.11.020
92. Munker R, Hill U, Jehn U, Kolb HJ, Schalhorn A. Renal complications in acute leukemias. Haematologica. 1998;83(5):416–421.
93. Liu C, Yang W, Devidas M, et al. Clinical and Genetic Risk Factors for Acute Pancreatitis in Patients With Acute Lymphoblastic Leukemia. J Clin Oncol. 2016;34(18):2133–2140. doi:10.1200/JCO.2015.64.5812
94. Catasus L, Vendrell J, Aviles FX, Carreira S, Puigserver A, Billeter M. The sequence and conformation of human pancreatic procarboxypeptidase A2. cDNA cloning, sequence analysis, and three-dimensional model. J Biol Chem. 1995;270(12):6651–6657. doi:10.1074/jbc.270.12.6651
95. Abaji R, Gagne V, Xu CJ, et al. Whole-exome sequencing identified genetic risk factors for asparaginase-related complications in childhood ALL patients. Oncotarget. 2017;8(27):43752–43767. doi:10.18632/oncotarget.17959
96. Cai L, Deng SL, Liang L, et al. Identification of genetic associations of SP110/MYBBP1A/RELA with pulmonary tuberculosis in the Chinese Han population. Hum Genet. 2013;132(3):265–273. doi:10.1007/s00439-012-1244-5
97. Rakonczay Z, Hegyi P, Takacs T, McCarroll J, Saluja AK. The role of NF-kappaB activation in the pathogenesis of acute pancreatitis. Gut. 2008;57(2):259–267. doi:10.1136/gut.2007.124115
98. Killander D, Dohlwitz A, Engstedt L, et al. Hypersensitive reactions and antibody formation during L-asparaginase treatment of children and adults with acute leukemia. Cancer. 1976;37(1):220–228. doi:10.1002/1097-0142(197601)37:1<220::aid-cncr2820370132>3.0.co;2-w
99. Fernandez CA, Stewart E, Panetta JC, et al. Successful challenges using native E. coli asparaginase after hypersensitivity reactions to PEGylated E. coli asparaginase. Cancer Chemother Pharmacol. 2014;73(6):1307–1313. doi:10.1007/s00280-014-2464-2
100. Kerner B, Jasinska AJ, DeYoung J, Almonte M, Choi OW, Freimer NB. Polymorphisms in the GRIA1 gene region in psychotic bipolar disorder. Am J Med Genet B Neuropsychiatr Genet. 2009;150B(1):24–32. doi:10.1002/ajmg.b.30780
101. Chen SH, Pei D, Yang W, et al. Genetic variations in GRIA1 on chromosome 5q33 related to asparaginase hypersensitivity. Clin Pharmacol Ther. 2010;88(2):191–196. doi:10.1038/clpt.2010.94
102. Fernandez CA, Smith C, Yang W, et al. Genome-wide analysis links NFATC2 with asparaginase hypersensitivity. Blood. 2015;126(1):69–75. doi:10.1182/blood-2015-02-628800
103. Kutszegi N, Yang X, Gezsi A, et al. HLA-DRB1*07:01-HLA-DQA1*02:01-HLA-DQB1*02:02 haplotype is associated with a high risk of asparaginase hypersensitivity in acute lymphoblastic leukemia. Haematologica. 2017;102(9):1578–1586. doi:10.3324/haematol.2017.168211
104. Yokoi T, Nakajima M. microRNAs as mediators of drug toxicity. Annu Rev Pharmacol Toxicol. 2013;53(1):377–400. doi:10.1146/annurev-pharmtox-011112-140250
105. Lopez-Lopez E, Gutierrez-Camino A, Pinan MA, et al. Pharmacogenetics of microRNAs and microRNAs biogenesis machinery in pediatric acute lymphoblastic leukemia. PLoS One. 2014;9(3):e91261. doi:10.1371/journal.pone.0091261
106. Gutierrez-Camino A, Oosterom N, den Hoed MAH, et al. The miR-1206 microRNA variant is associated with methotrexate-induced oral mucositis in pediatric acute lymphoblastic leukemia. Pharmacogenet Genomics. 2017;27(8):303–306. doi:10.1097/FPC.0000000000000291
107. Gutierrez-Camino A, Umerez M, Lopez-Lopez E, et al. Involvement of miRNA polymorphism in mucositis development in childhood acute lymphoblastic leukemia treatment. Pharmacogenomics. 2018;19(18):1403–1412. doi:10.2217/pgs-2018-0113
108. Gutierrez-Camino A, Umerez M, Martin-Guerrero I, et al. Mir-pharmacogenetics of Vincristine and peripheral neurotoxicity in childhood B-cell acute lymphoblastic leukemia. Pharmacogenomics J. 2018;18(6):704–712. doi:10.1038/s41397-017-0003-3
109. Iparraguirre L, Gutierrez-Camino A, Umerez M, et al. MiR-pharmacogenetics of methotrexate in childhood B-cell acute lymphoblastic leukemia. Pharmacogenet Genomics. 2016;26(11):517–525. doi:10.1097/FPC.0000000000000245
110. Zgheib NK, Akika R, Mahfouz R, et al. NUDT15 and TPMT genetic polymorphisms are related to 6-mercaptopurine intolerance in children treated for acute lymphoblastic leukemia at the Children’s Cancer Center of Lebanon. Pediatr Blood Cancer. 2017;64(1):146–150. doi:10.1002/pbc.26189
111. Kim HT, Choi R, Won HH, et al. NUDT15 genotype distributions in the Korean population. Pharmacogenet Genomics. 2017;27(5):197–200. doi:10.1097/FPC.0000000000000274
112. Yang JJ, Cheng C, Yang W, et al. Genome-wide interrogation of germline genetic variation associated with treatment response in childhood acute lymphoblastic leukemia. JAMA. 2009;301(4):393–403. doi:10.1001/jama.2009.7
113. Stanulla M, Schaeffeler E, Flohr T, et al. Thiopurine methyltransferase (TPMT) genotype and early treatment response to mercaptopurine in childhood acute lymphoblastic leukemia. JAMA. 2005;293(12):1485–1489. doi:10.1001/jama.293.12.1485
114. Perez-Andreu V, Roberts KG, Harvey RC, et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet. 2013;45(12):1494–1498. doi:10.1038/ng.2803
115. Aplenc R, Thompson J, Han P, et al. Methylenetetrahydrofolate reductase polymorphisms and therapy response in pediatric acute lymphoblastic leukemia. Cancer Res. 2005;65(6):2482–2487. doi:10.1158/0008-5472.CAN-04-2606
116. D’Angelo V, Ramaglia M, Iannotta A, et al. Methotrexate toxicity and efficacy during the consolidation phase in paediatric acute lymphoblastic leukaemia and MTHFR polymorphisms as pharmacogenetic determinants. Cancer Chemother Pharmacol. 2011;68(5):1339–1346. doi:10.1007/s00280-011-1665-1
117. El-Khodary NM, El-Haggar SM, Eid MA, Ebeid EN. Study of the pharmacokinetic and pharmacogenetic contribution to the toxicity of high-dose methotrexate in children with acute lymphoblastic leukemia. Med Oncol. 2012;29(3):2053–2062. doi:10.1007/s12032-011-9997-6
118. Takanashi M, Morimoto A, Yagi T, et al. Impact of glutathione S-transferase gene deletion on early relapse in childhood B-precursor acute lymphoblastic leukemia. Haematologica. 2003;88(11):1238–1244.
119. Leonardi DB, Abbate M, Riccheri MC, et al. Improving risk stratification of patients with childhood acute lymphoblastic leukemia: glutathione-S-Transferases polymorphisms are associated with increased risk of relapse. Oncotarget. 2017;8(1):110–117. doi:10.18632/oncotarget.8606
120. Yang JJ, Cheng C, Devidas M, et al. Genome-wide association study identifies germline polymorphisms associated with relapse of childhood acute lymphoblastic leukemia. Blood. 2012;120(20):4197–4204. doi:10.1182/blood-2012-07-440107
121. Lauten M, Matthias T, Stanulla M, Beger C, Welte K, Schrappe M. Association of initial response to prednisone treatment in childhood acute lymphoblastic leukaemia and polymorphisms within the tumour necrosis factor and the interleukin-10 genes. Leukemia. 2002;16(8):1437–1442. doi:10.1038/sj.leu.2402545
122. Rocha JC, Cheng C, Liu W, et al. Pharmacogenetics of outcome in children with acute lymphoblastic leukemia. Blood. 2005;105(12):4752–4758. doi:10.1182/blood-2004-11-4544
123. Schmiegelow K, Forestier E, Kristinsson J, et al. Thiopurine methyltransferase activity is related to the risk of relapse of childhood acute lymphoblastic leukemia: results from the NOPHO ALL-92 study. Leukemia. 2009;23(3):557–564. doi:10.1038/leu.2008.316
124. Relling MV, Pui CH, Cheng C, Evans WE. Thiopurine methyltransferase in acute lymphoblastic leukemia. Blood. 2006;107(2):843–844. doi:10.1182/blood-2005-08-3379
125. Zhai X, Wang H, Zhu X, et al. Gene polymorphisms of ABC transporters are associated with clinical outcomes in children with acute lymphoblastic leukemia. Arch Med Sci. 2012;8(4):659–671. doi:10.5114/aoms.2012.30290
126. Costea I, Moghrabi A, Krajinovic M. The influence of cyclin D1 (CCND1) 870A>G polymorphism and CCND1-thymidylate synthase (TS) gene-gene interaction on the outcome of childhood acute lymphoblastic leukaemia. Pharmacogenetics. 2003;13(9):577–580. doi:10.1097/00008571-200309000-00006
127. Krajinovic M, Labuda D, Mathonnet G, et al. Polymorphisms in genes encoding drugs and xenobiotic metabolizing enzymes, DNA repair enzymes, and response to treatment of childhood acute lymphoblastic leukemia. Clin Cancer Res. 2002;8(3):802–810.
128. Database of Single Nucleotide Polymorphisms (dbSNP). Available from: https://www.ncbi.nlm.nih.gov/snp/. Accessed July 21, 2020.
129. Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354(2):166–178. doi:10.1056/NEJMra052603
130. Cheok MH, Evans WE. Acute lymphoblastic leukaemia: a model for the pharmacogenomics of cancer therapy. Nat Rev Cancer. 2006;6(2):117–129. doi:10.1038/nrc1800
131. Tinhofer I, Marschitz I, Henn T, Egle A, Greil R. Expression of functional interleukin-15 receptor and autocrine production of interleukin-15 as mechanisms of tumor propagation in multiple myeloma. Blood. 2000;95(2):610–618. doi:10.1182/blood.V95.2.610
132. Cario G, Izraeli S, Teichert A, et al. High interleukin-15 expression characterizes childhood acute lymphoblastic leukemia with involvement of the CNS. J Clin Oncol. 2007;25(30):4813–4820. doi:10.1200/JCO.2007.11.8166
133. Zaza G, Cheok M, Yang W, et al. Gene expression and thioguanine nucleotide disposition in acute lymphoblastic leukemia after in vivo mercaptopurine treatment. Blood. 2005;106(5):1778–1785. doi:10.1182/blood-2005-01-0143
134. Heim S, Lage H. Transcriptome analysis of different multidrug-resistant gastric carcinoma cells. In Vivo. 2005;19(3):583–590.
135. Kim H, Kang HJ, Kim HJ, et al. Pharmacogenetic analysis of pediatric patients with acute lymphoblastic leukemia: a possible association between survival rate and ITPA polymorphism. PLoS One. 2012;7(9):e45558. doi:10.1371/journal.pone.0045558
136. Fujiwara T, O’Geen H, Keles S, et al. Discovering hematopoietic mechanisms through genome-wide analysis of GATA factor chromatin occupancy. Mol Cell. 2009;36(4):667–681. doi:10.1016/j.molcel.2009.11.001
137. Jeha S, Pei D, Choi J, et al. Improved CNS Control of Childhood Acute Lymphoblastic Leukemia Without Cranial Irradiation: st Jude Total Therapy Study 16. J Clin Oncol. 2019;37(35):3377–3391. doi:10.1200/JCO.19.01692
138. Gaudichon J, Jakobczyk H, Debaize L, et al. Mechanisms of extramedullary relapse in acute lymphoblastic leukemia: reconciling biological concepts and clinical issues. Blood Rev. 2019;36:40–56. doi:10.1016/j.blre.2019.04.003
139. Thummel KE, Brimer C, Yasuda K, et al. Transcriptional control of intestinal cytochrome P-4503A by 1alpha,25-dihydroxy vitamin D3. Mol Pharmacol. 2001;60(6):1399–1406. doi:10.1124/mol.60.6.1399
140. Kunkele A, Grosse-Lordemann A, Schramm A, Eggert A, Schulte JH, Bachmann HS. The BCL2-938 C > A promoter polymorphism is associated with risk group classification in children with acute lymphoblastic leukemia. BMC Cancer. 2013;13:452. doi:10.1186/1471-2407-13-452
141. McDonagh EM, Whirl-Carrillo M, Garten Y, Altman RB, Klein TE. From pharmacogenomic knowledge acquisition to clinical applications: the PharmGKB as a clinical pharmacogenomic biomarker resource. Biomark Med. 2011;5(6):795–806. doi:10.2217/bmm.11.94
142. Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414–417. doi:10.1038/clpt.2012.96
143. Weinshilboum RM, Sladek SL. Mercaptopurine pharmacogenetics: monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am J Hum Genet. 1980;32(5):651–662.
144. Andersen JB, Szumlanski C, Weinshilboum RM, Schmiegelow K. Pharmacokinetics, dose adjustments, and 6-mercaptopurine/methotrexate drug interactions in two patients with thiopurine methyltransferase deficiency. Acta Paediatr. 1998;87(1):108–111. doi:10.1080/08035259850158001
145. Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr. 1991;119(6):985–989. doi:10.1016/S0022-3476(05)83063-X
146. Krynetski EY, Schuetz JD, Galpin AJ, Pui CH, Relling MV, Evans WE. A single point mutation leading to loss of catalytic activity in human thiopurine S-methyltransferase. Proc Natl Acad Sci U S A. 1995;92(4):949–953. doi:10.1073/pnas.92.4.949
147. Lennard L, Gibson BE, Nicole T, Lilleyman JS. Congenital thiopurine methyltransferase deficiency and 6-mercaptopurine toxicity during treatment for acute lymphoblastic leukaemia. Arch Dis Child. 1993;69(5):577–579. doi:10.1136/adc.69.5.577
148. Otterness DM, Szumlanski CL, Wood TC, Weinshilboum RM. Human thiopurine methyltransferase pharmacogenetics. Kindred with a terminal exon splice junction mutation that results in loss of activity. J Clin Invest. 1998;101(5):1036–1044. doi:10.1172/JCI1004
149. Higgs JE, Payne K, Roberts C, Newman WG. Are patients with intermediate TPMT activity at increased risk of myelosuppression when taking thiopurine medications? Pharmacogenomics. 2010;11(2):177–188. doi:10.2217/pgs.09.155
150. Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther. 2013;93(4):324–325. doi:10.1038/clpt.2013.4
151. Adam de Beaumais T, Fakhoury M, Medard Y, et al. Determinants of mercaptopurine toxicity in paediatric acute lymphoblastic leukemia maintenance therapy. Br J Clin Pharmacol. 2011;71(4):575–584. doi:10.1111/j.1365-2125.2010.03867.x
152. Adam de Beaumais T, Jacqz-Aigrain E. Pharmacogenetic determinants of mercaptopurine disposition in children with acute lymphoblastic leukemia. Eur J Clin Pharmacol. 2012;68(9):1233–1242. doi:10.1007/s00228-012-1251-4
153. Yang SK, Hong M, Baek J, et al. A common missense variant in NUDT15 confers susceptibility to thiopurine-induced leukopenia. Nat Genet. 2014;46(9):1017–1020. doi:10.1038/ng.3060
154. Koutsilieri S, Caudle KE, Alzghari SK, Monte AA, Relling MV, Patrinos GP. Optimizing thiopurine dosing based on TPMT and NUDT15 genotypes: it takes two to tango. Am J Hematol. 2019;94(7):737–740. doi:10.1002/ajh.25485
155. Relling MV, Schwab M, Whirl-Carrillo M, et al. Clinical Pharmacogenetics Implementation Consortium Guideline for Thiopurine Dosing Based on TPMT and NUDT15 Genotypes: 2018 Update. Clin Pharmacol Ther. 2019;105(5):1095–1105. doi:10.1002/cpt.1304
156. Cargnin S, Genazzani AA, Canonico PL, Terrazzino S. Diagnostic accuracy of NUDT15 gene variants for thiopurine-induced leukopenia: a systematic review and meta-analysis. Pharmacol Res. 2018;135:102–111. doi:10.1016/j.phrs.2018.07.021
157. Moriyama T, Yang YL, Nishii R, et al. Novel variants in NUDT15 and thiopurine intolerance in children with acute lymphoblastic leukemia from diverse ancestry. Blood. 2017;130(10):1209–1212. doi:10.1182/blood-2017-05-782383
158. Yin D, Xia X, Zhang J, et al. Impact of NUDT15 polymorphisms on thiopurines-induced myelotoxicity and thiopurines tolerance dose. Oncotarget. 2017;8(8):13575–13585. doi:10.18632/oncotarget.14594
159. He HR, Chen SY, You HS, et al. Association between methylenetetrahydrofolate reductase polymorphisms and the relapse of acute lymphoblastic leukemia: a meta-analysis. Pharmacogenomics J. 2014;14(5):432–438. doi:10.1038/tpj.2014.10
160. Zhu C, Liu YW, Wang SZ, et al. Associations between the C677T and A1298C polymorphisms of MTHFR and the toxicity of methotrexate in childhood malignancies: a meta-analysis. Pharmacogenomics J. 2018;18(3):450–459. doi:10.1038/tpj.2017.34
161. Stock W, Diouf B, Crews KR, et al. An Inherited Genetic Variant in CEP72 Promoter Predisposes to Vincristine-Induced Peripheral Neuropathy in Adults With Acute Lymphoblastic Leukemia. Clin Pharmacol Ther. 2017;101(3):391–395. doi:10.1002/cpt.506
162. Gutierrez-Camino A, Martin-Guerrero I, Lopez-Lopez E, et al. Lack of association of the CEP72 rs924607 TT genotype with vincristine-related peripheral neuropathy during the early phase of pediatric acute lymphoblastic leukemia treatment in a Spanish population. Pharmacogenet Genomics. 2016;26(2):100–102. doi:10.1097/FPC.0000000000000191
163. Zgheib NK, Ghanem KM, Tamim H, et al. Genetic polymorphisms in candidate genes are not associated with increased vincristine-related peripheral neuropathy in Arab children treated for acute childhood leukemia: a single institution study. Pharmacogenet Genomics. 2018;28(8):189–195. doi:10.1097/FPC.0000000000000345
164. Fernandez CA, Smith C, Yang W, et al. HLA-DRB1*07:01 is associated with a higher risk of asparaginase allergies. Blood. 2014;124(8):1266–1276. doi:10.1182/blood-2014-03-563742
165. Abou Diwan E, Zeitoun RI, Abou Haidar L, Cascorbi I, Khoueiry Zgheib N. Implementation and obstacles of pharmacogenetics in clinical practice: an international survey. Br J Clin Pharmacol. 2019;85(9):2076–2088. doi:10.1111/bcp.13999
166. Roden DM, Van Driest SL, Mosley JD, et al. Benefit of Preemptive Pharmacogenetic Information on Clinical Outcome. Clin Pharmacol Ther. 2018;103(5):787–794. doi:10.1002/cpt.1035
167. Relling MV, Evans WE. Pharmacogenomics in the clinic. Nature. 2015;526(7573):343–350. doi:10.1038/nature15817
168. Hoffman JM, Haidar CE, Wilkinson MR, et al. PG4KDS: a model for the clinical implementation of pre-emptive pharmacogenetics. Am J Med Genet C Semin Med Genet. 2014;166C(1):45–55. doi:10.1002/ajmg.c.31391
169. van der Wouden CH, Cambon-Thomsen A, Cecchin E, et al. Implementing Pharmacogenomics in Europe: design and Implementation Strategy of the Ubiquitous Pharmacogenomics Consortium. Clin Pharmacol Ther. 2017;101(3):341–358. doi:10.1002/cpt.602
170. Volpi S, Bult CJ, Chisholm RL, et al. Research Directions in the Clinical Implementation of Pharmacogenomics: an Overview of US Programs and Projects. Clin Pharmacol Ther. 2018;103(5):778–786. doi:10.1002/cpt.1048
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.