Back to Journals » Drug Design, Development and Therapy » Volume 9
Impact of JAK2(V617F) mutation status on treatment response to anagrelide in essential thrombocythemia: an observational, hypothesis-generating study
Authors Cascavilla N, De Stefano V, Pane F, Pancrazzi A, Iurlo A, Gobbi M, Palandri F, Specchia G, Liberati AM, D'Adda M, Gaidano G, Fjerza R, Achenbach H, Smith J, Wilde P, Vannucchi AM ![]()
Received 19 December 2014
Accepted for publication 24 February 2015
Published 18 May 2015 Volume 2015:9 Pages 2687—2694
DOI https://doi.org/10.2147/DDDT.S79576
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Nicola Cascavilla,1 Valerio De Stefano,2 Fabrizio Pane,3 Alessandro Pancrazzi,4 Alessandra Iurlo,5 Marco Gobbi,6 Francesca Palandri,7 Giorgina Specchia,8 A Marina Liberati,9 Mariella D’Adda,10 Gianluca Gaidano,11 Rajmonda Fjerza,4 Heinrich Achenbach,12 Jonathan Smith,13 Paul Wilde,13 Alessandro M Vannucchi4
1Division of Hematology, Casa Sollievo della Sofferenza Hospital, IRCCS, San Giovanni Rotondo, Italy; 2Institute of Hematology, Catholic University, Rome, Italy; 3Department of Clinical Medicine and Surgery, University of Naples Federico II, Naples, Italy; 4Department of Experimental and Clinical Medicine, University of Florence, Florence, Italy; 5Oncohematology Unit, Fondazione IRCCS Ca’ Granda, Ospedale Maggiore Policlinico, Milan, Italy; 6IRCCS AOU San Martino, Genova, Italy; 7Department of Specialistic, Diagnostic and Experimental Medicine, St Orsola-Malpighi Hospital, University of Bologna, Bologna, Italy; 8Unit of Hematology with Transplantation, Department of Emergency and Organ Transplantation, University of Bari, Bari, Italy; 9Ospedale Santa Maria, Terni, Italy; 10Division of Hematology, Azienda Ospedaliera Spedali Civili di Brescia, Brescia, Italy; 11Division of Hematology, Department of Translational Medicine, Amedeo Avogadro University of Eastern Piedmont, Novara, Italy; 12Research and Development, Shire GmbH, Eysins, Switzerland; 13Shire Pharmaceutical Development Ltd, Basingstoke, United Kingdom
Abstract: A JAK2(V617F) mutation is found in approximately 55% of patients with essential thrombocythemia (ET), and represents a key World Health Organization diagnostic criterion. This hypothesis-generating study (NCT01352585) explored the impact of JAK2(V617F) mutation status on treatment response to anagrelide in patients with ET who were intolerant/refractory to their current cytoreductive therapy. The primary objective was to compare the proportion of JAK2-positive versus JAK2-negative patients who achieved at least a partial platelet response (≤600×109/L) after anagrelide therapy. Of the 47 patients enrolled, 46 were included in the full analysis set (JAK2-positive, n=22; JAK2-negative, n=24). At 12 months, 35 patients (n=14 and n=21, respectively) had a suitable platelet sample; of these, 74.3% (n=26) achieved at least a partial response. The response rate was higher in JAK2-positive (85.7%, n=12) versus JAK2-negative patients (66.7%, n=14) (odds ratio [OR] 3.00; 95% confidence interval [CI] 0.44, 33.97). By using the last observation carried forward approach in the sensitivity analysis, which considered the imbalance in patients with suitable samples between groups, the overall response rate was 71.7% (n=33/46), with 77.3% (n=17/22) of JAK2-positive and 66.7% (n=16/24) of JAK2-negative patients achieving at least a partial response (OR 1.70; 95% CI 0.39, 8.02). There was no significant change in median allele burden over 12 months in the 12 patients who achieved a response. In conclusion, the overall platelet response rate was high in both JAK2-positive and JAK2-negative patients; however, a larger study would be required to confirm the differences observed according to JAK2(V617F) mutation status.
Keywords: essential thrombocythemia, mutation, JAK2, anagrelide, treatment response, allele burden
Introduction
Essential thrombocythemia (ET) is a clonal myeloproliferative neoplasm (MPN) characterized by an overproduction of platelets and an increased risk of thrombohemorrhagic complications.1,2 A JAK2(V617F) gain-of-function mutation is found in approximately 55% of patients with ET.3–11 JAK2 encodes a cytoplasmic tyrosine kinase involved in normal hematopoiesis. Available data show the JAK2 mutation to be associated with an increased risk of arterial thrombosis in patients with ET.12,13 The risk of evolution to myelofibrosis may also be increased by the JAK2 mutation and appears to vary according to allele burden,9,13 although conflicting data are available.4 In 2008, the World Health Organization (WHO) identified the JAK2 mutation as a key diagnostic criterion for Philadelphia-negative MPNs.14 The JAK2 mutation does not differentiate between ET and other clonal MPNs (such as polycythemia vera and primary myelofibrosis), but is a molecular marker that distinguishes clonal MPNs from reactive thrombocytosis.14,15
Somatic mutations in the thrombopoietin receptor (MPL) and calreticulin (CALR) genes have also been reported in patients with ET. As with JAK2(V617F), the MPL mutation appears to have a phenotype-modifying effect in ET; however, this mutation is infrequent, occurring in only around 3% of patients.10,16,17 Most patients with ET who do not harbor a JAK2 or MPL alteration carry a CALR mutation,18 with an overall CALR mutational frequency in patients with ET up to 32% as reported in one recent study.10 The clinical course of ET in patients with mutated CALR appears more indolent than that in patients with mutated JAK2.19–21
Anagrelide (Xagrid®; 0.5 mg hard capsules, Shire Pharmaceutical Contracts Limited, Basingstoke, United Kingdom) is an orally active, quinazolone-derived platelet-lowering agent indicated for second-line treatment of high-risk patients with ET in Europe.15,22,23 Clinical studies in more than 4,000 patients have confirmed the safety and efficacy of anagrelide as a platelet-lowering agent in ET.22,24–26
Results from a previous study have suggested that anagrelide is similarly effective in controlling platelet levels in patients with ET irrespective of JAK2 mutation status.27 However, JAK2-positive patients have been reported to be more sensitive than JAK2-negative patients to hydroxycarbamide, as demonstrated by lower platelet counts at lower hydroxycarbamide dosages.27 This hypothesis-generating study was undertaken to further explore the potential impact of JAK2(V617F) mutation status and allele burden on the response to anagrelide in patients with ET.
Materials and methods
Study design
This was an exploratory, observational, multicenter study (clinicaltrials.gov registration: NCT01352585) conducted across eleven centers in Italy from July 2011 to September 2013. Anagrelide 0.5 mg was administered at doses determined by the treating physician and in accordance with the European Union Summary of Product Characteristics (SPC).22 Anti-aggregatory therapy was permitted at the discretion of the investigator. All evaluations were undertaken in accordance with routine clinical practice. No visits were imposed by the study outside of regularly scheduled visits for treatment purposes. Patients who discontinued anagrelide due to an adverse drug reaction (ADR) were followed throughout the study. All other patients who discontinued were followed according to local clinical practice. Patients were withdrawn from the study if they switched from anagrelide to another ET therapy or combination therapy.
Patients
Patients were eligible for enrollment if they had a confirmed diagnosis of ET, according to WHO 2008 diagnostic criteria,14 and were intolerant or refractory to their first-line or previous cytoreductive therapy due to the lack of efficacy or intolerance. Patients had either started anagrelide treatment in the 7 days prior to study entry or a decision had been documented to commence anagrelide. Patients were excluded from the study if they had a known or suspected intolerance to anagrelide or any of the stated ingredients, or closely related compounds. Other exclusion criteria were contraindications to anagrelide as listed in the anagrelide SPC,22 combination therapy with other cytoreductive agents, or participation in an interventional research study. Written informed consent was obtained from all patients prior to study entry. This observational study was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice (GCP) standards, and local ethical and legal requirements.
Assessments
Patient data were collected from medical records following routine visits at baseline and at 6±2 and 12±3 months after the initiation of anagrelide using electronic case report forms. As part of routine clinical practice, patients provided a 20 mL blood sample for hematological assessment at all visits. JAK2 mutation status was measured in all patients at baseline. Allele burden was tested at baseline and at 6 and 12 months in JAK2-positive patients; allele burden was measured in both granulocyte DNA and platelet RNA. JAK2 mutation status and allele burden were measured at a centralized laboratory (University of Florence, Italy) in order to standardize the assessments. JAK2 mutation status was determined by real-time polymerase chain reaction, and JAK2(V617F) allele burden was quantified using the ΔCt method as previously described.8,28 The safety of anagrelide was assessed by monitoring the incidence and severity of ADRs and changes in routine hematology parameters.
Statistical analysis
The primary objective of this study was to compare the proportion of JAK2-positive versus JAK2-negative patients with ET who achieved at least a partial platelet response (≤600×109/L) after anagrelide therapy. Secondary objectives were to compare the proportion of JAK2-positive versus JAK2-negative patients with ET who achieved a complete platelet response (≤400×109/L) after anagrelide therapy, to evaluate the relationship between platelet response and allele burden in the JAK2-positive group, and to observe the effect of anagrelide on routine hematological parameters.
The study was not statistically powered and was exploratory in nature. The target sample size was approximately 60 patients; enrollment was monitored to ensure that a minimum of ten patients were recruited into both the JAK2-positive and JAK2-negative groups. Efficacy was analyzed in all patients who received at least one dose of anagrelide and had at least one post-baseline platelet count and known JAK2 mutation status (full analysis set). Safety was analyzed in all enrolled patients who received at least one dose of anagrelide (safety set).
For the primary endpoint and other treatment response endpoints, the odds ratio (OR) and 95% confidence interval (CI) were calculated for the difference in the proportion of patients achieving a response between the JAK2-positive and JAK2-negative groups. Patients without an available platelet sample within the specified time window were excluded from the analysis. The efficacy results from the last observation carried forward (LOCF) sensitivity analysis are also reported here as this approach considers the imbalance in patients with suitable platelet samples between the JAK2-positive and JAK2-negative groups. For JAK2-positive patients, platelet counts were cross-tabulated with the allele burden at 6 and 12 months; separate summaries were produced for the allele burden in granulocyte DNA and platelet RNA. ADRs were recorded using the Medical Dictionary for Regulatory Activities (MedDRA), version 15.1 or newer.
Role of the funding source
The study was funded by Shire Pharmaceutical Development Ltd. iMed Comms was funded by Shire for support in writing and editing this manuscript.
Results
Patients
Forty-seven patients were enrolled and received at least one dose of anagrelide. Of these, 23 (48.9%) were JAK2-positive and 24 (51.1%) were JAK2-negative. Forty-six patients had at least one post-baseline platelet count and known JAK2 status (22 [47.8%] JAK2-positive and 24 [52.2%] JAK2-negative) and were included in the analysis of efficacy. Fifteen patients in the JAK2-positive group and 21 patients in the JAK2-negative group completed the study. The most common reason for withdrawal was an adverse event (AE) not related to anagrelide (n=6, 12.8%; JAK2-positive, n=4 [including three due to transformation to myelofibrosis]; JAK2-negative, n=2). Three patients (6.4%; JAK2-positive, n=2; JAK2-negative, n=1) withdrew from the study due to an ADR.
Patient demographics and baseline characteristics were generally well balanced between the JAK2-positive and JAK2-negative groups (Table 1), except for median (range) time since ET diagnosis (8.2 years [0–22] versus 5 years [0–16]) and median (range) platelet count (584.0×109/L [310–1,158] versus 752.5×109/L [323–2,126]), respectively. Mean (standard deviation) age was 58.0 (14.87) years, 59.6% of patients were female, and most were Caucasian (97.9%).
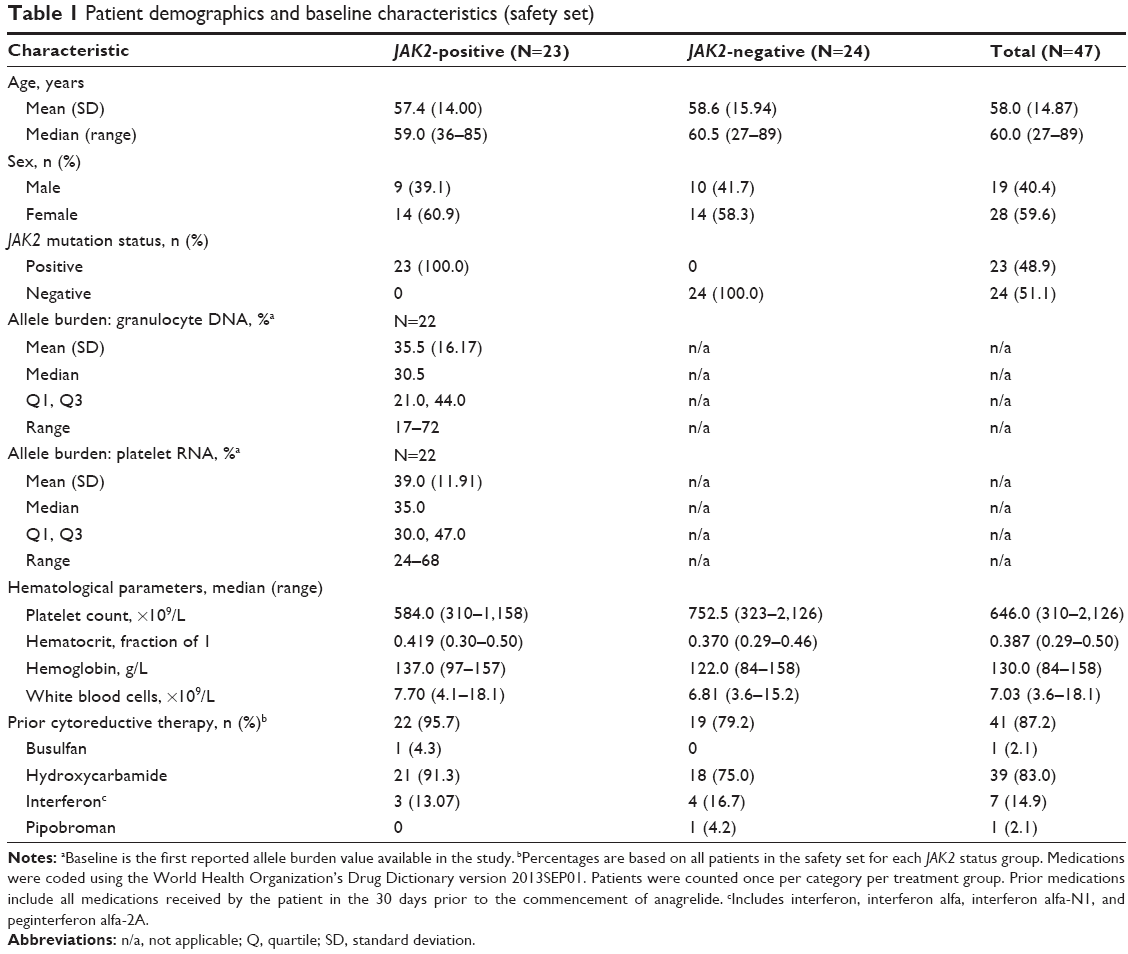 | Table 1 Patient demographics and baseline characteristics (safety set) |
At baseline, most JAK2-positive patients had a low (<50%) granulocyte DNA and platelet RNA allele burden (each n=18 [78.3%]). The distribution of allele burden differed between granulocyte DNA and platelet RNA. Approximately half of JAK2-positive patients (n=11 [47.8%]) had a granulocyte DNA allele burden of 25% to <50%, with less than a third (n=7 [30.4%]) having a granulocyte DNA allele burden <25%. In contrast, the majority of JAK2-positive patients (n=17 [73.9%]) had a platelet RNA allele burden of 25% to <50%, and only one (4.3%) JAK2-positive patient had a platelet RNA allele burden <25%.
Treatment response
At 12 months in the safety analysis set, the median (range) platelet count was 416.6×109/L (235–854) in the JAK2-positive group and 425.0×109/L (165–1,279) in the JAK2-negative group. At 12 months, 35 patients (14 patients in the JAK2-positive group and 21 patients in the JAK2-negative group) had an available platelet sample for evaluation. Of these 35 patients, 26 (74.3%) achieved at least a partial platelet response (≤600×109/L) (95% CI 57.9, 85.8). The proportion of patients who achieved at least a partial response was higher in the JAK2-positive group (n=12 [85.7%]; 95% CI 60.1, 96.0) than in the JAK2-negative group (n=14 [66.7%]; 95% CI 45.4, 82.8). The odds of patients in the JAK2-positive group achieving at least a partial platelet response were 3.00 times higher than in the JAK2-negative group, although this difference was not statistically significant (95% CI 0.44, 33.97) (Figure 1A).
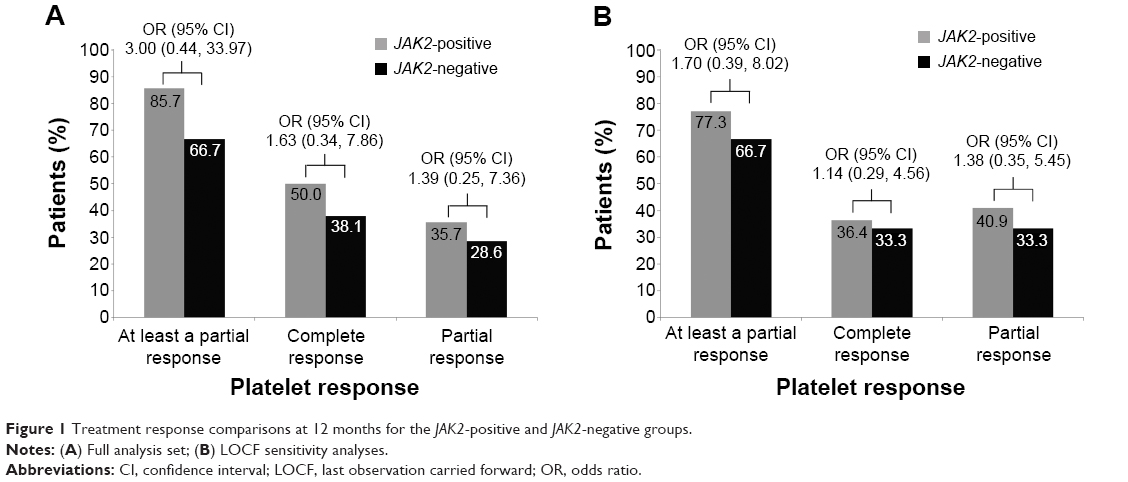 | Figure 1 Treatment response comparisons at 12 months for the JAK2-positive and JAK2-negative groups. |
By using the LOCF approach in the sensitivity analysis, 33 (71.7%) patients achieved at least a partial response (95% CI 57.5, 82.7). The response rate in the JAK2-positive group was more similar to the JAK2-negative group than in the main analysis. The proportion of patients with at least a partial response was higher in the JAK2-positive group (n=17 [77.3%]; 95% CI 56.6, 89.9) than in the JAK2-negative group (n=16 [66.7%]; 95% CI 46.7, 82.0). The odds of patients in the JAK2-positive group achieving at least a partial response in the LOCF sensitivity analysis were 1.7 times higher than in the JAK2-negative group (Figure 1B). However, this difference was not statistically significant (95% CI 0.39, 8.02).
At 12 months, 15 patients (42.9%) had achieved a complete platelet response (≤400×109/L) (95% CI 28.0, 59.1). A higher proportion of JAK2-positive patients (n=7 [50.0%]; 95% CI 26.8, 73.2) achieved a complete platelet response than JAK2-negative patients (n=8 [38.1%]; 95% CI 20.8, 59.1). The odds of patients in the JAK2-positive group achieving a complete platelet response were 1.63 times higher than in the JAK2-negative group, although this difference was not statistically significant (95% CI 0.34, 7.86) (Figure 1A).
In the sensitivity analysis using the LOCF approach, 16 (34.8%) patients achieved a complete platelet response at 12 months (95% CI 0.29, 4.56). The proportion of patients who achieved a complete response was higher in the JAK2-positive group (n=8 [36.4%]; 95% CI 19.7, 57.0) than in the JAK2-negative group (n=8 [33.3%]; 95% CI 18.0, 53.3). The odds of patients in the JAK2-positive group achieving a complete platelet response in the LOCF sensitivity analysis were 1.14 times higher than in the JAK2-negative group, although this difference was not statistically significant (95% CI 0.29, 4.56) (Figure 1B).
Platelet responses to anagrelide treatment were also observed at 6 months, although the difference in platelet responses between the JAK2-positive and JAK2-negative groups at this time was not as pronounced as that observed at 12 months (Figure 2).
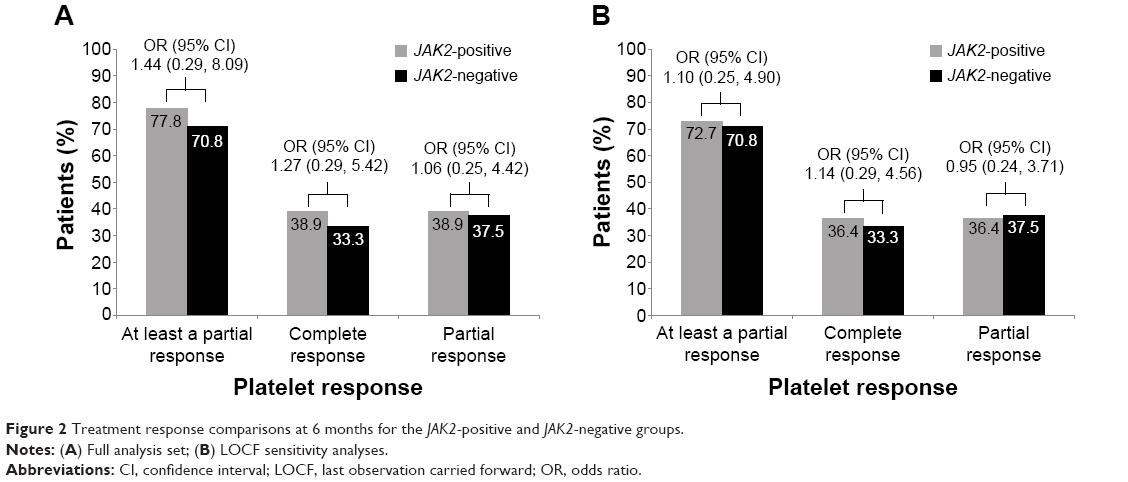 | Figure 2 Treatment response comparisons at 6 months for the JAK2-positive and JAK2-negative groups. |
Allele burden
In JAK2-positive patients, no clear relationship was identified between platelet response and baseline allele burden. The median allele burden for the 12 patients who achieved at least a partial response at 12 months and had a suitable platelet sample was 41.0% (range: 17–47) for granulocyte DNA and 49.5% (range: 31–58) for platelet RNA. There was no clinically relevant change in allele burden from baseline over the 12 months of treatment with anagrelide (data not shown).
Exposure and safety
The mean (standard deviation) daily dose of anagrelide was 1.40 (0.482) mg/day overall, 1.32 (0.378) mg/day in the JAK2-positive group, and 1.47 (0.563) mg/day in the JAK2-negative group. Total exposure was similar in JAK2-positive (19.6 person-years) and JAK2-negative patients (22.8 person-years).
A total of 15 (31.9%) patients experienced 20 ADRs (Table 2). Fewer patients in the JAK2-positive group had an ADR than in the JAK2-negative group (26.1% versus 37.5%). The most common ADR was headache (12.8%). All of the ADRs reported were of mild or moderate severity. None of the ADRs were fatal, serious, or severe and no deaths occurred during the study. Three patients were withdrawn due to an ADR (dermatitis and pruritus in the JAK2-positive group and headache in the JAK2-negative group). There were no notable changes from baseline to 12 months in any hematological parameters other than platelets.
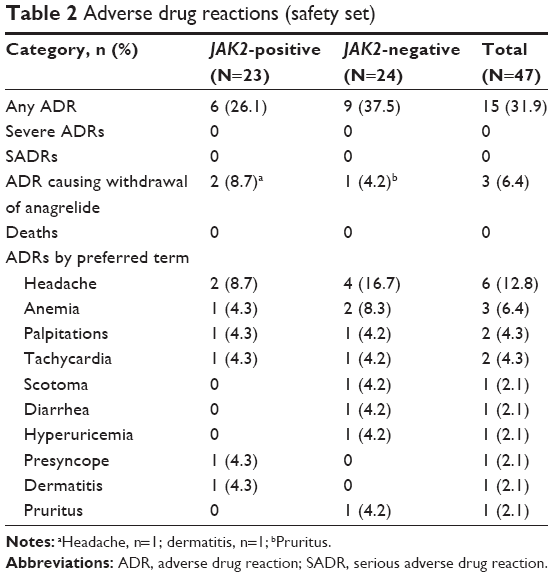 | Table 2 Adverse drug reactions (safety set) |
Discussion
In this hypothesis-generating study, the overall platelet response rate to anagrelide in patients with ET was found to be high irrespective of JAK2 mutation status. Our findings are in line with the efficacy of anagrelide observed in other studies in patients with ET.22,24–26 Previous studies have shown that responses to anagrelide are unaffected by JAK2 mutation status.27,29 In the current study, we found the likelihood of having at least a partial response to anagrelide to be higher in JAK2-positive patients than in those who were JAK2-negative. However, the sensitivity analyses using LOCF showed more comparable treatment response findings between the JAK2-positive and JAK2-negative groups and lower ORs, indicating a similar sensitivity to anagrelide independent of JAK2 mutation status.
No clear relationship or clinically relevant difference was identified between JAK2 allele burden and platelet treatment response in this study, and there was no significant change in allele burden over the 12 months of anagrelide therapy. The majority of JAK2-positive patients had an allele burden of ≥25%–50%, regardless of whether this was measured using granulocyte DNA or platelet RNA methodology. JAK2 allele burden appears to play a role in clinical phenotype and disease evolution in ET.9,13
Anagrelide was found to be well tolerated in this study, with a safety profile consistent with the SPC22 and current literature.26,30,31 The frequency of ADRs was lower than that reported in the current literature, but there is no obvious explanation for this finding. The most commonly reported ADR was headache. All of the ADRs reported were of mild or moderate severity, no severe or serious ADRs occurred, and few patients were withdrawn due to an ADR. Three patients were withdrawn due to transformation to myelofibrosis, but these cases were not considered to be related to anagrelide because of the short time interval between anagrelide initiation and myelofibrosis diagnosis. No thrombotic or hemorrhagic complications were reported over the duration of this study. Furthermore, no differences in ADRs were evident between the JAK2-positive and JAK2-negative groups.
It should be noted that results from this study were not statistically significant (CIs were wide) and data should be interpreted with caution due to the small study size. In addition, the imbalance of patients who remained in the study at 12 months between groups could have added potential bias to the results. This is supported by the sensitivity analyses using LOCF, which showed more comparable treatment response findings between the two groups.
Conclusion
In conclusion, the overall platelet response rate was high in both JAK2-positive and JAK2-negative patients with ET in this study. The odds of achieving at least a partial response were found to be slightly higher if patients had a JAK2(V617F) mutation than if they did not. However, no firm conclusions can be made and a larger well-controlled study would be needed to confirm the findings of this hypothesis-generating study.
Acknowledgments
The authors thank the patients and acknowledge the contribution of all investigators who participated in this study. Jaideep Purkayastha, a Shire employee, provided statistical support to the manuscript and contributed to the data check. Under the direction of the authors, Emma Burke, employee of iMed Comms, provided writing assistance for this publication. Editorial assistance in formatting, proofreading, copy-editing, and fact checking was also provided by iMed Comms. iMed Comms was funded by Shire for support in writing and editing this manuscript.
Disclosure
The University of Florence (AMV) received funding from Shire for the conduct of this study. The Catholic University, Rome (VDS) received research funding outside of this study from Shire. VDS, AI, and AMV received speaker funding outside of this study from Shire. The Amedeo Avogadro University of Eastern Piedmont, Novara (GG) received funding from Pfizer, Amgen, and Celgene outside of this study. GG also received personal fees from Roche, Onyx, Novartis, Amgen, Celgene, and GlaxoSmithKline outside of this study. HA is a Shire employee. PW is a former employee and JS is a former contractor of Shire. NC, F Pane, AP, MG, F Palandri, GS, AML, MDA, and RF declare that they have no conflicts of interest.
References
Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haemopoietic and Lymphoid Tissues. 4th ed. Lyon: IARC Press; 2008. | ||
Tefferi A. Polycythemia vera and essential thrombocythemia: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol. 2013;88:507–516. | ||
Baxter EJ, Scott LM, Campbell PJ, et al; Cancer Genome Project. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. | ||
Barbui T, Thiele J, Passamonti F, et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: an international study. J Clin Oncol. 2011;29:3179–3184. | ||
Jones AV, Kreil S, Zoi K, et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood. 2005;106:2162–2168. | ||
Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. | ||
Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. | ||
Lippert E, Boissinot M, Kralovics R, et al. The JAK2-V617F mutation is frequently present at diagnosis in patients with essential thrombocythemia and polycythemia vera. Blood. 2006;108:1865–1867. | ||
Passamonti F, Rumi E. Clinical relevance of JAK2 (V617F) mutant allele burden. Haematologica. 2009;94:7–10. | ||
Tefferi A, Wassie EA, Lasho TL, et al. Calreticulin mutations and long-term survival in essential thrombocythemia. Leukemia. 2014;28(12):2300–2303. | ||
Vizmanos JL, Ormazabal C, Larrayoz MJ, Cross NC, Calasanz MJ.JAK2 V617F mutation in classic chronic myeloproliferative diseases: a report on a series of 349 patients. Leukemia. 2006;20:534–535. | ||
Carobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood. 2011;117:5857–5859. | ||
Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. 2007;110:840–846. | ||
Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937–951. | ||
Barbui T, Barosi G, Birgegard G, et al; European LeukemiaNet. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. 2011;29:761–770. | ||
Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1,182 patients. Blood. 2006;108:3472–3476. | ||
Vannucchi AM, Antonioli E, Guglielmelli P, et al. Characteristics and clinical correlates of MPL 515W>L/K mutation in essential thrombocythemia. Blood. 2008;112:844–847. | ||
Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369:2391–2405. | ||
Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369:2379–2390. | ||
Rumi E, Harutyunyan AS, Pietra D, et al; Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative Investigators. CALR exon 9 mutations are somatically acquired events in familial cases of essential thrombocythemia or primary myelofibrosis. Blood. 2014;123:2416–2419. | ||
Rotunno G, Mannarelli C, Guglielmelli P, et al; Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative Investigators. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood. 2014;123:1552–1555. | ||
European Medicines Agency. Xagrid Summary of Product Characteristics [Internet]. Shire Pharmaceuticals Ltd; 2014 [updated 2014]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000480/WC500056557.pdf. Accessed February 5, 2014. | ||
Barbui T, Barosi G, Grossi A, et al. Practice guidelines for the therapy of essential thrombocythemia. A statement from the Italian Society of Hematology, the Italian Society of Experimental Hematology and the Italian Group for Bone Marrow Transplantation. Haematologica. 2004;89:215–232. | ||
Fruchtman SM, Petitt RM, Gilbert HS, Fiddler G, Lyne A. Anagrelide: analysis of long-term efficacy, safety and leukemogenic potential in myeloproliferative disorders. Leuk Res. 2005;29:481–491. | ||
Gisslinger H, Gotic M, Holowiecki J, et al; ANAHYDRET Study Group. Anagrelide compared to hydroxyurea in WHO-classified essential thrombocythemia: the ANAHYDRET Study, a randomized controlled trial. Blood. 2013;121:1720–1728. | ||
Harrison CN, Campbell PJ, Buck G, et al; United Kingdom Medical Research Council Primary Thrombocythemia 1 Study. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med. 2005;353:33–45. | ||
Campbell PJ, Scott LM, Buck G, et al; United Kingdom Myeloproliferative Disorders Study Group; Medical Research Council Adult Leukaemia Working Party; Australasian Leukaemia and Lymphoma Group. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet. 2005;366:1945–1953. | ||
Jovanovic JV, Ivey A, Vannucchi AM, et al. Establishing optimal quantitative-polymerase chain reaction assays for routine diagnosis and tracking of minimal residual disease in JAK2-V617F-associated myeloproliferative neoplasms: a joint European LeukemiaNet/MPN&MPNr-EuroNet (COST action BM0902) study. Leukemia. 2013;27:2032–2039. | ||
Cacciola E, Di Francesco E, Pezzella F, Tibullo D, Cacciola R. Effect of anagrelide on JAK2 mutational status in patients with essential thrombocythemia. Clin Leukemia. 2008;2:272–274. | ||
Birgegård G, Björkholm M, Kutti J, et al. Adverse effects and benefits of two years of anagrelide treatment for thrombocythemia in chronic myeloproliferative disorders. Haematologica. 2004;89:520–527. | ||
Okamoto S, Miyakawa Y, Smith J, et al. Open-label, dose-titration and continuation study to assess efficacy, safety, and pharmacokinetics of anagrelide in treatment-naive Japanese patients with essential thrombocythemia. Int J Hematol. 2013;97:360–368. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.