Back to Journals » Drug Design, Development and Therapy » Volume 9
Impact of enzyme replacement therapy and hematopoietic stem cell transplantation in patients with Morquio A syndrome
Authors Tomatsu S, Sawamoto K, Alméciga-Díaz CJ, Shimada T, Bober M, Chinen Y, Yabe H, Montano A, Giugliani R ![]() , Kubaski F, Yasuda E, Rodríguez-López A, Espejo-Mojica A, Sanchez O, Mason R, Barrera L, Mackenzie W, Orii T
, Kubaski F, Yasuda E, Rodríguez-López A, Espejo-Mojica A, Sanchez O, Mason R, Barrera L, Mackenzie W, Orii T
Received 1 December 2014
Accepted for publication 12 January 2015
Published 1 April 2015 Volume 2015:9 Pages 1937—1953
DOI https://doi.org/10.2147/DDDT.S68562
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Supplementary video showing activities of daily living in a 25-year-old patient with MPS IVA 10 years post-HSCT and successive osteotomies. The patient was completely wheelchair bound prior to HSCT.
Views: 668
Shunji Tomatsu,1,2 Kazuki Sawamoto,1 Carlos J Alméciga-Díaz,3 Tsutomu Shimada,1 Michael B Bober,1 Yasutsugu Chinen,4 Hiromasa Yabe,5 Adriana M Montaño,6 Roberto Giugliani,7 Francyne Kubaski,1,8 Eriko Yasuda,1 Alexander Rodríguez-López,3 Angela J Espejo-Mojica,3 Oscar F Sánchez,9 Robert W Mason,1 Luis A Barrera,3 William G Mackenzie,1 Tadao Orii2
1Nemours/Alfred I duPont Hospital for Children, Wilmington, DE, USA; 2Department of Pediatrics, Gifu University, Gifu, Japan; 3Institute for the Study of Inborn Errors of Metabolism, School of Sciences, Pontificia Universidad Javeriana, Bogotá, Colombia; 4Department of Pediatrics, Faculty of Medicine, University of the Ryukyus, Okinawa, 5Department of Cell Transplantation and Regenerative Medicine, Tokai University School of Medicine, Isehara, Japan; 6Department of Pediatrics, Saint Louis University, St Louis, MO, USA; 7Medical Genetics Service/HCPA and Department of Genetics/UFRGS, Porto Alegre, Brazil; 8Department of Biological Sciences, University of Delaware, Newark, DE, 9School of Chemical Engineering, Purdue University, West Lafayette, IN, USA
Abstract: Patients with mucopolysaccharidosis IVA (MPS IVA) can present with systemic skeletal dysplasia, leading to a need for multiple orthopedic surgical procedures, and often become wheelchair bound in their teenage years. Studies on patients with MPS IVA treated by enzyme replacement therapy (ERT) showed a sharp reduction on urinary keratan sulfate, but only modest improvement based on a 6-minute walk test and no significant improvement on a 3-minute climb-up test and lung function test compared with the placebo group, at least in the short-term. Surgical remnants from ERT-treated patients did not show reduction of storage materials in chondrocytes. The impact of ERT on bone lesions in patients with MPS IVA remains limited. ERT seems to be enhanced in a mouse model of MPS IVA by a novel form of the enzyme tagged with a bone-targeting moiety. The tagged enzyme remained in the circulation much longer than untagged native enzyme and was delivered to and retained in bone. Three-month-old MPS IVA mice treated with 23 weekly infusions of tagged enzyme showed marked clearance of the storage materials in bone, bone marrow, and heart valves. When treatment was initiated at birth, reduction of storage materials in tissues was even greater. These findings indicate that specific targeting of the enzyme to bone at an early stage may improve efficacy of ERT for MPS IVA. Recombinant N-acetylgalactosamine-6-sulfate sulfatase (GALNS) in Escherichia coli BL21 (DE3) (erGALNS) and in the methylotrophic yeast Pichia pastoris (prGALNS) has been produced as an alternative to the conventional production in Chinese hamster ovary cells. Recombinant GALNS produced in microorganisms may help to reduce the high cost of ERT and the introduction of modifications to enhance targeting. Although only a limited number of patients with MPS IVA have been treated with hematopoietic stem cell transplantation (HSCT), beneficial effects have been reported. A wheelchair-bound patient with a severe form of MPS IVA was treated with HSCT at 15 years of age and followed up for 10 years. Radiographs showed that the figures of major and minor trochanter appeared. Loud snoring and apnea disappeared. In all, 1 year after bone marrow transplantation, bone mineral density at L2–L4 was increased from 0.372 g/cm2 to 0.548 g/cm2 and was maintained at a level of 0.48±0.054 for the following 9 years. Pulmonary vital capacity increased approximately 20% from a baseline of 1.08 L to around 1.31 L over the first 2 years and was maintained thereafter. Activity of daily living was improved similar to the normal control group. After bilateral osteotomies, a patient can walk over 400 m using hip–knee–ankle–foot orthoses. This long-term observation of a patient shows that this treatment can produce clinical improvements although bone deformity remained unchanged. In conclusion, ERT is a therapeutic option for MPS IVA patients, and there are some indications that HSCT may be an alternative to treat this disease. However, as neither seems to be a curative therapy, at least for the skeletal dysplasia in MPS IVA patients, new approaches are investigated to enhance efficacy and reduce costs to benefit MPS IVA patients.
Keywords: mucopolysaccharidosis IVA, ERT, HSCT, skeletal dysplasia, keratan sulfate
Introduction
Mucopolysaccharidosis type IVA (MPS IVA; Morquio A syndrome) is an autosomal recessive lysosomal storage disorder caused by deficiency of N-acetylgalactosamine-6-sulfate sulfatase (GALNS). Deficiency of the enzyme results in a progressive accumulation of the glycosaminoglycans (GAGs): chondroitin 6-sulfate (C6S) and keratan sulfate (KS). C6S and KS are produced mainly in cartilage, and successively, these GAGs accumulate primarily in lysosomes of chondrocytes, associated ligaments, and the neighboring extracellular matrix (ECM).1–4 Accumulation of C6S and KS causes systemic skeletal dysplasia, such as short stature, cervical instability with subsequent cord compression, pectus carinatum, kyphoscoliosis, genu valgum, hypermobile joints (hands), and an abnormal gait.5–8
Patients with MPS IVA are usually asymptomatic at birth. Main signs and symptoms in most patients are observed before/around their first birthday, including kyphosis and pectus carinatum. Patients with MPS IVA are usually evaluated and diagnosed during the second year of life for unique skeletal features, including genu valgum (knock-knee), poor growth, hypermobile joints, abnormal gait with a tendency to fall, kyphosis, and pectus carinatum. Patients with MPS IVA are distinguished from patients with other MPS by preservation of intelligence and unique hypermobile joints. Odontoid hypoplasia in combination with ligamentous laxity and extradural GAGs deposition results in atlantoaxial subluxation and cervical stenosis with cord compression, cervical myelopathy, or even death.4,9,10
Other potential complications include pulmonary compromise, muscle weakness, valvular heart disease, hearing loss, corneal clouding, and widely spaced teeth with abnormally thin enamel.4,9,10 Patients with a severe phenotype often do not survive beyond the second or third decade of life because of cervical instability, pulmonary compromise, and heart valvular disease. Most patients with MPS IVA have difficulty with anesthesia because of the narrow airway and a small, restrictive lung. Difficulty of administration in both upper and lower airways increases with the risk of anesthesia.4,9–11
A wide spectrum of disease progression among individuals with MPS IVA is observed. Less severe forms of MPS IVA have been known as mild12,13 or attenuated phenotypes. An intermediate subtype of MPS IVA has been defined.12–15 The onset of disease symptoms occurs as late as the second decade of life, in less severely affected (mild) patients.9
A recent autopsied case revealed the accumulation of foam cells and macrophages in multiple tissues, including bones, cartilages, ligaments, heart valves, aorta, lung, liver, and kidney. These findings indicate that chronic inflammation leads to worsening clinical features in patients with MPS IVA.16 Accumulation of KS in multiple tissues appears to be the primary contributing factor to the clinical phenotype of MPS IVA. However, it is noteworthy that C6S accumulates in aorta and heart valves and is elevated in blood,16,17 which means that both KS and C6S can play a significant role in the clinical manifestations of MPS IVA.
Therapies for MPS have been established and expanded clinically. These comprise enzyme replacement therapy (ERT), gene therapy, hematopoietic stem cell transplantation (HSCT), and substrate reduction therapy, although none of them totally cure the disorders. ERT is approved in many countries around the world for use in patients with MPS I,7 MPS II,8,18 MPS IVA,11,19 and MPS VI.20–23 Patients treated with ERT showed clinical improvement of somatic manifestations and improved quality of life. Conventional ERT for MPS IVA was approved in Europe and USA, following results of a clinical trial that showed an improvement in a 6-minute walk test (6MWT) and a reduction of urinary KS.19,24
However, there are several limitations with conventional ERT for MPS IVA: 1) it has a limited effect on skeletal symptoms;25,26 2) the enzyme has a short half-life and is rapidly cleared from the circulation; 3) there are immunological issues; and 4) it is very expensive (estimated cost in USA: the enzyme is priced at $1,068 per vial, which for a typical 22.5 kg patient requiring nine vials per weekly infusion equates to approximately $380,000 per patient per year) (Table 1). These limitations are also observed for ERT for other forms of MPS.7,27–29 To address the above issues, an improved long circulating ERT and a bone-targeting enzyme are proposed,30,31 as well as the use of microorganism for the production of recombinant GALNS,32,33 which could potentially help to lower the cost.
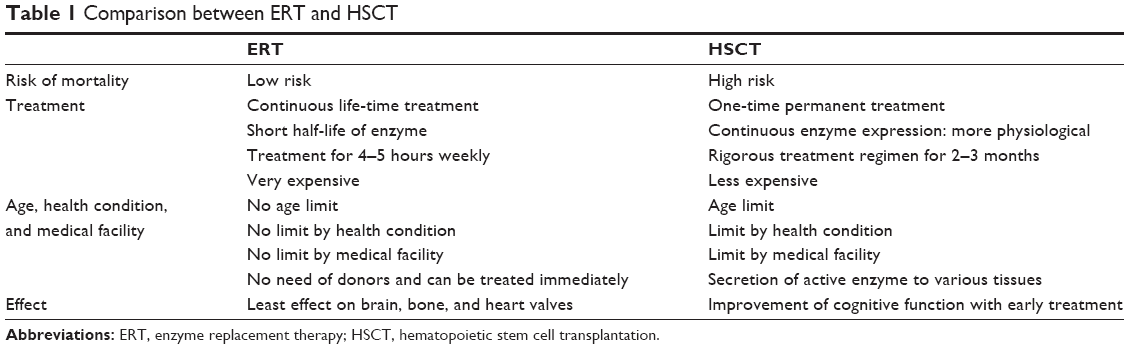 | Table 1 Comparison between ERT and HSCT |
HSCT for MPS shows benefits in physical activity and bone mineral density (BMD) of treated mice, and early interventions provide further benefits.34–36 ERT and HSCT provide a compatible impact on growth in patients with MPS II.37 HSCT in patients with MPS II improves more activities of daily living (ADL) than conventional ERT, but therapeutic effect remains limited in bone lesions.38
Thus, therapies to fix bone lesions remain an unmet challenge. Specific delivery of drugs to avascular cartilage tissue has not been attained yet, although several promising results with targeting drugs to bone have been demonstrated to be effective experimentally and clinically.30,31,39–46
In this review, we describe current outcomes of ERT and HSCT for MPS IVA and their expectations, limitations, and future prospects. These treatments may provide a better quality of life by ameliorating the underlying disease progression of MPS IVA, leading to prevention of further damage of targeted organs and reduction in risks associated with additional surgical procedures. While current treatments have limited beneficial effects on bone pathology of MPS IVA, new approaches to address limitations of current therapies are emerging and discussed in this review.
Pathology in bone
Skeletal development of MPS patients is similar to unaffected babies at birth, although some already have a mild sign of skeletal dysplasia, such as gibbus or pectus carinatum.47,48 However, accumulation of GAGs in articular and epiphyseal cartilage can be observed in the fetus or at birth. Most skeletal manifestations are progressive and irreversible unless treated before signs and symptoms appear. Therefore, the first months of life represent the best window of possibility for preventing bone deformities in patients with MPS. Development of newborn screening programs for patients with MPSs may help identify patients so that they can receive therapy in the first week of their lives.49–52
Evidence that lysosomal GAGs accumulate in chondrocytes before birth was reported in mouse models and in the tissue obtained from the postmortem of a human fetus affected by MPS IVA. An MPS IVA-affected fetus showed storage vacuoles in chondrocytes at 18–20 weeks gestation.53 Initial clinical signs and symptoms for skeletal dysplasia in newborn patients with MPS IVA are seen as sacral dimple, gibbus, and abnormal shape of vertebrae.54 In newborn mice with IVA, vacuolated chondrocytes were observed.55 Thus, substantial storage materials have already been accumulated in chondrocytes during the prenatal period in both animal models and patients affected with MPS IVA.56
Bone formation (endochondral ossification) occurs by three coordinated processes: the production of osteoid matrix, its maturation, and its subsequent mineralization.39 To initiate mineralization in growth plate cartilage, high local concentrations of Ca2+ and PO43− ions are required to induce their precipitation into amorphous calcium phosphate, leading to hydroxyapatite (HA) crystal formation: Ca10(PO4)6(OH)2. HA is a major inorganic component in bone that binds tightly to proteins via the calcium sites.57 In MPS IVA, this process is hampered due to storage of C6S and KS in the cartilage tissue, leading to disruption of bone mineralization. Since bone is remodeled by resorption and formation throughout life,57 drugs that bind to HA should be released in the bone resorption process. Targeting of estradiol to HA has been shown to specifically improve bone pathology in ovariectomized rats,39 and consequently, HA targeting of enzymes is a potential strategy for selective delivery to cartilage.
Enzyme replacement therapy
Conventional ERT
Background
ERT is known as one of the most important therapies for MPS, and studies on its effectiveness were initiated over 30 years ago. ERT has been performed for MPS I,7 MPS II,8,18 and MPS VI.20–23 Recombinant human GALNS (elosulfase alfa, Vimizim®) was approved as ERT for patients with MPS IVA by the US Food and Drug Administration and the European Medicines Agency in 2014.
The enzyme has oligosaccharide chains containing mannose-6-phosphate (M6P) residues that facilitate uptake of the infused enzyme into lysosomes of cells via the M6P receptor. The enzyme can then catabolize the GAGs that have accumulated in the lysosome.58 Most infused enzymes are delivered to the visceral organs, such as liver, kidney, and spleen. The infused enzyme has a short half-life in the circulation due to rapid binding to M6P receptors and delivery into these visceral organs. The enzyme cannot be delivered directly to tissues with poor vascular flow (such as cartilage) in patients with MPS. Therefore, current conventional ERT is expected to have a limited effect for bone and cartilage lesions in patients with MPS, even after long-term treatment.
Clinical trial results
Two clinical trials of ERT for patients with MPS IVA have been performed. A phase 1/2 trial included 20 patients in open-label, dose escalation trial and a phase 3 trial included 176 patients in a double-blind, randomized, placebo-controlled trial at multiple clinical sites.59,60
The phase 3 trial was conducted for 24 weeks in patients aged between 5 years and 57 years. Patients were randomized to three treatment groups: 2 mg/kg once per week (n=58), 2 mg/kg once every other week (n=59), or placebo (n=59). At baseline, all enrolled patients could walk more than 30 m but less than 325 m in 6 minutes (average 210 m); all patients were treated with antihistamines prior to each infusion to decrease immune response toward the administered enzyme. After 24-week administration of elosulfase alfa, the primary clinical endpoint of a 6MWT showed that patients treated with 2 mg/kg/week could walk 22.5 m further compared to patients in the placebo group. The patients treated with ERT every 2 weeks did not show a significant improvement in this test. Furthermore, patients in either ERT group showed no statistically significant improvement in any other tests, including the 3-minute stair climb test, pulmonary function testing (% maximum voluntary ventilation, % forced vital capacity, % forced expiratory volume in 1 second), growth rate, or ADL. There were some signs of improvement in some of these measures, but the study was not sufficiently powered to show statistical significance.
Compared with the placebo group, patients who received elosulfase alfa 2 mg/kg weekly showed a small improvement in the caregiver assistance and mobility domains, but not in the self-care domain, using the Mucopolysaccharidosis Health Assessment Questionnaire. The Mucopolysaccharidosis Health Assessment Questionnaire measures self-care (eating/drinking, dressing, bathing, grooming, tooth brushing, and toileting), mobility skills (dexterity, mobility, walking, stair climbing, and gross motor skills), and the extent of caregiver assistance required to perform these activities. There were no apparent treatment effects, at least in the short term, on hearing, echocardiogram, corneal clouding, or lower extremity long bone length.24
The most common adverse event of elosulfase alfa was an infusion-associated reaction. In all, 22.4% patients in the weekly administration group had adverse events, leading to a temporary infusion interruption/discontinuation for required medical intervention. However, no patients had adverse events that led to permanent discontinuation of the study drug. Thus, ERT appears to be a safe treatment for patients with MPS IVA.
Although this clinical trial was successful according to its primary endpoint and is now an approved treatment, more long-term studies are required to determine the extent to which this treatment can improve outcomes for MPS IVA patients. The primary endpoint of the clinical trial, the 6MWT, may be affected subjectively by multiple factors. There is a wide range of variability in how the 6MWT is administered despite availability of guidelines. The test is effort dependent, which could be particularly problematic in pediatric patients. Performance is often influenced by the level of training, stage in development, understanding of instructions, and willingness to cooperate (motivation). These factors could also vary across the clinical sites participating in this trial. The 6MWT has been shown to vary by the type of chronic pediatric disease, and therefore, should be interpreted with caution.61 Experts have recommended using additional outcome measures to help interpret the clinical meaningfulness of the 6MWT results in each chronic disease population.24
Spirometry is also an effort-dependent test that requires cooperation between a subject and an examiner, limiting this measurement to specific cohorts of patients. Patients with MPS IVA require more effort to conduct the 3-minute stair climb test due to the need for coordination of multiple joints, including those in the hip, knee, and foot, and therefore, this test could be a better measure of true improvement of skeletal dysplasia. The clinical trial was not sufficiently powered to show an improvement in this test, and longer term studies are needed to determine whether ERT will improve this outcome for MPS IVA patients. The physical assessments in the phase III clinical trial required exclusion of patients younger than 5 years, those wheelchair bound, and post-operative patients with severe muscle weakness that prevents them from performing physical assessments. Non-invasive tests will need to be employed to determine whether ERT is beneficial for these younger patients.
Thus, clinical studies have not yet shown whether ERT can reverse pre-existing skeletal damage.24,62,63
Bone was not comprehensively assessed in the clinical trial (range of motion: hyperlaxity of joints, BMD), despite its importance in MPS IVA. It is expected that a trial that lasted just 24 weeks does not show any significant impact on bone pathology. We have investigated cartilage tissues from patients with MPS IVA, who underwent 6–12 months of ERT, and did not observe any reduction of vacuoles in chondrocytes or improvement of abnormal ECM in any patient (Figure 1).
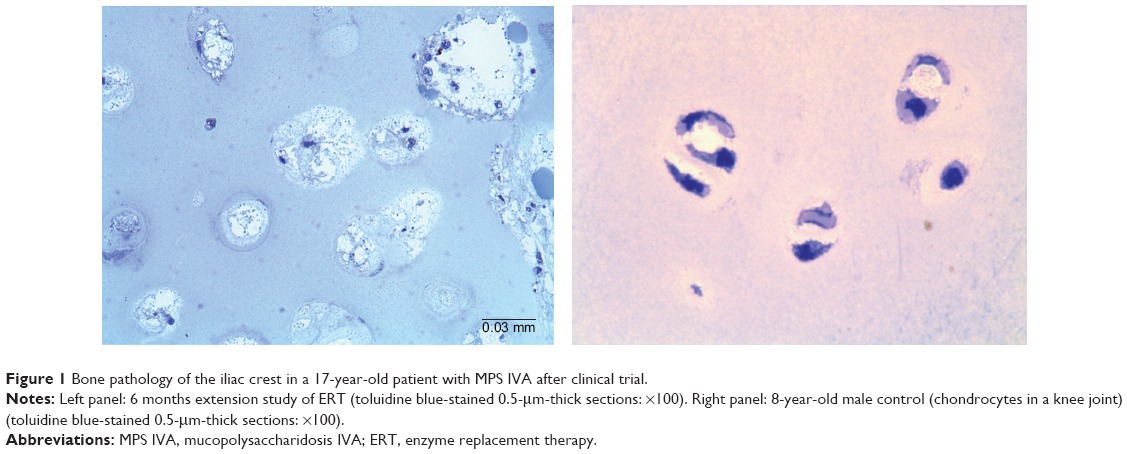 | Figure 1 Bone pathology of the iliac crest in a 17-year-old patient with MPS IVA after clinical trial. |
Thus, ERT provides a limited impact on established skeletal lesions, at least during the early stages of treatment. Long-term follow-up of ERT is required to clarify the effect of ERT on bone pathology.
ERT in MPS IVA mouse models
A preclinical trial was performed by using a native GALNS enzyme in a 3-month-old MPS IVA mouse model.64 After 12 weeks of intravenous treatment with GALNS, lysosomal storage vacuoles were eliminated from visceral organs, such as liver, spleen, and heart. However, the storage materials in articular and epiphyseal cartilages did not show improvements, even using high doses of GALNS (2.5 mg/kg). The column structure of the growth plate region remained disorganized under ERT.64 Thus, although this ERT preclinical trial in adult MPS mice resulted in a marked reduction of storage material in the visceral organs, it provided a limited effect in hyaline and fibrous cartilage cells in the femur, ligaments, and synovium.
When ERT was started at birth in MPS IVA mice, clearance of storage materials from bone was better than seen in MPS IVA mice treated at 3 months. The chondrocytes were still vacuolated, but the column structure was organized.65 Thus, conventional ERT alone, even if started at birth, cannot prevent bone pathology completely. Most human patients may already have progressive vacuolated chondrocytes prior to treatment. Overall, the clinical and mouse data indicate that current conventional ERT using enzymes that bind to carbohydrate-recognizing receptors does not function efficiently to improve established bone and cartilage lesions.
Targeting ERT
An alternative approach has been developed to combine ERT with a bone-targeting strategy. HA [Ca10(PO4)6(OH)2] is a positively charged, major inorganic component in a hard tissue (bone) that is absent in soft tissues. Bone matrix proteins (osteopontin, bone sialoprotein, etc), which bind to HA, have a repetitive sequence of negatively charged acidic amino acids (aspartic acid or glutamic acid), which are proposed as possible HA-binding sites.66,67 In osteoblastic cell culture, osteopontin and bone sialoprotein rapidly bind to HA after they are secreted by osteoblasts.68 A drug attached to bisphosphonate is targeted to HA and released during the bone resorption process, showing that targeting a drug to HA is a potential strategy for a selective drug delivery to bone.39,69 Targeting to bone could also be achieved by attaching the drug to six Glu (E6) residues.40,41 We and other groups have recently applied this new bone-targeting system to a large molecule, an enzyme (tissue-nonspecific alkaline phosphatase), showing that the tagged enzymes are delivered more efficiently to bone42–44 and that clinical and pathological improvements in the systemic bone disease, hypophosphatasia, are better using the tagged enzyme when compared with native enzyme.43 Human GALNS has also been bioengineered to add an E6 tag. The tagged enzyme had a markedly reduced rate of clearance from the circulation, resulting in blood levels of the enzyme that were 10–20 times higher than those of the native enzyme in MPS IVA mice.30 The bone-targeting enzyme was retained longer in bone, with substantial residual enzyme activity 48 hours after treatment. Pathological examination of MPS IVA mice after treatment with the targeting enzyme showed more clearance of storage materials in bones than mice treated with unmodified enzyme. Thus, the tagged enzyme enhances delivery and reduces pathological effects in bones of MPS IVA mice. We have not seen any immunological response in MPS IVA mice treated with the tagged enzyme.
Overall, these results indicate that targeting of the enzyme to bone and/or treating at the newborn stage provide improvements in clinical disease manifestations and pathological bone lesions in mouse models. Despite these improvements in efficacy, no therapies that completely remove storage vacuoles from chondrocytes have been reported to date.
Recombinant GALNS in microorganisms
One of the main concerns about the use of recombinant proteins as drugs in humans is the very high price. These prices are mostly due to the cost of using mammalian cells to produce lysosomal enzymes.70 More recently, several studies have shown that active lysosomal enzymes can be produced by other expression systems, including plant and insect cells, and microorganisms.32,71–76 Production of enzymes in microorganisms is considerably less expensive than in mammalian cells and, consequently, may significantly reduce the cost of ERT in addition to producing recombinant proteins with improved stability, and pharmacokinetic and pharmacodynamic profiles (Figure 2).72,77
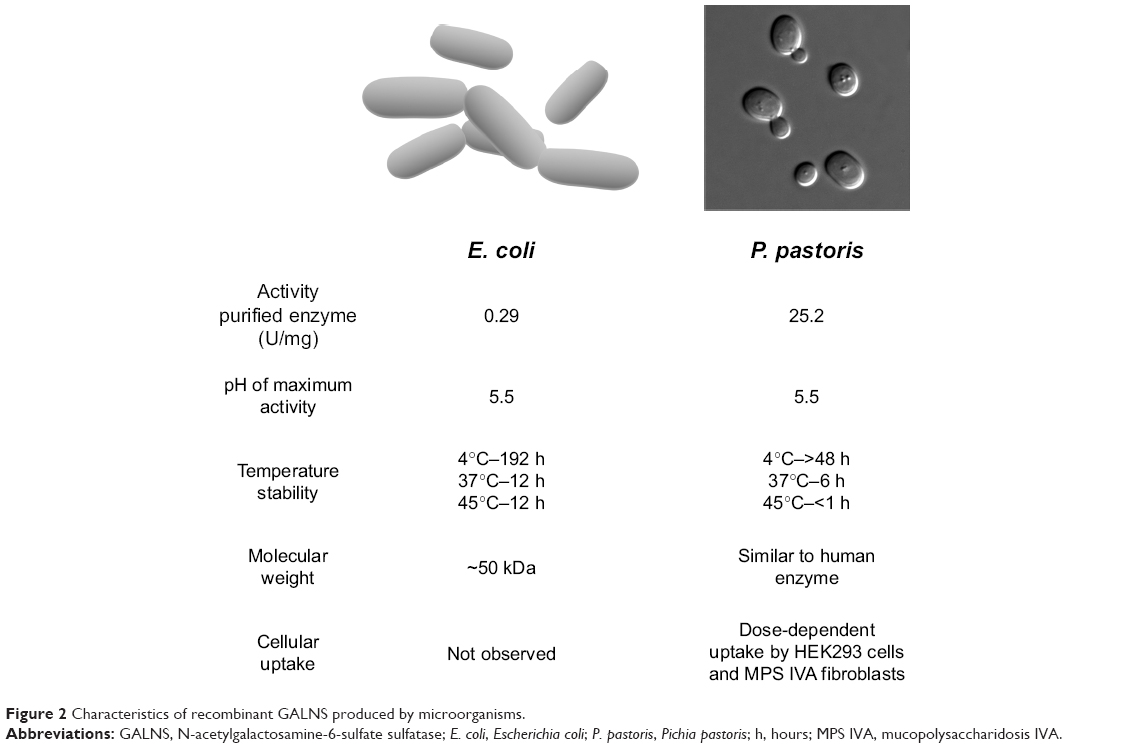 | Figure 2 Characteristics of recombinant GALNS produced by microorganisms. |
Recombinant GALNS has been produced in Chinese hamster ovary (CHO) cells, with enzyme activities of approximately 170,000 nmol/h/mg78 and 120,000 nmol/h/mg;79 however, these activity values are not equivalent because they have been obtained using substantially different methods. Production of recombinant GALNS in E. coli BL21 (DE3) (erGALNS)32,80,81 and in the methylotrophic yeast Pichia pastoris (prGALNS)33,82 has emerged as an alternative to conventional production in CHO cells. In E. coli, as well as in other prokaryotes, cysteine- and serine-sulfatase types have been identified, which are activated by a formylglycine-generating enzyme and anaerobic sulfatase-maturating enzyme (AtsB), respectively, in these microorganisms.83 The first report related to recombinant GALNS production in E. coli BL21 (DE3) evaluated the cell growth conditions and protein induction in a batch process at shake (100 mL) and bioreactor (3 L) scales.32 The soluble enzyme extracted from cells had a specific activity of between 0.054 U/mg and 0.071 U/mg, much lower than that of the enzyme produced in mammalian cells. The majority of the erGALNS was obtained as inclusion bodies (~71%), and most of this form of the enzyme was inactive. Western-blot analysis of the erGALNS showed a ~50 kDa protein that was smaller than recombinant GALNS produced in CHO cells. This reduction in molecular weight is due to lack of N-glycosylation in E. coli. Although further development is needed to produce a correctly folded enzyme, these results do show that the N-glycosylation is not necessary for producing active GALNS.32 It is noteworthy that prokaryotic formylglycine-generating enzyme can activate human sulfatases, reflecting high conservation of this mechanism and the possibility of producing active sulfatases in prokaryotic systems.
Semi-continuous culture of transformed E. coli BL21 (DE3) using an expression vector containing the native signal peptide of GALNS favored the secretion of erGALNS.80 Enzyme purified from the extracellular crude extract had a specific activity of 0.29 U/mg and a production yield of 0.78 mg/L. The purified enzyme was optimally active at pH of 5.5, and it was stable at 4°C for 8 days and in human serum for 6 hours.81 A cellular assay was unable to show uptake of erGALNS by HEK293 cells or by Morquio A skin fibroblasts. These results suggest that the N-glycosylation of GALNS is not required for producing an active and stable enzyme but is required for efficient protein cellular uptake.81 Current studies are focusing on the modification of this enzyme to include specific glycosylation to favor its cell uptake.
Production of recombinant enzymes in E. coli BL21(DE3) allowed identification of a relationship between enzyme activity and the presence of the native signal peptide.80 The results showed that the deletion of native signal peptide caused a 7.6-fold reduction in enzyme specific activity. It is proposed that the signal peptide participates in the cysteine-to-formylglycine conversion at the active site by a prokaryotic cytosolic enzyme, since GALNS concentration, as determined by an Enzyme-Linked ImmunoSorbent Assay (ELISA), was similar with or without signal peptide.80 In mammalian cells, this essential conversion is performed by an enzyme in the endoplasmic reticulum, which is highly conserved among organisms, and it has been described in E. coli as well as in other prokaryotes.83 These findings showed for the first time that regardless of the mechanism by which the signal peptide is associated with sulfatase activation in prokaryotes, this E. coli system can recognize a eukaryotic signal peptide and mediate the activation of a human sulfatase. In addition, it was observed that the presence of the signal peptide prevented the production of inclusion bodies, which could be the result of reduced hydrophobic interactions and enzyme secretion.80
In order to produce a recombinant glycosylated version GALNS in a microorganism, its production has been evaluated in the methylotrophic yeast P. pastoris (prGALNS).34,82 In this expression system, extracellular enzyme activity values were up to 0.26 U/mg in the crude extract. In addition, the co-expression with SUMF1 cDNA allowed a 2.3-fold increase in enzyme activity. These results showed for the first time the advantage of sulfatase-SUMF1 co-expression within a yeast expression system.34,82 The purified prGALNS showed a specific activity of 25.3 U/mg. Western-blot analysis showed that prGALNS has a protein pattern similar to that of human GALNS from leukocytes, as well as similar temperature and pH stability profiles to those observed for the enzyme produced in CHO cells.34 Furthermore, prGALNS was taken up by HEK293 cells and Morquio A skin fibroblasts. These results, coupled with the high specific activity of the recombinant enzyme, show the potential of yeast systems for development for ERT of MPS IVA.
An important issue regarding a biopharmaceutical is immunogenicity. However, it is not possible to predict the immunogenicity of a recombinant protein.84 Although both erGALNS and prGALNS are produced using the human GALNS cDNA that is currently used to produce enzyme in mammalian cells for ERT, there are some specific factors that might enhance the immunogenicity of recombinant proteins expressed in non-mammalian systems.84 Among those factors, changes in glycosylations, both absence (ie, E. coli) and altered patterns (ie, P. pastoris), can expose cryptic B- or T-cell epitopes and cause the enzyme to appear foreign to the immune system.84 Other factors include the presence of aggregates, degradation products, or process-related impurities. Nevertheless, preclinical studies of lysosomal acid lipase produced in P. pastoris (phLAL) showed that the development of low titers of anti-phLAL antibodies did not inhibit the phLAL activity or cellular capture, and were associated with the enzyme N-glycosylations.85 Immune response against any recombinant form of GALNS should be evaluated carefully prior to development of ERT.
It is critical to evaluate the therapeutic effect on the bone and joint lesions, such as BMD and range of motion. KS is synthesized mainly in chondrocytes, and blood and urinary KS levels are normalized naturally after the growth plate is closed or destroyed. It is also important to determine how long ERT should continue if the effect does not meet cost–benefit once the storage materials are removed from the soft tissues.
Hematopoietic stem cell transplantation
Background of HSCT
A possible benefit of HSCT for treating MPS is that marrow-derived donor cells provide a secreting source of enzyme that has access to bone and cartilage close to the bone marrow. By contrast, it has been proposed that the cells produced by HSCT may not penetrate into bone and cartilage.86–89 HSCT has shown efficacy in correcting the disease course in patients with MPS I, MPS II, MPS VI, and MPS VII.37,38,90–94 The clinical outcome of HSCT depends on 1) the age of the patient at the transplantation; 2) the clinical phenotype and the stage of the disease (clinical condition) at the transplantation; 3) the type of donor; and 4) the preparative regimen.95
Hobbs et al first treated a patient with MPS I using bone marrow transplantation (BMT) and showed the possibility of therapeutic effect for this disorder.96 Subsequent studies on additional MPS I patients have demonstrated substantial clinical improvements of hearing impairment, psychomotor regression, neurocognitive performance, joint mobility, coarse facial features, growth, claw hands, and somatic features following transplantation.89,97–99 HSCT has also been shown to ameliorate and slow the disease progression in patients with MPS II, MPS VI, and MPS VII.37,38,92–94,100
Over 500 patients with MPS I, over 160 patients with MPS II, and over 60 patients with MPS VI have been treated with HSCT to date.38,89,92,101–107 Early HSCT is the gold standard of care for patients with a severe phenotype (MPS I), preferably treating under 2 years of age before neurological signs and symptoms appear.37,38,92,93 However, skeletal diseases, including genu valgum, thoracolumbar kyphosis, and hip dysplasia, still develop in HSCT-treated patients with MPS I. Skeletal deformity is not as responsive to HSCT as other important clinical outcome parameters that produce favorable effects, including CNS involvement.108–116 These skeletal abnormalities lead to the need for orthopedic corrective surgeries. We are following a 15-year-old patient with MPS I who had received successful HSCT at the age of 16 months. His ADL has remained normal, but bone deformities have developed, requiring hemiepiphysiodesis of bilateral medial proximal tibia at 12 years and successive arthrodesis of thoracolumbar spine at 13 years. It is noteworthy that skeletal pathological examination of surgical remnants showed almost complete clearance of storage materials in chondrocytes and normal levels of blood heparan sulfate and dermatan sulfate, even though the incomplete correction of the skeletal phenotype still continues. The bone abnormalities may have been irreversible at the time of the transplant, and the structure of ECM (collagen fiber) may still remain abnormal since the enzyme cannot access directly to ECM at neutral pH.
These data suggest that the first months of life represent the best opportunity for preventing bone deformities in patients with a severe form of MPS I. A retrospective analysis supports superior long-term clinical outcome of patients with MPS I when HSCT is performed early in life.101
One of the disadvantages of HSCT in MPS patients is the high mortality rate of this procedure. Mortality rates of HSCT were high during initial transplantation from 1980 to 1990 because patients treated at this time were already at an advanced stage of disease progression. Patients with an advanced stage of disease would not be good candidates for HSCT if they are not able to withstand the rigorous regimen of this treatment; however, with the advanced technology and awareness of MPS, early diagnosis is more feasible, and consequently, patients with MPS can receive HSCT when they are young and healthy.
Recent data indicate that 5-year survival rate from HSCT is 88.5% for those with MPS II92 and over 90% for those with MPS I.117,118 ERT and HSCT provide a comparable impact on growth in MPS II, but the average ADL score in HSCT-treated patients is higher than in ERT-treated patients, and HSCT before 5 years of age provides a better ADL score than HSCT performed at later ages.38
HSCT for MPS IVA
There is only one case report detailing a patient with MPS IVA who has received HSCT treatment to date.4,10,93,119 This patient with a severe form of Morquio A received successful allogeneic BMT at 15 years and 8 months of age. Two years post-BMT, the enzyme activity of GALNS in white blood cells of the recipient was approximately 50% of normal levels, and this has been preserved for over 9 years. In all, 1 year post-BMT, BMD at L2–4 increased from 0.372 g/cm2 to 0.548 g/cm2 and was kept at the level of 0.48±0.054 g/cm2 for the following 9 years. Radiographs showed that figures of major and minor trochanter emerged, while the epiphyseal dysplasia in the femoral cap remained unchanged (Figure 3). His loud snoring and shortness of breath stopped, coinciding with remission of pulmonary dysfunction. With correction osteotomies for genu valgum, the patient can walk for 100 m by ankle–foot orthoses and for 400 m by hip–knee–ankle–foot orthoses, although after walking a long distance, the patient has pain at the ankles (see Videos S1 and S2).
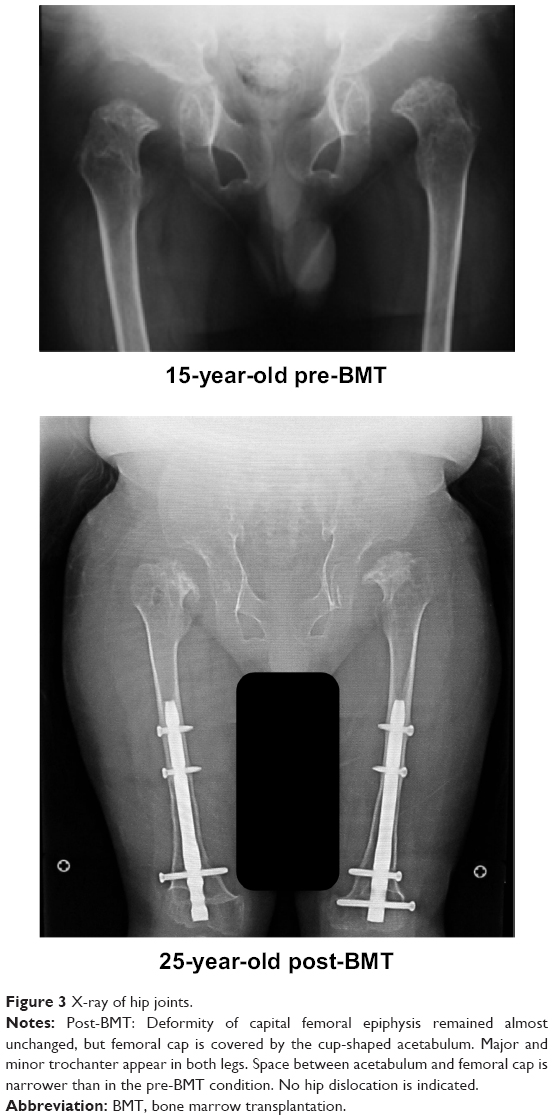 | Figure 3 X-ray of hip joints. |
ADL score in this patient also improved in areas of work/study, sleep, joint mobility, pain, respiratory status, and infection, indicating that HSCT has the potential to be of therapeutic benefit to help improve the quality of life for Morquio A patients. This patient is working as a graphic designer. However, problems such as restriction of physical activity, hypermobile joints (hands and fingers), and faint corneal clouding remain unsolved. These problems might have been less of a concern if HSCT had been performed when the patient was younger. In this particular case, no serious HSCT-associated side effects or graft-versus host disease were observed. The substantial clinical correction post-HSCT for this MPS IVA patient indicates that HSCT may be a therapeutic option for Morquio A patients. Several additional patients with MPS IVA have been treated with HSCT in Japan without serious complication post-HSCT. Follow-up assessment of these patients is now underway.
Newborn HSCT
The effect of treatment with HSCT at birth on skeletal features has been examined in an MPS animal model.34,35 Newborn mice with MPS VII receiving ablative BMT lived longer than untreated mice. Treated mice had less severe facial dysmorphism, better mobility, and showed pathological and clinical improvements, including clearance of lysosomal storage in bones, joints, and visceral organs, even though engraftment efficiency was low (15%–20%).34 Nonablative neonatal BMT showed that 12 months after BMT, several structural features of femurs were more similar to those of normal mice than untreated MPS VII mice. Periosteal circumference and bone cortical thickness were significantly improved, and cortical density did not differ significantly from values in normal mice. A significant reduction in lysosomal GAG storage corresponded with β-glucuronidase (GUS) enzyme activity and the percentage of histochemically GUS positive cells in visceral organs and hematopoietic tissues.
We have tested the hypothesis that newborn HSCT can prevent skeletal dysplasia in MPS I mice.120 Neonatal BMT was effective at restoring α-l-iduronidase activity and clearing elevated GAGs in blood and multiple organs. At 37 weeks of age, bone tissue parameters assessed by radiographic, micro-computer tomography, biochemical, and histological analyses were nearly normalized. The extent of improvements correlated with the extent of hematopoietic engraftment. Moreover, improvements in bone parameters correlated with levels of bone marrow-derived cell engraftment in multiple hematopoietic compartments, suggesting that the early and complete restoration of normal hematopoiesis provides a significant impact on bone development of newborn MPS I mice. This proof of concept study advocates newborn BMT as a highly effective therapeutic approach for MPS I, indicating that earlier treatment should have a greater impact on clinical outcomes for these patients. This emphasizes the importance of implementation of newborn screening procedures to allow an early diagnosis and immediate treatment of affected children.
These mouse models indicate that HSCT should be performed as early as possible to ameliorate skeletal deformities and impaired growth development in MPS patients. For MPS IVA, data need to be accumulated from more HSCT-treated cases to determine whether a regimen of HSCT will be effective and safe. Regimens for HSCT have been dramatically improved in recent years, and well-established institutions with trained staff show a low mortality rate caused by the HSCT procedure. Some risks of mortality caused by infection, graft-versus host disease, and additional complications remain, so a decision to treat with HSCT will still need to be considered carefully with pre-transplantation counseling, clinical evaluation, and systemic longitudinal monitoring of outcomes.
Biomarkers for MPS IVA
Biomarker requirements
A surrogate endpoint is deemed as a biomarker intended to substitute for a clinical endpoint. Biomarkers are used for diagnosing, staging, and monitoring the progress of a disease and its response to therapy. In general, biomarkers are cheaper and easier to measure than true endpoints and can be assayed over a shorter period.121,122 In clinical trials, an ideal biomarker is a measure of effect of a specific treatment that may correlate with a real clinical endpoint (Figures 4 and 5). The first step in identifying suitable biomarkers is to understand the pathophysiology of the disease and to find factors that determine it. The next step is to identify potential biomarkers based on the mechanism of action of the intervention related to the pathophysiology of the disease. The last step is to determine the extent to which the putative marker correlates with the process and how useful it is in predicting the outcome.
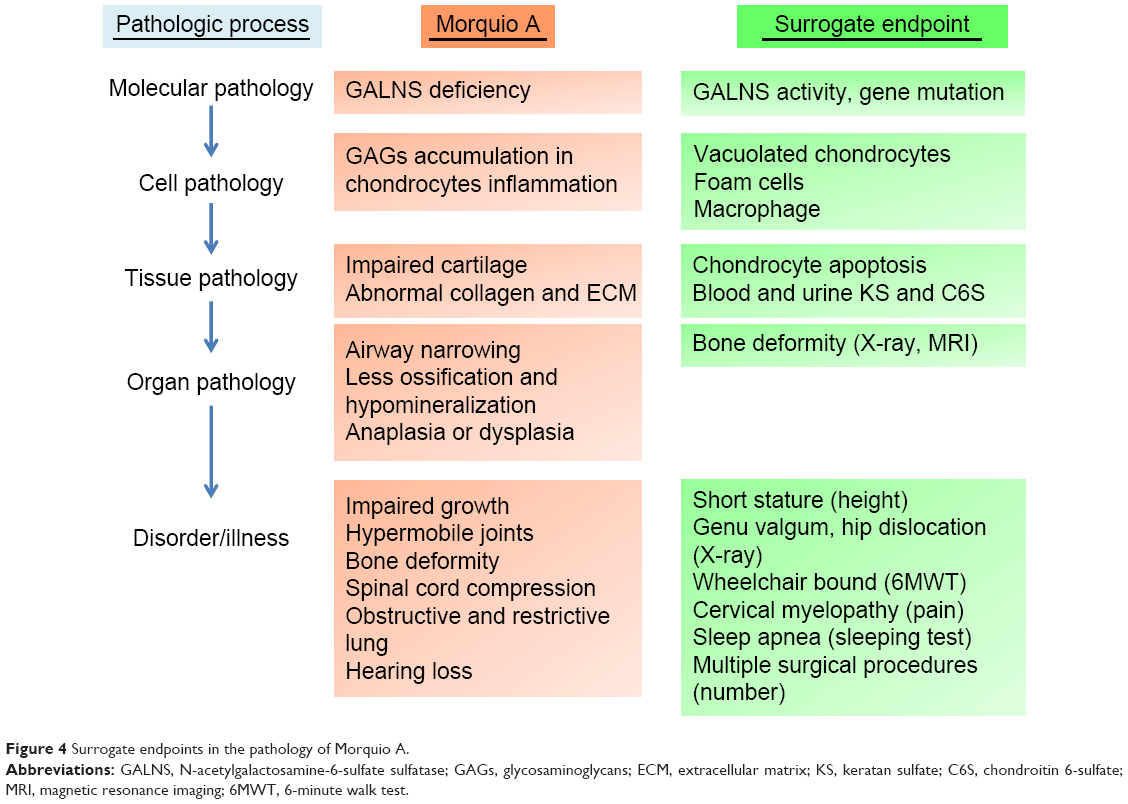 | Figure 4 Surrogate endpoints in the pathology of Morquio A. |
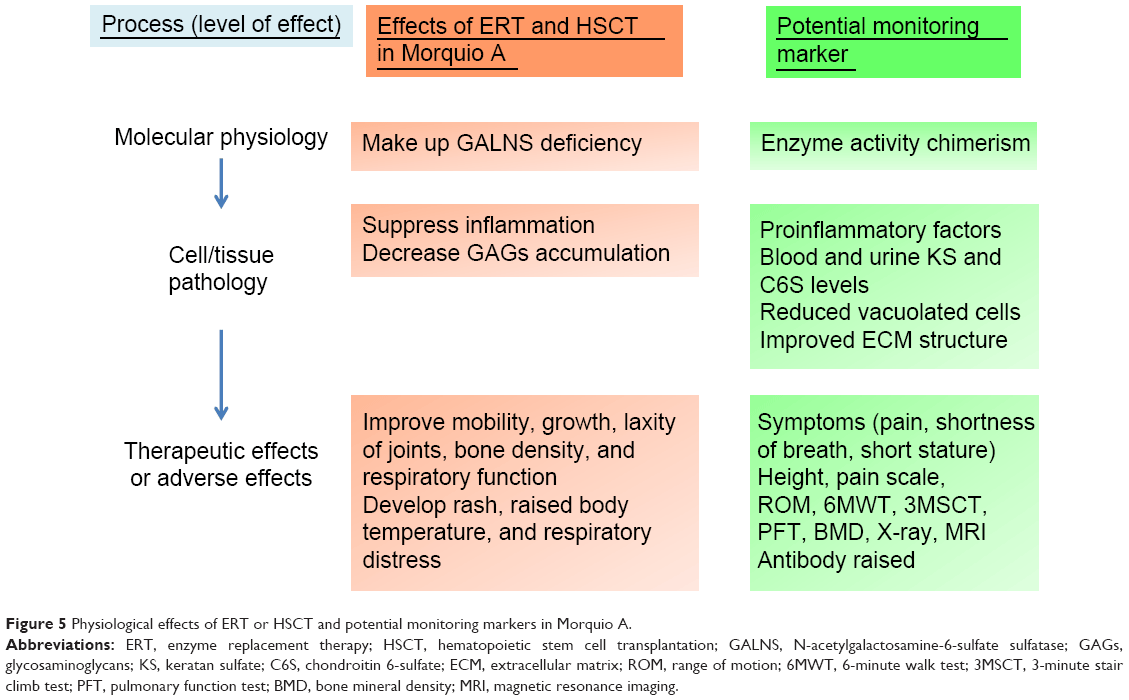 | Figure 5 Physiological effects of ERT or HSCT and potential monitoring markers in Morquio A. |
The US Institute of Medicine recommends evaluating biomarkers by the following steps: 1) analytical validation to ensure that biomarker tests are reliable, reproducible, and adequately sensitive and specific; 2) qualification to ensure the biomarker is associated with the clinical outcome of concern; and 3) utilization analysis to determine that the biomarker is appropriate for the proposed use.
GALNS is the enzyme required for the breakdown of C6S and KS by removing the sulfate (moiety from the glycosaminoglycan substrates). Deficiency of GALNS activity results in the accumulation of undegraded primary substrates, C6S and KS, in lysosomes of these cells, which leads to progressive skeletal dysplasia. Excessive undegraded C6S and KS is released into the circulation where it can be examined as an important biomarker for characterizing MPS IVA. C6S and KS syntheses occur mainly in chondrocytes. As a result, C6S and KS represent potential biomarkers for MPS IVA.
KS
Assay methods for blood and urinary KS in MPS IVA were developed by ELISA and liquid chromatography tandem mass spectrometry (LC-MS/MS).123,124 Blood and urinary KS in MPS IVA was found to be age dependent,125,126 declining with age and becoming near normal by the age of 20 years. Urinary KS levels have been related to clinical severity in patients with MPS IVA.123,126,127 In a phase 3 trial, in patients treated with 2.0 mg/kg/week of elosulfase alfa, urinary KS levels decreased by 41% from the baseline.19 However, decreases in urinary KS levels did not reflect the clinical effects measured by 6MWT or other clinical endpoints after 24-week administration. Patients treated with 2.0 mg/kg/every other week with elosulfase alfa also had a 30% reduction in urinary KS level from the baseline even though none of the clinical endpoints were improved. Thus, in this study, measurement of urinary KS is of limited value as a biomarker for clinical endpoints. Since many clinical outcomes of MPS IVA (short stature, hypermobile joints [cervical instability, genu valgum, floppy hands, and double fingers]) are due to skeletal dysplasia that results from vacuolated chondrocytes that cause abnormal ECM formation, it is very critical to define whether a measured biomarker is correlated with improvement of these clinical outcomes. Urinary KS may be useful to differentiate MPS IVA from other forms of MPS and does demonstrate pharmacodynamic effects of ERT treatment, but does not appear to be valuable for either determining therapeutic efficacy or predicting outcomes of ERT treatment for MPS IVA. In MPS IVA patients, blood KS levels correlate with clinical severity of MPS IVA.123,125–128 Blood KS peaks between 5 years and 15 years, while urinary KS is the highest in newborns.123,125–127 Shortly after the start of ERT, most patients show a decrease in urinary KS level, but blood KS levels after ERT have not yet been reported. Urinary KS is derived from small KS fragments that filter through the kidney from the circulation, and therefore, total urinary KS is not necessarily related to total blood KS that includes both small and large fragments of KS. Thus, blood KS should be a better indicator of improvement in true clinical endpoints, including bone pathology or any other skeletal signs and symptoms. A limitation of both blood and urinary KS analysis is that it is only of value in younger patients before closure or destruction of growth plates because after that time, synthesis of KS decreases dramatically so that levels of KS in MPS IVA patients are indistinguishable from unaffected individuals of the same age.
Ratio of mono-sulfated and di-sulfated KS
GALNS is also known as galactose-6-sulfatase because the enzyme hydrolyzes the sulfated galactose of KS and can convert di-sulfated KS to mono-sulfated KS. Deficiency of GALNS activity results in the accumulation of di-sulfated KS, and consequently, proportion of di-sulfated KS in total KS should increase.126,129,130
We have measured both mono-sulfated and di-sulfated KS using LC-MS/MS. The mean levels of both mono-sulfated and di-sulfated KS in blood from patients with IVA were elevated compared with age-matched controls. The increase in di-sulfated KS in MPS IVA patients was more significant than the increase in mono-sulfated KS. The proportion of di-sulfated KS in total KS in plasma/serum increased with age in control subjects but was age independent in MPS IVA patients. Consequently, the proportion of di-sulfated KS is better at discriminating younger MPS IVA patients from controls than older patients. The levels of mono- and di-sulfated KS in urine of MPS IVA patients were all higher than age-matched controls for all studied ages.
In conclusion, a significant difference in sulfation levels of KS between control subjects and patients with MPS IVA demonstrates that di-sulfated KS is a potential biomarker for this disease.
C6S
C6S is distributed mainly in the growth plates, aorta, and cornea. We developed a method to digest polymeric C6S and measure resultant disaccharides using LC-MS/MS.17 C6S levels were measured in blood and urine from control subjects and patients with MPS IVA and VII aged from 0 year to 58 years. Levels of C6S in blood and urine decreased with age in both MPS IVA patients and control subjects, but were significantly elevated in patients compared with age-matched controls. Combining KS and C6S data discriminated patients with MPS IVA from age-matched control subjects better than either C6S or KS levels alone.
Overall, it would be of great interest to understand: 1) whether blood KS levels decrease over time during treatment with ERT; 2) the extent to which blood and urinary KS levels are correlated; 3) how clinical improvements are correlated with reduction in blood KS; 4) the extent to which the ratio of mono-sulfated KS to di-sulfated KS changes during treatment of MPS IVA; 5) whether blood and urine C6S is a better biomarker than KS; and 6) whether a combination of C6S and KS can lead to a better biomarker for MPS IVA.
C6S and KS assays by LC-MS/MS satisfy the first step. C6S and KS are closely correlated with the clinical outcome of skeletal dysplasia (second step). Further study will be required to clarify whether KS and/or C6S levels correlate with the response to therapy (third step).
Conclusion
Resolution of bone and cartilage lesions remains an unmet challenge for patients with MPS IVA. Patients with MPS IVA have severe progressive skeletal dysplasia that leads to significant morbidity and handicap with poor ADL. Management should be a multidisciplinary approach to care for patients, particularly those who have serious issues, such as spinal cord compression, ambulatory problems, and restrictive and obstructive lung issues. A comprehensive assessment of the individual patient at initial diagnosis is required with continuous follow-up by experienced clinicians. Supportive management, physiotherapy, and appreciation of possible complications can also improve the quality of life of patients with MPS IVA and their families. Families of the patients should become aware of management measures, including genetic counseling, ERT (already approved), HSCT, future gene therapy or anti-inflammatory drug, supportive therapies, physiotherapies, and orthopedic interventions (Figure 6). Physicians who take care of patients with MPS IVA should be familiar with the most common complications, diagnosis of the disease, and locations of expert centers, as well as available therapies. Hopefully, this will lead to earlier diagnosis for patients, resulting in better comprehensive therapy and avoidance of progression to irreversible damage. Although the current treatments will not cure the disease, they provide the potential to rescue most patients from consequences of the disease and to improve the quality of life if treatment starts at an early stage. Therapy for established systemic bone dysplasia remains a serious challenge, and robust, innovative approaches, such as bone targeting, should be further developed. Longitudinal observation of patients with MPS IVA under current optional therapies provides more precise and valuable information regarding the appropriate assessment, including biomarkers, physical activity, supportive treatment, efficacy of therapy, and the clinical endpoints.
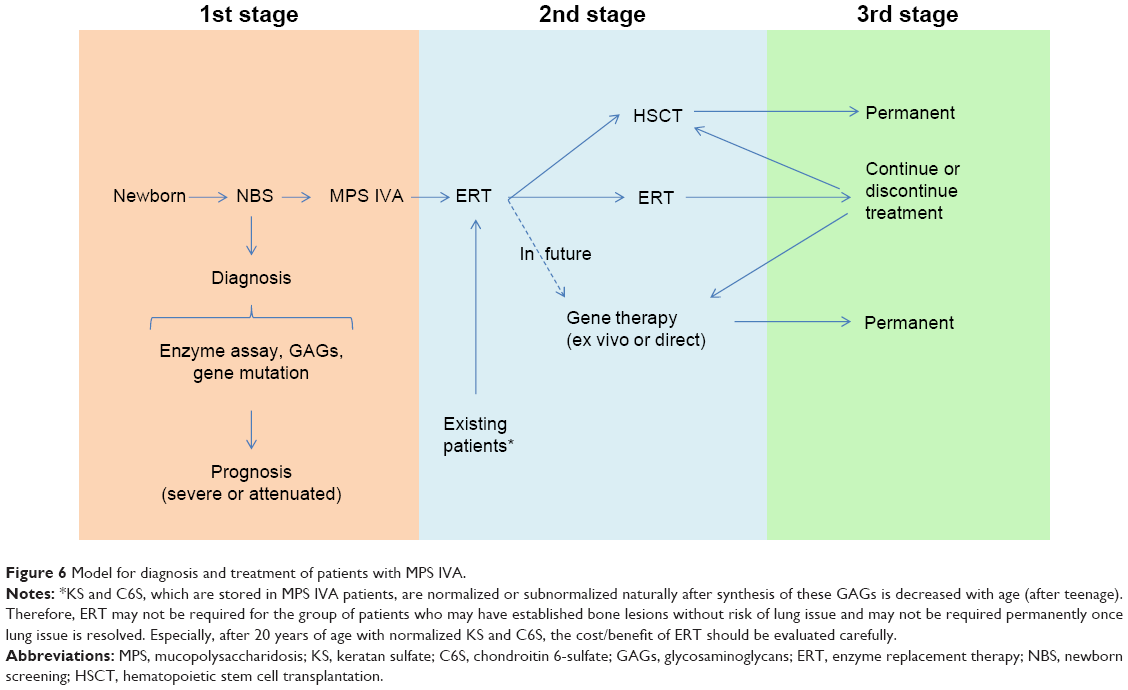 | Figure 6 Model for diagnosis and treatment of patients with MPS IVA. |
Acknowledgments
This review article was supported by grants from the Austrian MPS Society, the Bennett Foundation, and the International Morquio Organization (Carol Ann Foundation). This work was also supported by Japanese MPS Family Society. RWM and ST were supported by an Institutional Development Award from the National Institute of General Medical Sciences of National Institutes of Health (NIH) under grant number P20GM103464. ST and AMM were supported by NIH grant R01HD065767. FK was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico from Brazil (CNPq). AJEM and OFS were supported by Administrative Department of Science, Technology and Innovation (Colciencias) from Colombia. ARL was supported by Pontificia Universidad Javeriana. CJAD was supported by Pontificia Universidad Javeriana (grant IDs 3635, 5501, and 5596) and Colciencias (grant no 120356933205). The content of the article is solely the responsibility of the authors and does not necessarily represent the official views of NIH or other sponsors. Editorial assistance to the manuscript was provided by Michelle Stofa at Nemours/Alfred I duPont Hospital for Children.
Author Contributions
Shunji Tomatsu is the principal investigator for this review article and has contributed to the concept and planning of the article, collection of data, and reporting of the work described. Kazuki Sawamoto contributed to the planning of the article, collection of data on ERT and HSCT, and reporting of the work described. Carlos J Alméciga-Díaz, Alexander Rodríguez-López, Angela J Espejo-Mojica, Oscar F Sánchez and, Luis A Barrera contributed to the planning of the article, collection of data on production of the enzyme in microorganisms, and reporting of the work described. Tsutomu Shimada contributed to the planning of the article, collection of data on GAG assay, and reporting of the work described. Michael B Bober and Roberto Giugliani contributed to the planning of the article, treating MPS IVA patients by ERT, and reporting of the work described. Yasutsugu Chinen, Hiromasa Yabe, Francyne Kubaski, and Robert W Mason contributed to the planning of the article, collection of data on HSCT, and reporting of the work described. Adriana M Montaño contributed to the planning of the article, collection of data on ERT, and reporting of the work described. Eriko Yasuda and William G Mackenzie contributed to the planning of the article, collection of data, and reporting of the work described. Tadao Orii is the other principal investigator for this review article and has contributed to the concept of the manuscript, planning of the article, collection of data, and reporting of the work described. All authors contributed to drafting the article or revising it critically for important intellectual content.
Disclosure
All the authors contributed to the review article and had no conflicts of interest with any other party.
References
Neufeld E, Muenzer J. The mucopolysaccharidoses. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill; 2001:3421–3452. | ||
Tomatsu S, Montaño AM, Oikawa H, et al. Impairment of body growth in mucopolysaccharidoses. In: Preedy VR, editor. Handbook of Growth and Growth Monitoring in Health and Disease 1. New York: Springer; 2012:2091–2117. | ||
Patel P, Suzuki Y, Maeda M, et al. Growth charts for patients with Hunter syndrome. Mol Genet Metab Rep. 2014;1:5–18. | ||
Tomatsu S, Montaño AM, Oikawa H, et al. Mucopolysaccharidosis type IVA (Morquio A disease): clinical review and current treatment. Curr Pharm Biotechnol. 2011;12(6):931–945. | ||
Rodriguez M, Miller TL, Mackenzie WG, Ditro C, Chidekel AS, Shaffer TH. Characteristics of impulse oscillometry and thoracoabdominal motion in children with thoracic cage disorders. Pediatr Pulmonol. 2010;45(7):679–686. | ||
Theroux MC, Nerker T, Ditro C, Mackenzie WG. Anesthetic care and perioperative complications of children with Morquio syndrome. Paediatr Anaesth. 2012;22(9):901–907. | ||
Kakkis ED, Muenzer J, Tiller GE, et al. Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med. 2001;344(3):182–188. | ||
Muenzer J, Wraith JE, Beck M, et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med. 2006;8:465–473. | ||
Montaño AM, Tomatsu S, Gottesman GS, Smith M, Orii T. International Morquio A Registry: clinical manifestation and natural course of Morquio A disease. J Inherit Metab Dis. 2007;30(2):165–174. | ||
Tomatsu S, Mackenzie WG, Theroux MC, et al. Current and emerging treatments and surgical interventions for Morquio A syndrome: a review. Res Rep Endocr Disord. 2012;2:65–77. | ||
Hendriksz CJ, Al-Jawad M, Berger KI, et al. Clinical overview and treatment options for non-skeletal manifestations of mucopolysaccharidosis type IVA. J Inherit Metab Dis. 2012;36(2):309–322. | ||
Beck M, Glössl J, Grubisic A, Spranger J. Heterogeneity of Morquio disease. Clin Genet. 1986;29(4):325–331. | ||
Orii T, Kiman T, Sukegawa K, et al. Late onset N-acetylgalactosamine-6-sulfate sulfatase deficiency in two brothers. Connect Tissue Res. 1981;13(3):169–175. | ||
Fukuda S, Tomatsu S, Masue M, et al. Mucopolysaccharidosis type IVA. N-acetylgalactosamine-6-sulfate sulfatase exonic point mutations in classical Morquio and mild cases. J Clin Invest. 1992;90(3):1049–1053. | ||
Orii T, Minami R, Chiba T, et al. Study on Morquio syndrome. Bone Metab. 1971;5(1):72–78. | ||
Yasuda E, Fushimi K, Suzuki Y, et al. Pathogenesis of Morquio A syndrome: an autopsied case reveals systemic storage disorder. Mol Genet Metab. 2013;109(3):301–311. | ||
Shimada T, Tomatsu S, Yasuda E, et al. Chondroitin 6-sulfate as a novel biomarker for mucopolysaccharidosis IVA and VII. JIMD Rep. 2014;16:15–24. | ||
Muenzer J, Lamsa JC, Garcia A, Dacosta J, Garcia J, Treco DA. Enzyme replacement therapy in mucopolysaccharidosis type II (Hunter syndrome): a preliminary report. Acta Paediatr Suppl. 2002;91(439):98–99. | ||
Hendriksz CJ, Burton B, Fleming TR, et al; STRIVE Investigators. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study. J Inherit Metab Dis. 2014;37(6):979–990. | ||
Harmatz P, Whitley CB, Waber L, et al. Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). J Pediatr. 2004;144(5):574–580. | ||
Harmatz P, Ketteridge D, Giugliani R, et al; MPS VI Study Group. Direct comparison of measures of endurance, mobility, and joint function during enzyme-replacement therapy of mucopolysaccharidosis VI (Maroteaux-Lamy syndrome): results after 48 weeks in a phase 2 open-label clinical study of recombinant human N-acetylgalactosamine 4-sulfatase. Pediatrics. 2005;115(6):e681–e689. | ||
Harmatz P, Giugliani R, Schwartz I, et al; MPS VI Phase 3 Study Group. Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or rhASB) and follow-on, open-label extension study. J Pediatr. 2006;148(4):533–539. | ||
Harmatz P, Giugliani R, Schwartz IV, et al; MPS VI Study Group. Long-term follow-up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: final results of three clinical studies of recombinant human N-acetylgalactosamine 4-sulfatase. Mol Genet Metab. 2008;94(4):469–475. | ||
FDA Advisory Committee Briefing Document Elosulfase alfa for Mucopolysaccharidosis Type IVA; 2013. Available from: http://www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/endocrinologicandmetabolicdrugsadvisorycommittee/ucm375126.pdf. Accessed November 19, 2013. | ||
Connock M, Juarez-Garcia A, Frew E, et al. A systematic review of the clinical effectiveness and cost-effectiveness of enzyme replacement therapies for Fabry’s disease and mucopolysaccharidosis type I. Health Technol Assess. 2006;10(20):1–6. | ||
Rohrbach M, Clarke JT. Treatment of lysosomal storage disorders: progress with enzyme replacement therapy. Drugs. 2007;67(18):2697–2716. | ||
Chirino AJ, Mire-Sluis A. Characterizing biological products and assessing comparability following manufacturing changes. Nat Biotechnol. 2004;22(11):1383–1391. | ||
Parveen S, Sahoo SK. Nanomedicine: clinical applications of polyethylene glycol conjugated proteins and drugs. Clin Pharmacokinet. 2006;45(10):965–988. | ||
Dickson P, Peinovich M, McEntee M, et al. Immune tolerance improves the efficacy of enzyme replacement therapy in canine mucopolysaccharidosis I. J Clin Invest. 2008;118(8):2868–2876. | ||
Tomatsu S, Montaño AM, Dung VC, et al. Enhancement of drug delivery: enzyme replacement therapy for murine Morquio A syndrome. Mol Ther. 2010;18(6):1094–1102. | ||
Rowan DJ, Tomatsu S, Grubb JH, et al. Long circulating enzyme replacement therapy rescues bone pathology in mucopolysaccharidosis VII murine model. Mol Genet Metab. 2012;107(1–2):161–172. | ||
Rodríguez A, Espejo AJ, Hernández A, et al. Enzyme replacement therapy for Morquio A: an active recombinant N-acetylgalactosamine-6-sulfate sulfatase produced in Escherichia coli BL21. J Ind Microbiol Biotechnol. 2010;37(11):1193–1201. | ||
Rodriguez A, Moreno J, Sánchez J, Moreno J, Díaz D. Production of recombinant human N-acetylgalactosamine-6-sulfate sulfatase enzyme in Pichia pastoris. Mol Genet Metab. 2013;108(2):S79–S80. | ||
Sands MS, Barker JE, Vogler C, et al. Treatment of murine mucopolysaccharidosis type VII by syngeneic bone marrow transplantation in neonates. Lab Invest. 1993;68(6):676–686. | ||
Soper BW, Lessard MD, Vogler CA, et al. Nonablative neonatal marrow transplantation attenuates functional and physical defects of beta-glucuronidase deficiency. Blood. 2001;97(5):1498–1504. | ||
Lau AA, Shamsani NJ, Winner LK, et al. Neonatal bone Marrow transplantation in MPS IIIA mice. JIMD Rep. 2013;8:121–132. | ||
Patel P, Suzuki Y, Tanaka A, et al. Impact of enzyme replacement therapy and hematopoietic stem cell therapy on growth in patients with Hunter syndrome. Mol Genet Metab Rep. 2014;1:184–196. | ||
Tanjuakio J, Suzuki Y, Patel P, et al. Activities of daily living in patients with Hunter syndrome: impact of enzyme replacement therapy and hematopoietic stem cell transplantation. Mol Genet Metab. 2015;114(2):161–169. | ||
Fujisaki J, Tokunaga Y, Takahashi T, Shimojo F, Kimura S, Hata T. Osteotropic drug delivery system (ODDS) based on biphosphonic prodrug. I.v. effects of osteotropic estradiol on bone mineral density and uterine weight in ovariectomized rats. J Drug Target. 1998;5(2):129–138. | ||
Sekido T, Sakura N, Higashi Y, et al. Novel drug delivery system to bone using acidic oligopeptide: pharmacokinetic characteristics and pharmacological potential. J Drug Target. 2001;9(2):111–121. | ||
Yokogawa K, Miya K, Sekido T, et al. Selective delivery of estradiol to bone by aspartic acid oligopeptide and its effects on ovariectomized mice. Endocrinology. 2001;142(3):1228–1233. | ||
Nishioka T, Tomatsu S, Gutierrez MA, et al. Targeted drug delivery to bone: characterization of human tissue-nonspecific alkaline phosphatase tagged with an acidic oligopeptide. Mol Genet Metab. 2006;88(3):244–255. | ||
Millán JL, Narisawa S, Lemire I, et al. Enzyme replacement therapy for murine hypophosphatasia. J Bone Miner Res. 2008;23(6):777–787. | ||
Whyte MP, Greenberg CR, Salman NJ, et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med. 2012;8(10):904–913. | ||
Oikawa H, Tomatsu S, Haupt B, Montaño AM, Shimada T, Sly WS. Enzyme replacement therapy on hypophosphatasia mouse model. J Inherit Metab Dis. 2014;37(2):309–317. | ||
Montaño AM, Oikawa H, Tomatsu S, et al. Acidic amino acid tag enhances response to enzyme replacement in mucopolysaccharidosis type VII mice. Mol Genet Metab. 2008;94(2):178–189. | ||
Cleary MA, Wraith JE. The presenting features of mucopolysaccharidosis type IH (Hurler syndrome). Acta Paediatr. 1995;84(3):337–339. | ||
Chakrapani A, Cleary MA, Wraith JE. Detection of inborn errors of metabolism in the newborn. Arch Dis Child Fetal Neonatal Ed. 2001;84(3):F205–F210. | ||
Paciotti S, Persichetti E, Pagliardini S, et al. First pilot newborn screening for four lysosomal storage diseases in an Italian region: identification and analysis of a putative causative mutation in the GBA gene. Clin Chim Acta. 2012;413(23–24):1827–1831. | ||
Scott CR, Elliott S, Buroker N, et al. Identification of infants at risk for developing Fabry, Pompe, or mucopolysaccharidosis-I from newborn blood spots by tandem mass spectrometry. J Pediatr. 2013;163(2):498–503. | ||
Lin SP, Lin HY, Wang TJ, et al. A pilot newborn screening program for Mucopolysaccharidosis type I in Taiwan. Orphanet J Rare Dis. 2013;8:1–8. | ||
Tomatsu S, Fujii T, Fukushi M, et al. Newborn screening and diagnosis of mucopolysaccharidoses. Mol Genet Metab. 2013;110(1–2):42–53. | ||
Beck M, Braun S, Coerdt W, Merz E, Young E, Sewell AC. Fetal presentation of Morquio disease type A. Prenat Diagn. 1992;12(12):1019–1029. | ||
Ohashi A, Montaño AM, Colón JE, Oguma T, Luisiri A, Tomatsu S. Sacral dimple: incidental findings from newborn evaluation. Mucopolysaccharidosis IVA disease. Acta Paediatr. 2009;98(5):768–769. | ||
Tomatsu S, Orii KO, Vogler C, et al. Mouse model of N-acetylgalactosamine-6-sulfate sulfatase deficiency (Galns-/-) produced by targeted disruption of the gene defective in Morquio A disease. Hum Mol Genet. 2003;12(24):3349–3358. | ||
Tomatsu S, Alméciga-Díaz CJ, Montaño AM, et al. Therapies for the bone in mucopolysaccharidoses. Mol Genet Metab. 2015;114(2):94–109. | ||
Bilezikian JP, Raisz LG, Martin J. Principles of Bone Biology. San Diego, CA: Academic Press; 2008. | ||
Wraith JE, Scarpa M, Beck M, et al. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr. 2008;167(3):267–277. | ||
Hendriksz C, Vellodi A, Jones S, et al. Long term outcomes of a phase1/2, multicenter, open-label, dose-escalation study to evaluate the safety, tolerability, and efficacy of BMN 110 in patients with mucopolysaccharidosis IVA (Morquio A syndrome). Mol Genet Metab. 2012;105(2):S35. | ||
Hendriksz CJ, Giugliani R, Harmatz P, et al; STRIVE Investigators. Multi-domain impact of elosulfase alfa in Morquio A syndrome in the pivotal phase III trial. Mol Genet Metab. 2015;114(2):178–185. | ||
Shah MR, Hasselblad V, Gheorghiade M, et al. Prognostic usefulness of the six-minute walk in patients with advanced congestive heart failure secondary to ischemic or nonischemic cardiomyopathy. Am J Cardiol. 2001;88(9):987–993. | ||
Stokowski LA, Pariser A, Mulberg A. FDA’s rare disease program:a rare opportunity to help kids. Medscape. 2014. Available from: http://www.medscape.com/viewarticle/821918_2. Accessed: March 18, 2014. | ||
Brooks M. FDA clears the first drug for rare Morquio A Syndrome. Medscape. 2014. Available from: http://www.medscape.com/viewarticle/820677. Accessed: February 14, 2014. | ||
Tomatsu S, Montaño AM, Ohashi A, et al. Enzyme replacement therapy in a murine model of Morquio A syndrome. Hum Mol Genet. 2008;17(6):815–824. | ||
Tomatsu S, Montaño AM, Oikawa H, et al. Enzyme replacement therapy in newborn mucopolysaccharidosis IVA mice: early treatment rescues bone lesions? Mol Genet Metab. 2015;114(2):195–202. | ||
Oldberg A, Franzen A, Heinegard D. The primary structure of a cell-binding bone sialoprotein. J Biol Chem. 1988;263(36):19430–19432. | ||
Kasugai S, Fujisawa R, Waki Y, Miyamoto K, Ohya K. Selective drug delivery system to bone: small peptide (Asp)6 conjugation. J Bone Miner Res. 2000;15(5):936–943. | ||
Nagata T, Bellows CG, Kasugai S, Butler WT, Sodek J. Biosynthesis of bone proteins [SPP-1 (secreted phosphoprotein-1, osteopontin), BSP (bone sialoprotein) and SPARC (osteonectin) in association with mineralized-tissue formation by fetal-rat calvarial cells in culture. Biochem J. 1991;274(pt2):513–520. | ||
Butler WT. The nature and significance of osteopontin. Connect Tissue Res. 1989;23(2–3):123–136. | ||
Desnick RJ, Schuchman EH. Enzyme replacement therapy for lysosomal diseases: lessons from 20 years of experience and remaining challenges. Annu Rev Genomics Hum Genet. 2012;13:307–335. | ||
Grabowski GA, Golembo M, Shaaltiel Y. Taliglucerase alfa: an enzyme replacement therapy using plant cell expression technology. Mol Genet Metab. 2014;112(1):1–8. | ||
Tiels P, Baranova E, Piens K, et al. A bacterial glycosidase enables mannose-6-phosphate modification and improved cellular uptake of yeast-produced recombinant human lysosomal enzymes. Biotechnology. 2012;30(12):1225–1231. | ||
Wendeler M, Lemm T, Weisgerber J, et al. Expression of recombinant human GM2-activator protein in insect cells: purification and characterization by mass spectrometry. Protein Expr Purif. 2003;27(2):259–266. | ||
Poutou-Piñales RA, Vanegas Niño A, Landázuri P, et al. Human sulfatase transiently and functionally active expressed in E. coli K12. Electron J Biotechnol. 2010;13:5–6. | ||
Morales-Álvarez ED, Rivera-Hoyos CM, Baena-Moncada AM, et al. Laboratory scale production of the human recombinant iduronate 2-sulfate sulfatase-Like from Pichia pastoris. Afr J Biotechnol. 2009;8:1786–1792. | ||
Espejo-Mojica A, Mosquera AR, Díaz SA, Rodríguez EA, Alméciga-Díaz CJ, Barrera LA. Production of human recombinant alpha-N-acetylglucosaminidase enzymes in two Pichia pastoris strains. Mol Genet Metab. 2013;108(2):S38–S39. | ||
Demain AL, Vaishnav P. Production of recombinant proteins by microbes and higher organisms. Biotechnol Adv. 2009;27(3):297–306. | ||
Tomatsu S, Montaño AM, Gutierrez M, et al. Characterization and pharmacokinetic study of recombinant human N-acetylgalactosamine-6-sulfate sulfatase. Mol Genet Metab. 2007;91(1):69–78. | ||
Dvorak-Ewell M, Wendt D, Hague C, et al. Enzyme replacement in a human model of mucopolysaccharidosis IVA in vitro and its biodistribution in the cartilage of wild type mice. PLoS One. 2010;5(8):e12194. | ||
Hernández A, Velásquez O, Leonardi F, et al. Effect of culture conditions and signal peptide on production of human recombinant N-acetylgalactosamine-6-sulfate sulfatase in Escherichia coli BL21. J Microbiol Biotechnol. 2013;23(5):689–698. | ||
Rodríguez A, Espejo AJ, Hernández A, et al. Characterization of a recombinant N-acetylgalactosamine-6-sulfate sulfatase produced in E. coli for enzyme replacement therapy of Morquio A disease. Process Biochem. 2012;47(12):2097–2102. | ||
Almeciga CJ, Rodríguez-López A, Sánchez J, et al. Production of an active recombinant human N-acetylgalactosamine-6-sulfate sulfatase enzyme in Pichia pastoris. Mol Genet Metab. 2014;111(2):S19. | ||
Landgrebe J, Dierks T, Schamidt B, von Figura K. The human SUMF1 gene, required for posttranslational sulfatase modification, defines a new gene family which is conserved from pro- to eukaryotes. Gene. 2003;316:47–56. | ||
Tovey MG, Lallemand C. Immunogenicity and other problems associated with the use of biopharmaceuticals. Ther Adv Drug Saf. 2011;2(3):113–128. | ||
Du H, Schiavi S, Levine M, Mishra J, Heur M, Grabowski GA. Enzyme therapy for lysosomal acid lipase deficiency in the mouse. Hum Mol Genet. 2001;10(16):1639–1648. | ||
Wang C, Hwu W, Lin K. Long-term follow-up of a girl with Maroteaux-Lamy syndrome after bone marrow transplantation. World J Pediatr. 2008;4(2):152–154. | ||
Boelens J. Trends in haematopoietic cell transplantation for inborn errors of metabolism. J Inherit Metab Dis. 2006;29(2–3):413–420. | ||
Orchard P, Blazar B, Wagner J, Charnas L, Krivit W, Tolar J. Hematopoietic cell therapy for metabolic disease. J Pediatr. 2007;151(4):340–346. | ||
Rovelli AM. The controversial and changing role of haematopoietic cell transplantation for lysosomal storage disorders: an update. Bone Marrow Transplant. 2008;41(suppl 2):S87–S89. | ||
Vellodi A, Young E, Cooper A, Lidchi V, Winchester B, Wraith JE. Long-term follow-up following bone marrow transplantation for Hunter disease. J Inherit Metab Dis. 1999;22(5):638–648. | ||
Tolar J, Grewal SS, Bjoraker KJ, et al. Combination of enzyme replacement and hematopoietic stem cell transplantation as therapy for Hurler syndrome. Bone Marrow Transplant. 2008;41(6):531–535. | ||
Tanaka A, Okuyama T, Suzuki Y, et al. Long-term efficacy of hematopoietic stem cell transplantation on brain involvement in patients with mucopolysaccharidosis type II: a nationwide survey in Japan. Mol Genet Metab. 2012;107(3):513–520. | ||
Tomatsu S, Yasuda E, Patel P, et al. Morquio A syndrome: diagnosis and current and future therapies. Pediatr Endocrinol Rev. 2014;12(suppl 1):141–151. | ||
Yamada Y, Kato K, Sukegawa K, et al. Treatment of MPS VII (Sly disease) by allogeneic BMT in a female with homozygous A619V mutation. Bone Marrow Transplant. 1998;21(6):629–634. | ||
Khanna G, Van Heest AE, Agel J, et al. Analysis of factors affecting development of carpal tunnel syndrome in patients with Hurler syndrome after hematopoietic cell transplantation. Bone Marrow Transplant. 2007;39(6):331–334. | ||
Hobbs JR, Hugh-Jones K, Barrett AJ, et al. Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet. 1981;2(8249):709–712. | ||
Guffon N, Souillet G, Maire I, Straczek J, Guibaud P. Follow-up of nine patients with Hurler syndrome after bone marrow transplantation. J Pediatr. 1998;133(1):119–125. | ||
Vellodi A, Young EP, Cooper A, et al. Bone marrow transplantation for mucopolysaccharidosis type I: experience of two British centres. Arch Dis Child. 1997;76(2):92–99. | ||
Staba SL, Escolar ML, Poe M, et al. Cord-blood transplants from unrelated donors in patients with Hurler’s syndrome. N Engl J Med. 2004;350(19):1960–1969. | ||
Krivit W, Pierpont ME, Ayaz K, et al. Bone-marrow transplantation in the Maroteaux-Lamy syndrome (mucopolysaccharidosis type VI). Biochemical and clinical status 24 months after transplantation. N Engl J Med. 1984;311(25):1606–1611. | ||
Boelens JJ, Aldenhoven M, Purtill D, et al; Inborn Errors Working Party of European Blood and Marrow Transplant group; Duke University Blood and Marrow Transplantation Program; Centre for International Blood and Marrow Research. Outcomes of transplantation using various hematopoietic cell sources in children with Hurler syndrome after myeloablative conditioning. Blood. 2013;121:3981–3987. | ||
Herskhovitz E, Young E, Rainer J, et al. Bone marrow transplantation for Maroteaux-Lamy syndrome (MPS VI): long-term follow-up. J Inherit Metab Dis. 1999;22:50–62. | ||
Malatack J, Consolini D, Bayever E. The status of hematopoietic stem cell transplantation in lysosomal storage disease. Pediatr Neurol. 2003;29:391–403. | ||
Beck M. New therapeutic options for lysosomal storage disorders: enzyme replacement, small molecules and gene therapy. Hum Genet. 2007;121:1–22. | ||
Turbeville S, Nicely H, Rizzo JD, et al. Clinical outcomes following hematopoietic stem cell transplantation for the treatment of mucopolysaccharidosis VI. Mol Genet Metab. 2011;102:111–115. | ||
Guffon N, Bertrand Y, Forest I, Fouilhoux A, Froissart R. Bone marrow transplantation in children with Hunter syndrome: outcome after 7 to 17 years. J Pediatr. 2009;154:733–737. | ||
Araya K, Sakai N, Mohri I, et al. Localized donor cells in brain of a Hunter disease patient after cord blood stem cell transplantation. Mol Genet Metab. 2009;98:255–263. | ||
Field RE, Buchanan JA, Copplemans MG, Aichroth PM. Bone-marrow transplantation in hurler’s syndrome: effect on skeletal development. J Bone Joint Surg Br. 1994;76(6):975–981. | ||
Stoop FJ, Kruyt MC, van der Linden MH, Sakkers RJ, van Hasselt PM, Castelein RM. Prevalence and development of orthopaedic symptoms in the dutch hurler patient population after haematopoietic stem cell transplantation. JIMD Rep. 2013;9:17–29. | ||
Vellodi A, Young EP, Cooper A, et al. Bone marrow transplantation for mucopolysaccharidosis type I: experience of two British centres. Arch Dis Child. 1997;76(2):92–99. | ||
Souillet G, Guffon N, Maire I, et al. Outcome of 27 patients with hurler’s syndrome transplanted from either related or unrelated haematopoietic stem cell sources. Bone Marrow Transplant. 2003;31(12):1105–1117. | ||
Masterson EL, Murphy PG, O’Meara A, Moore DP, Dowling FE, Fogarty EE. Hip dysplasia in hurler’s syndrome: orthopaedic management after bone marrow transplantation. J Pediatr Orthop. 1996;16(6):731–733. | ||
Taylor C, Brady P, O’Meara A, Moore D, Dowling F, Fogarty E. Mobility in Hurler syndrome. J Pediatr Orthop. 2008;28(2):163–168. | ||
Weisstein JS, Delgado E, Steinbach LS, Hart K, Packman S. Musculoskeletal manifestations of Hurler syndrome: long-term follow-up after bone marrow transplantation. J Pediatr Orthop. 2004;24(1):97–101. | ||
Aldenhoven M, Sakkers RJ, Boelens J, de Koning TJ, Wulffraat NM. Musculoskeletal manifestations of lysosomal storage disorders. Ann Rheum Dis. 2009;68(11):1659–1665. | ||
Langereis EJ, Borgo A, Crushell E, et al. Treatment of hip dysplasia in patients with mucopolysaccharidosis type I after hematopoietic stem cell transplantation: results of an international consensus procedure. Orphanet J Rare Dis. 2013;8:1–9. | ||
Prasad VK, Kurtzberg J. Transplant outcomes in mucopolysaccharidoses. Semin Hematol. 2010;47(1):59–69. | ||
Mitchell R, Nivison-Smith I, Anazodo A, et al. Outcomes of haematopoietic stem cell transplantation for inherited metabolic disorders: a report from the Australian and New Zealand Children’s Haematology Oncology Group and the Australasian Bone Marrow Transplant Recipient Registry. Pediatr Transplant. 2013;17(6):582–588. | ||
Chinen Y, Higa T, Tomatsu S, Suzuki Y, Orii T, Hyakuna N. Long-term therapeutic efficacy of allogenic bone marrow transplantation in a patient with mucopolysaccharidosis IVA. Mol Genet Metab Rep. 2014;1:31–41. | ||
Pievani A, Azario I, Antolini L, et al. Neonatal bone marrow transplantation prevents bone pathology in a mouse model of mucopolysaccharidosis type I. Blood. Epub 2014 Oct 8. | ||
Aronson JK. Biomarkers and surrogate endpoints. Br J Clin Pharmacol. 2005;59(5):491–494. | ||
Aronson JK. Research priorities in biomarkers and surrogate end-points. Br J Clin Pharmacol. 2012;73(6):900–907. | ||
Tomatsu S, Okamura K, Taketani T, et al. Development and testing of new screening method for keratan sulfate in mucopolysaccharidosis IVA. Pediatr Res. 2004;55(4):592–597. | ||
Oguma T, Tomatsu S, Okazaki O. Analytical method for determination of disaccharides derived from keratan sulfates in human serum and plasma by high-performance liquid chromatography/turbo-ionspray ionization tandem mass spectrometry. Biomed Chromatogr. 2007;21(4):356–362. | ||
Tomatsu S, Montaño AM, Oguma T, et al. Validation of keratan sulfate level in mucopolysaccharidosis IVA by liquid tandem mass spectrometry method. J Inherit Metab Dis. 2010;33(suppl 3):S35–S42. | ||
Hintze JP, Tomatsu S, Fujii T, et al. Comparison of liquid chromatography-tandem mass spectrometry and sandwich ELISA for determination of keratan sulfate in plasma and urine. Biomark Insights. 2011;6:69–78. | ||
Martell LA, Cunico RL, Ohh J, Fulkerson W, Furneaux R, Foehr ED. Validation of an LC-MS/MS assay for detecting relevant disaccharides from keratan sulfate as a biomarker for Morquio A syndrome. Bioanalysis. 2011;3(16):1855–1866. | ||
Dũng VC, Tomatsu S, Montaño AM, et al. Mucopolysaccharidosis IVA: correlation between genotype, phenotype and keratan sulfate levels. Mol Genet Metab. 2013;110(1–2):129–138. | ||
Shimada T, Tomatsu S, Mason RW, et al. Di-sulfated keratan sulfate as a novel biomarker for mucopolysaccharidosis IVA. J Inherit Metab Dis Reports. In press 2014. | ||
Harmatz P, Mengel KE, Giugliani R, et al. The Morquio A Clinical Assessment Program: baseline results illustrating progressive, multisystemic clinical impairments in Morquio A subjects. Mol Genet Metab. 2013;109(1):54–61. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.