Back to Journals » Drug Design, Development and Therapy » Volume 9
Impact of crystal polymorphism on the systemic bioavailability of rifaximin, an antibiotic acting locally in the gastrointestinal tract, in healthy volunteers
Authors Blandizzi C, Viscomi GC, Scarpignato C
Received 11 August 2014
Accepted for publication 26 September 2014
Published 16 December 2014 Volume 2015:9 Pages 1—11
DOI https://doi.org/10.2147/DDDT.S72572
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Corrado Blandizzi,1 Giuseppe Claudio Viscomi,2 Carmelo Scarpignato3
1Division of Pharmacology and Chemotherapy, Department of Clinical and Experimental Medicine, University of Pisa, Pisa, 2Research and Development Division, Alfa Wassermann SpA, Bologna, 3Clinical Pharmacology and Digestive Pathophysiology Unit, Department of Clinical and Experimental Medicine, University of Parma, Parma, Italy
Background: Rifaximin is an antibiotic, acting locally in the gastrointestinal tract, which may exist in different crystal as well as amorphous forms. The current marketed rifaximin formulation contains polymorph alpha, the systemic bioavailability of which is very limited. This study compared the pharmacokinetics of this formulation with those of the amorphous form.
Methods: Amorphous rifaximin was specifically prepared for the study and formulated as the marketed product. Two doses (200 mg and 400 mg) of both formulations were given to two groups of 12 healthy volunteers of either sex according to a single-blind, randomized, two-treatment, single-dose, two-period, cross-over design. Plasma and urine samples were collected at preset times (for 24 hours or 48 hours, respectively) after dosing, and assayed for rifaximin concentrations by high-performance liquid chromatography-mass spectrometry.
Results: For both dose levels, peak plasma concentration, area under the concentration-time curve, and cumulative urinary excretion were significantly higher after administration of amorphous rifaximin than rifaximin-α. Ninety percent confidence intervals for peak plasma concentration, area under the concentration-time curve, and urinary excretion ratios were largely outside the upper limit of the accepted (0.80–1.25) range, indicating higher systemic bioavailability of the amorphous rifaximin. The few adverse events recorded were not serious and not related to the study medications.
Conclusion: Rifaximin-α, a crystal polymorph, does differ from the amorphous form, the latter being systemically more bioavailable. In this regard, care must be taken when using – as a medicinal product – a formulation containing even small amounts of amorphous form, which may alter the peculiar pharmacologic properties of this poorly absorbed antibiotic.
Keywords: rifaximin, crystal polymorphism, systemic bioavailability, gastrointestinal tract
Introduction
Locally acting drugs are applied locally and are assumed to exert their effect at the site of application.1,2 Examples include, amongst others, orally administered formulations designed to achieve a local effect within the gastrointestinal tract. In these cases, a systemic action, if any, would be considered undesirable and could actually elicit adverse effects.1,2
Through variations in local and/or systemic bioavailability, changes in formulation or dosage form may influence the efficacy and/or safety of locally acting medicines. In addition to formulation, the physicochemical characteristics of the active ingredient are also relevant to local and/or systemic bioavailability. Indeed, the chemical structure of the molecule, its polar surface area, pH partition, particle size, salt form, drug complexation, as well as crystal forms or amorphous state can all affect dissolution and absorption rates.3,4 While most of the above parameters have been extensively studied for locally acting compounds, crystal polymorphism has often been overlooked.
Polymorphism, which is very common in pharmaceuticals,5–8 is the ability of a molecule to assemble into multiple crystal structures. Different polymorphs have different arrangements of atoms within the unit cell, and this can quite often have a remarkable impact on the physicochemical properties of the crystallized compound.
Different polymorphs of a drug can display different chemicophysical properties, including stability and reactivity, dissolution rate and solubility, which can affect pharmacokinetics (PKs) and pharmacodynamics (PDs).5,7 Relevant examples of the impact of polymorphism on systemic bioavailability have been previously reported.9–12 In extreme cases, a polymorph can even be ineffective, as occurred with polymorph II of ritonavir.13
In contrast with polymorphs, amorphous forms consist of disordered molecular arrangements and do not display three-dimensional crystalline lattices.14 In general, compared with crystalline polymorphs, amorphous forms tend to have a higher dissolution rate and solubility, which may increase the rate and extent of their oral absorption.15
Rifaximin (4-deoxy-4′-methylpyrido[1′,2′-1,2]imidazo [5,4-c]rifamycin SV) is a synthetic derivative of rifamycin, with very low gastrointestinal absorption, but still displaying a broad spectrum of antibacterial activity.16–18 Being virtually nonabsorbed, its gastrointestinal bioavailability is high, with fecal concentrations largely exceeding minimum inhibitory concentrations against pathogenic enterobacteria, while its limited impact on extragastrointestinal sites minimizes the risk of antimicrobial resistance and systemic adverse events.19 With appreciation of the pathogenic role of gut flora in several gastrointestinal diseases, the use of rifaximin has been extended from gastrointestinal infections to hepatic encephalopathy, small intestine bacterial overgrowth (SIBO), inflammatory bowel disease, and colonic diverticular disease.19,20
According to the European Pharmacopoeia, rifaximin shows crystal polymorphism,21 and several polymorphs (α, β, γ, δ, ε) have been described.22 In vitro studies show different dissolution and solubility rates for these polymorphs, and in vivo investigations in dogs found different PK patterns, with δ and γ polymorphs displaying the highest systemic bioavailability.22
Previous PK studies in healthy volunteers and patients with inflammatory bowel disease or intestinal infections showed minimal absorption of rifaximin after single and repeated doses.20 Of note, these studies were performed with the rifaximin product marketed before the discovery of rifaximin polymorphism, while the current formulation contains only polymorph-α,23 which is widely recognized as a poorly absorbed antibiotic.20,24–27
Preclinical studies have shown that polymorphs α and β of rifaximin are the least absorbed ones. In vitro dissolution tests suggested also that the PK behavior of amorphous rifaximin is similar to that of γ polymorph, thus implying a higher systemic bioavailability than polymorph-α.22 Indeed, preliminary animal studies have shown that this is the case (Viscomi, unpublished observations).
Generic formulations of rifaximin are available in some countries. In a previous paper,28 we compared the PKs of a generic rifaximin formulation with those of the branded product (the latter containing only polymorph-α) and found that most PK parameters were significantly higher after administration of generic rifaximin. In particular, the differences for the highest concentration achieved in plasma (Cmax), area under the concentration-time curve (AUC), and cumulative urinary excretion between the generic formulation and the branded product ranged from 165% to 345%. X-ray power diffraction analysis of the generic formulation showed the presence of both rifaximin-α and amorphous rifaximin, which we thought could have contributed to the increased systemic bioavailability of the generic formulation.28 Since the key feature of this broad-spectrum antibiotic is to be poorly absorbable, the increased absorption could well have clinical consequences, especially in long-term use.
The present investigation was therefore carried out to prove (or disprove) our hypothesis that the presence of amorphous rifaximin is responsible for the increased systemic absorption. To this end, we prepared ad hoc an amorphous form of rifaximin, formulated as film-coated tablets, using the same composition and manufacturing procedure employed to produce the reference branded product, and compared its plasma PKs with those of polymorph-α in healthy volunteers. Since data concerning the systemic bioavailability of amorphous rifaximin in humans are lacking, this study will also bridge this gap.
Materials and methods
Volunteers
Twenty-four healthy Caucasian adult volunteers of either sex and aged 18–55 years participated in this study. At the time of enrolment, they were informed of the purpose, methods, and potential hazards of the study, and were requested to sign their written informed consent. They were also informed of the ability to withdraw from the study at any time. The clinical evaluations performed to assess the health status of volunteers (both before and after participation in the study) included medical history, physical examination, body mass index, vital signs (heart rate, systolic and diastolic blood pressure, body temperature), electrocardiography, and laboratory investigations. The results of clinical evaluations were documented in individual case report forms. Women were screened for β-human chorionic gonadotropin to rule out pregnancy, and were requested to use contraception throughout the study period. Exclusion criteria included body mass index ≤18 or ≥30 kg/m2, positive urinary drug screening, positive tests for human immunodeficiency virus or hepatitis B/C, history of major diseases, allergies, intolerance to any ingredients in the study drugs, use of medicinal products within 4 weeks of starting the study, history of alcohol or drug abuse, drinking excessive amounts of beverages containing caffeine or wine (>0.5 L/day) or spirits (>50 mL/day), and abnormal diet or eating habits. The volunteers were requested to report any abnormality occurring during and after the study.
Design of study
The study was performed in accordance with a single-blind, randomized, two-treatment, single-dose, two-period, cross-over design, with a washout period of 7 days. It was approved by the ethics committee of Canton Ticino and Swissmedic (Switzerland), and was conducted in accordance with International Conference on Harmonisation guidelines for Good Clinical Practice. The study procedures were in compliance with the Declaration of Helsinki in its last amendment (Seoul 2008).
Since the amorphous rifaximin was being tested in humans for the first time, volunteers were allocated to six cohorts as a safety measure, in accordance with current regulatory recommendations.29,30 Subjects within each cohort received the study medication only in the absence of unexpected serious adverse events or safety concerns related to the study medication in the preceding cohort. A total of 12 volunteers were allocated to the first four cohorts (three subjects each), and received a single 200 mg dose (one tablet) of the test drug (amorphous rifaximin) and the reference drug (rifaximin polymorph-α; Normix®) on different days according to the cross-over design. The remaining 12 volunteers were allocated to the last two cohorts (six subjects each), and received a single 400 mg dose (2×200 mg tablets) of the test drug (amorphous rifaximin) and reference drug (rifaximin polymorph-α; Normix®), on different days according to the cross-over design. Tablets were administered with 250 mL of water at 8 am under fasting conditions. About 4, 8, and 12 hours after drug intake, a lunch (1,200 kcal), a snack (150 kcal), and a dinner (900 kcal) were served, respectively, with water ad libitum. After drug intake, subjects were requested to drink water in accordance with the following schedule: 500 mL (one glass of 125 mL approximately every hour) of plain mineral water at room temperature during each of the 4-hour intervals from drug administration time until 12 hours after dosing, and then ad libitum until the end of urine collection (ie, 48 hours post-dosing).
Ten milliliter venous blood samples were collected into tubes containing sodium heparin at preset time intervals of 0 (predosing) and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, and 24 hours post-dosing and kept on ice. Plasma was separated from blood within 20 minutes by centrifugation at 2,000 g for 10 minutes at 4°C. Each plasma sample was split into two aliquots and stored at −80°C±10°C. Urine was collected predosing and at intervals of 0–4, 4–8, 8–12, 12–24, 24–48 hours post-dosing into weighed flasks and refrigerated at 4°C–6°C. The weight of each urine fraction was recorded, and a 100 mL sample was split into two aliquots and stored at −20°C±5°C.
Preparation and characterization of amorphous rifaximin
Amorphous rifaximin was prepared in a GPCG 60 fluid bed dryer (Glatt, Ramsey, NJ, USA), equipped with a 1.8 mm spraying nozzle. Forty kg of rifaximin-α were charged and added with 457.2 L of 96% ethanol (v/v). The suspension was stirred continuously until complete dissolution of rifaximin. The ethanol solution was sprayed into the fluid bed with a pressure of 1–1.5 bar through the nozzle under a flow of warm air. At the end of the spraying phase, the solid rifaximin powder was further dried to remove the excess solvent and then characterized by X-ray power diffraction.31
X-ray powder diffraction analysis
X-ray powder diffraction data were collected on a Panalytical X’Pert automated diffractometer in a Bragg-Brentano geometry, equipped with a graphite monochromator. The Cu anode was used as an X-ray source at 40 kV and 40 mA. Data were collected at room temperature, start angle 2q 1/4 3″, stop angle 2q 1/4 30″. The instrument was configured with 1/2″ divergence and 0.1 mm receiving slits, respectively. Samples, stored in stoppered glass bottles, were prepared immediately before analysis, avoiding manipulation as much as possible to reduce the risk of water uptake.
Preparation of amorphous rifaximin tablets and dissolution test
Amorphous rifaximin was used to formulate a single batch of 200 mg film-coated tablets, according to the same composition and manufacturing procedure employed to produce Normix®.32 The dissolution test was performed according to the European Pharmacopoeia,33 dissolving each tablet in 900 mL of aqueous solution (phosphate buffer at pH 7.4). In one set of experiments, sodium lauryl sulfate was added to the aqueous solution up to 0.225% (v/v) in order to reach the sink conditions.
Study medications
The 200 mg film-coated tablets containing amorphous rifaximin were prepared specifically in one batch for this study as reported above. The tablets of Normix® employed in the study came from the very same batch (number 6188).
Safety evaluation
Volunteers were asked about the occurrence of any adverse event after their admission to the clinical unit, both before administration of the study drugs and throughout the study period, every 4 hours, until their discharge. Radial pulse, blood pressure, respiratory rate, and body temperature were monitored as vital signs. Clinical evaluations, including electrocardiography, blood tests, and urine analysis, were performed before enrolment and on the last day of the study.
Rifaximin assay
Rifaximin concentrations in plasma and urine were measured by liquid chromatography-tandem mass spectrometry, as previously described;28 the lower limit of quantification being 0.5 ng/mL in both biological fluids. Each sample was assayed in duplicate. Intra-assay precision (coefficient of variation, CV%) for the quality control samples was ≤7.2 and mean accuracy ranged from −5.3 to −0.3% of the nominal concentration. Interassay precision (CV%) was ≤5.1 and ACC% ranged from −1.1 to +2.3 of the nominal concentration. No significant interfering peaks were found at retention times of rifaximin and of internal standard.
Pharmacokinetic evaluations
Noncompartmental analysis was used to calculate the PK parameters and was performed using WinNonLin™ software (Pharsight, Mountain View, CA, USA). The Cmax, the time needed to achieve Cmax, the area under the drug plasma concentration-time curve from time 0 to infinity (AUC0–∞), the AUC from time 0 to the time of the last quantifiable drug concentration (AUC0–t), the plasma drug elimination half-life (t1/2), and cumulative urinary excretion (Ae0–48h) were estimated from individual concentration-time curves. The first order elimination rate constant was estimated by linear regression of time versus the log of drug concentration using the terminal (log-linear) portion of the curve. AUC0–t was calculated by the linear trapezoidal rule. Extrapolation to infinity for estimation of AUC0–∞ was obtained by dividing the last quantifiable drug concentration by the elimination rate constant and adding this value to AUC0–t.34 The AUC0–∞ has a considerable (>30%) extrapolated portion, which somewhat weakens its value and interpretation. However, this parameter is almost always calculated in PK studies.
Statistical analysis
The results are presented as the mean ± standard error of mean. The parametric general linear model for statistical analysis included factors accounting for period, sequence, and rifaximin formulations. Comparisons for systemic bioavailability of the test drug versus the reference drug were performed according to European Medicines Agency recommendations2 and were defined as the 90% confidence intervals of Cmax, AUC0–∞ (or AUC0–t) and Ae0–48h ratios falling within the range of 0.80–1.25. For this purpose, Cmax, AUC0–∞, AUC0–t, and Ae0–48h values were log-natural transformed and used to calculate the ratios of each test drug over the respective reference drug. Analysis of variance was applied to these values, and the residual variance was used in Schuirmann’s test35 and for computing the 90% confidence intervals of mean ratios.
Results
Characterization of amorphous rifaximin
Conversely from rifaximin-α, X-ray diffractogram of the amorphous rifaximin preparation showed only two halo-peaks, with a maximum at 7.75°±0.2°, 14.54°±0.2°, and 18.33°±0.2° 2θ and the absence of signals of crystallinity (Figure 1).
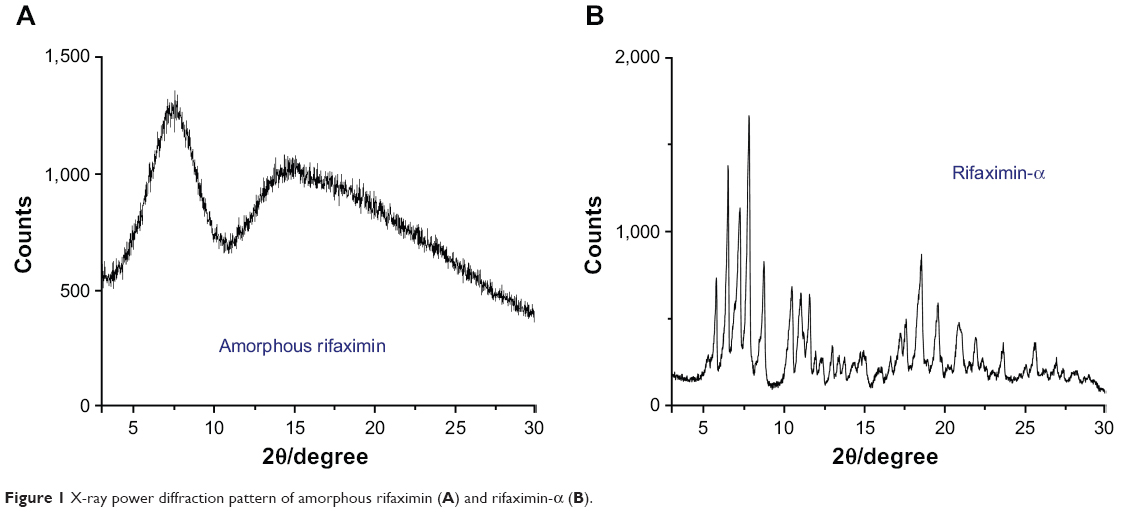
|
Figure 1 X-ray power diffraction pattern of amorphous rifaximin (A) and rifaximin-α (B). |
Dissolution test of amorphous rifaximin
Dissolution profiles of rifaximin tablets (with or without sodium lauryl sulfate) are shown in Figure 2. To compare properly the two profiles, the so-called f1 (difference) and f2 (similarity) factors were calculated.36,37 It is generally accepted that an f2 value ranging from 50 to 100 indicates similarity.36,37 In the absence of sodium lauryl sulfate, f1 and f2 values were 40.1 and 36.6, respectively, while in the presence of 0.225% sodium lauryl sulfate f1 and f2 values were 387.4 and 44.5, respectively.
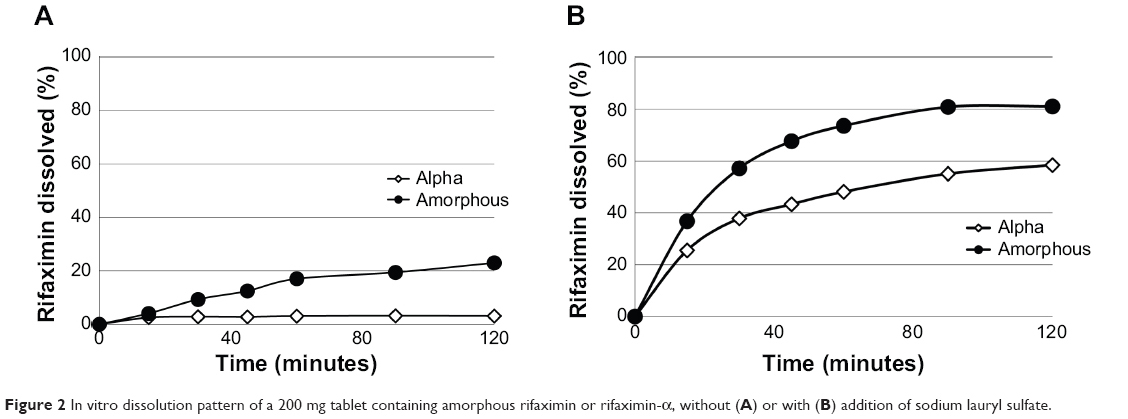
|
Figure 2 In vitro dissolution pattern of a 200 mg tablet containing amorphous rifaximin or rifaximin-α, without (A) or with (B) addition of sodium lauryl sulfate. |
Demographic characteristics of healthy volunteers
The characteristics of healthy subjects, who participated to the present studies, are displayed in Table 1.
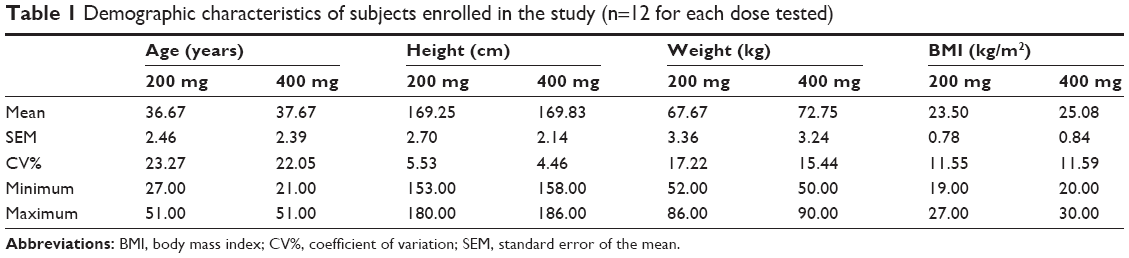
|
Table 1 Demographic characteristics of subjects enrolled in the study (n=12 for each dose tested) |
Plasma and urinary PK profiles
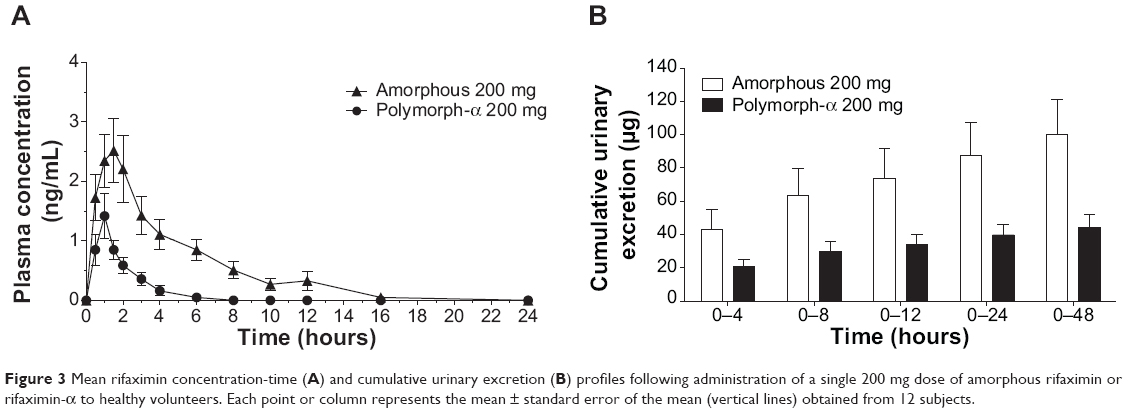
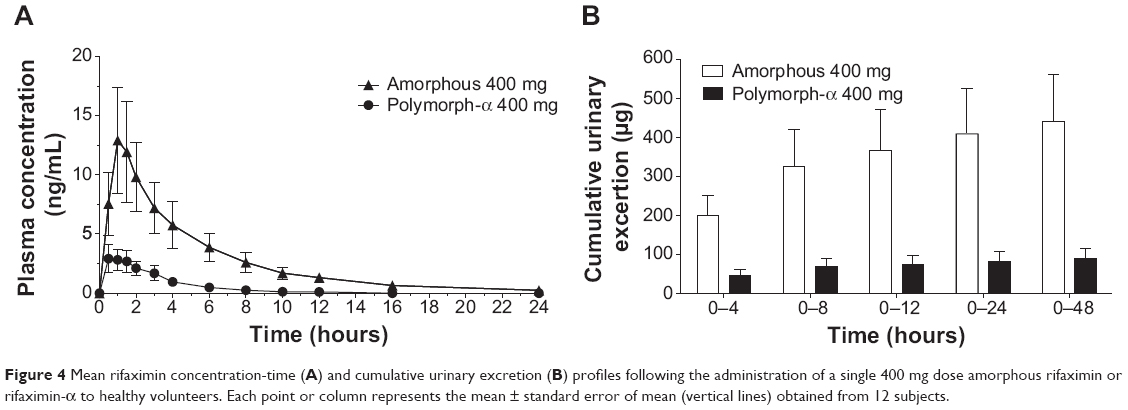
Mean plasma and urinary profiles of the two subgroups (n=12 each), treated with amorphous rifaximin or rifaximin-α, are shown in Figures 3 and 4 for the doses of 200 mg and 400 mg, respectively. The respective PK parameters are reported in Table 2. In most subjects, the first plasma concentration of rifaximin was detected at the first blood sampling (30 min after dosing) for both rifaximin formulations and doses. Concentrations then increased up to peak, which, in subjects treated with amorphous rifaximin, was achieved at 1.96 (range 0.50–12.0) hours and 1.71 (range 0.50–4.0) hours for the dose of 200 mg and 400 mg, respectively. In subjects receiving rifaximin-α, the peak was reached at 1.04 (range 0.50–2.0) hours and 1.21 (0.50–4.0) hours after dosing with 200 mg and 400 mg, respectively.
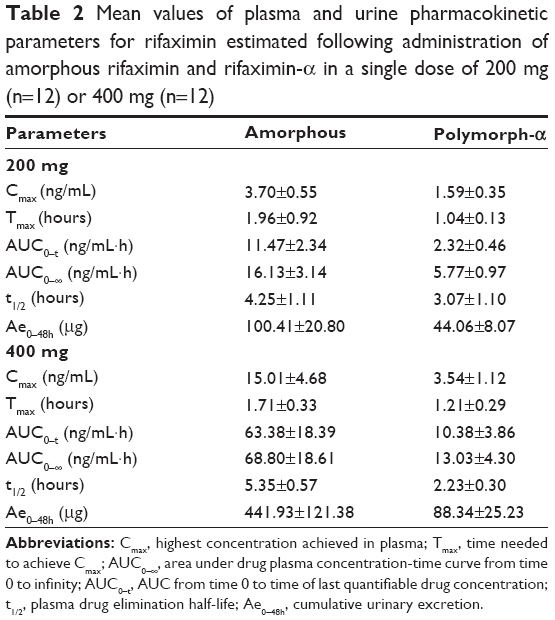
|
Table 2 Mean values of plasma and urine pharmacokinetic parameters for rifaximin estimated following administration of amorphous rifaximin and rifaximin-α in a single dose of 200 mg (n=12) or 400 mg (n=12) |
|
Figure 3 Mean rifaximin concentration-time (A) and cumulative urinary excretion (B) profiles following administration of a single 200 mg dose of amorphous rifaximin or rifaximin-α to healthy volunteers. Each point or column represents the mean ± standard error of the mean (vertical lines) obtained from 12 subjects. |
|
Figure 4 Mean rifaximin concentration-time (A) and cumulative urinary excretion (B) profiles following the administration of a single 400 mg dose amorphous rifaximin or rifaximin-α to healthy volunteers. Each point or column represents the mean ± standard error of mean (vertical lines) obtained from 12 subjects. |
In subjects treated with 200 mg, rifaximin plasma concentrations were lower after administration of rifaximin-α than amorphous rifaximin. In this setting, rifaximin concentrations were never above the lower limit of quantification in plasma samples collected after 8 hours from dosing with rifaximin-α, while rifaximin could be detected in plasma up to 16 hours from dosing with amorphous rifaximin. Likewise, in volunteers treated with 400 mg, detectable rifaximin plasma concentrations were lower after administration of rifaximin-α than amorphous rifaximin. With this dose, rifaximin plasma concentrations was detected in 2/12 subjects after 12 hours and 0/12 after 24 hours from dosing with rifaximin-α, while rifaximin plasma concentrations could be measured in 10/12 subjects after 12 hours and 4/12 subjects 24 hours after administration of amorphous rifaximin.
After administration of rifaximin-α and amorphous rifaximin at both 200 mg and 400 mg, urine rifaximin concentrations were detected at all collection intervals in all subjects, with the exception of one (rifaximin concentration at the 24–48 hour interval was below the lower limit of quantification after 200 mg of rifaximin-α). For both rifaximin forms and doses, the highest urine concentration was measured at the first interval (0–4 hours), with the exception of three volunteers, who displayed the highest concentration at the 4–8 hour interval.
Rifaximin plasma and urine concentration-time profiles and PK parameters showed relevant differences when amorphous rifaximin and rifaximin-α were compared. As shown in Table 2, most PK parameters were significantly higher after administration of amorphous rifaximin than rifaximin-α. In particular, the differences for Cmax, AUC0–t, AUC0–∞, and Ae0–48h between the amorphous form and polymorph-α ranged from 128% to 394% at the dose of 200 mg, and from 324% to 510% at the dose of 400 mg. Moreover, the PK parameters estimated after administration of amorphous rifaximin 400 mg were from 4.1-fold to 5.5-fold higher than those obtained with 200 mg, while the PK parameters estimated for rifaximin-α 400 mg were from 2.0-fold to 4.5-fold higher.
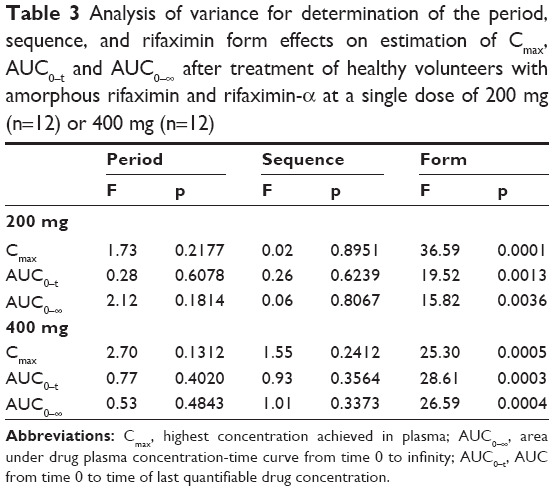
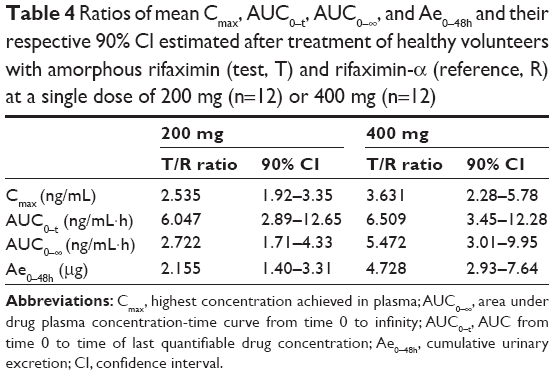
The statistical evaluation for cross-over design by means of analysis of variance indicated the lack of period and sequence effects both for the dose of 200 mg and 400 mg, while it showed significant effects of the rifaximin forms at both doses, indicating significant differences in systemic bioavailability (Table 3). Consistently with this observation, the 90% confidence interval of the ratios of mean Cmax, AUC0–t, AUC0–∞, and Ae0–48h were largely outside the upper limit of the 0.80–1.25 interval recommended by the European Medicines Agency guideline2 (Table 4). The higher systemic bioavailability of amorphous rifaximin 200 mg and 400 mg, as compared with rifaximin-α, was also confirmed by the Schuirmann’s test.
|
Table 3 Analysis of variance for determination of the period, sequence, and rifaximin form effects on estimation of Cmax, AUC0–t and AUC0–∞ after treatment of healthy volunteers with amorphous rifaximin and rifaximin-α at a single dose of 200 mg (n=12) or 400 mg (n=12) |
|
Table 4 Ratios of mean Cmax, AUC0–t, AUC0–∞, and Ae0–48h and their respective 90% CI estimated after treatment of healthy volunteers with amorphous rifaximin (test, T) and rifaximin-α (reference, R) at a single dose of 200 mg (n=12) or 400 mg (n=12) |
Safety
No serious adverse events occurred during the study. Six subjects developed a total of eight adverse events (six mild, two moderate). Headache occurred in six subjects (one after amorphous rifaximin 200 mg, two after amorphous rifaximin 400 mg, and three after rifaximin-α 400 mg). Vomiting occurred in two of the above six subjects (one after amorphous rifaximin 200 mg and one after rifaximin-α 400 mg). All adverse events resolved spontaneously, and were not considered to be related to the study medications upon application of Naranjo et al’s algorithm.38
Discussion
Crystalline polymorphism exhibited by pharmacologically active compounds has received widespread attention since the early report by Aguiar et al.9 Given that polymorphic forms can display different physicochemical properties, which in turn may affect chemical processing and/or manufacturing of medicinal products, as well as PK/PD profiles,5,8 it has been recommended that attention be paid to solid-state forms of compounds from the very beginning of their development. All long-lived forms of a drug should indeed be revealed as early as possible.39,40 Unfortunately, this is not always the case, since some compounds, such as rifaximin, had been employed in clinical practice for decades before the discovery of their polymorphism.
After the case of ritonavir polymorph II,13 regulatory authorities started to pay more attention to polymorphism.1,2,41 Since differences between polymorphs and the amorphous form of a drug might even be more relevant than those among different crystalline forms, the issue can become very critical from a therapeutic standpoint when the amorphous form is administered instead of its crystalline counterpart. Provided this happens, it might translate into remarkable bioavailability changes, which could affect the efficacy and safety of systemic drugs, while impacting mostly on safety when locally acting compounds are considered.
Dissolution tests, performed with identical drug formulations containing amorphous rifaximin and polymorph-α, respectively, showed that the different water solubility of the two rifaximin forms persists in the product formulations, even in the presence of sodium lauryl sulfate, a surfactant that improves the solubility of rifaximin. Calculation of f1 (difference) and f2 (similarity) factors demonstrated no similarity between the dissolution profiles of tablets containing amorphous rifaximin and rifaximin-α.
Our PK investigations clearly show that, in comparison with polymorph-α, amorphous rifaximin displays higher bioavailability, reflected by higher Cmax, AUC, and urinary excretion. While this difference was evident with both doses, the higher the dose the higher the ratios observed. Since rifaximin is usually given at daily doses of 800–1,200 mg, it is conceivable that, with the current regimens used in clinical practice, systemic absorption of the amorphous form would become considerable.
During the 48 hours after its administration, less than 50% of rifaximin was excreted in the urine. Rifaximin belongs to the rifamycin family, all members of which display considerable biliary excretion, reaching minimum inhibitory concentrations high enough to allow their use in the treatment of biliary infections.42 Although at lower concentrations, also rifaximin is excreted through the bile.43 This biliary excretion can account for the amount of rifaximin not recovered in the urine.
All the above results agree with previous preclinical findings and, to the best of our knowledge, represent the first evidence of the impact of polymorphism on the bioavailability of locally acting oral drugs. In this study, only rifaximin-α was investigated, but additional studies should be performed to evaluate the PKs of the other polymorphs.
Drug polymorphs occur during the synthesis and purification of drugs. The occurrence of a given polymorph depends on the conditions of synthesis and crystallization.7,8 The production process, which must be strictly standardized, generally leads to formation of a given polymorph rather than the amorphous form of a drug. The increased absorption of amorphous rifaximin could translate into changes in local (ie, intraluminal) bioavailability, which might affect clinical efficacy. In addition, the increased systemic bioavailability of amorphous rifaximin raises some clinically relevant concerns. Indeed, for a poorly absorbed antibiotic, the antimicrobial activity of which is intended to be exerted within the gastrointestinal tract, enhanced systemic absorption might increase the risk of adverse events.44,45 In the case of rifaximin with the polymorph-α contained in the marketed formulation, both the short-term and long-term tolerability are extremely good,19,25–27 most likely because of the lack of systemic absorption. However, after a single 400 mg dose of amorphous rifaximin, the Cmax increased by over 300%. When translating these findings to the clinical setting, blood levels of rifaximin would be expected to increase even more in patients with liver disease, in whom blood concentrations of rifaximin-α are already 10–20 times higher, depending on disease severity.26,46
Rifaximin is very effective and widely used for the treatment of SIBO.47 The recent appreciation of an association between SIBO and functional gut symptoms has renewed interest in this mimicry. SIBO indeed can often be present in different functional (eg, irritable bowel syndrome, chronic constipation, diarrhea) or organic (eg, inflammatory bowel disease, celiac disease, diverticular disease) conditions, where bacterial proliferation (and consequent minimal inflammation) may trigger similar patterns of abdominal symptoms. In all these conditions, especially the organic ones, rifaximin is used long-term to eradicate pathogenic bacteria, reduce the inflammatory response, and achieve symptom relief.48,49 As for every drug used on a long-term basis, safety, besides efficacy, is of paramount importance. Rifaximin-α proved to be extremely safe, even when given continuously for 6 months, and its minimal, if any, systemic absorption accounts for its favorable adverse event profile, which overlaps that of placebo.26 Use of amorphous rifaximin, systemic absorption of which is higher, might not have the excellent tolerability of rifaximin-α.
A peculiar and potentially serious adverse event related to systemic rifaximin exposure is the development of extragastrointestinal cross-resistance. Rifamycins are pivotal drugs for the treatment of Mycobacterium tuberculosis infection,42,50 and the activity of rifaximin in the treatment of this micro-organism has been carefully investigated. Development of resistance to rifaximin in different strains of M. tuberculosis isolated from patients with pulmonary and renal disease has been studied under stringent conditions.51 Since the results of these assays showed concentration-dependent subinhibitory effects of rifaximin on mycobacterial growth, it was speculated that circulating, albeit very low, levels of rifaximin could represent a stimulus for the selection of resistant mycobacterial strains.52 However, the evaluation of rifaximin and rifampicin minimum inhibitory concentrations did not vary in all tested Mycobacterium strains, both before and after exposure to rifaximin,51 suggesting that rifaximin therapy in gastrointestinal patients harboring M. tuberculosis should not represent a hazard to treatment of tuberculosis. A 10-year survey53 in Italy showed that mycobacterial resistance to rifampicin has remained quite stable over time, despite the widespread consumption of rifaximin in Italy. It is also worth mentioning that rifampicin is currently used to eradicate pharyngeal Neisseria meningitidis, thereby lowering the potential risk of meningitis.54–56 In this setting, the selection of resistant Neisseria mutants could theoretically result from use of rifaximin for gastrointestinal infections. Here again, a recent Italian study57 found that all meningococci isolated from asymptomatic carriers were susceptible to rifampicin, thus arguing against this possibility. Overall, while the above-mentioned examples of bacterial cross resistance are very unlikely to occur with the polymorph-α, taking into account the present data on systemic bioavailability, it is conceivable that the risk of resistance with amorphous rifaximin might become clinically relevant.
In summary, the results of the present PK investigations show different systemic bioavailability of different solid-state forms of rifaximin. Rifaximin-α, a crystal polymorph, does differ from the amorphous form, the latter being systemically bioavailable. In this regard, care must be taken in using – as a drug product – a formulation not containing even small amounts of the amorphous form, which may alter the peculiar pharmacologic properties of this poorly absorbed antibiotic.
Acknowledgments
We are indebted to Drs Antonio Marzo and Shevqet Ismaili for managing the experiments in healthy volunteers at the Institute for Pharmacokinetic and Analytical Studies SA (Ligornetto, Switzerland) and to Professor Fabrizia Grepioni (Department of Chemistry G Ciamician, University of Bologna, Italy) for performing and interpreting the X-ray powder diffraction analysis.
Disclosure
CB has been occasionally involved as a speaker in satellite symposia supported by Alfa Wassermann, the manufacturer of rifaximin. GCV is an employee of Alfa Wassermann.
CS is a member of the speakers’ bureau and scientific advisory board for Alfa Wassermann.
References
European Medicines Agency. The European Agency for the Evaluation of Medicinal Products. Note for guidance on the clinical requirements for locally applied, locally acting products containing known constituents. CPMP/EWP/239/95, 1995. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_ guideline/2009/09/WC500003687.pdf. Accessed September 15, 2014. | ||
European Medicines Agency. The European Agency for the Evaluation of Medicinal Products. Guideline on the investigation of bioequivalence. CPMP/EWP/QWP/1401/98 Rev. 1, 2010. Available from: http://www.emea.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf. Accessed September 15, 2014. | ||
Thomas VH, Bhattachar S, Hitchingham L, et al. The road map to oral bioavailability: an industrial perspective. Expert Opin Drug Metab Toxicol. 2006;2(4):591–608. | ||
Hurst S, Loi CM, Brodfuehrer J, El-Kattan A. Impact of physiological, physicochemical and biopharmaceutical factors in absorption and metabolism mechanisms on the drug oral bioavailability of rats and humans. Expert Opin Drug Metab Toxicol. 2007;3(4):469–489. | ||
Singhal D. Drug polymorphism and dosage form design: a practical perspective. Adv Drug Deliv Rev. 2004;56(3):335–347. | ||
Huang LF, Tong WQ. Impact of solid state properties on developability assessment of drug candidates. Adv Drug Deliv Rev. 2004;56(3):321–334. | ||
Peterson ML, Hickey MB, Zaworotko MJ, Almarsson O. Expanding the scope of crystal form evaluation in pharmaceutical science. J Pharm Pharm Sci. 2006;9(3):317–326. | ||
Lee AY, Erdemir D, Myerson AS. Crystal polymorphism in chemical process development. Annu Rev Chem Biomol Eng. 2011;2:259–280. | ||
Aguiar AJ, Krc J, Kinkel AW, Samyn JC. Effect of polymorphism on the absorption of chloramphenicol from chloramphenicol palmitate. J Pharm Sci. 1967;56(7):847–853. | ||
Pandit JK, Gupta SK, Gode KD, Mishra B. Effect of crystal form on the oral absorption of phenylbutazone. Int J Pharm. 1984;21(1):129–132. | ||
Kato Y, Kohketsu M. Relationship between polymorphism and bioavailability of amobarbital in the rabbit. Chem Pharm Bull. 1981;29(1):268–272. | ||
Kimura N, Fukui H, Takagaki H, Yonemochi E, Terada K. Characterisation of polymorphs of a novel quinolinone derivative, TA-270 (4-hydroxy-1-methyl-3-octyloxy-sinapinoylamino-2(1H)-quinolinone). Chem Pharm Bull. 2001;49(10):1321–1325. | ||
Bauer J, Spanton S, Henry R, et al. Ritonavir: an extraordinary example of conformational polymorphism. Pharm Res. 2001;18(6):859–866. | ||
Grant DJW. Theory and origin of polymorphism. In: Brittain HG, editor. Polymorphism in Pharmaceutical Solids. New York, NY, USA: Marcel Dekker; 1999. | ||
Rodriguez-Spong B, Price CP, Jayasankar A, Matzger AJ, Rodriguez-Hornedo N. General principles of pharmaceutical solid polymorphism: a supramolecular perspective. Adv Drug Deliv Rev. 2004;56(3):241–274. | ||
Marchi E, Montecchi L, Venturini AP, Mascellani G, Brufani M, Cellai L. 4-Deoxypyri- do[1),2):1,2]imidazo[5,4-c]rifamycin SV derivatives. A new series of semisynthetic rifamycins with high antibacterial activity and low gastroenteric absorption. J Med Chem. 1985;28(7):960–963. | ||
Jiang ZD, DuPont HL. Rifaximin: in vitro and in vivo antibacterial activity – review. Chemotherapy. 2005;51 Suppl 1:67–72. | ||
Adachi JA, DuPont HL. Rifaximin: a novel nonabsorbed rifamycin for gastrointestinal disorders. Clin Infect Dis. 2006;42(4):541–547. | ||
Scarpignato C, Pelosini I. Rifaximin, a poorly absorbed antibiotic: pharmacology and clinical potential. Chemotherapy. 2005;51 Suppl 1:36–66. | ||
Scarpignato C, Pelosini I. Experimental and clinical pharmacology of rifaximin, a gastrointestinal selective antibiotic. Digestion. 2006;73 Suppl 1:13–27. | ||
European Pharmacopoeia. Rifaximin (revised monograph). 2011;Suppl 7.1:2362. Available from: [http://www.edqm.eu/en/european-pharmacopoeia-8th-edition-1563.html. Accessed September 15, 2014]. | ||
Viscomi GC, Campana M, Barbanti M, et al. Crystal forms of rifaximin and their effect on pharmaceutical properties. Cryst Eng Comm. 2008;10:1074–1081. | ||
Normix® Summary of Product Characteristics. Revision September 1, 2007. Section 5.1. Available from: [https://farmaci.agenziafarmaco.gov.it/bancadatifarmaci/farmaco?farmaco=025300#. Accessed September 15, 2014]. | ||
Center for Drug Evaluation and Research. Xifaxan® (rifaximin) Summary review – June 24, 2009. Available from: http://www.accessdata. fda.gov/drugsatfda_docs/nda/2010/022554Orig1s000SumR.pdf. Accessed September 15, 2014. | ||
Ericsson CD. Safety and tolerability of the antibacterial rifaximin in the treatment of travellers’ diarrhoea. Drug Saf. 2006;29(3):201–207. | ||
Bass NM, Mullen KD, Sanyal A, et al. Rifaximin treatment in hepatic encephalopathy. N Engl J Med. 2010;362(12):1071–1081. | ||
Menees SB, Maneerattannaporn M, Kim HM, Chey WD. The efficacy and safety of rifaximin for the irritable bowel syndrome: a systematic review and meta-analysis. Am J Gastroenterol. 2012;107(1):28–35. | ||
Blandizzi C, Viscomi GC, Marzo A, Scarpignato C. Is generic rifaximin still a poorly absorbed antibiotic? A comparison of branded and generic formulations in healthy volunteers. Pharmacol Res. 2014;85:39–44. | ||
European Medicines Agency. Guideline on strategies to identify and mitigate risks for first-in-human clinical trials with investigational medicinal products, 2009. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf. Accessed September 15, 2014. | ||
Milton MN, Horvath CJ. The EMEA guideline on first-in-human clinical trials and its impact on pharmaceutical development. Toxicol Pathol. 2009;37(3):363–371. | ||
Viscomi GC, Maffei P, Lauro V, Barbanti M, Confortini D, Braga D. Rifaximin powder, process for preparing the same and controlled release compositions containing said rifaximin useful for obtaining a long-lasting effect. European Patent W02011107970. Available from: https://register.epo.org/espacenet/application?number=EP11714663. Accessed September 15, 2014. | ||
Italian Medicines Agency. Modification of marketing authorization of medicinal products for human use, Normix™, 2007. Available from: http://gazzette.comune.jesi.an.it/2007/43/index.htm. Accessed September 15, 2014. | ||
European Pharmacopoeia. 6th ed. Dissolution test for solid dosage forms. 2007. Available from: http://www.edqm.eu/en/european-pharmacopoeia-8th-edition-1563.html. Accessed September 15, 2014]. | ||
Marzo A. Clinical pharmacokinetic registration file for NDA and ANDA procedures. Pharmacol Res. 1997;36(6):425–450. | ||
Schuirmann DJ. A comparison of the two one-sided tests procedure and the power approach for assessing the equivalence of average bioavailability. J Pharmacokinet Biopharm. 1987;15(6):657–680. | ||
Moore JW, Flanner HH. Mathematical comparison of curves with an emphasis on in vitro dissolution profiles. Pharm Tech. 1996;20(6):64–74. | ||
Shah VP, Tsong Y, Sathe P. In vitro dissolution profile comparison – statistics and analysis of the similarity factor, f2. Pharm Res. 1998;15(6):889–896. | ||
Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239–245. | ||
Snider DA, Addicks W, Owens W. Polymorphism in generic drug product development. Adv Drug Deliv Rev. 2004;56(3):391–395. | ||
Raw AS, Furness MS, Gill DS, Adams RC, Holcombe FO Jr, Yu LX. Regulatory considerations of pharmaceutical solid polymorphism in Abbreviated New Drug Applications (ANDAs). Adv Drug Deliv Rev. 2004;56(3):397–414. | ||
Food and Drug Administration. Guidance for Industry. ANDAs: Pharmaceutical Solid Polymorphism. Chemistry, Manufacturing and Controls Information. Center for Drug Evaluation and Research, 2007. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072866.pdf. Accessed September 15, 2014. | ||
Burman WJ, Gallicano K, Peloquin C. Comparative pharmacokinetics and pharmacodynamics of the rifamycin antibiotics. Clin Pharmacokinet. 2001;40(5):327–341. | ||
Verardi S, Verardi V. Bile rifaximin concentration after oral administration in patients undergoing cholecystectomy. Farmaco. 1990;45(1):131–135. | ||
Cohen JS. Avoiding adverse reactions. Effective lower-dose drug therapies for older patients. Geriatrics. 2000;55(2):54–56. | ||
Hickey E. Tools to define the relevance of PK/PD parameters to the efficacy, toxicity and emergence of resistance of antimicrobials. Curr Opin Drug Discov Devel. 2007;10(1):49–52. | ||
Salix Pharmaceuticals Inc. Xifaxan™ 550 Prescribing Information. Available from: http://www.xifaxan550.com/assets/pdfs/xifaxan550-pi.pdf. Accessed September 15, 2014. | ||
Scarpignato C, Gatta L. Commentary: towards an effective and safe treatment of small intestine bacterial overgrowth. Aliment Pharmacol Ther. 2013;38(11–12):1409–1410. | ||
Scarpignato C, editor. Rifaximin, A Poorly Absorbed Antibiotic. Pharmacology and Clinical Use. Basel, Switzerland: Karger AG; 2005. | ||
Scarpignato C, Lanas A, editors. Bacterial Flora in Digestive Disease. Focus on Rifaximin. Basel, Switzerland: Karger AG; 2006. | ||
Frieden TR, Sterling TR, Munsiff SS, Watt CJ, Dye C. Tuberculosis. Lancet. 2003;362(9387):887–899. | ||
Soro O, Pesce A, Raggi M, Debbia EA, Schito GC. Selection of rifampicin-resistant Mycobacterium tuberculosis does not occur in the presence of low concentrations of rifaximin. Clin Microbiol Infect. 1997;3(1):147–151. | ||
Rice LB, Bonomo LA. Genetic and biochemical mechanisms of bacterial resistance to antimicrobial agents. In: Lorian V, editor. Antibiotics in Laboratory Medicine. 5th ed. Baltimore, MD, USA: Williams and Wilkins; 2005. | ||
Fietta A, Cascina A, Meloni F, et al. A 10-year survey of Mycobacterium tuberculosis isolates in Pavia and their drug resistance: a comparison with other Italian reports. J Chemother. 2002;14(1):33–40. | ||
Girgis N, Sultan Y, Frenck RW Jr, El-Gendy A, Farid Z, Mateczun A. Azithromycin compared with rifampin for eradication of nasopharyngeal colonization by Neisseria meningitidis. Pediatr Infect Dis J. 1998;17(9):816–819. | ||
Cuevas LE, Kazembe P, Mughogho GK, Tillotson GS, Hart CA. Eradication of nasopharyngeal carriage of Neisseria meningitidis in children and adults in rural Africa: a comparison of ciprofloxacin and rifampicin. J Infect Dis. 1995;171(3):728–731. | ||
Schwartz B, Al-Tobaiqi A, Al-Ruwais A, et al. Comparative efficacy of ceftriaxone and rifampicin in eradicating pharyngeal carriage of group A Neisseria meningitidis. Lancet. 1988;1(8597):1239–1242. | ||
Cresti S, Giordano I, Donati E, Giaccherini R, Barberi A, Cellesi C. Prevalence and chemosusceptibility of Neisseria meningitidis and Haemophilus influenzae in a population of central Italy. New Microbiol. 2003;26(3):281–288. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.