Back to Archived Journals » Virus Adaptation and Treatment » Volume 7
Impact and differential clinical utility of cobicistat-boosted darunavir in HIV/AIDS
Authors Cossu MV, Astuti N, Capetti A, Rizzardini G
Received 28 February 2015
Accepted for publication 30 March 2015
Published 21 August 2015 Volume 2015:7 Pages 47—56
DOI https://doi.org/10.2147/VAAT.S83680
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Jonathan Dinman
Maria Vittoria Cossu, Noemi Astuti, Amedeo Capetti, Giuliano Rizzardini
1st Division of Infectious Diseases, Luigi Sacco University Hospital, Milan, Italy
Abstract: Cobicistat (Cobi) is a pharmacoenhancer, without anti-HIV activity, that optimizes systemic exposures of protease inhibitors (PIs) such as atazanavir (ATV) and darunavir (DRV). In particular, Cobi is a potent inhibitor of cytochrome P450 (CYP) 3A enzymes, the main metabolizing pathway of this class of antiretrovirals. Due to its more selective inhibition of CYP3A, Cobi has a lower potential for off-target drug interactions than the standard boosting agent ritonavir (RTV), and lower likelihood for enzymatic induction. In pharmacokinetic studies of healthy volunteers, DRV boosted with Cobi attains plasma concentrations that are comparable to those achieved with 100 mg RTV as booster. In Phase III clinical trials, conducted in naïve subjects, the coformulation yielded high rates of suppression of viral replication, on average 82%–83% of treated patients reaching undetectable viremia after 48 weeks of therapy. The selection of resistance associated with mutations was a rare event and no phenotypic resistance to DRV emerged in viral failures. Generally, Cobi was well tolerated; however, it has been shown to decrease estimated creatinine clearance (ClCr) due to inhibition of tubular secretion of creatinine. Cobi, therefore, should not be initiated in patients with ClCr less than 70 mL/min, if any coadministered agent (eg, emtricitabine, lamivudine, tenofovir disoproxil fumarate, or adefovir) requires dose adjustment based on ClCr.DRV/Cobi fixed-dose combination is part of an important evolution of antiretroviral therapy toward simpler yet potent formulations. In this setting, it is important to combine potency, simplicity, safety, and high genetic barrier, as in this case.
Keywords: HIV, darunavir, Cobicistat, fixed dose, pharmacoenhancer
Introduction
On September 26, 1997, the US Food and Drug Administration (FDA) approved the first combined formulation (FDC, fixed-dose combination) of zidovudine plus lamivudine;1
Seven years passed until, on August 2, 2004, two more nucleoside analog FDCs, tenofovir dipivoxil/emtricitabine (FTC) and abacavir/lamivudine, were licensed, and on July 12, 2006, 2 years after the first coformulation of a full antiretroviral regimen, tenofovir dipivoxil/FTC/efavirenz was approved by FDA.2 However, it was necessary to wait until September 2011 to have a new single-tablet regimen (STR) released, tenofovir dipivoxil/FTC/rilpivirine,3 followed 1 year later by tenofovir/dipivoxil/FTC/elvitegravir (EVG)/Cobi,4 and lastly, on August 2014, by abacavir/lamivudine/dolutegravir.5
Today, therefore, the new antiretroviral regimens in naïve subjects shall be mainly taken once a day, with a limited number of tablets (1–3), high antiviral potency, and limited drug–drug interactions. The class that is being less involved in such progress is that of protease inhibitors (PIs), which until recently suffered from the need of ritonavir (RTV) as a booster (one additional pill with metabolic and gastrointestinal side effects in some patients, even at low doses). RTV 100 mg twice daily, a dose commonly used to boost PIs, is associated with an increase in the concentration of total cholesterol, LDL (low-density lipoprotein) cholesterol, triglycerides, and total/HDL (high-density lipoprotein) cholesterol ratio, as well as a decrease in HDL cholesterol concentration.6,7
However, several limitations are still present, which constitute the unmet needs up to date.
STRs, except the latest, are based on tenofovir dipivoxil, a drug that tends to accumulate in the cells of the renal proximal convoluted tubule, interacting with phosphate and calcium reabsorption and potentially causing interstitial damage.8,9 The presence, in the coformulations, of drugs interacting with basolateral and/or apical transporters in the same cells10 causes a rise in serum creatinine, not related to renal damage, but potentially confounding in case a tenofovir (TFV)-related damage may occur. However, such phenomenon may mask possible true renal damage, ie: induced by tenofovor. Moreover, the rise in serum creatinine, a problem shared by all the most recent STRs, is also a confounder of treatment-unrelated medical events that are often reported in our prematurely aging population, and this is not a desirable effect when the time and resources for evaluating each subject are limited.
STRs, except the latest, are characterized by a low genetic barrier to resistance by HIV in case of poor compliance by the patients. The compounds that showed the highest genetic barrier up to date are darunavir (DRV), among PIs, and dolutegravir, among integrase inhibitors. Both appear to be therefore essential compounds in a future where no subject should develop resistance mutations to antivirals in case of mistakes or drop of adherence. Indeed, subjects harboring resistant strains need more complicated and expensive solutions, and sometimes predictable yet inevitable toxicities.
In such a scenario, the development of coformulated PIs with a new booster, to be used in diverse combinations, appears a very much welcome novelty.
Overview of pharmacology
PIs, important components of the antiretroviral therapy, were historically associated with poor oral bioavailability and high pill burden. However, because the PIs are metabolized by cytochrome P450 (CYP) 3A enzymes, intentional inhibition of these enzymes leads to higher drug exposure, lower pill burden, and therefore simplified dosing schedules with this class of drug. This is the basis of pharmacokinetic enhancement. In HIV therapy, two pharmacokinetic enhancers or boosting agents are used: RTV and Cobicistat (Cobi). Cobi is a pharmacoenhancer, without anti-HIV activity, that optimized systemic exposures of PIs such as atazanavir (ATV) and DRV. In particular, Cobi is a potent inhibitor of CYP3A enzymes, the main metabolizing pathway of this class of antiretrovirals.11 Like RTV, Cobi inactivates CYP3A in a concentration- and time-dependent fashion, exhibiting a CYP3A inactivation rate of 0.44 min–1 and an inhibition constant of 939 nmol/L in vitro (corresponding values for RTV were 0.23 min–1 and 256 nmol/L). Potent inhibition of CYP3A activity has also been demonstrated with Cobi in a Phase I study in healthy volunteers (n≤60).12 Upon coadministration, oral Cobi at doses of 100 and 200 mg increased the median half-life of oral midazolam 2.1- and 3.7-fold and reduced its mean apparent clearance by 93% and 95%, respectively. Similarly, RTV 100 mg increased the midazolam half-life 4.9-fold and reduced the midazolam clearance by 96%. However, due to its more selective inhibition of CYP3A, Cobi has a lower potential for off-target drug interactions than the standard boosting agent RTV, and lower likelihood for enzymatic induction. Cobi, unlike RTV, is devoid of anti-HIV activity in vitro, displaying a mean IC50 of >30 mmol/L in a HIV-1 protease enzymatic assay (vs 0.0006 mmol/L with RTV), a mean half maximal effective concentration (EC50) of >30 mmol/L in a cell-based HIV infection assay (vs 0.0133 mmol/L with RTV),11 and no activity against primary HIV isolates.13
Absorption of Cobi is rapid after oral administration, with the maximum plasma concentration (Cmax) of the drug being reached within 4 hours (Table 1).14 As may be expected for a drug that is both a mechanism-based inhibitor of CYP3A and a CYP3A substrate, exposure to Cobi was nonlinear across oral dosages of 50–300 mg/day in healthy volunteers, with exposure increasing more than proportionally with increasing dose (up to 47-fold).12
 | Table 1 Pharmacokinetic properties of Cobi in patients with HIV-1 infection |
Comparing DRV/Cobi to DRV/RTV by figuring the geometric mean ratio of the two regimens, the two treatments were equivalent for area under the plasma concentration (AUC) (102%, 90% CI, 97.4–106) and Cmax (103%, 90% CI, 100–106),15 as shown in Table 2.
 | Table 2 Relative efficacy of Cobi 150 mg vs RTV 100 mg OD in boosting DRV in a crossover study of healthy volunteers |
Systemic exposure to Cobi is higher in the fed than in the fasted state. Following the administration of a single dose of Cobi 150 mg plus DRV 800 mg to healthy volunteers under fed (n=38) and fasted (n=72) conditions, Cobi had a mean Cmax of 823 and 664 ng/mL, a mean AUC from the time of administration to the last measurable concentration (AUClast) of 6,401 and 4,962 ng · h/mL, and a median time to Cmax of 4.0 and 2.5 hours.16 These data suggest that Cobi should be taken with food.14
Steady-state DRV pharmacokinetic parameters were consistent with those observed in previous Phase I studies in healthy volunteers;15,16 a summary of the impact of Cobi on DRV bioavailability in patients with HIV-1 infection is given in Table 3.17
 | Table 3 Impact of Cobi on DRV bioavailability in patients with HIV-1 infection |
Cobi is not extensively metabolized, with unchanged drug accounting for 99% of the radioactivity circulating in plasma following oral administration of radiolabelled Cobi.14 Metabolism of Cobi is via oxidation (primarily via CYP3A and to a minor degree via CYP2D6) and does not involve glucuronidation; the resultant metabolites do not contribute to the inhibitory activity of the drug against CYP3A.14
Elimination of Cobi is mainly via the feces (86%), with only 8.2% being excreted via the urine.14
Cobi is not to be recommended for patients with creatinine clearance (ClCr) <70 mL/L, as it increases serum creatinine levels (possibly via inhibition of proximal renal tubular cell transporters) and thus reduces the estimated glomerular filtration rate (eGFR), although it does not appear to affect the actual glomerular filtration rate (GFR).18
Efficacy studies
Only few studies have evaluated the efficacy of Cobi-boosted DRV. The efficacy studies available concerning the use of Cobi-boosted DRV are summarized in Table 4.
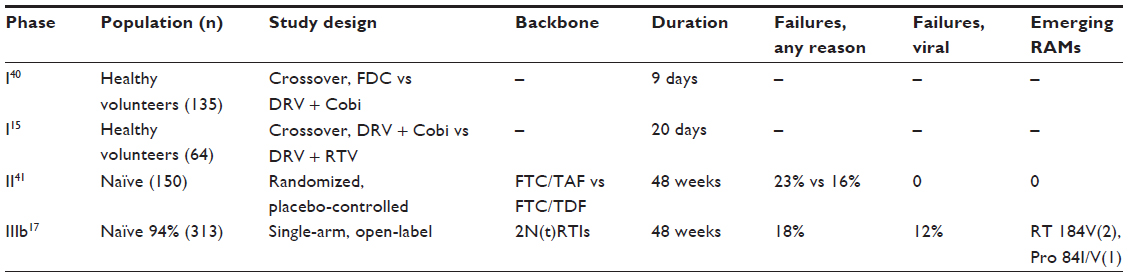 | Table 4 Summary of clinical trials of Cobi-boosted DRV |
A Phase III, noncomparative, open-label clinical trial has evaluated the efficacy and safety of switching RTV to Cobi in patients with ClCr 50–89 mL/min, virologically suppressed on a stable regimen containing RTV-boosted ATV or DRV. Seventy-three patients were enrolled and received at least one dose of Cobi; 52 patients were taking an ATV-based regimen and 21 patients a DRV-based regimen. Considering the overall population (n=73), the rate of virologic success (HIV-1 RNA <50 copies/mL) at week 24 was 90.4% (66/73; 95% CI, 81.2%–96.1%) and at week 48 was 82.2% (60/73; 95% CI, 71.5%–90.2%). Of the 13 patients who did not have virologic success at week 48, two had HIV-1 RNA >50 copies/mL and eleven had discontinued the study drug prior to week 48 (five due to adverse events and six due to nonvirologic reasons). The proportion of patients with HIV-1 RNA <50 copies/mL at week 48 was 87.7% (64/73 patients; intention-to-treat, missing equals failure [ITT: M = F] analysis) and 97% (64/66 patients; missing equals excluded, ITT: M = E). No change was observed in the CD4 cell count (mean [SD]) from baseline at week 48 (–7 [103.5] cells/μL). One patient with confirmed virologic rebound was analyzed for resistance and did not develop resistance mutations to any components of the PI + Cobi-containing regimen.19
A Phase IIIb, single-arm, US multicenter study evaluated safety, efficacy, and pharmacokinetics of DRV/Cobi 800/150 mg once daily (as single agents) + two investigator-selected nucleoside/nucleotide reverse transcriptase inhibitors (N[t]RTIs) in HIV-1-infected adults. Patients had no DRV resistance-associated mutations (RAMs), and had a plasma viral load ≥1,000 HIV-1 RNA copies/mL, eGFR ≥80 mL/min, and full genotypic sensitivity to the two N[t]RTIs. Of the 313 enrolled patients, 295 were treatment-naïve and 18 were treatment-experienced, with no DRV RAMs. The virologic response rate was 82% (258/313) (95% CI, 78%–87%) at week 24 and was sustained and durable through week 48 (81%; 95% CI, 76%–85%). Virologic failure occurred in 11% of the patients at week 48, and 9% had no virologic data in the week 48 window. In treatment-naïve patients, the week 48 virologic response rate was 83% (244/295; 95% CI, 78%–87%). Twenty-four treatment-naïve patients (8%) were classified as virologic failures, and 27 treatment-naïve patients (9%) had no virologic data in the week 48 window. The week 48 rates of virologic response were comparable in patients who had a baseline viral load ≤100,000 HIV-1 RNA copies/mL or >100,000 HIV-1 RNA copies/mL, both overall (81% vs 80%, respectively) and in treatment-naïve patients (84% vs 81%). Sensitivity analyses showed that the week 48 virologic response rate was 81% (253/313) overall and 83% (245/295) in treatment-naïve patients, using the time-to-loss of virologic response analysis, and was 83% (260/313) and 85% (250/295), respectively, using the ITT: M = F method. As for immunologic response, there was an increase in the CD4 cell count from baseline at all time points following the initiation of study drugs. At weeks 24 and 48 in the overall population, the median (range) increases from baseline in CD4 cell count (ITT: M = E) were 131 (–47 to 596) cells/mm3 and 167 (−193 to 1,086) cells/mm3, respectively. In treatment-naïve patients, the median (range) CD4 cell count increased by 169 (−193 to 1,086) cells/mm3 from baseline to week 48. As for the development of resistance, of the 15 patients with samples eligible for resistance analysis, three had suboptimal virologic response, eight had virologic rebound, and four who discontinued with viral load ≥400 HIV-1 RNA copies/mL were analyzed at their last visit. Only one of these 15 patients who were treatment-experienced developed a resistance mutation to DRV (at position I84 as a mixture with wild-type, I84I/V), which was not associated with phenotypic resistance to DRV or other PIs. Two patients (one treatment-experienced and one treatment-naïve) developed the M184V N[t]RTI RAM in reverse transcriptase while receiving FTC that was associated with phenotypic resistance to FTC and lamivudine. One patient showed a transient development of the N[t]RTI RAM L74I/L and the non-nucleoside reverse transcriptase inhibitor (NNRTI) RAM P225H/P at the week 16 visit, which were not detected at the subsequent week 48 visit analysis. These mutations were not associated with resistance to the agents in the patient’s regimen (FTC/TFV/zidovudine), and may reflect the drug history. New primary RAMs were not detected in the eleven remaining patients.17
Drug–drug interactions
Cobi is metabolized by CYP3A and CYP2D6; therefore, its plasma concentration may be altered by drugs that induce or inhibit such enzymes. Azole antifungals and clarithromycin may increase serum Cobi concentrations (and Cobi may alter their serum levels at the same time). In particular, concomitant itraconazole should not exceed 200 mg/day, while voriconazole should be avoided unless benefits clearly overcome the risk of adverse events. Rifabutin, some antiepileptic medications (ie, carbamazepine and phenytoin), and NNRTIs, with the exception of rilpivirine, well-known inducers of CYP3A, are expected to reduce the AUC of Cobi, and hence of the associated PI, and are therefore not indicated for the association with Cobi. The coadministration of rifabutin with DRV boosted with Cobi is feasible, provided that rifabutin dose is reduced to 150 mg three times per week, with particular attention to the risk of uveitis and neutropenia, since the AUC of the main metabolite, 25-O-desacetyl-rifabutin, increases by more than 5-folds. By enzymatic inhibition, Cobi may increase the plasma levels of some calcium channel blockers (nifedipin, nicardipin, felodipin, amlodipin), β-blockers (metoprolol, thymolol, but not atenolol), HMG-CoA reductase inhibitors (rosuvastatin should be preferred), antiarrhythmics (verapamil, diltiazem, digoxin, disopyramide, mexiletine, systemic lidocaine, but with a weaker interaction potential than RTV-boosted DRV toward flecainide and propafenone), sedative-hypnotics, erectile dysfunction agents, inhaled corticosteroids, immune suppressants, and norgestimate. Also, the side effects of the anticancer agents dasatinib, nilotinib, vinblastine, and vincristine are likely to increase, as observed with conventionally boosted PIs, and the plasma concentrations of fluticasone and metformin may be increased. On the contrary, methadone levels are not affected by coadministration,20 and only minor interaction exists with morphin and its derivatives, as with buprenorphine and naloxone.21 Cobi might mitigate the interaction potential of boosted DRV as compared with RTV toward 22 therapeutic compounds, while 14 drug classes, in the Liverpool classification, do not show any difference as compared with RTV-boosted DRV.22 In Table 5, all clinically significant drug interactions, demonstrated or predicted to occur, between Cobi and agents likely to be coadministered, are summarized.
 | Table 5 Drug interactions of potential clinical significance demonstrated or predicted to be associated with Cobi |
Safety and tolerability
Preclinical data
A single oral dose of Cobi at 50 mg/kg had no effects on the central nervous system in the rat, on the respiratory rate, tidal volume, or derived minute volume. The Cmax at the no-effect level was 4.2-fold higher than that observed clinically.14
Cardiac safety
During a radioligand binding assay at a panel of receptor and ion channels, Cobi showed significant binding to the benzodiazepine-sensitive L-type calcium channel, hERG potassium channel, and sodium channel, at a concentration of 10 μM, which is 105-fold higher than the proposed clinical Cmax. In electrophysiology studies, Cobi inhibited the hERG potassium current and the hCav1.2 L-type calcium channel (with IC50 values of 1.8–1.9 and 6 μM, respectively). Given that the safety margin between the IC50 value for hERG inhibition and the observed clinical Cmax was below 30, additional studies were performed to further evaluate the potential for QT prolongation. The effects on heart rate and PR interval appeared to be more pronounced when ATV (1.5 μM) and Cobi (1.5 μM) were administered in combination, although the observed differences were not considered to be clinically significant. In vitro data indicate that when compared to RTV, Cobi shows a lower potential to inhibit host proteases (such as cathepsin D), affect adipocyte functions (such as lipid accumulation and inhibition of glucose uptake), and cause cytotoxicity in MT-2 lymphoblastoid T-cells. The effects of Cobi on the inhibition of chymotryptic-like activity of the 26S proteasome and the viability of HepG2 hepatoma cells were similar to that observed with RTV.14
Renal safety
The recent studies on renal safety of boosted PIs suffer from the fact that most were performed with a TFV-containing backbone. Indeed TFV accumulates in the proximal tubular cells of the kidney, where it is imported by basolateral anionic transporters OAT1 and OAT3 and expelled in the lumen by apical transporters of the multidrug resistance-related family. The latter is shared by PIs, which compete with TFV for excretion, increasing intracellular and plasma TFV concentrations and proximal tubular toxicity.23 The coadministration of RTV-boosted PIs with TFV increased the frequency of proteinuria development by 7-fold, generally in a reversible manner, compared to TFV treatment not containing PIs.24
Serum creatinine is 90% filtrated at the glomerular level and 10% excreted in the proximal convoluted tubule. Cobi inhibits SLC47A1 multidrug and toxin extrusion protein 1 (MATE-1) and dolutegravir and rilpivirine inhibit SLC22A2 organic cation transporter 2 (OCT2), both of which transport creatinine through to the lumen.25
To investigate the potential for a renal drug–drug interaction between TFV and Cobi in vitro, the uptake of TFV in the presence and absence of Cobi was determined in fresh human renal cortex tissue and in cells expressing the relevant renal transporters. At concentrations exceeding clinical protein-unbound plasma levels, Cobi did not significantly inhibit the transport of TFV by the anion transporters OAT1, OAT3, and MRP4 (50% inhibitory concentrations [IC50s] of >15, 6.6, and 8.5 μM, respectively). Conversely, TFV had little or no effect on the cation transporters OCT2 and MATE1 (IC50 >100 μM). Consistent with studies using individual transporters, no increase in the accumulation of TFV in freshly isolated human renal cortex tissue or renal proximal tubule cells was observed in the presence of Cobi.26
In a Phase I study,27 conducted to compare the pharmacokinetics of Cobi and RTV in 36 subjects with eGFR ≥80 mL/min and in 18 subjects with eGFR 50–79 mL/min, who took 150 mg of Cobi, 100 mg RTV and placebo, each for 7 days, small increases in serum ClCr (10 mL/min or mL/min/1.73 m2) with corresponding mean decreases in estimated GFR were observed on day 7 relative to day 0 in subjects receiving Cobi (P<0.05), but not with RTV or placebo. This did not reflect in significant changes in actual GFR (measured by iohexol clearance). The decreases were reversible on Cobi discontinuation: mean eGFR values returned to baseline on day 14 (P>0.05). The time to onset, magnitude, and time to resolution of changes in eGFR are consistent with altered proximal tubular secretion of creatinine through inhibition of drug transporters by Cobi. Another study, performed by the same group in 12 subjects with eGFR <30 vs 11 with eGFR ≥90 who took EVG and Cobi for 14 days,28 showed a reduction of the eGFR (either by Cockroft–Gault and modification of diet in renal disease equations) by −8.40 mL/min at day 7 and further by −1.20 mL/min at day 14 in the ≥90 group, while in the <30 group, it decreased by −10.50 mL/min at day 7 and further by −4.00 mL/min at day 14, with a difference of 4.90 mL/min increased damage in already impaired kidneys.
These data resulted in the indication not to use Cobi in subjects whose baseline ClCr is <70 if they have concomitant medications that require renal dose adjustments, and to stop the drug if the patients’ ClCr declines below 50 mL/min.
In a noncomparative switch study, from RTV-boosted DRV plus two nucleoside/nucleotide analogs,19 a mild initial (2 weeks), nonprogressive decay in the eGFR was observed, the mildest among all studies conducted to date. This may be due to the fact that the backbone was not necessarily TFV-based, but the choice of the PI may also have contributed. Indeed, in a recent systematic review, ATV and lopinavir have been associated with a nonprogressive reduction in eGFR and a decrease in the risk of chronic kidney disease after discontinuation, while data on DRV did not allow conclusions.29
The mean variations in GRF, observed in clinical trials with Cobi, are summarized in Table 6.
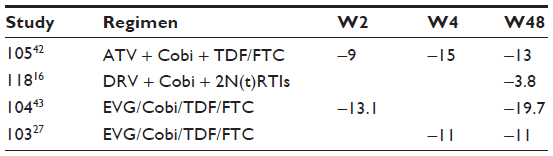 | Table 6 Mean eGFR reduction (m/min) in clinical trials of Cobi |
Bone safety
In study 103, a dual-energy X-ray absorptiometry (DEXA) comparison was performed, which showed a significantly lesser impact of EVG/Cobi/TDF (tenofovir disoproxil fumarate)/FTC vs the comparator arm, ATV plus RTV plus TDF/FTC, especially on the trabecular bone. However, after 96 weeks, subjects taking EVG/Cobi/TDF/FTC lost 1.96% in spine bone mineral density (vs 3.54% for ATV + RTV) and 3.16% in hip bone mineral density (vs 4.19% for ATV + RTV). This suggests that the renal impact of the whole compound makes it less bone friendly than the other commercial integrase inhibitor-based regimens.30
Although the etiology is considered to be multifactorial (including corticosteroid use, alcohol consumption, severe immunosuppression, higher body mass index), cases of osteonecrosis have been reported particularly in patients with advanced HIV disease and/or long-term exposure to combination antiretroviral therapy.14
Hepatotoxicity and safety in hepatic impairment
DRV and Cobi undergo primarily hepatic biotransformation, with minimal renal elimination, and are highly protein-bound. During the DRV/RTV clinical development program (n=3,063), hepatitis was reported in 0.5% of patients receiving combination antiretroviral therapy with DRV/RTV.
The safety and efficacy of DRV have not been established in patients with severe underlying liver disorders, and DRV is therefore contraindicated in patients with severe hepatic impairment.14
The pharmacokinetics and safety of Cobi boosted-EVG were studied in ten HIV uninfected subjects with moderate liver impairment (CPT class B) as well as in ten healthy controls matched for age, sex, and body mass index.31 Each subject received EVG 150 mg plus Cobi 150 mg once daily for 10 days, followed by a follow-up period of 11 days. No clinically relevant pharmacokinetic changes were observed. Treatment-emergent adverse events incidence was comparable between the two groups. The authors concluded that Cobi does not require dose adjustment in moderate or mild liver impairment.
In the study TMC114-C211 (DRV/RTV), 5% grade 3–4 ALT (alanine transaminase)/AST (aspartate transaminase) elevations were observed, as in the comparator arm (lopinavir/RTV).32 In the Phase IIIb study of DRV/Cobi in association with two nucleosides or nucleotides,17 3% had grade 3–4 ALT/AST elevations. Hepatitis B or C virus coinfection, however, was rare in this study.
Diabetes mellitus/hyperglycemia
New-onset diabetes mellitus, hyperglycemia, or exacerbation of existing diabetes mellitus has been reported in patients receiving antiretroviral therapy, including PIs, in some cases with severe hyperglycemia and occasionally associated with ketoacidosis. Many patients had confounding medical conditions, some of which required therapy with agents that have been associated with the development of diabetes mellitus or hyperglycemia (ie, steroids).33
Fat redistribution and metabolic disorders
Combination antiretroviral therapy has been associated with the redistribution of body fat (lipodystrophy) in HIV-infected patients. Knowledge about the mechanism is incomplete. A connection between visceral lipomatosis and PIs has been hypothesized. A higher risk of lipodystrophy has been associated with individual factors such as older age and with drug-related factors such as longer duration of antiretroviral treatment and associated metabolic disturbances.
Skin rash
During the DRV/RTV clinical development program (n=3,063), severe skin reactions, with or without fever and/or elevations of transaminases, have been reported in 0.4% of patients. DRESS (drug rash with eosinophilia and systemic symptoms) and Stevens–Johnson syndrome have been rarely (<0.1%) reported, and during postmarketing experience, toxic epidermal necrolysis and acute generalized exanthematous pustulosis have been reported. DRV contains a sulfonamide moiety and should be used with caution in patients with a known sulfonamide allergy.33
In a pharmacokinetic study on healthy volunteers taking DRV/Cobi, five subjects (13.9%) prematurely discontinued because of rashes, while in the Phase III study of DRV/COBI in association with two nucleosides or nucleotides17 only 0.9% of subjects had rashes.
Bleeding (in hemophiliacs only)
There have been reports of increased bleeding, including spontaneous skin hematomas and hemarthrosis, in patients with hemophilia types A and B treated with PIs. In some patients, additional factor VIII was given. In more than half of the reported cases, treatment with PIs was continued or reintroduced if it had been discontinued. A causal relationship has been suggested, although the mechanism of action has not been elucidated.33
Final considerations on safety
It should be clear that safety data are rarely given for the combination of Cobi and DRV, and most of the data presented in this section refer to either single agent within other combinations. Moreover, long-term safety data on the combination are still insufficient to draw clear conclusions, although much can be hypothesized from the aforementioned evidences.
Patient-focused perspectives
The availability of a FDC of DRV and Cobi represents certainly a step forward in the management of HIV-1 infection, because it reduces the pill burden, attenuates the gastrointestinal and metabolic side effects attributed to RTV-boosted PIs, and finally, confers less drug–drug interactions.
In the future of antiretroviral therapies, STRs, integrase inhibitors, and once-daily regimens based on a modest number of pills are likely to dominate the market. When long-term parenteral therapy will be available, thanks to its attraction potential, it will probably gain much field to the other options.
A new STR is under study, containing DRV plus Cobi plus tenofovir alafenamide, a precursor of TFV that achieves high intracellular antiviral activity with a 10-fold lower dose than tenofovir dipivoxil (25 vs 245 mg). In a Phase II study on naïve subjects,34 the STR failed in 6/103 participants with no resistance mutations being detected at failure. The renal and bone profile of the combination was better than that observed in the tenofovir dipivoxil arm.
Another chance is to switch stably suppressed patients toward a boosted PI plus lamivudine regimen, a very cost-effective and toxicity-sparing strategy that has been studied up to date in a complex population (63% coinfected with hepatitis C virus, 42% CDC stage C) with RTV35 but needs confirmation with the new booster.
Finally, in complex subjects, the DRV/Cobi FDC can be associated with a variety of other agents, such as rilpivirine, dolutegravir, or maraviroc, although these fields require pharmacokinetic studies followed by randomized, controlled clinical studies. With RTV as booster, the first of such combinations (rilpivirine) is currently under evaluation in an Italian multicenter trial,36 while the modern study, assessing maraviroc vs TFV/FTC plus DRV/RTV, showed lower efficacy of the dual combination in naïve subjects with high viral loads37 but comparable effects in those who had low baseline viraemia, and the combination is still being studied by others in different settings.38
For the patients’ safety, particular care should be paid to the subject’s baseline situation and to the follow-up analyses, including 24-hour urine evaluation of differential proteinuria and bone markers when associate in the DRV/Cobi FDC with tenofovir dipivoxil, as the potential renal and bone toxicities raise some concern. Finally, the Rating Scheme for Recommendations of the International Guidelines would probably attribute a level of evidence II to the combination, and the Risk Management Plan of the commercial formulation admits that “[t]here is limited information about the long-term use of RezolstaTM, and about use in certain subgroups of patients: the elderly (65 years and above), children, pregnant and breastfeeding women and patients with reduced liver or kidney function.” However, several studies are planned or ongoing in an attempt to fill the gaps, such as GS-US-216-0128 in children less than 18 years of age, GS-US-236-0118 in subjects with kidney impairment, and a phase of study TMC114HIV3015 in pregnant women.39
Conclusion
This new fixed-dose coformulation fits very well in a field, antiretroviral therapy, that is evolving toward conjugating efficacy with simplicity. With a single pill, a high genetic barrier to resistance, and a good tolerability profile, DRV/Cobi FDC will be a very helpful tool for physicians.
Disclosure
The authors report no conflicts of interest in this work.
References
FDA. HIV/AIDS Historical Time Line 1995–1999. Available from: http://www.fda.gov/ForPatients/Illness/HIVAIDS/History/ucm151079.htm. Accessed January 12, 2015. | |
FDA. HIV/AIDS Historical Time Line 2000–2010. Available from: http://www.fda.gov/ForPatients/Illness/HIVAIDS/History/ucm151081.htm. Accessed January 12, 2015. | |
FDA. Gilead resolves warning letter, gets FDA Complera approval. Available from: http://www.fdanews.com/articles/139884-gilead-resolves-warning-letter-gets-fda-complera-approval?v=preview. Accessed January 12, 2015. | |
Stribild™ 150mg/150 mg/200 mg/245 mg film-coated tablets [FDA summary of product characteristics]. Foster City, CA: Gilead Sciences Inc.; 2012. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203100s000lbl.pdf. Accessed January 12, 2015. | |
NDA Approval for TRIUMEQ. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2014/205551Orig1s000ltr.pdf. Accessed January 12, 2015. | |
Shafran SD Mashinter LD, Roberts SE. The effect of low-dose ritonavir monotherapy on fasting serum lipid concentrations. HIV Med. 2005; 6(6):421–425. | |
Samaras K, Richardson R, Carr A. Postprandial lipid effects of low-dose ritonavir vs raltegravir in HIV-uninfected adults. AIDS. 2010;24(11):1727–1731. | |
Quinn KJ. Incidence of proximal renal tubular dysfunction in patients on tenofovir disoproxil fumarate. Int J STD AIDS. 2010;21(2):150–151. | |
Herlitz LC, Mohan S, Stokes MB, et al. Tenofovir nephrotoxicity: acute tubular necrosis with distinctive clinical, pathological, and mitochondrial abnormalities. Kidney Int. 2010;78(11):1171–1177. | |
Gutiérrez F, Fulladosa X, Barril G, et al. Renal tubular transporter-mediated interactions of HIV drugs: implications for patient management. AIDS Rev. 2014;16(4):199–212. | |
Xu L, Liu H, Murray BP, et al. Cobicistat (GS-9350): a potent and selective inhibitor of human CYP3A as a novel pharmacoenhancer. ACS Med Chem Lett. 2010;1(5):209–213. | |
Mathias AA, German P, Murray BP, et al. Pharmacokinetics and pharmacodynamics of GS-9350: a novel pharmacokinetic enhancer without anti-HIV activity. Clin Pharmacol Ther. 2010;87(3):322–329. | |
Callebaut C, Tsai L, Stray K, et al. Biological profiling of GS-9350, a novel pharmacoenhancer that lacks anti-HIV activity and exhibits low potential for metabolic adverse effects in vitro [abstract]. Antivir Res. 2010;86(1):A31. | |
Tybost 150 mg film-coated tablets [EU summary of product characteristics]. Foster City, CA: Gilead Sciences International Ltd; 2013. Available from: http://ec.europa.eu/health/documents/community-register/2015/20150210131020/anx_131020_en.pdf. Accessed February 28, 2015. | |
Mathias A, Liu HC, Warren D, et al. Relative bioavailability and pharmacokinetics of darunavir when boosted with the pharmacoenhancer GS-9350 versus ritonavir [abstract 28]. Presented at: 11th International Workshop on Clinical Pharmacology of HIV Therapy, April 7–9, 2010, Sorrento, Italy. | |
Kakuda TN, van de Casteele T, Petrovic R, et al. Bioequivalence of darunavir (800 mg) co-administered with Cobicistat (150 mg) as either a fixed-dose combination tablet or as single agents under fasted and fed conditions in healthy volunteers [abstract no MOPE029 plus poster]. Presented at: 7th International AIDS Society Conference on HIV Pathogenesis and Treatment, June 30–July 3, 2013, Kuala Lumpur, Malaysia. | |
Tashima K, Crofoot G, Tomaka FL, et al. Cobicistat-boosted darunavir in HIV-1-infected adults: week 48 results of a Phase IIIb, open-label single-arm trial. AIDS Res Ther. 2014;11:39. | |
Coffey S. Cobicistat as a booster of atazanavir. 2012. Available from: http://hivinsite.ucsf.edu/InSite?page=hmq-1210-02. Accessed January 28, 2015. | |
McDonald CK, Martorell C, Ramgopal M, et al. Cobicistat-boosted protease inhibitors in HIV-infected patients with mild to moderate renal impairment. HIV Clin Trials. 2014;15(6):269–273. | |
Bruce RD, Winkle P, Custodio JM, et al. Investigation of the interactions between methadone and elvitegravir-cobicistat in subjects receiving chronic methadone maintenance. Antimicrob Agents Chemother. 2013; 57(12):6154–6157. | |
Bruce RD, Winkle P, Custodio JM, et al. The phamacokinetic and pharmacodynamic interactions between buprenorphine/naloxone and elvitegravir/cobicistat in subjects receiving chronic buprenorphine/naloxone treatment. J Acquir Immune Defic Syndr. 2013;63(4):480–484. | |
HIV drug interactions. Interactions with Protease Inhibitors. Available from: http://www.hiv-druginteractions.org/data/PrintableCharts/PI_col.pdf. Accessed January 12, 2015. | |
Moss DM, Neary M, Owen A. The role of drug transporters in the kidney: lessons from tenofovir. Front Pharmacol. 2014;5:248. | |
Kelly MD, Gibson A, Bartlett H, et al. Tenofovir-associated proteinuria. AIDS. 2013;27(3):479–481. | |
Gutiérrez F, Fulladosa X, Barril G, et al. Renal tubular transporter-mediated interactions of HIV drugs: implications for patient management. AIDS Rev. 2014;16(4):199–212. | |
Stray KM, Bam RA, Birkus G, et al. Evaluation of the effect of cobicistat on the in vitro renal transport and cytotoxicity potential of tenofovir. Antimicrob Agents Chemother. 2103;57(10):4982–4989. | |
German P, Liu HC, Szwarcberg J, et al. Effect of cobicistat on glomerular filtration rate in subjects with normal and impaired renal function. J Acquir Immune Defic Syndr. 2012;61(1):32–40. | |
German P, Wei X, Mathias A, et al. Pharmacokinetics of elvitegravir and cobicistat in subjects with severe renal impairment. Presentated at: 13th HIV Clinical Pharmacology International Workshop, April 16–18, 2012, Barcelona, Spain. | |
Bagnis CI, Stellbrink H. Protease inhibitors and renal function in patients with HIV infection: a systematic review. Infect Dis Ther. Epub January 8, 2015. | |
Rockstroh JK, DeJesus E, Henry K, et al. A randomized, double-blind comparison of coformulated elvitegravir/Cobicistat/emtricitabine/tenofovir DF vs ritonavir-boosted atazanavir plus coformulated emtricitabine and tenofovir DF for initial treatment of HIV-1 infection: analysis of week 96 results. J Acquir Immune Defic Syndr. 2013;62(5):483–486. | |
Custodio JM, Rhee M, Shen G, et al. Pharmacokinetics and safety of boosted elvitegravir in subjects with hepatic impairtment. Antimicrob. Agents Chemother. 2014;58(5):2564–2569. | |
Orkin C, DeJesus E, Khanlou H, et al. Final 192-week efficacy and safety of once-daily darunavir/ritonavir compared with lopinavir/ritonavir in HIV-1 infected treatment-naïve patients in the ARTEMIS trial. HIV Med. 2013;14(1):49–59. | |
Prezista 400/600 mg tablets [EU summary of product characteristics]. Latina, Italy: Janssen-Cilag; 2013. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000707/WC500041756.pdf. Accessed January 12, 2015. | |
Mills A, Ortiz R, Crofoot G Jr, et al. 48 week study of the first PI-based Single Tablet-Regimen (STR) Darunavir/Cobicistat/Emtricitabine/Tenofovir Alafenamide (D/C/F/TAF) vs Cobicistat (COBI)-boosted Darunavir (DRV) and Emtricitabine/Tenofovir Disoproxil Fumarate (F/TDF) in treatment-naïve (TN) adults [abstract H-647c]. Presented at: 54th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC), September 5–9, 2014, Washington, DC, USA. | |
Casado JL, Bañón S, Moreno A, et al. Lamivudine plus darunavir boosted with ritonavir as simplification dual regimen in HIV-infected patients. J Int AIDS Soc. 2014;17(4 Suppl 3):19801. | |
DRV/r + RPV QD: Efficacy and Toxicity Reduction. Available from: https://clinicaltrials.gov/ct2/show/NCT01792570. NLM identifier: NCT01792570. Accessed January 20, 2015. | |
Stellbrink HJ, Pulik P, Szlavik J, et al. Maraviroc (MVC) dosed once daily with darunavir/ritonavir (DRV/r) in a 2 drug-regimen compared to emtricitabine/tenofovir (TDF/FTC) with DRV/r; 48-week results from MODERN (Study A4001095) [abstract TUAB0101]. Presented at: 20th International AIDS Conference (AIDS 2014), July 20–25, 2014, Melbourne, Australia. | |
EU Clinical Trials Register. Available from: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2012-003501-10. Accessed January 20, 2015. | |
European Medicines Agency. Risk Management for Rezolsta (darunavir/cobicistat). Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Risk-management-plan_summary/human/002819/WC500173519.pdf. Accessed March 23, 2015. | |
Kakuda TN, Van De Casteele T, Petrovic R, et al. Bioequivalence of a darunavir/cobicistat fixed-dose combination tablet versus single agents and food effect in healthy volunteers. Antivir Ther. 2014;19(6):597–606. | |
Mills A, Ortiz R, Crofoot G, et al. 48 week study of the first PI-based Single Tablet-Regimen (STR) Darunavir/Cobicistat/Emtricitabine/Tenofovir Alafenamide (D/C/F/TAF) vs Cobicistat (COBI)-boosted Darunavir (DRV) and Emtricitabine/Tenofovir Disoproxil Fumarate (F/TDF) in Treatment-naïve (TN) Adults [abstracts, H-647c]. Presented at: Interscience Conference of Antimicrobial Agents and Chemotherapy, September 2015, San Diego, CA, USA. | |
Elion R, Cohen C, Gathe J, et al. Phase 2 study of cobicistat versus ritonavir each with atazanavir plus fixed-dose emtricitabine/tenofovir DF in the initial treatment of HIV infection. AIDS. 2011;25(15):1881–1886. | |
Cohen C, Elion R, Ruane P, et al. Randomized, phase 2 evaluation of two single-tablet regimens elvitegravir/Cobicistat/emtricitabine/tenofovir disoproxil fumarate versus efavirenz/emtricitabine/tenofovir disoproxil fumarate for the initial treatment of HIV infection. AIDS. 2011;25(6):F7–F12. | |
Deeks ED. Cobicistat: A Review of Its Use as a Pharmacokinetic Enhancer of Atazanavir and Darunavir in Patients with HIV-1 Infection. Drugs. 2014; 74(2):195–206. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.