Back to Journals » International Journal of Nanomedicine » Volume 13
Immunotoxicity of titanium dioxide nanoparticles via simultaneous induction of apoptosis and multiple toll-like receptors signaling through ROS-dependent SAPK/JNK and p38 MAPK activation
Authors Dhupal M ![]() , Oh JM, Tripathy DR
, Oh JM, Tripathy DR ![]() , Kim SK
, Kim SK ![]() , Koh SB
, Koh SB ![]() , Park KS
, Park KS
Received 1 June 2018
Accepted for publication 3 August 2018
Published 23 October 2018 Volume 2018:13 Pages 6735—6750
DOI https://doi.org/10.2147/IJN.S176087
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Madhusmita Dhupal,1,2 Jae-Min Oh,3 Dipti Ranjan Tripathy,4 Soo-Ki Kim,2 Sang Baek Koh,5 Kyu-Sang Park6
1Department of Global Medical Science, Wonju College of Medicine, Yonsei University, Wonju, Republic of Korea; 2Department of Microbiology, Wonju College of Medicine, Yonsei University, Wonju, Republic of Korea; 3Department of Chemistry and Medical Chemistry, College of Science and Technology, Yonsei University, Wonju, Republic of Korea; 4Department of Neurology, Gauhati Medical College and Hospital, Gauhati, India; 5Department of Preventive Medicine, Wonju College of Medicine, Yonsei University, Wonju, Republic of Korea; 6Department of Physiology, Wonju College of Medicine, Yonsei University, Wonju, Republic of Korea
Background: Titanium dioxide nanoparticles (TiO2 NPs) represent a scientific breakthrough in the areas of biological and medicinal applications. Interaction of TiO2 NPs with components of innate immune system remains elusive.
Aim: This study explored in vitro immunotoxicity of murine macrophage RAW 264.7 to TiO2 NPs (20 nm, negative charge) and its underlying molecular mechanism by way of immunoredox profiling.
Materials and methods: In this study, chemically synthesized BSA-functionalized TiO2 NPs (20 nm, negative charge) were characterized and immunotoxicity was investigated on RAW 264.7 cells.
Results: We found that reactive oxygen species levels significantly increased with increasing nitric oxide production, whereas depleting endogenous antioxidant super oxide dismutase as well as nuclear factor erythroid 2-related factor 2 (Nrf2) protein levels. Furthermore, NPs exposure increased the expression of apoptotic factors such as BAX, BIM, and PUMA with disruption of mitochondrial membrane potential (ΔΨm) that lead to decrease in immunocytes. Molecular immune profiling revealed the activation of multiple toll-like receptors (TLRs) 4/9/12/13 simultaneously with the phosphorylation of p-p38MAPK and p-SAPK/c-Jun N-terminal kinase (JNK) compared to untreated control.
Conclusion: Collectively, this study shows that the molecular nature of TiO2SA20(-) NP-induced immunotoxicity in RAW 264.7 macrophage is simultaneous induction of immunocyte apoptosis and multiple TLRs signaling through oxidative stress-dependent SAPK/JNK and p38 mitogen-associated protein kinase activation. This is the first study to address newer molecular mechanism of TiO2SA20(-) NP-induced immunotoxicity.
Keywords: titanium dioxide, toll-like receptors, MAPK pathways, apoptosis, nanoparticles, oxidative stress
Plain language summary
TiO2SA20(−) NPs induced immunocyte apoptosis via disrupting mitochondrial membrane potential (Δψm) as well as upregulating proapoptotic BIM, PUMA, and BAX. TiO2SA20(−) NPs induced molecular redox imbalance. Reactive oxygen species levels significantly increased with increase in nitric oxide production, whereas depleting endogenous antioxidant super oxide dismutase and nuclear factor erythroid 2-related factor 2 (Nrf2) protein levels. TiO2SA20(−) NPs induced simultaneous activation of selective toll-like receptors (TLRs) such as TLR4, TLR9, TLR12, and TLR13. We found that the activation of p-p38MAPK and p-SAPK/c-Jun N-terminal kinas mitogen-associated protein kinases in TiO2SA20(−) NPs induced immunotoxicity.
Introduction
Metal oxide nanoparticles (NPs), such as titanium dioxide (TiO2), have sparked increasing interest recently with its wide range of application in the areas of nanomedicine, medical implants, drug delivery, medical imaging, biomedical nanodevices, food industry, and cosmetics; ranking it as one among the two top most commercially produced and extensively used NPs on a global scale.1 At the same time, its complex biosurface reactivity and dynamic physiochemical properties are often challenged by growing concerns on its safety and undesirable intracellular interactions that lead to potential health hazards for humans. The International Agency for Research on Cancer has reported TiO2 as group 2B carcinogen and possibly carcinogenic to humans.2,3 However, there is still no official safety regulation for the assessment and evaluation of NPs toxicity. Moreover, since the effects of TiO2 NPs on immune function have been poorly documented, further study for the underlying molecular mechanism of NP-induced immunotoxicity has been highly demanded.
Apoptosis is a significant and distinct form of “programmed cell death,” which is genetically determined to digest intracellular components and eliminate damaged cells from the surrounding as a defense mechanism to stress or pathophysiological stimuli.4 Overproduction of ROS is generally counteracted and neutralized by endogenous antioxidant enzymes such as super oxide dismutase (SOD), catalase, and GPx.5 In addition, nuclear factor erythroid 2-related factor 2 (Nrf2) acts as an important transcription factor for an array of detoxification antioxidant genes and protects cellular organelles like mitochondria from oxidative decay.6 Once the generation of ROS supersedes the capacity of the antioxidant defense, cells undergo severe cellular dysfunction or cell death resulting from “oxidative stress”. Oxidative stress plays a pivotal role in activating apoptosis and upregulating proapoptotic PUMA and BIM that recruits BAX on mitochondrial membrane to release apoptotic factors, such as cytochrome c and caspases, lowering mitochondrial membrane potential (MMP, Δψm) delivering “self-killing” signals to the cells.7 On the other hand, apoptosis could be regulated by oxidative stress or toll-like receptors (TLRs)-mediated activation of c-Jun N-terminal kinase (JNK) and p38 mitogen-associated protein (MAP) kinases pathway.8 Several studies suggested cytotoxic effects of TiO2 NPs decreasing cellular viability that lead to potential pathophysiological conditions in animal model and in human9–12 demanding essentiality to gauze underlying cell death mechanism.
TLRs are subfamily of pattern recognition receptors, placed strategically on the cell surface and endosome of primary immune cell macrophages. TLRs at the cell surface specifically recognize pathogen-associated molecular patterns (PAMPs) like lipopolysaccharide and intracellular endosomal TLRs recognize self-derived damage-associated molecular patterns (DAMPs) such as nucleic acids, ROS, and reactive nitrogen species (RNS).13 In human, the TLR family comprises 10 members from TLR1 to TLR10, and in mouse 12 members from TLR1 to TLR9 and TLR11 to TLR13 that have been discovered till date. Surface plasma membrane TLRs include TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10, whereas endosomal TLRs include TLR3, TLR7, TLR8, TLR9, TLR11, TLR12, and TLR13, which are located inside the cell.13 The pathway from TLR downstream initiating macrophage survival/death also recruits the intracellular Toll/interleukin-1 receptor (TIR) homology domain-containing MyD88 to elicit expression of the immune response.14 Unfortunately, the effects of nanotoxicity-related innate immune response, specifically TLR involvement of metal oxide-induced immunotoxicity has been poorly documented.
Although TiO2 NPs have burgeoning applications in the areas of environmental protection, food industry, cosmetics, and nano-biomedical purposes, macrophages are the primary sentinel to sense nonself through TLRs. We attempted to explore the underlying molecular mechanism by which TiO2SA20(−) NPs (20 nm, negative charge) induce immune cell death, in relation to TLRs involvement by way of immune-redox profiling.
Materials and methods
Materials
Manufactured TiO2 NPs were purchased from Sigma-Aldrich (Lot No 718467, Merck KGaA, Darmstadt, Germany). Potassium bromide (KBr, Lot No 221864) and BSA (Lot No A9418) were purchased from Sigma-Aldrich (Sigma Chemical Co., St Louis, MO, USA). DMEM and FBS were purchased from Hyclone Laboratories Incorporated (South Logan, UT, USA). The antibiotic–antimycotic was received from Gibco Invitrogen Corporation (Auckland, New Zealand). 2,7-dichlorodihydrofluorescein diacetate (H2DCF-DA) fluorescent probe was acquired from Sigma-Aldrich (Sigma Chemical Co.). Antioxidants such as SOD activity assay kits were purchased from BioVision (BioVision Inc., Milpitas, CA, USA). Remaining chemicals mentioned in the Methods section were purchased from Sigma-Aldrich (Sigma Chemical Co.) unless otherwise stated.
Preparation and characterization of TiO2 NPs
The TiO2 NPs used in this study were of 20 nm (TiO2SA20, Sigma-Aldrich, Merck KGaA), with 99.5% approximate purity, spherical, white, odorless powder. As described in Figure 1, the surface charge (zeta potential) was modified with coating reagents such as BSA solution, to make the TiO2 NPs negative charge (TiO2SA20(−)). Briefly, 10% wt/v of TiO2 NPs was suspended in deionized water for 30 minutes on a magnetic vortex rotor and mixed with 8% wt/v BSA solution with the same volume. The suspension was then vigorously stirred for 24 hours. NPs suspension was vortexed and sonicated for 30 minutes before treatment and acute toxicity was evaluated according to standard Organisation for Economic Co-operation and Development (OECD) nanosafety guidelines (OECD 64, 83 2016–17) on immune-redox parameter.24,34
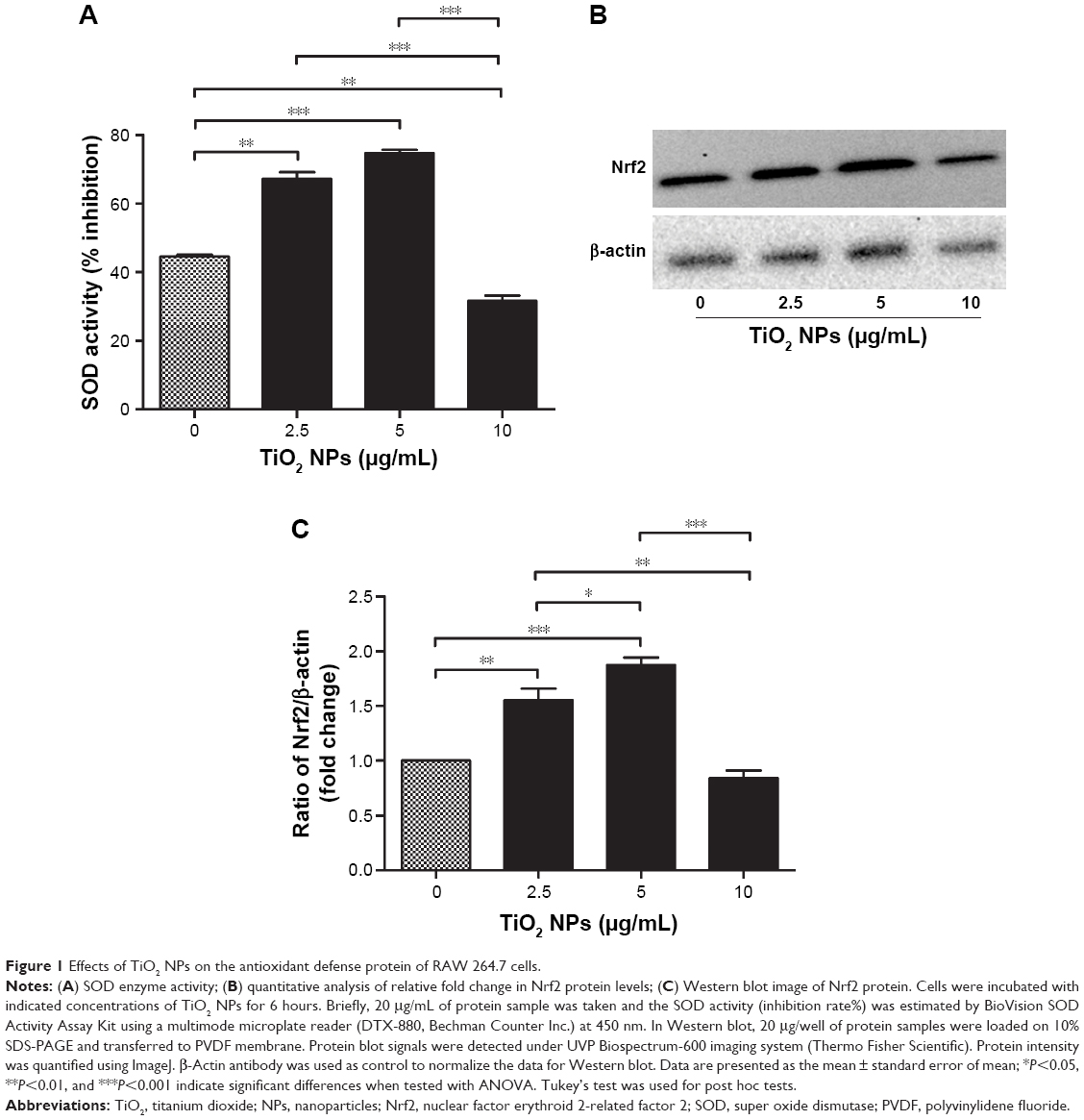 | Figure 1 Effects of TiO2 NPs on the antioxidant defense protein of RAW 264.7 cells. |
Morphological characteristics were observed using a predictable field-emission scanning electron microscope (FE-SEM, JEOL JSM-7500F, Tokyo, Japan) for prepared TiO2SA20(−) NPs sample with drop casting 5–10 μL suspension onto silica substrate surface (1 × 1 cm3). The hydrodynamic size and surface charge (TiO2SA20(−) NPs: 400 μg/mL) were verified by evaluating the dynamic light scattering and zeta potential with ELSZ-1000 particle size analyzer (Otsuka Electronics Co., Ltd, Osaka, Japan). X-ray diffraction pattern was determined using AXS D2 Phaser X-ray diffractometer (Brucker Optik GmbH, Ettlingen, Germany) with Ni-filtered Cu-Kα radiation (λ=1.5418 Å) in the range of 2 θ from 0° to 80° angels of X-ray scan. Fourier-transform infrared spectroscopic spectra of TiO2SA20(−) NPs were obtained on a Fourier-transform infrared spectroscope (FT-IR) spectrometer (Nicolet™6700, Thermo Fisher Scientific, Waltham, MA, USA), using KBr pellet method, in the range of 4,000 from 500 at 4 cm−1 resolution.
Cell culture and cytotoxicity study
RAW 264.7 murine leukemic monocyte macrophages (~1 × 106 cells, American Type Culture Collection, Manassa, VA, USA) were cultured using DMEM culture medium supplemented with 10% FBS and 1% antibiotic–antimycotic in 6-well plate at 37° in 5% CO2 incubator. After reaching 70% confluence, media was discarded and replaced with TiO2SA20(−) NP suspensions with concentrations (2.5, 5, 10, 20, 40, and 80 μg/mL) and plates were incubated for 24 hours at 37°C in 5% CO2 incubator. Cells that were untreated with TiO2SA20(−) NPs were considered as normal control group for all in vitro experiments. After 24 hours of NPs exposure, macrophage cells were analyzed to detect cytotoxicity under digital inverted microscope (EVOS® XL, Life Technologies Co., Ltd., Carlsbad, CA, USA). Then Cell Counting Kit-8 (CCK-8, Dojindo Lab., Kumamoto, Japan) was used to investigate cell proliferation and cytotoxicity. Approximately 1 × 105 murine macrophage cells (100 μL/well) were plated in a 96-well plate with desired concentrations of TiO2SA20(−) NPs suspensions and incubated for 24 hours at 37°C in 5% CO2 incubator. Afterward, 10 μL/well CCK-8 solutions were added, incubated for 1 hour and absorbance was measured at 450 nm using DTX-880 multimode microplate reader (Beckman Coulter, Inc., Brea, CA, USA). Percent viable cells were calculated using the below formula. All experiments were repeated three times in triplicate.
|
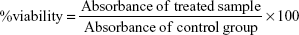
Intracellular total ROS detection
The generation of total ROS was detected using 2,7-dichlorodihydrofluorescein diacetate (H2DCF-DA) fluorescent probe (Sigma Chemical Co.). Approximately 1 × 106 murine macrophage cells (100 μL/well) were plated in 96-well black plate (Costar® 96-Well Black Polystyrene Plate) with flat bottom wells. After overnight incubation media was discarded and replaced with desired concentrations of TiO2SA20(−) NP suspensions. Then, media was discarded and cells washed with 1 × PBS and incubated with 100 μL/well of H2DCF-DA (10 μmol/L) for 30 minutes at 37°C in the dark. Then, cells were washed and analyzed by a multimode microplate reader (DTX-880, Bechman Counter Inc., Fullerton, CA, USA) at an excitation wavelength of 488 nm and an emission wavelength of 525 nm.
Measurement of intracellular nitric oxide (NO) production
Intracellular NO production was measured using Griess reagent system (Cat. # G2930, Promega Corporation, Fitchburg, WI, USA) according to the manufacturer’s instruction. Briefly, after 24 hours of NPs exposure, absorbance was measured every 5 minutes by incubating with required reagent using ELx800 microplate reader (BioTek, Thermo Fisher Scientific) at 540 nm.
MMP (Δψm) assay
Mitochondrial transmembrane potential (Δψm) was measured using the fluorochrome cationic dye JC-1 (5,5,6,6-tetrachloro-1, 1,3,3-tetraethylbenzimidazolcarbocyanineiodide) with NucleoCounter® NC-3000™ system (ChemoTec, Allerod, Denmark). Briefly, 1 × 106 cells/mL were suspended in PBS after TiO2SA20(−) NPs for 24 hours. Then, cells were stained with 5 μM of JC-1 dye (Lot: 755414, Invitrogen™, Molecular Probes™, OR, USA) and incubated for 15 minutes in room temperature in dark. Then, media was discarded and stained cells were washed twice with 1 mL of PBS by centrifugation at 400 rpm for 5 minutes at room temperature. Pellets were resuspended in PBS and 10 μL of sample were loaded in each chambers of the NC-slide A8 (ChemoTec). Cellular red and green fluorescence intensity was visualized using NC-3000™ system.
Muse Annexin V and Dead Cell assay
Apoptosis was investigated using Muse™ flowcytometric method with Muse™ Annexin V and Dead Cell Kit (Cat. No: MCH100105, Merck Millipore, KGaA, Darmstadt, Germany). Briefly, 3 × 105 cells/mL were plated in 6-well plate with TiO2SA20(−) for 24 hours and 100 μL of cell suspension was mixed thoroughly with 100 μL of Muse Annexin V and dead cell and incubated for 20 minutes in room temperature in dark. The samples were then analyzed using MUSE™ cell analyzer (Merck Millipore, KGaA). Percentage of events was categorized in dot plot after gating.
Reverse transcriptase (RT-PCR) and quantitative real-time polymerase chain reaction (Q-PCR)
Total RNA was extracted from cells using easy-BLUE(™) Total RNA Extraction Kit (Cat. #17061, iNtRON Biotechnology, Sungnam, South Korea) and reverse transcribed to first strand of c-DNA using Hyperscript RT Premix (w/oligo dT; Cat. #601–632, GeneAll Biotechnology Co. Ltd., Seoul, South Korea). The reverse transcriptase PCR reactions were performed using AmpONE™ Taq premix (Cat. #521–200, GeneAll Biotechnology Co. Ltd., Seoul, South Korea) on cDNA aliquots using GeneAmp® PCR System 2400 (Thermo Fisher Scientific). The PCR reactions were performed under PCR conditions of 95°C for 15 minutes and 35 cycles of denaturation at 95°C for 5 seconds, annealing at 60°C for 45 seconds, and elongation at 72°C for 1 minute.
The Q-PCR reactions were performed using QuantiTect SYBR Green PCR Kit (Qiagen, Basel, Switzerland) on cDNA aliquots in 7900HT fast real-time PCR system (Thermo Fisher Scientific) under PCR conditions of 95°C for 15 minutes and 40 cycles of denaturation at 95°C for 5 seconds, annealing at 60°C for 45 seconds, and elongation at 72°C for 1 minute. The primer sequences used to amplify all TLRs, TIR (IDT, Coralville, IA, USA) BAX, BIM, and PUMA (Bioneer, Seoul, South Korea) gene fragments are listed in Table 1. Housekeeping gene glyceraldehyde-3-phosphate (GAPDH) used as control. The gene expressions were quantified by comparative Ct method (ΔΔCt), and the relative quantification was calculated as 2−ΔΔCt. PCR products were separated by 1% agarose gel electrophoresis and bands were visualized under UVP Biospectrum-600 imaging system (Thermo Fisher Scientific) after ethidium bromide staining (EtBr; Sigma-Aldrich).
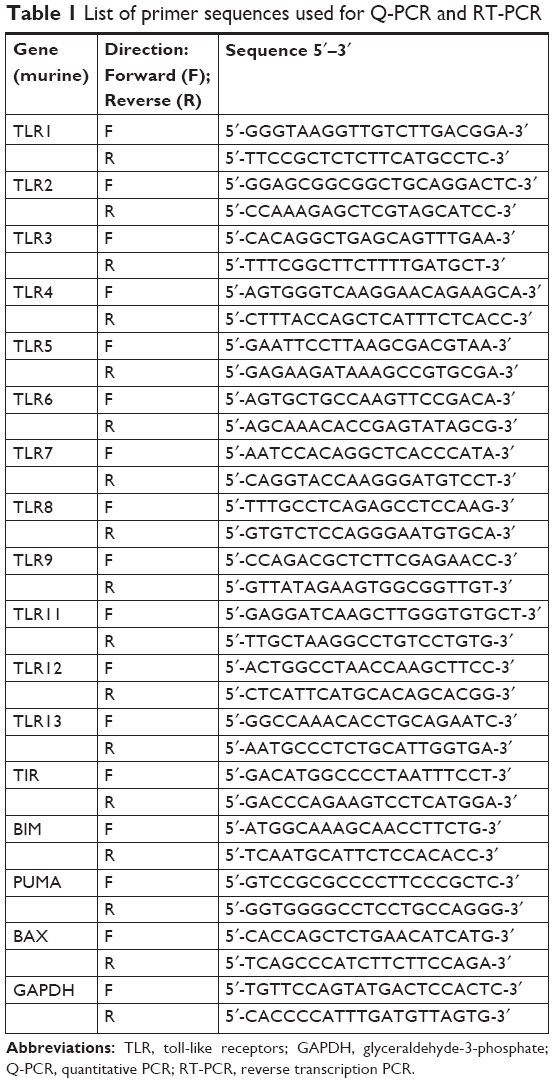 | Table 1 List of primer sequences used for Q-PCR and RT-PCR |
Quantification of total cytosolic and mitochondrial SOD activity
Macrophage cells were lysed using RIPA lysis buffer (Thermo Fisher Scientific, Inc.) supplemented with 1% proteinase inhibitor and phosphatase inhibitor cocktail (Roche Diagnostics GmbH, Mannheim, Germany). Total protein was quantified using Pierce™ BCA Protein Assay Kit (Pierce™, Thermo Fisher Scientific). The total cytosolic and mitochondrial SOD activity in murine macrophage upon TiO2SA20(−) NPs exposure for 24 hours was measured by BioVision SOD Activity Assay Kit (Cat. #K335-100, Milpitas, CA, USA). Briefly, 20 μL of protein sample (20 μg/mL) was pipetted into each sample wells, and 200 μL of the WST-1 working solution and 20 μL of the SOD enzyme working solution were added to the sample wells and mixed thoroughly. The solution was incubated at 37°C for 20 minutes. The inhibition/reduction activity of SOD was estimated by using multimode microplate reader DTX-880 (Bechman Counter Inc.) at 450 nm. The SOD activity (inhibition rate %) was calculated according to the manufacturer’s instruction.
Western blot analysis
At 24 hours postexposure of TiO2SA20(−) NPs, equal volume (20 μg/well) of RAW 246.7 cells protein sample were loaded on 10% SDS-PAGE. Then the separated protein was electrophoretically transferred to a (0.2-μm pore size) hydrophobic polyvinylidene fluoride (PVDF) transfer membrane (Millipore, Bedford, MA, USA) via wet-transfer procedure at 100 V, 200 mA for 2 hours. To check for phospho-protein, blocking of protein was performed using 5% w/v BSA for 1 hour at 4°C with agitation. Primary antibodies against Phospho-SAP/JNK (Thr 183/Tyr 185), Phospho-p38MAPK (Thr 180/Tyr 182), and Nrf2 were diluted in tris-buffered Saline-Tween (TBST) with 5% w/v BSA and incubated overnight at 4°C with agitation. Anti-β-actin antibody was used as loading control protein. Horseradish peroxide-linked anti-rabbit IgG secondary antibody was diluted with TBST and membrane was incubated for 2 hours at 4°C with agitation. Desired protein blot signals were detected using enhanced chemiluminescence solution (Thermo Fisher Scientific, Pierce™) and bands were visualized under UVP Biospectrum-600 imaging system (Thermo Fisher Scientific). Protein intensity was quantified using image processing software ImageJ (National Institutes of Health, Bethesda, MD, USA). Detailed information on antibody specification and dilution are shown in Table 2.
 | Table 2 List of antibodies used in Western blot |
Statistical analysis
Data are represented as mean ± standard error of mean. ANOVA followed by subsequent Tukey’s multiple comparison test were considered for comparing mean values among groups, while for Q-PCR analysis mean ± SD value was considered. For this, GraphPad Prism version 5.0 software packages (GraphPad Software, La Jolla, CA, USA) were used. Statistically significant differences were considered at *P<0.05, **P<0.01, and ***P<0.001 vs untreated control.
Results
Physiochemical characterization of TiO2 NPs
Preparation and surface charge modification of TiO2SA20(−) NPs has been schematically depicted in Figure 2A. The morphological characterization based on FE-SEM images (Figure 2B) showed that particles having irregular shape were homogenously distributed without significant agglomerate formation. The NPs did not show clear grain boundaries which is a typical morphology of inorganic NPs coated with polymeric organic moiety.35 Further, surface charge and hydrodynamic radius of TiO2SA20(−) NPs was estimated from zeta potential and dynamic light scattering methods. According to the incorporation information, the TiO2SA20(−) NPs used in this study have actual size of 21±3 nm. Hydrodynamic radii of the NPs (Figure 2C) increased and strong negative surface charge was detected to be zeta potential of average −32.5 mV initially from −6.7 mV and polydispersity index of 0.235 from 1.001 (nonfabricated) indicating NPs suspension has good dispersion stability with BSA surface coating. Powder X-ray diffraction study showed that TiO2SA20(−) NPs is a mixture of anatase and rutile phase (Figure 2D) and did not alter its crystallography structure with surface modification. Strong diffraction peaks at 2θ=25.3° and 48.1° corresponding to (101) and (200) reflection, respectively, indicate TiO2 NPs in the anatase phase (JCPDS No 78-2486). Several small peaks at 2θ=27.4° and 36° attributed to (110) and (101), respectively, revealed that TiO2 NPs also partially included rutile phase (JCPDS No 65–0191). FT-IR spectra showed both peaks for TiO2 at 740 cm−1 (Ti−O−Ti stretching) and 3,384 cm−1 (surface hydroxyl or H2O) and BSA at 1,684 (N−H stretching from amine moiety), suggesting the coexistence of both TiO2 NP and BSA in TiO2SA20(−) NPs (Figure 2E).
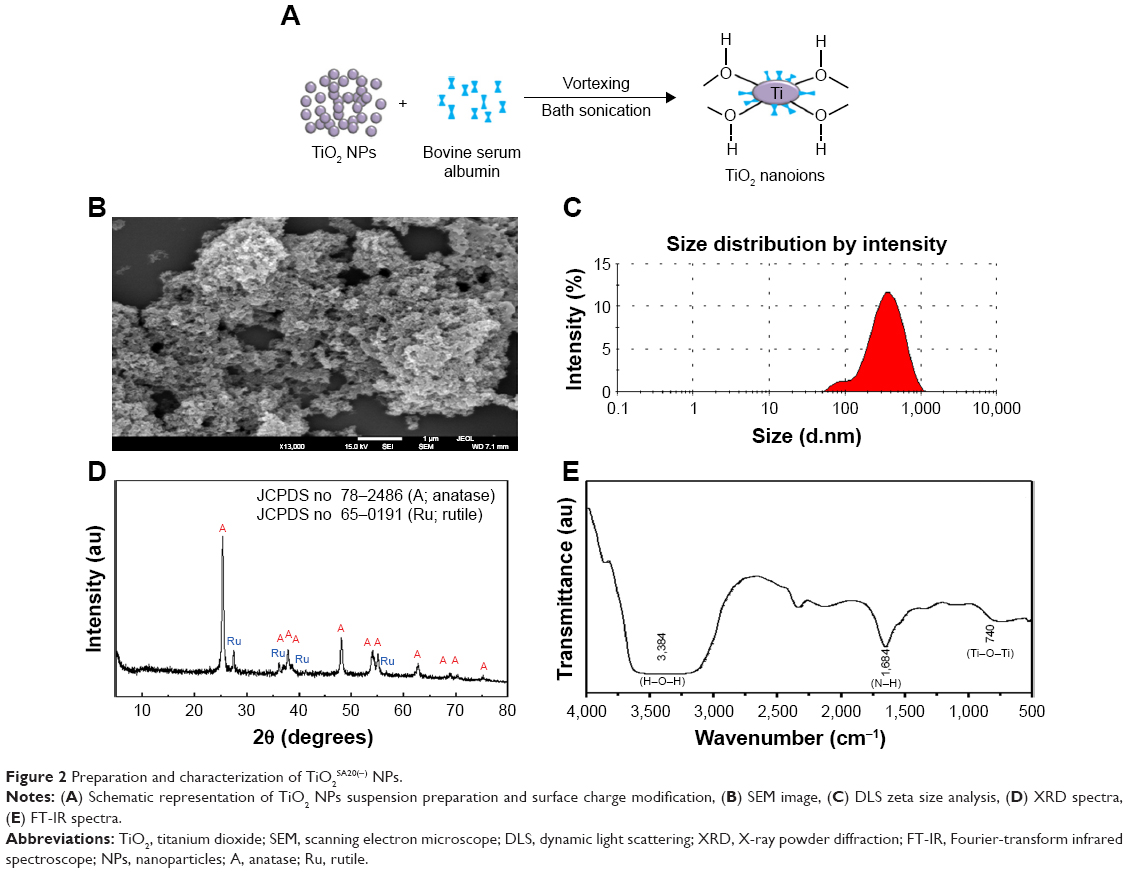 | Figure 2 Preparation and characterization of TiO2SA20(−) NPs. |
TiO2SA20(−) NPs induce cytotoxicity in murine macrophages
To examine the potential toxicity of TiO2SA20(−) NPs on the macrophage, RAW 264.7 cells were exposed to indicated concentrations of NPs for 24 hours. Cytotoxicity was observed under inverted microscope and viability was assessed using CCK-8 Kit. Results were compared to untreated control. Results indicated that there was a dose-dependent increase in cytotoxicity as evidenced by cell shrinkage, cell death, and apoptotic body formation (Figure 3A) leading to significant decrease (P<0.001) in cell viability (Figure 3B) compared to untreated control. The half maximal effective concentration (EC50) value of TiO2SA20(−) NPs on 24 hours growth inhibition of murine macrophage was 20.04 μg/mL.
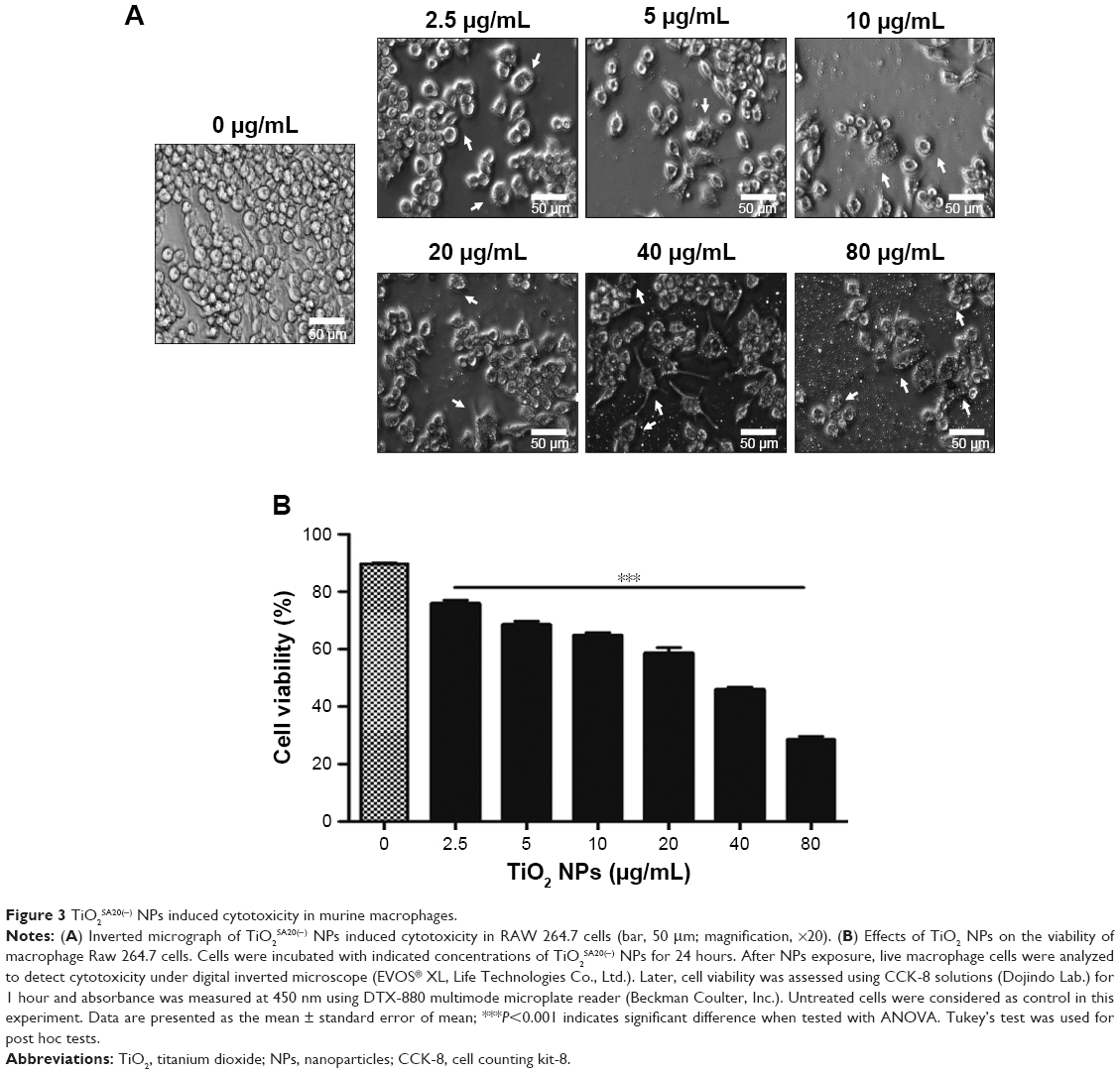 | Figure 3 TiO2SA20(−) NPs induced cytotoxicity in murine macrophages. |
TiO2 NPs induce oxidative stress via ROS generation and intracellular NO production
To test whether ROS is involved in TiO2SA20(−) NPs induced cytotoxicity, RAW 264.7 cells were exposed to TiO2SA20(−) NPs suspensions. We found that at initial phase (up to 10 μg/mL) ROS level remained unaffected but suddenly increased significantly with higher dose-concentration as compared to untreated cells (Figure 4A).
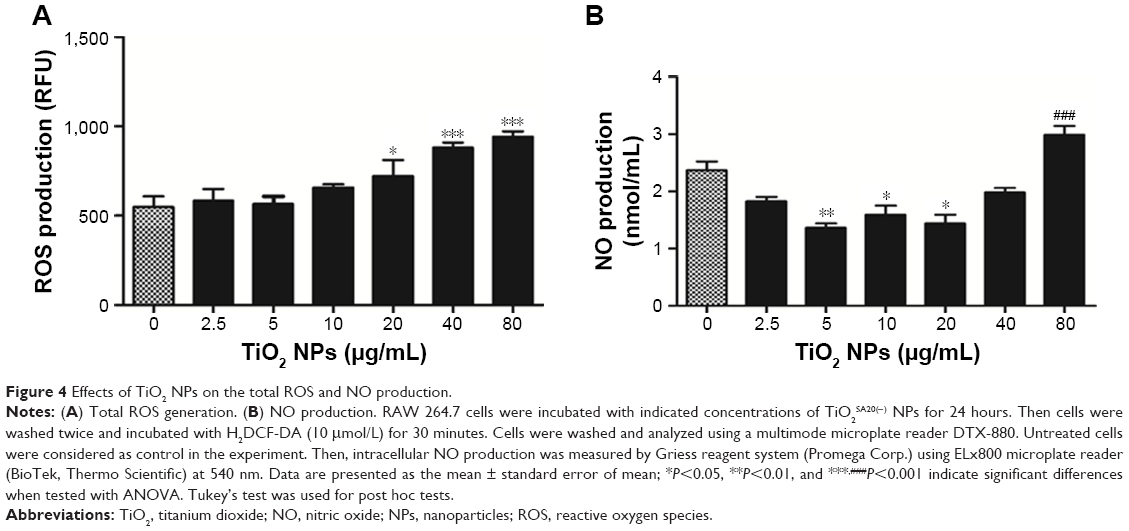 | Figure 4 Effects of TiO2 NPs on the total ROS and NO production. |
NO is a well-known indicator of macrophagic activity, inflammation, and tissue damage15 and to check whether NO generation is involved in TiO2 NP-induced cytotoxicity. Intracellular NO production was measured. Figure 4B shows that at initial stage of TiO2 NPs (5 μg/mL, 10 μg/mL, 20 μg/mL) exposure significantly reduced NO level (P<0.05, P<0.01, P<0.01, respectively) albeit gradually with higher concentration of NPs (80 μg/mL). NO generation significantly increased (P<0.001) as compared to untreated control group.
Effects of TiO2 NPs on endogenous antioxidant response
Modulation of oxidative stress by cellular endogenous antioxidant enzymes was determined by measuring the activities of SOD and Nrf2 protein levels in RAW 264.7 cells after 6 hours exposure to different concentrations of TiO2SA20(−) NPs. Figure 1A showed that SOD activity was significantly higher in cells treated with TiO2SA20(−) NPs suspensions at 2.5 μg/mL (P<0.01) and 5 μg/mL (P<0.001), but eventually with higher dose at 10 μg/mL SOD activity decreased significantly (P<0.01) than in control. Conversely, Nrf2 activity was significantly higher in cells treated at 2.5 μg/mL (P<0.01) and 5 μg/mL (P<0.001), and in later stage at higher dose TiO2SA20(−) NPs suspensions at 10 μg/mL Nrf2 activity depleted significantly (P<0.01) as compared to control (Figure 1B and C).
TiO2 NPs induce the disruption of MMP
Apart from being the energy source of life, mitochondria can also act as the source of apoptotic cell death signals.16 During apoptotic process, the MMP is collapsed, but whether this phenomenon occurs in TiO2SA20(−) NP-induced immune cell death has not been identified. We observed in this study that TiO2 NPs trigger mitochondrial depolarization, which was detected as a decrease in the red/green ratio of JC-1 fluorescence intensity. Due to the depolarization of MMP by TiO2 NPs, mitochondrial JC-1 aggregates (red fluorescence) decrease with potential-dependent manner along with increased JC-1 monomers (green fluorescence; Figure 5). It is conceivable that the depolarization of MMP indicates the early apoptosis mechanism of cell death by TiO2 NPs.
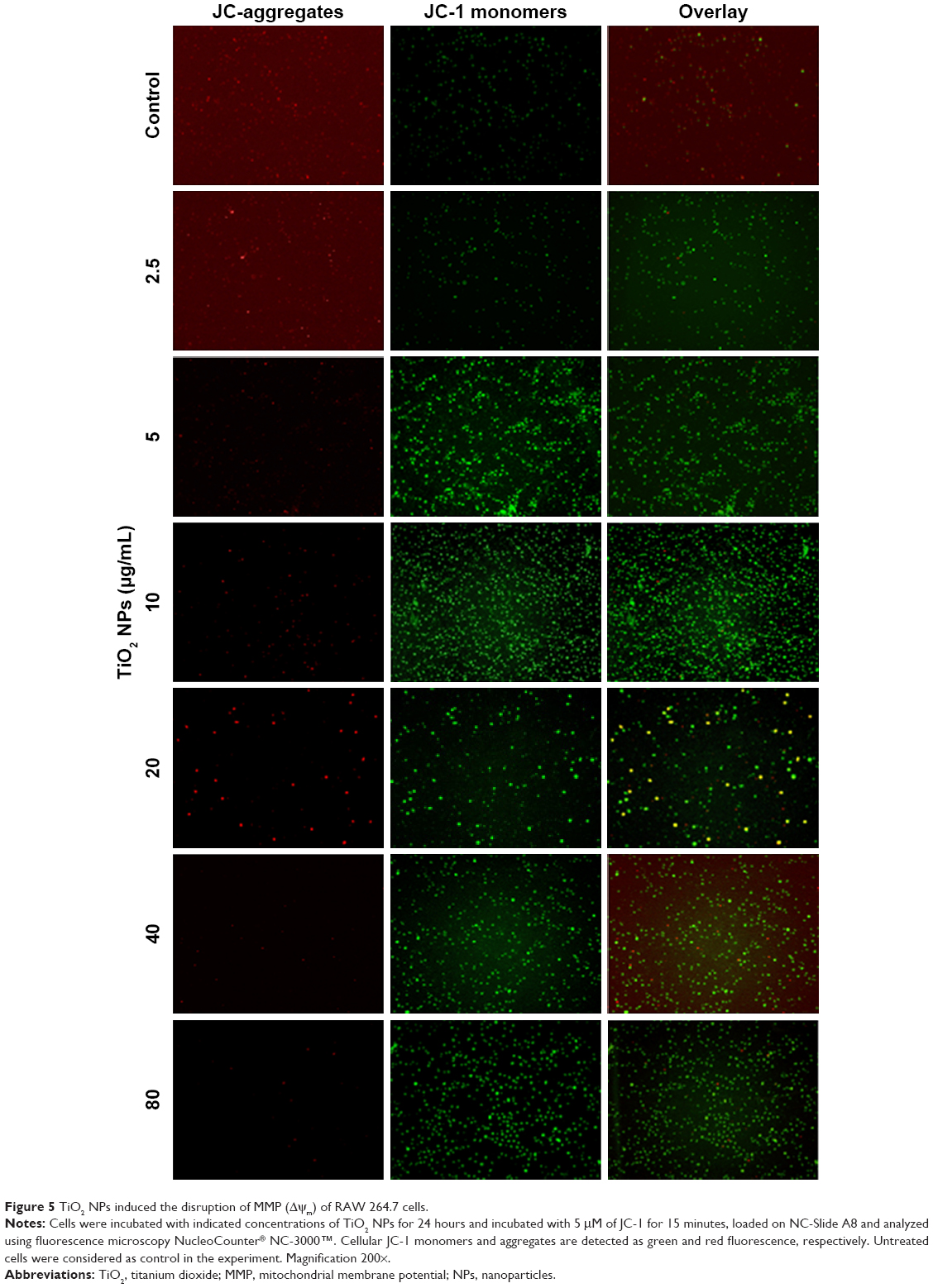 | Figure 5 TiO2 NPs induced the disruption of MMP (Δψm) of RAW 264.7 cells. |
TiO2 NPs induce mitochondrial proapoptotic factors and subsequent apoptosis in RAW 264.7 cells
Whether TiO2 NPs induce immune cell death via mitochondria-mediated apoptotic pathway enforcing BAX expression is a matter of conjecture. Herein, we proposed a possible mechanism depicting PUMA and BIM could activate mitochondrial BAX to execute apoptosis upon TiO2 NPs exposure (Figure 6A). RT-PCR analysis (Figure 6B) depicted that a 24 hours exposure of murine macrophage cells to TiO2 NPs led to a dose-independent increase in expression of BIM, PUMA, and BAX in comparison to untreated control group. To explore whether apoptosis is the form of cell death involved in TiO2SA20(−) NP-induced immunotoxicity on 24 hours exposure, early/late apoptosis stages were investigated using Muse™ Annexin V and Dead Cell assay, and analyzed using flowcytometric method (Muse cell analyzer, Merck Millipore, KGaA). We found that there was a significant increase (P<0.001) in early stages of apoptotic cells [Annexin V-PE (+) and dead cell marker (−)] and late apoptotic/dead cells [Annexin V-PE (+) and dead cell marker (+)] in concentration-dependent manner as compared to untreated control (Figure 6C and D).
 | Figure 6 TiO2 NPs induced mitochondrial proapoptotic factors and subsequent apoptosis in RAW 264.7 cells. |
TiO2 NPs activate multiple TLRs
TLRs are innate sensors of foreign particles that lead to innate immune response, and cell survival/death is partly regulated by TLRs activation.17 To uncover underlying molecular immune mechanism on exposure of TiO2SA20(−) NP-induced immunotoxicity, we have investigated mRNA expression levels of all discovered murine TLRs and adaptor TIR (Figure 7). Screening of TLRs via real-time PCR showed that mRNA expression levels of multiple TLRs were activated simultaneously. Specifically, TLR4, TLR9, TLR12, and TLR13 mRNA expression was upregulated compared to control group (Figure 7B). Unexpectedly, newly discovered TLR12 and TLR13 has been significantly upregulated upon TiO2SA20(−) NPs exposure. In line, adaptor TIR mRNA expression was increased in the experimental group (2.5, 5, and 10 μg/mL) compared to untreated control group. By contrast, TLR5 and TLR11 m-RNA was not expressed (Figure 7A). However, TLRs response was not clearly dose-dependent by TiO2SA20(−) NPs.
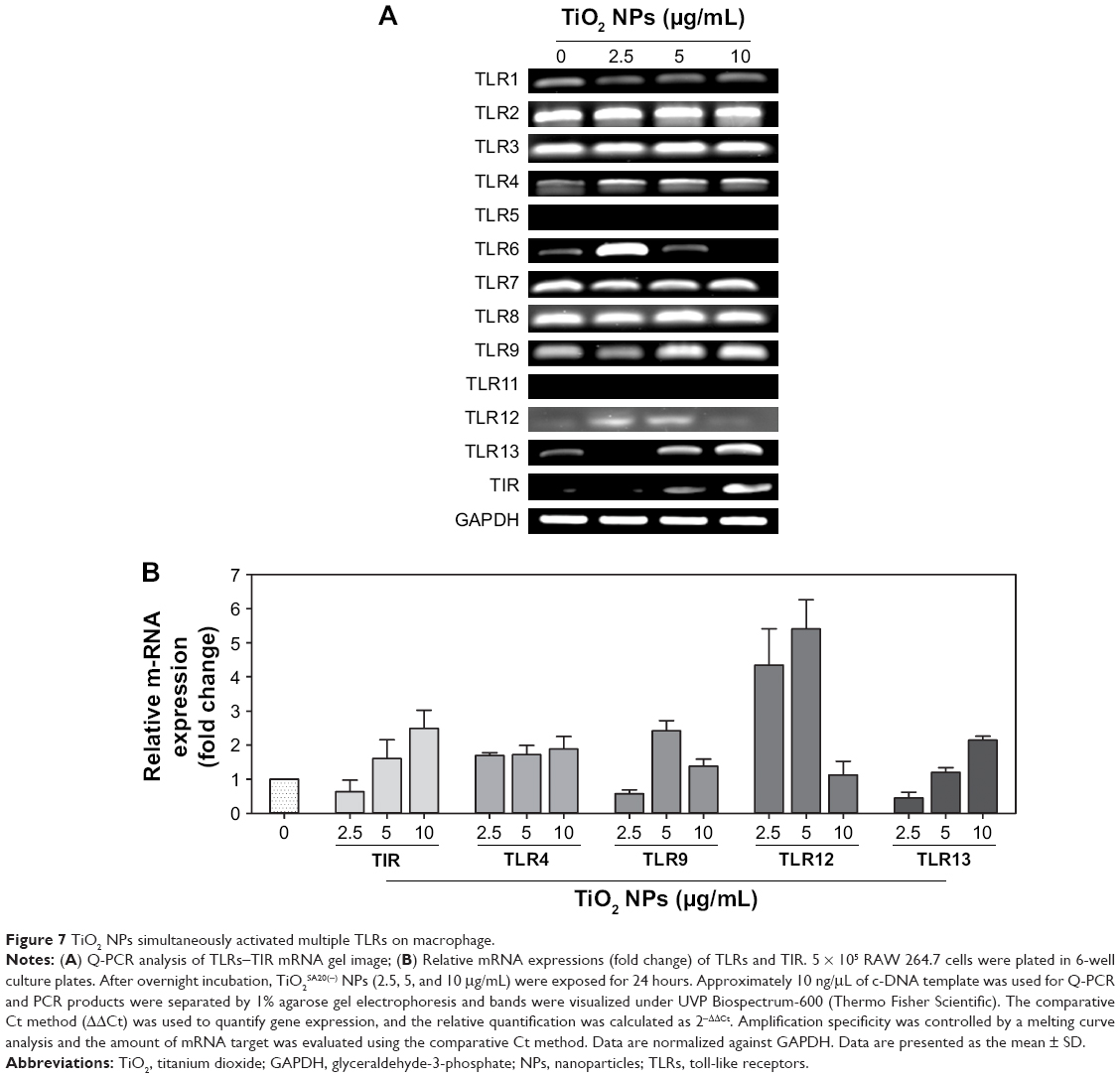 | Figure 7 TiO2 NPs simultaneously activated multiple TLRs on macrophage. |
Role of MAP kinase signaling pathway in TiO2 NP-induced immunotoxicity against macrophage
To unveil the downstream signaling targets of activated TLRs and/or ROS/RNS on TiO2 NP-induced immunotoxicity in murine macrophage, Western blot on mitogen-associated protein kinases (MAPK) was done. We found that there was an increase in phosphorylation of tyrosine and threonine regions of p38MAPK (Figure 8A and B) and SAPK/JNK (Figure 8C and D) at 6 hours NPs incubation. Protein expression of p-p38MAPK significantly increased at initial phase of NPs incubation at 2.5 μg/mL (P<0.01) and 5 μg/mL (P<0.05). Importantly, with higher concentration of TiO2 NPs (10 μg/mL) phosphorylation was increased by twofold (P<0.001). Simultaneously, p-SAPK/JNK protein expression was significantly (P<0.001) upregulated at higher concentration of TiO2 NPs (10 μg/mL). Moreover, MAPKs response was dose-independent by TiO2SA20(−) NPs.
 | Figure 8 TiO2 NPs triggered activation of MAP kinase signaling cascade. |
Discussion
In this study, we offered a new insight into the molecular mechanism of TiO2 NP-induced immunotoxicity, involving simultaneous activation of multiple TLRs, such as TLR4, TLR9, TLR12, and TLR13 triggering p-p38MAPK and p-SAPK/JNK MAP kinases pathway-mediated apoptosis and inflammatory response via mitochondrial dysfunction and immuno-redox imbalance (Figure 9). Since the molecular nature of TiO2 NP-induced immunotoxicity has been controversial, first we checked in vitro cytotoxicity in RAW 264.7 macrophage, thus confirming increase of dose-dependent cytotoxicity (Figure 3A and B). Next, we examined whether this cell death might be ascribed from mitochondrial dysfunction because of insufficient evidences from few, known reports. Clearly, this was evidenced by loss of Δψm (Figure 5), upregulating proapoptotic BIM, PUMA, and BAX (Figure 6B). Mitochondria plays an explicit role in regulating ROS-triggered apoptosis upon xenobiotic stress stimuli.16,27 MMP (Δψm) is a crucial parameter of mitochondrial function and integrity.28,29 To unveil TiO2SA20(−) NP-induced mitotoxicity, it is crucial to estimate Δψm that can predict mitochondrial damage. For this, the advanced cytofluorimetric, cationic lipophilic dye JC-1 probe was used, which can selectively enter into mitochondria and remain in the monomeric form changing color from red fluorescence to green as the Δψm decreases in depolarized apoptotic or unhealthy mitochondria.30 In this study, we observed that TiO2 NPs trigger mitochondrial depolarization, which was detected as a decrease in mitochondrial JC-1 aggregates (red fluorescence) in potential-dependent manner along with increased JC-1 monomers (green fluorescence; Figure 5). It is conceivable that the depolarization of MMP indicates the early apoptosis mechanism of cell death by TiO2 NPs. Mitochondrial apoptosis initiated through a complicated cascade signaling where PUMA and BIM that can directly activate BAX-mediated permeabilization of the outer mitochondrial membrane to release cytochrome c and apoptotic factors.31,32 To delineate a key event in the commitment to apoptosis which is mostly elicited upon activation of the proapoptotic BAX translocating to mitochondria, we employed RT-PCR analysis to observe BAX expression. Consistent with BIM and PUMA, the expression of proapoptotic BAX remarkably increased (Figure 6B). To further delineate the kinetics of apoptosis, we employed Muse™ Annexin V and Dead Cell assay for the quantitative analysis of live cells, early and late apoptosis/death on TiO2SA20(−) NPs exposure for 24 hours. This can allow for highly quantitative single-cell analysis compared to conventional Western blot method by laser-based fluorescence detection with microcapillary cytometry method. In line with other evidences, there was a significant increase in early stages of apoptotic cells (P<0.001) and late apoptotic/dead cells (P<0.001) in concentration-dependent manner as compared to untreated control (Figure 6C and D).
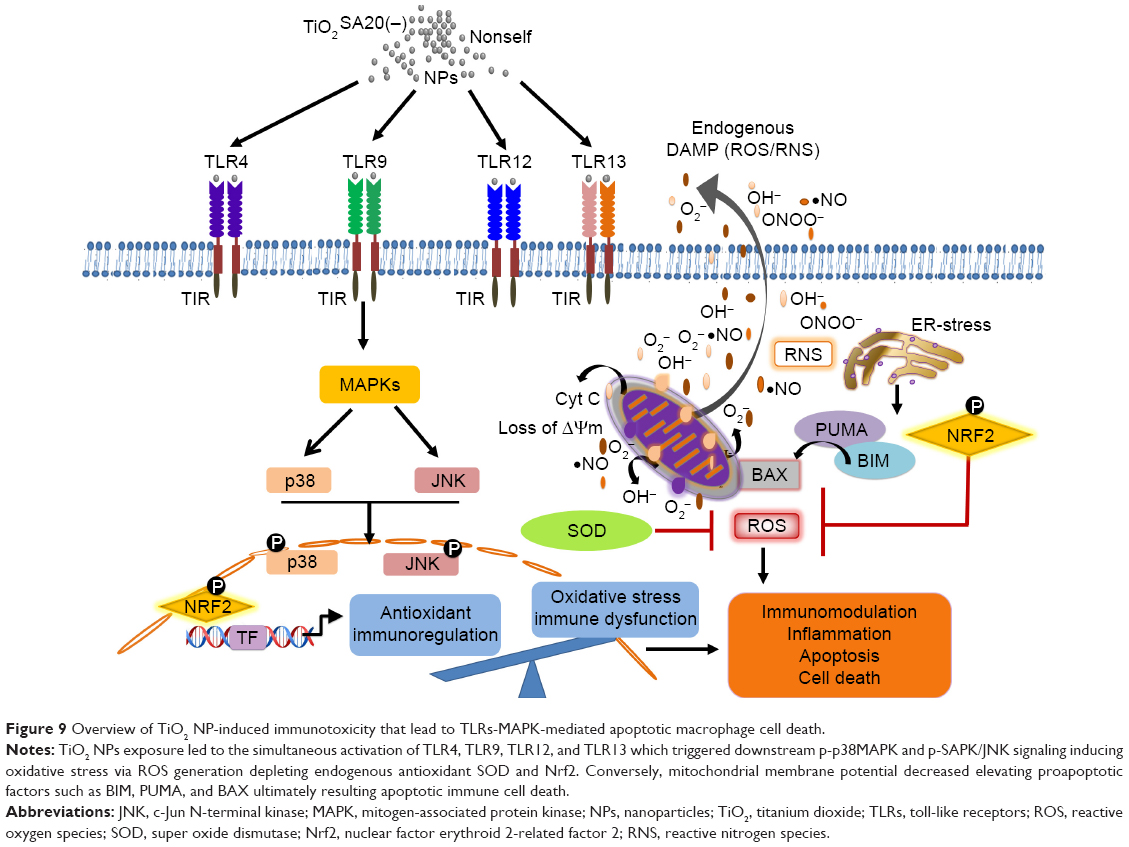 | Figure 9 Overview of TiO2 NP-induced immunotoxicity that lead to TLRs-MAPK-mediated apoptotic macrophage cell death. |
Third, while in metal oxide NP-induced immunotoxicity, the role of oxidative stress such as ROS/RNS and NO has been well known, there has been a scanty report on the molecular redox milieu. Our redox hypothesis is that molecular redox imbalance might culminate in cell death. To prove this, we adopted molecular redox profiling such as oxidative stress marker (ROS, NO) and endogenous antioxidant (SOD, Nrf2). ROS generation significantly increased with higher concentration of TiO2SA20(−) NPs (Figure 4A) which is indicative of oxidative stress. NO reacts with ROS superoxide ion (O2•−) to produce RNS-like peroxynitrite (ONOO−).5,9 NPs-mediated ROS and RNS act together to damage cells to induce pathophysiological conditions in various inflammatory diseases.3,5,9,23 Interestingly, in this study, NO production initially decreased significantly at lower doses (5–20 μg/mL) and later increased significantly with higher dose (80 μg/mL) of TiO2SA20(−) NPs on 24 hours exposure (Figure 4B). Oxidative burst is primarily counteracted by SOD, an antioxidant enzyme that selectively removes cytotoxic ROS super oxide ion (O2•−) and converts it into less toxic H2O2 and O2.5,24 In this study, SOD enzyme levels were found to be initially activated at lower concentration (2.5–5 μg/mL) to counteract the ROS production. However, ROS levels supersede the antioxidant defense, hence SOD enzyme level eventually degraded (Figure 1A) with increase in TiO2SA20(−) NPs concentration (10 μg/mL). Essentially, Nrf2, an antioxidant protein that is central to the transactivation of important detoxification antioxidants, is expressed in various tissue types, and considered major cytoprotective regulator in defense to oxidative stress.6,25 In chronic pathological conditions, Nrf2 is found to be degraded and cannot initiate antioxidant defense. Conversely with SOD enzyme, antioxidant Nrf2 also found to be activated at initial phage (2.5–5 μg/mL) of TiO2SA20(−) NPs exposure but at 10 μg/mL concentration Nrf2 protein degraded significantly (Figure 1B and C). In conclusion, ROS and RNS supersede SOD and Nrf2 antioxidant defense system resulting in reduced viability of macrophages. This implies that immune cells suffer from oxidative stress and imbalance in redox homeostasis upon TiO2SA20(−) NPs exposure. In support, several reports unanimously opined that ultrafine and NPs of TiO2 would induce oxidative stress.9,19,26 One of the new finding in this study is in consistency with degradation of major cytoprotective antioxidant Nrf2, mitochondrial integrity, and functions were found to be impaired on exposure to TiO2SA20(−) NPs for 24 hours, depletion of cytoprotective antioxidant enzyme SOD and Nrf2 (Figure 1) that lead to reduced macrophage RAW 264.7 cell viability (Figure 3A and B) with increased percent cell death (Figure 6D). Interestingly, MAPK-mediated apoptotic-signaling cascade was (Figures 7 and 8) activated by oxidative stress as evidenced by increase in ROS/RNS (Figure 4A and B).
Fourth, TLRs engagement in metal oxide-induced immunotoxicity is unclear, and only fewer studies reported specifically TLR4 involvement in NPs intracellular uptake into organelles, mitochondria, endosome, and the nucleus18–21 and immune response to TiO2 NP exposure in human cell lines and mouse embryonic cell lines.18,21,22 Unexpectedly, we discovered TiO2SA20(−) NPs induced selective activation of TLRs such as TLR4, TLR9, TLR12, and TLR13 (Figure 7A and B). Of those TLRs, except TLR4 on the cell surface, TLR9, TLR12, and TLR13 are expressed on intracellular endosome. This might imply the possibility of potential interaction between TiO2 NPs and endosomal TLRs. Owing to the absence of the precedent report, further meticulous study is required to clarify this. Our study might be partly supported by previous reports about TLR4 stimulation on different TiO2 NPs treatment.18,19,22 In contrast, the present study reports the activation of TLR9 and newly discovered TLR12 and TLR13 upon TiO2SA20(−) NPs treatment for the first time. It is likely that TiO2SA20(−) NPs act as TLR ligand, were sensed as PAMP/DAMP by macrophages, and internalized through TLR-mediated endocytosis that lead to apoptosis for elimination of NPs from the surrounding in order to protect the neighboring immune cells from the TiO2SA20(−) NP-induced immunotoxicity. Immune function of recently discovered murine TLR12 and TLR13 is unknown, which is functionally unidentified in human yet. In that context, this finding has an important implication to gain insights into the molecular mechanism of metal oxides NP-induced immunotoxicity.
Finally, to solve the linkage between the bottom path, mitochondrial apoptosis and the upper paths, redox, and TLR, we attempted to excavate the plausible common pathway – MAPK. MAPK family members importantly JNKs and p38-MAPKs are called stress responsive kinases thus known to regulate oxidative stress-mediated apoptosis.33 In accordance, recent studies reported that involvement of MAPK/NFkB in TLRs downstream signaling cascade resulted in NPs immune toxicity.9,18,21 It was reported that cellular uptake of Degussa TiO2 p25 NPs through TLR4 and activation of p38MAPK to induce phagocytosis in sea urchin.21 Given these, we examined TLRs downstream apoptosis signaling involving p38 and JNK MAPK pathways via Western blotting on TiO2SA20(−) NPs exposure for 6 hours. Acquired results showed that phosphorylation of p-p38MAPK increased significantly (P<0.001) by twofold in response to TiO2SA20(−) NPs exposure (Figure 8A and B). In the same line, the expression of phosphorylated p-SAPK/JNK protein was significantly (P<0.001) enhanced only at higher concentration of NPs (Figure 8C and D). However, both stress-activated proteins were phosphorylated in dose-independent manner. Taken together, these might imply the involvement of p38MAPK and SAPK/JNK signaling pathways in triggering TiO2SA20(−) NP-induced apoptosis in murine macrophage. In this regard, further morphological evidences provided by inverted microscopy observations (Figure 3A), which showed cell shrinkage and apoptotic body formation (arrowhead) might validate TiO2SA20(−) NP-induced macrophage death. Despite the elucidation of this MAPK path, whether this would be the downstream of TLR and/or redox path remains elusive. However, in vivo experimental confirmation using inhibitors would further validate this hypothesis.
Conclusion
Collectively, this study shows that the molecular nature of TiO2SA20(−) NP-induced immunotoxicity in RAW 264.7 macrophage is simultaneous induction of immunocyte apoptosis and multiple TLRs signaling through ROS-dependent SAPK/JNK and p38MAPK activation. This is the first study to address newer molecular mechanism of TiO2SA20(−) NP-induced immunotoxicity. This study might be an instrument to extrapolate new nanoimmunotoxicity biomarkers for risk assessment and mitigation in special reference to unveil nano-immunotoxic TLRs–MAPK signaling pathway.
Acknowledgment
This research was supported by the Basic Science Research Program through the National Research Foundation (NRF) funded by the Ministry of Education (NRF-2018R1D1A1B07048194) and the Medical Research Center (MRC) Program, Ministry of Science, ICT and Future Planning (2017R1A5A2015369), Republic of Korea.
Disclosure
The authors report no conflicts of interest in this work.
References
Bondarenko O, Juganson K, Ivask A, Kasemets K, Mortimer M, Kahru A. Toxicity of Ag, CuO and ZnO nanoparticles to selected environmentally relevant test organisms and mammalian cells in vitro: a critical review. Arch Toxicol. 2013;87(7):1181–1200. | ||
Iafro C. Titanium dioxide group 2B. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. 2006;93:193–214. | ||
Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, Cogliano V. Carcinogenicity of carbon black, titanium dioxide, and talc. Lancet Oncol. 2006;7(4):295–296. | ||
Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. | ||
Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012;5(1):9–19. | ||
Strom J, Xu B, Tian X, Chen QM. Nrf2 protects mitochondrial decay by oxidative stress. FASEB J. 2016;30(1):66–80. | ||
Liu FT, Newland AC, Jia L. Bax conformational change is a crucial step for PUMA-mediated apoptosis in human leukemia. Biochem Biophys Res Commun. 2003;310(3):956–962. | ||
Son Y, Kim S, Chung HT, Pae HO. Reactive oxygen species in the activation of MAP kinases. Methods Enzymol. 2013;528:27–48. | ||
Iavicoli I, Leso V, Bergamaschi A. Toxicological effects of titanium dioxide nanoparticles: a review of in vivo studies. J Nanomater. 2012;2012:1–36. | ||
Warheit DB, Brock WJ, Lee KP, Webb TR, Reed KL. Comparative pulmonary toxicity inhalation and instillation studies with different TiO2 particle formulations: impact of surface treatments on particle toxicity. Toxicol Sci. 2005;88(2):514–524. | ||
Scuri M, Chen BT, Castranova V, et al. Effects of titanium dioxide nanoparticle exposure on neuroimmune responses in rat airways. J Toxicol Environ Health A. 2010;73(20):1353–1369. | ||
Wang J, Chen C, Liu Y, et al. Potential neurological lesion after nasal instillation of TiO2 nanoparticles in the anatase and rutile crystal phases. Toxicol Lett. 2008;183(1–3):72–80. | ||
Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461. | ||
Aliprantis AO, Yang RB, Weiss DS, Godowski P, Zychlinsky A. The apoptotic signaling pathway activated by toll-like receptor-2. EMBO J. 2000;19(13):3325–3336. | ||
Petrarca C, Clemente E, Amato V, et al. Engineered metal based nanoparticles and innate immunity. Clin Mol Allergy. 2015;13(1):1313. | ||
Gottlieb RA. Mitochondria: execution central. FEBS Lett. 2000;482(1–2):6–12. | ||
Dowling DJ, Scott EA, Scheid A, et al. Toll-like receptor 8 agonist nanoparticles mimic immunomodulating effects of the live BCG vaccine and enhance neonatal innate and adaptive immune responses. J Allergy Clin Immunol. 2017;140(5):1339–1350. | ||
Kim AS, Chae CH, Kim J, Choi JY, Kim SG, Băciut G. Silver nanoparticles induce apoptosis through the toll-like receptor 2 pathway. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;113(6):789–798. | ||
Chen GY, Yang HJ, Lu CH, et al. Simultaneous induction of autophagy and toll-like receptor signaling pathways by graphene oxide. Biomaterials. 2012;33(27):6559–6569. | ||
Breunig M, Bauer S, Goepferich A. Polymers and nanoparticles: intelligent tools for intracellular targeting? Eur J Pharm Biopharm. 2008;68(1):112–128. | ||
Pinsino A, Russo R, Bonaventura R, Brunelli A, Marcomini A, Matranga V. Titanium dioxide nanoparticles stimulate sea urchin immune cell phagocytic activity involving TLR/p38 MAPK-mediated signalling pathway. Sci Rep. 2015;5:14492. | ||
El-Said KS, Ali EM, Kanehira K, Taniguchi A. Molecular mechanism of DNA damage induced by titanium dioxide nanoparticles in toll-like receptor 3 or 4 expressing human hepatocarcinoma cell lines. J Nanobiotechnology. 2014;12:48. | ||
Hu R, Gong X, Duan Y, et al. Neurotoxicological effects and the impairment of spatial recognition memory in mice caused by exposure to TiO2 nanoparticles. Biomaterials. 2010;31(31):8043–8050. | ||
Dhupal M, Kim C-S, Ignacio RMC, Tripathy D, Kim S-K. Effects of small black soybean product germinated with sulfur on immuno-redox status in C57BL/6 mice. Mol Cell Toxicol. 2017;13(1):115–124. | ||
Jung KA, Kwak MK. The Nrf2 system as a potential target for the development of indirect antioxidants. Molecules (Basel, Switzerland). 2010;15(10):7266–7291. | ||
Zhao L, Zhu Y, Chen Z, et al. Cardiopulmonary effects induced by occupational exposure to titanium dioxide nanoparticles. Nanotoxicology. 2018;12(2):169–184. | ||
Sharma V, Anderson D, Dhawan A. Zinc oxide nanoparticles induce oxidative DNA damage and ROS-triggered mitochondria mediated apoptosis in human liver cells (HepG2). Apoptosis. 2012;17(8):852–870. | ||
Wang C, Youle RJ. The role of mitochondria in apoptosis. Annu Rev Genet. 2009;43:95–118. | ||
Gottlieb E, Armour SM, Harris MH, Thompson CB. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Death Differ. 2003;10(6):709–717. | ||
Perry SW, Norman JP, Barbieri J, Brown EB, Gelbard HA. Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. Biotechniques. 2011;50(2):98–115. | ||
Letai A. Puma strikes Bax. J Cell Biol. 2009;185(2):189–191. | ||
Kim H, Tu HC, Ren D, et al. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell. 2009;36(3):487–499. | ||
Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23(16):2838–2849. | ||
OECD. Series on the Safety of Manufactured Nanomaterials. 2017;64(83):2016. | ||
Kim TH, Lee JA, Choi SJ, Oh JM, Jm O. Polymer coated CaAl-layered double hydroxide nanomaterials for potential calcium supplement. Int J Mol Sci. 2014;15(12):22563–22579. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.