Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 17
Immunotherapy in Glioblastoma: An Overview of Current Status
Authors Sarfraz Z, Maharaj A, Venur VA, Lathia JD, Odia Y ![]() , Ahluwalia MS
, Ahluwalia MS
Received 27 February 2025
Accepted for publication 25 June 2025
Published 24 July 2025 Volume 2025:17 Pages 185—209
DOI https://doi.org/10.2147/CPAA.S497903
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Arthur E. Frankel
Zouina Sarfraz,1 Arun Maharaj,1 Vyshak Alva Venur,2 Justin D Lathia,3 Yazmin Odia,1 Manmeet S Ahluwalia1,4
1Miami Cancer Institute, Baptist Health South Florida, Miami, FL, USA; 2Seattle Cancer Care Alliance, Fred Hutchinson Cancer Center, University of Washington, Seattle, WA, USA; 3Cleveland Clinic Research, Cleveland, OH, USA; 4Herbert Wertheim College of Medicine, Florida International University, Miami, FL, USA
Correspondence: Manmeet S Ahluwalia MD, MBA, FASCO, Fernandez Family Foundation Endowed Chair in Cancer Research, Chief of Medical Oncology, Chief Scientific Officer & Deputy Director,Miami Cancer Institute, Baptist Health South Florida, Miami, FL, USA, Tel +1-786-527-8010, Fax +1-786-533-9416, Email [email protected]
Abstract: Glioblastoma (GB) is an aggressive brain tumor with standard therapies offering limited but measurable survival benefit. Immunotherapy is expanding the treatment landscape for GB. Immune checkpoint inhibitors (ICIs), including nivolumab and pembrolizumab, have shown benefit in several cancers and are being studied in GB, with ongoing efforts to address the tumor’s immunosuppressive environment. Chimeric Antigen Receptor (CAR) T-cell therapies are also being explored, with new approaches designed to overcome antigen variability and improve access across the blood-brain barrier. Cancer vaccines, especially dendritic cell-based platforms like DCVax-L, have shown promising survival outcomes in clinical trials. Advances in biomarker analysis and genomics are supporting more personalized immunotherapy approaches. In addition, combination strategies involving ICIs, CAR T-cells, vaccines, and oncolytic viruses are being developed to enhance immune response. This review outlines current immunotherapy approaches in GB, focusing on their mechanisms, clinical development, and future directions.
Plain Language Summary: Glioblastoma (GB) is an aggressive and common type of brain cancer, with current treatments offering modest survival benefits. Standard treatments like surgery, chemotherapy, and radiation therapy are not enough to stop the tumor from coming back. Because of this, researchers are exploring new approaches, including immunotherapy, which uses the body’s immune system to fight cancer. Immunotherapy has shown success in treating other cancers, but GB presents unique challenges. The tumor creates an environment that weakens the immune response and prevents treatments from working effectively. This review explores different types of immunotherapy being tested for GB:Immune checkpoint inhibitors (ICIs): Drugs like nivolumab and pembrolizumab help the immune system recognize and attack GB cells. However, clinical trials show only modest improvements in survival.CAR T-cell therapy: This treatment modifies a patient’s immune cells to better target the tumor. While promising, barriers like tumor diversity and the brain’s natural defenses make it difficult for CAR T-cells to work effectively.Cancer vaccines: These aim to train the immune system to recognize GB cells as threats, but results have been mixed, with some showing potential for improving survival.Oncolytic viruses: These viruses selectively infect and kill cancer cells while stimulating an immune response. Early trials suggest they may help when combined with other treatments.
Keywords: glioblastoma, immunotherapy, tumor microenvironment, antigen heterogeneity, personalized treatment, blood-brain barrier
Introduction
Glioblastoma (GB) is a prevalent primary malignant brain tumor in the United States (US) with an average annual age-adjusted incidence of 3.19 per 100,000 persons.1–3 Over 10,000 individuals in the US succumb to GB each year.4,5 The estimated median survival is 14–16 months with a 5-year survival rate of 6.9%.6,7 Although GB was first identified in the early 20th century, limited Food and Drug Administration (FDA)-approved treatments are available.6,8 Based on the results of a landmark clinical trial by Stupp et al (2005), temozolomide (TMZ), an alkylating antineoplastic agent, was approved in 2005 for GB.9–11 Bevacizumab, a humanized monoclonal antibody, was granted US-FDA accelerated approval in 2009 for recurrent GB based on improvements in objective response rates in two single-arm phase II trials (AVF3708g and NCI 06-C-0064E).12,13 Despite their respective FDA approvals and incorporation into standard care, both TMZ and bevacizumab yield modest survival benefits, largely due to the intrinsic resistance mechanisms of GB, such as DNA repair systems that counteract the effects of TMZ, and the activation of alternate angiogenic pathways that undermine the efficacy of bevacizumab.14
GB is characterized by rapid growth, tumor cell infiltration into the brain, and high recurrence rates.15 Genetic and epigenetic alterations disrupt key pathways regulating proliferation, differentiation, and apoptosis, complicating treatment.16 Common mutations involve tumor suppressor genes such as p53, RB1, PTEN, and activation of the PI3K/AKT/mTOR pathway.17 The immunosuppressive microenvironment of GB features cytokines like interleukin-10 (IL-10) and transforming growth factor-β (TGF-β), which inhibit T-cell function and expand regulatory T cells (Tregs), contributing to immune evasion.18 Elevated programmed death-ligand 1 (PD-L1) expression suppresses T-cell cytotoxicity, further facilitating tumor escape.19–22 The immunosuppressive tumor microenvironment (TME) drives resistance to therapy. Tumor-associated macrophages (TAMs), including brain-resident microglia and bone marrow-derived macrophages, adopt an anti-inflammatory phenotype that promotes tumor growth, angiogenesis, immune evasion, and T-cell suppression.23,24 Glioma stem cells (GSCs), a therapy-resistant, self-renewing subpopulation, further remodel the TME by recruiting immunosuppressive cells such as Tregs, myeloid-derived suppressor cells (MDSCs), and dysfunctional dendritic cells (DCs), which collectively inhibit effector T cells, promote exhaustion, and impair antigen presentation.25,26 Hypoxia in GB induces vascular endothelial growth factor (VEGF), enhancing angiogenesis, tumor proliferation, and invasion.27,28 The blood-brain barrier (BBB) limits therapeutic penetration and immune cell access.29 Moreover, the immune-privileged nature of the central nervous system (CNS) limits anti-tumor immunity, compounding the difficulty of effective immunotherapy for CNS tumors.30 At present, there are significant challenges in the current immunotherapy landscape, providing context to the low survival rates and difficulty in developing effective treatment strategies for GB.
In clinical practice, molecular biomarkers such as isocitrate dehydrogenase (IDH) mutation status and O(6)-methylguanine-DNA methyltransferase (MGMT) promoter methylation are used to stratify patients and predict response to therapy.31,32 IDH-mutant GB is typically less aggressive and has a more immune-permissive microenvironment compared to IDH-wildtype tumors, which are often associated with poor prognosis and higher immunosuppressive signaling.33 MGMT promoter methylation correlates with increased sensitivity to TMZ and may also reflect broader epigenetic changes that influence immune responses.34 These biomarkers are being increasingly considered in immunotherapeutic trial designs and may help identify patients most likely to benefit from specific interventions. In addition to IDH and MGMT, the 1p/19q co-deletion is an important molecular marker associated with enhanced therapeutic sensitivity in gliomas, particularly oligodendrogliomas. However, current detection methods are invasive. Recent advances in radiogenomics have enabled non-invasive prediction of 1p/19q status through magnetic resonance imaging (MRI)-based radiomic features, achieving high predictive accuracy and offering a potential alternative to surgical sampling.35,36
Overall survival (OS) and progression-free survival (PFS) rates remain poor for CNS tumors such as GB, owing to their inherent biological resistance and immunosuppressive characteristics, a challenge further compounded by factors such as age- and sex- based differences.37–39 Age-stratified differences in comorbidities and medication responses seem to affect survival in younger GB patients.40 In older GB patients, senescence-driven immunosuppression and age-related brain alterations may impair immunotherapeutic efficacy, adversely affecting OS and PFS.41,42
Recent advances in GB immunotherapy have expanded into four key areas: immune checkpoint inhibitors (ICI), CAR T-cell therapy, cancer vaccines, and oncolytic viruses. Immune checkpoint blockade, targeting PD-1/PD-L1 and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) pathways, remains under active investigation at present. CAR T-cell therapies, particularly those directed against antigens such as epidermal growth factor receptor variant III (EGFRvIII) and IL13Rα2, have shown potential in early-phase trials. Cancer vaccines like rindopepimut and DCVax-L aim to stimulate GB-specific T-cell responses and are also being explored in multiple clinical settings. Oncolytic viruses such as DNX-2401 not only lyse tumor cells directly but also initiate systemic immune activation, with somewhat encouraging responses. Integrating these modalities with existing treatments may improve therapeutic efficacy and help overcome inherent resistance mechanisms seen in GB. As the field of oncology research delves deeper into the landscape of immunotherapy, it is all the more important to explore how novel immunotherapeutic approaches can be integrated with existing treatment modalities to improve outcomes for patients with GB. This review aims to collate the latest advances in the immunotherapy landscape of GB.
Current Immunotherapies for Glioblastoma
Immunotherapy holds premise for GB despite the inherent challenges of the disease. Interventions consist of ICIs such as nivolumab and pembrolizumab, which improve T-cell activity by targeting PD-1 and CTLA-4.43 CAR T-cell therapy uses engineered T-cells against antigens including EGFRvIII, providing a more targeted approach. Cancer vaccines such as the DCVax-L, seek to trigger immune memory against GB. Another line of immunotherapy includes oncolytic viruses like DNX-2401 (Delta-24-RGD; tasadenoturev) which selectively infects tumor cells to active immune responses. Table 1 provides an overview of these therapies.
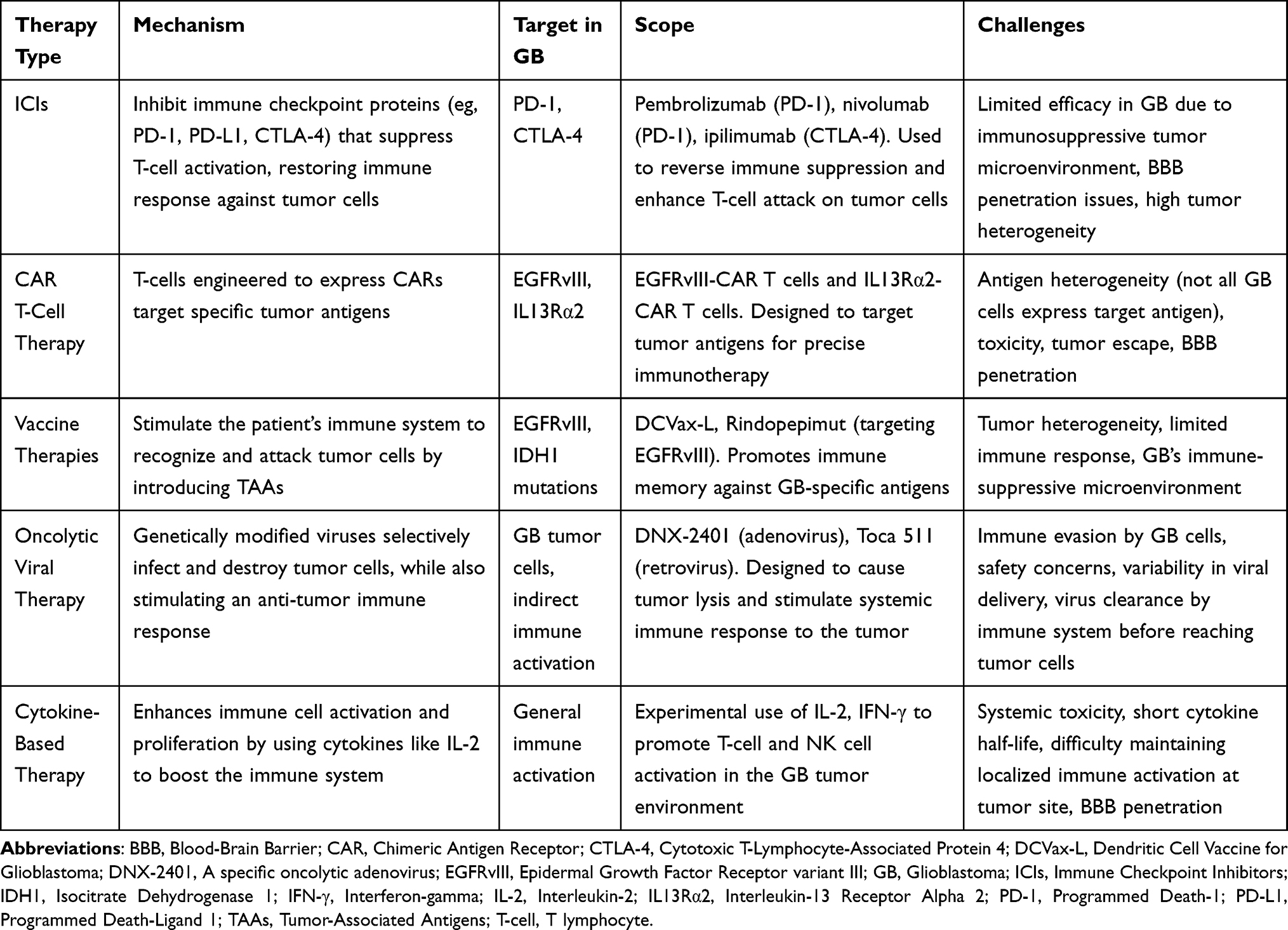 |
Table 1 Overview of Immunotherapy Approaches for Glioblastoma: Mechanisms, Targets, and Challenges |
Immune Checkpoint Inhibitors
PD-1, a T-cell receptor, binds to PD-L1, an inhibitory ligand overexpressed by tumors, suppressing the immune response and enabling tumor evasion.44 ICIs such as nivolumab and pembrolizumab restore T-cell activity by blocking PD-1/PD-L1 interactions, which is particularly relevant in GB due to its immunosuppressive microenvironment.45–47 Additionally, the LAG-3 checkpoint, upregulated on exhausted T cells, is being explored as a complementary target, with monoclonal antibodies like BMS-986016 enhancing T-cell activation by inhibiting LAG-3 and PD-1.48–51 Combination regimens, such as DNX-2401 with pembrolizumab, may further boost immune responses by directly targeting tumor cells and releasing tumor antigens.52,53 Table 2 provides information on key trials investigating ICIs in GB.
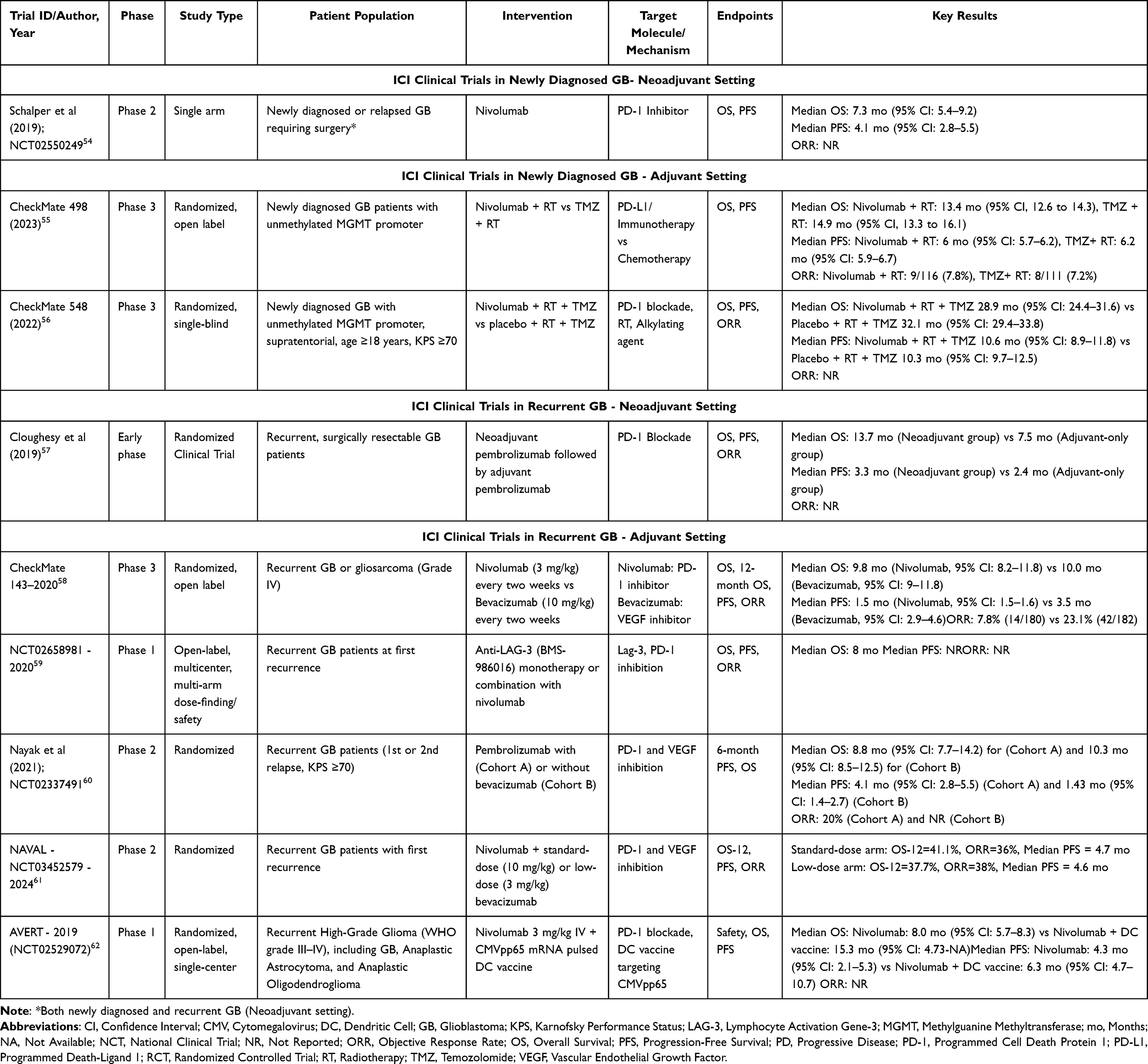 |
Table 2 Summary of Clinical Trials Investigating Immune Checkpoint Inhibitors in Glioblastoma |
ICI Clinical Trials in Newly Diagnosed GB
Neoadjuvant Setting
In 2019, Schalper and colleagues conducted a phase II single-arm trial (NCT02550249).54 Nivolumab was given to newly diagnosed or relapsed GB patients requiring surgical resection. Of the 30 patients enrolled, 27 underwent salvage therapy for recurrent disease and 3 had primary tumor resection. An OS of 7.3 months (95% confidence interval, CI: 5.4–9.2) and PFS of 4.1 months was noted (95% CI: 2.8–5.5). Although the overall efficacy was modest, this trial provided crucial insight into the potential immunomodulatory effects of nivolumab on GB patients in a surgical setting.54
Adjuvant Setting
CheckMate 498 was a phase III randomized trial reported in 2023 across the US and Europe.55 The trial enrolled 560 newly diagnosed GB patients who had unmethylated MGMT promoter status. Nivolumab and radiation therapy (RT) as compared to TMZ and RT was tested in 280 patients per group. The median OS was 13.4 months (95% CI: 12.6–14.3) in the standard of care arm and it was 14.9 months (95% CI: 13.3–16.1) in the nivolumab and RT arm (P=0.0037). At 6 months, there were no differences in median PFS between treatment arms (nivolumab: 6 months, 95% CI: 5.7–6.2; TMZ: 6.2 months, 95% CI: 5.9–6.7, P=0.25). Investigator-assessed Response Assessment in Neuro-Oncology (RANO) criteria demonstrated an objective response rate (ORR) of 7.8% in the nivolumab subset and 7.2% in the TMZ group. This trial did not meet its primary endpoint of improving OS.55 CheckMate 548 was a phase III randomized trial conducted in 2022 in the US, Europe, and Canada.56 The trial enrolled 716 newly diagnosed and histologically confirmed supratentorial GB patients with methylated MGMT promoter status who had received surgical intervention, had a Karnofsky Performance Scale (KPS) score of ≥70, and were eligible to receive RT with TMZ. Patients in Arm 1 were administered nivolumab, RT, and TMZ, while patients in Arm 2 were treated with a placebo, RT, and TMZ. In Arm 1, the median OS was 28.9 months (95% CI: 24.4–31.6), and 32.1 months (95% CI: 29.4–33.8) in Arm 2. There were no differences in median PFS between arms (Arm 1: 10.6 months, 95% CI: 8.9–11.8; Arm 2: 10.3 months, 95% CI: 9.7–12.5). Nivolumab, when combined with RT and TMZ, did not improve survival in patients and no new safety signals were noted.56
These findings suggest that while ICIs like nivolumab are well-tolerated, they do not provide significant survival benefits when combined with standard therapies in newly diagnosed GB. Future studies should explore combination strategies with other immunotherapeutic agents or biomarkers to better identify patients who may benefit from checkpoint inhibition.
ICI Clinical Trials in Recurrent GB
Neoadjuvant Recurrent GB Trials
Cloughesy and colleagues (2019) conducted an early-phase randomized clinical trial with neoadjuvant and adjuvant-only pembrolizumab.57 Thirty-two of the 35 patients with resectable recurrent GB were included in the analysis. The median OS in the neoadjuvant group was 13.7 months compared to 7 months in the adjuvant-only pembrolizumab group (P=0.04). The median PFS was 3.3 months in the neoadjuvant group and 2.4 months in the adjuvant arm (P=0.03). Molecular analysis of neoadjuvant PD-1 blockade reveals enhanced immune activation associated with increased PD-L1 expression and greater tumor CD8+ T-cell infiltration.57
Another phase II single-arm trial in 2019 tested neoadjuvant nivolumab in recurrent GB patients (NCT02550249).54 Of 30 patients, 27 underwent surgery for recurrent GB while 3 newly diagnosed patients were administered nivolumab before and after primary surgery. Tumor samples were taken before and after nivolumab administration to test molecular and cellular changes. Neoadjuvant nivolumab had local immune-stimulating effects reflected in increased chemokine expression, T-cell receptor diversity in tumor tissues, and immune cell infiltration. While salvage surgeries typically have limited success, two patients who received neoadjuvant nivolumab experienced prolonged survival. These findings suggest that nivolumab may enhance immune responses in recurrent GB, highlighting the need for further research into its potential when combined with other therapies.54
Adjuvant Recurrent GB Trials
CheckMate 143, a phase III randomized, open-label study was conducted in 2020 across 57 international locations.58 Bevacizumab or nivolumab was given to 369 patients with gliosarcoma or recurrent GB. No differences in median OS were seen between groups (bevacizumab: 10 months vs nivolumab: 9.8 months, P=0.76). PFS findings favored bevacizumab (3.5 months vs 1.5 months, P<0.001). The ORR was higher with bevacizumab compared to nivolumab (23.1% vs 7.8%). High rates of pseudoresponse were associated with anti-VEGF therapy while the responses seen with nivolumab were more durable (11.1 months vs 5.3 months); both treatments had comparable safety profiles.58 Bevacizumab improved short-term control, while nivolumab had more durable responses, highlighting the need for better treatment strategies in recurrent GB.
A 2020 phase I multicenter trial (NCT02658981) enrolled 33 patients with recurrent GB.59 Anti-LAG-3 (BMS-986016) as monotherapy or combined with nivolumab was administered. The trial sought to ascertain the maximum tolerated dose (MTD) and dose-limiting toxicities (DLTs). BMS-986016 dose ranged from 80–800mg with no reported DLTs, and thus, the MTD was set at 800mg. Grade 3 hypertension and grade 4 edema were reported in the combination arm. Notably, the median OS was 8 months in the combination arm; 44% of patients survived long-term (>20 months) and 3 patients surpassed 20 months. Results from this trial suggest that there may be a potential survival benefit with the combination of PD-1 and LAG-3 inhibition treatment in GB patients.59
Nayak and colleagues conducted a phase II trial in 2021 enrolling 80 patients with recurrent GB (NCT02337491).60 In Arm 1, 30 patients received pembrolizumab and 50 were given pembrolizumab and bevacizumab in Arm 2. At 6 months, PFS was 6.7% in Arm 1 versus 26% in Arm 2 (P=0.003), with a median OS of 10.3 months and 8.8 months, respectively (0.87). ORR was 0% in Arm 1 compared to 20% in Arm 2. ORR and PFS may have been impacted by the high pseudoresponse rates associated with bevacizumab. Poorer outcomes were seen in those with increased plasma VEGF and prior dexamethasone use. Pembrolizumab was well-tolerated, but the efficacy was limited either alone or with bevacizumab.60
The phase II NAVAL trial (NCT03452579) enrolled 90 patients with recurrent GB.61 The researchers examined the impact of standard-dose nivolumab (10mg, biweekly) or low-dose bevacizumab (2.5mg/2 weeks) on OS. OS at 12 months (OS-12) was higher in the standard-dose nivolumab group compared to the low-dose bevacizumab group (41.1% vs 37.7%). Patients aged ≥60 years in the standard-dose arm experienced significant survival benefits with standard dosing. They also showed notable immune activity, which was characterized by increased systemic inflammatory activity and fewer immunosuppressive myeloid-derived suppressor cells in the standard-dose bevacizumab group. Common adverse events in the standard-dose nivolumab arm included fatigue (13.3%), proteinuria (33.3%), and hypertension (33.3%). Bevacizumab was associated with stronger systemic inflammation and potential survival benefit in older age groups.61
The phase I randomized, open-label AVERT trial (NCT02529072) was conducted in 2019 enrolling 6 patients with recurrent high-grade gliomas, including four with GB.62 Three patients were administered nivolumab combined with a CMVpp65 mRNA-pulsed DC vaccine whereas the other 3 patients were administered nivolumab alone. The combination therapy yielded a median OS of 15.3 months (95% CI: 4.73–N/A) whereas nivolumab alone had a median OS of 8 months (95% CI: 5.7–8.3). This preliminary trial was limited by a small sample size but lends promise for future, larger-scale trials combining PD-1 blockage with a DC vaccine in high-grade gliomas.62
Among ICI trials, two studies investigated the combination of oncolytic viral therapy with PD-1 blockade in recurrent GB. The CAPTIVE (KEYNOTE-192) trial, an adjuvant therapy study, evaluated DNX-2401 with pembrolizumab, reporting a median OS of 12.5 months (95% CI: 10.7–13.5).63 Meanwhile, Chiocca et al conducted a neoadjuvant and adjuvant study assessing nivolumab with Ad-RTS-hIL-12 gene therapy and veledimex, demonstrating a median OS of 9.8 months (95% CI: 5.2–17.4), extending to 16.9 months (95% CI: 3.8–24.0) in a subset of patients.64 Further details on these trials are described in Section 6.
CAR-T Cell Therapy in GB
CAR-T cell therapy focused on tumor-associated antigens commonly found in GB. These antigens include EGFRvIII, IL13 receptor subunit alpha-2 (IL13Rα2), GD2, Human epidermal growth factor receptor 2 (HER2), B7-H3 (CD276), Roundabout Guidance Receptor 1 (ROBO1), CD133, and Ephrin type-A receptor 2 (EphA2).65–69 Typically, IL13Rα2 is present in around 50–80% of CAR-T cases, whereas EGFRvIII is seen in nearly 25–30% of CAR-T cases, and thus, are of special interest as they are present within the tumor cells.70–72 EphA2 and HER2, which are involved in tumor growth and invasion, are also key targets for CAR-T cell therapy.73 Some of the challenges for CAR-T therapy in GB include tumor heterogeneity and changes in antigen expression over time.74,75 Most CAR-T cell therapies for GB are in very early phases of development. Table 3 depicts key trials investigating the role of CAR T-cell therapy in GB.
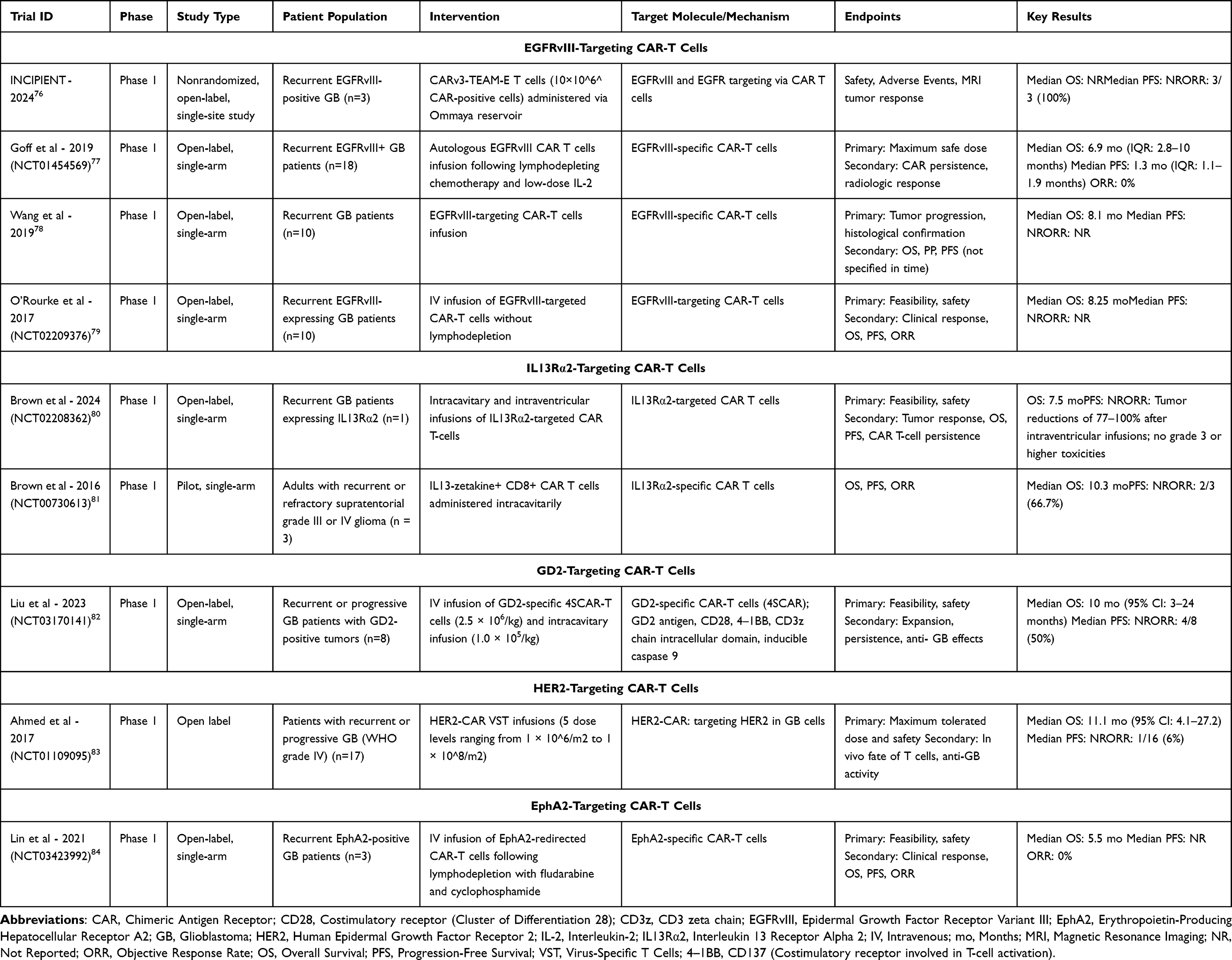 |
Table 3 Summary of Clinical Trials Investigating CAR-T Cell Therapy in Glioblastoma |
EGFRvIII-Targeting CAR-T Cells
The INCIPIENT study was a phase I, open-label trial reported in 2024 within the US.76 The trial enrolled 3 patients with EGFRvIII-positive GB. The patients were administered an intraventricular infusion of 10 million CARv3-TEAM-E T-cells via an Ommaya reservoir, but MRIs revealed tumor progression in patient 1, showing temporary regression but later died due to gastrointestinal perforation 63 days post-study completion, which was unrelated to CAR-T therapy. Patient 2 presented a 60.7% reduction in tumor cross-sectional area which was sustained for over 150 days. Patient 3 saw a near-complete regression, but recurrence was seen 1 month after treatment. No DLTs were seen, although 2 of 3 patients experienced fatigue and grade 3 encephalopathy. An ORR of 100% was seen as all patients showed tumor regression following infusion of CARv3-TEAM-E T-cells.76 These results suggest that CARv3-TEAM-E T-cell therapy demonstrates promising initial antitumor activity in EGFRvIII-positive GB, with a 100% ORR, but the durability of response remains a challenge, warranting further investigation.
Goff and colleagues conducted a phase I trial (NCT01454569) in 2019 enrolling 18 patients with recurrent EGFRvIII-positive GB seeking to establish a safe maximum dose along with determining CAR-T cell persistence and tumor response.77 Patients had a history of RT, surgery, and TMZ. As part of the trial, they underwent lymphodepleting chemotherapy with fludarabine and cyclophosphamide before receiving CAR-T cell therapy. In total, 4 patients had MGMT methylated tumors whereas 10 had been previously treated with bevacizumab. From GB diagnosis to trial screening, the median time was 15.7 months (interquartile range, IQR: 11.6–20.6). Following CAR-T cell infusion, patients received low-dose IL-2 to support CAR-T cell persistence, which was initiated within 24 hours of cell transfer and continued every eight hours based on tolerance. The median OS was 6.9 months (IQR: 2.8–10) but no objective responses were seen. It must be noted that radiologic progression was seen in 16 of the 17 evaluable patients within 3 months; the median PFS was 1.3 months (IQR: 1.1–1.9). Of note, 1 patient was alive at 59 months whereas 2 patients had survived for over 1 year. There was 1 death associated with pulmonary toxicity due to a high CAR-T dose. There was no clinically meaningful impact among patients with GB in this phase-1 pilot trial.77
In 2019, Wang and colleagues led a phase I trial in China where they enrolled 10 patients with recurrent GB. This trial focused on progression as a surrogate marker for response assessment as well as the role of imaging in assessing response and safety/feasibility. Patients were intervened with EGFRvIII-targeted CAR-T infusions and monitored with relative cerebral blood volume (rCBV).78 Pseudoprogression was confirmed using histological methods in patients undergoing early or late surgery. The median OS was 8.1 months. Three patients died within 6 months, and 6 survived longer than 6 months. Only 1 patient was alive at 34 months.78
Another phase I trial led by O’Rourke and colleagues in 2017 sought to assess the safety and feasibility of EGFRvIII-targeted CAR-T cells. Ten patients were enrolled who had recurrent GB and EGFRvIII-positive.79 Patients underwent CAR-T intravenous (IV) infusions without lymphodepletion. The median OS was 8.25 months, no DLTs were noted, and patients did not report any EGFR-related adverse events. Notably, 3 patients had neurological events most likely owing to localized T-cell activity. Although tumor control benefits were inconclusive, this trial provided quantifiable proof for the tolerability and safety of EGFRvIII-targeted CAR-T cells for EGFRvIII-positive recurrent GB patients.79
IL13Rα2-Targeting CAR-T Cells
A phase I trial by Brown and colleagues was conducted in the US (NCT02208362),80,81 investigating IL13Rα2-targeted CAR-T cell therapy for GB. While the trial included 65 patients, only 1 patient had recurrent multifocal GB. The target patient underwent various infusions of CAR-T cells that were delivered intracranially over 220 days (16 total CAR-T cell infusions). Of these, 10 were intraventricular and 6 were intracavitary. Significant reductions in tumor size after intraventricular infusions (77–100%). No ≥ grade 3 toxicities were noted, and the treatment was well-tolerated. The clinical response duration was 7.5 months for the target patient. This trial highlighted the potential efficacy of IL13Rα2-targeted CAR-T cell therapy. A significant limitation of this study is that only one patient had recurrent multifocal GB, highlighting a prominent literature gap given the robust results of this study.80,81
Another phase I trial (NCT00730613) in 2015 evaluated the safety, efficacy, and feasibility of IL13Rα2-specific CAR-T cell therapy in 3 patients with recurrent high-grade gliomas (World Health Organization, WHO grades III/IV).85 The patients received intracavitary infusions and the treatment was well-tolerated with only 2 grade-3 side effects noted; headache and neurological symptoms, both managed successfully. One patient had a survival post-relapse of 10.3 months whereas tumor responses were observed in the other 2 patients, who experienced no recurrences and had an ORR of 66.7%. This trial showcased the feasibility and potential efficacy of IL13Rα2-specific CAR-T cell therapy in patients with recurrent high-grade gliomas.85
GD2-Targeting CAR-T Cells
In 2023, a phase I open-label trial (NCT03170141) was conducted by Liu et al in China. The authors evaluated GD2-specific 4SCAR-T cell therapy in eight patients with recurrent GD2-positive GB.82 Patients received multiple IV infusions of GD2-specific 4SCAR-T cells (2.5 × 10⁶/kg), whereas some patients received a single intracavitary dose (1.0 × 10⁵/kg) after lymphodepletion. Primary endpoints included safety and feasibility with secondary endpoints examined cell persistence and anti-tumor effects. The median OS was 10 months (range: 3–24). In addition, the ORR was 50% and only 4 out of 8 patients achieved a partial response. Side effects included 1 grade-3 headache and 1 grade-2 seizure. There were no severe toxicities, suggesting that GD2-specific 4SCAR-T therapy could be a promising and tolerable option.82
HER2-Targeting CAR-T Cells
In 2017, Ahmed and colleagues led a phase I trial in the US (NCT01109095) seeking to evaluate the safety, MTD and anti-tumor effects of HER-2 targeted CAR virus-specific T cells (VSTs) in 17 patients with HER2-positive GB.83 The patient population previously underwent extensive treatments prior including RT, surgery, and TMZ, and were now given escalating doses of HER2-CAR VSTs at a median of 12.9 months post-diagnosis. While no DLTs were reported, 2 patients had grade-2 seizures/headaches. In total, 7 patients had stable disease that lasted between 8 weeks to 29 months, although 1 patient had a partial response that lasted 9.2 months. The median OS was 11.1 months post-infusion, and PFS was 3.5 months. HER2-CAR VSTs were tolerated well, and there was some evidence that anti-tumoral activity occurred for up to 12 months.83
EphA2-Targeting CAR-T Cells
A 2021 phase I trial was conducted in China by Lin et al (NCT03423992).84 The authors enrolled 3 patients with recurrent EphA2-positive GB to assess the efficacy of EphA2-targeted CAR-T cells. All patients had lymphodepletion using fludarabine and cyclophosphamide followed by a single IV infusion of CAR-T cells. The median OS was reported as 5.5 months and only 1 patient achieved stable disease at the 4-week timepoint, with 2 having progressive disease. Two of 3 patients developed grade-2 cytokine release syndrome that was characterized by cytokine elevations, fever, and pulmonary edema, which was resolved with dexamethasone. Overall, clinical responses had limitations with an ORR of 0%, therefore future studies are necessitated to understand the efficacy and persistence of CAR-T cells in GB.84
Cancer Vaccines
Mechanism of Action of Cancer Vaccines
Cancer vaccines target the immune system by introducing tumor-associated or tumor-specific antigens (TAAs/TSAs).86 These mechanisms vary based on the type of vaccine. Peptide-based vaccines work by presenting small tumor-specific protein fragments (peptides) via Major Histocompatibility Complex (MHC) molecules to cytotoxic T lymphocytes (CTLs), leading to T-cell activation and targeted tumor destruction.87 DC vaccines are unique in that they collect and load patient-derived DCs along with tumor antigens, and are subsequently reintroduced in the patient to elicit a CD8+ T-cell response against the tumor.88 Heat shock protein (HSP) vaccines deliver tumor antigens to antigen-presenting cells that improve the cytotoxic T-cell response.89 In addition, autologous tumor vaccines use modified patient-derived tumor cells that stimulate immune recognition and attack residual cancer cells.90 All of these cancer vaccines eventually aim to counteract immune evasion and provide anti-tumor immunity. A summary of key trials investigating cancer vaccines in GB is given in Table 4.
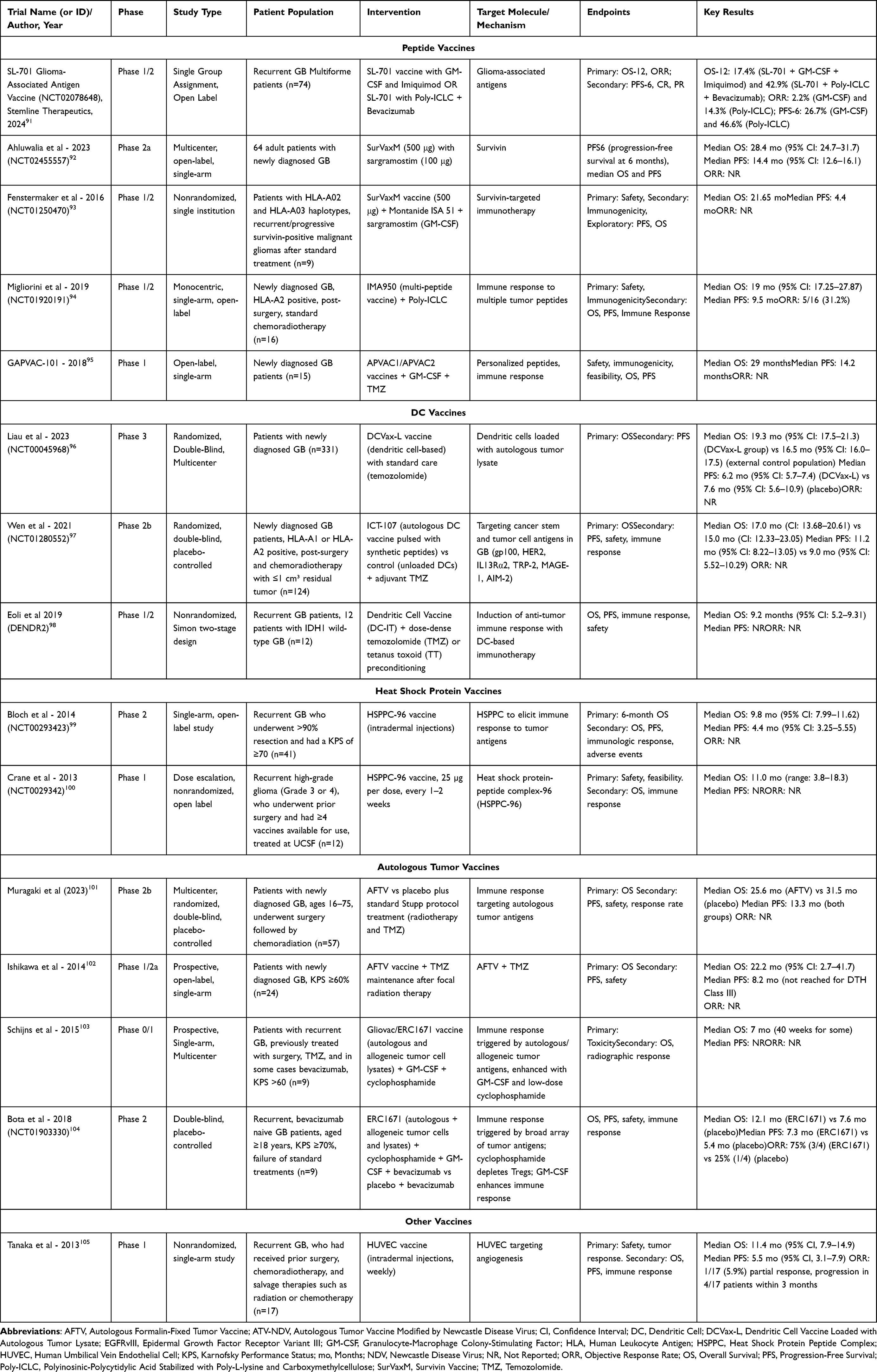 |
Table 4 Summary of Clinical Trials Investigating Vaccines in Glioblastoma |
Clinical Trial Applications of Vaccines in GB
Peptide Vaccines
Stemline Therapeutics led a phase I/II two-stage trial in 2024 where they evaluated a glioma-associated antigen vaccine (SL-701) among 74 patients with recurrent GB.91 One arm was injected with SL-701 and GM-CSF with imiquimod cream, which was followed by re-application in 24 hours. The second arm was given poly-ICLC with SL-701, which was followed by bi-weekly infusions of bevacizumab. The 12-month OS rate was 17.4% in the GM-CSF/imiquimod arm and 42.9% in the poly-ICLC/bevacizumab arm. The 6-month PFS was 26.7% and 42.9%, respectively. It may be possible that the PFS and ORR were enhanced due to bevacizumab. The entire cohort had good tolerance to treatment with adverse events mostly being reported as headaches, injection site reactions, and fatigue. Overall, SL-701 had modest survival benefits in patients with recurrent GB.91
Ahluwalia and colleagues led a phase IIa, open-label trial in 2023 where they assessed SurVaxM, a survivin-targeting peptide vaccine. Montanide was used as an adjuvant.92 The trial enrolled 64 newly diagnosed GB patients across 5 sites in the US. Patients were given 4 priming doses of SurVaxM every 2 weeks followed by maintenance doses of SurvaxM, administered every 8 weeks, along with standard adjuvant doses of TMZ given at least 28 days post-chemoradiation. At the 6-month time point, 95.2% of patients were progression-free. The median OS was 28.4 months (95% CI: 24.7–31.7), and PFS was 14.4 months (95% CI: 12.6–16.1). There was acceptable tolerance to treatment with no severe adverse events. Notably, patients that had methylated MGMT status had longer OS as compared to unmethylated patients, suggesting that newly diagnosed patients may be an applicable target.92
Fenstermaker et al conducted a 2016 phase I study evaluating SurVaxM in 9 recurrent GB patients post-standard therapy, administering it with Montanide ISA 51 and GM-CSF in a prime-boost regimen.93 The median OS was 21.7 months, PFS was 4.4 months, and adverse events were mostly mild (grade 1–2), with no serious vaccine-related events reported.93
In 2019, Migliorini and colleagues evaluated the safety and immunogenicity of a multi-peptide vaccine termed IMA-950 with poly-ICLC in Italy among 16 patients with newly diagnosed GB (NCT01920191) in a phase I/II trial.94 Patients were intervened after standard chemoradiotherapy regimens. The median OS was 19 months (95% CI: 17.25–27.87) and median PFS was 9.5 months. Treatment response was documented in 5 of the 16 patients. Overall, the IMA-950 vaccine depicted favorable safety profiles with no major adverse events.94
In 2018, the phase I, open-label GAPVAC-101 trial was conducted in Germany, Switzerland, Denmark, and the Netherlands among 15 patients with newly diagnosed GB.95 Two vaccines (APVAC1 and APVAC2) were administered in combination with GM-CSF and TMZ. The intervention was targeted at tumor antigens that were identified using whole-exome sequencing and HLA-ligandome analyses. The median OS was 29 months and the median PFS was 14.2 months. The GAPVAC-101 trial provides preliminary evidence for safety and immunogenicity.95
Dendritic Cell Vaccines
Liau and colleagues conducted a phase III randomized, double-blind trial across multiple sites in the US, Canada, United Kingdom, and Germany in 2023 (NCT00045968).96 DCVax-L, an autologous tumor lysate-loaded DC vaccine, was administered to 331 patients with newly diagnosed GB. The median OS was 19.3 months (95% CI: 17.5–21.3) in the intervention group, whereas it was 16.5 months (95% CI: 16–17.5) in the control group (P=0.002). The median PFS in the DCBVax-L group was 6.2 months (95% CI: 5.7–7.4) while it was 7.6 months (95% CI: 5.6–10.9) in the placebo group (P=0.47). The survival benefits were modest among patients with newly diagnosed GB, confounded by cross-over.96
Wen and colleagues led a phase IIb trial in 2021 on ICT-107, an autologous DC vaccine to target tumor cell antigens and cancer stem cells in 124 newly diagnosed GB patients (NCT01280552).97 HLA-A1 or HLA-A2 positive patients who underwent subtotal or complete resection with ≤1 cm³ residual tumor were included. In total, 81 patients were given ICT-107 and 43 were given a placebo with adjuvant TMZ. In the ICT-107 group, the median OS was 17.0 months (95% CI: 13.68–20.61), whereas it was 15.0 months (95% CI: 12.33–23.05) in the placebo group (P=0.58). The median PFS did show significant improvement in the ICT-107 group at 11.2 months (95% CI: 8.22–13.05) as compared to 9.0 months (95% CI: 5.52–10.29) in the placebo group (P=0.011). Greater benefit was seen in HLA-A2+ patients with MGMT promoter methylation status. Overall, 50% of patients intervened with ICT-107 exhibited immune responses compared to 33% in the placebo group, with the treatment group maintaining a longer quality of life.97
Eoli et al (2019) conducted a phase I/II trial called DENDR2 in Italy where the safety and efficacy of DC vaccines was assessed in 16 patients with recurrent GB.98 Of these, 12 patients received autologous tumor lysate-loaded DC vaccines with dose-dense TMZ, whereas 8 patients received DC vaccines with tetanus toxoid (TT) pre-conditioning without TMZ. In the entire cohort, the median OS was 7.4 months (95% CI: 5.2–9.31). At 9 months, 33% of patients in the TMZ group had survived, whereas in the TT precondition group, 62.5% survived. The TT group additionally had improved T-cell activation along with interferon-gamma (IFN-γ) secretion. Overall, the study depicted the safety and feasibility of TMZ-DC vaccines combined, but with limitations in recurrent GB when compared to TT preconditioning.98
Heat Shock Protein Vaccines
In 2014, Bloch et al conducted a phase II, single-arm, open-label trial where they assessed the safety and efficacy profile of the HSPPC-96 vaccine (NCT00293423).99 A total of 41 patients with recurrent GB were included who had undergone ≥90% tumor resection prior and had a postoperative KPS score of ≥70. The HSPPC-96 vaccine was given weekly for the first 4 weeks and then every 2 weeks until dose depletion or progression. The median OS was 9.8 months (95% CI: 7.99–11.62) and the median PFS was 4.4 months (95% CI: 3.25–5.55). Notably, 6-month OS was achieved by 90.2% of patients whereas the 6-month PFS rate was 29.3%.99
In 2013, Crane et al led a phase I dose-escalation trial where they enrolled 12 patients with recurrent high-grade gliomas (grades 3/4).100 Patients were given the autologous HSPPC-96 vaccine that was derived from their tumor tissue and dosed at 25µg per injection every 1–2 weeks. The median OS was 11 months (range: 3.8–18.3). Of the 12 patients, 11 had immune responses (increases in IFNγ production). There were no severe toxicities and the study provided early-stage evidence towards the use of HSPPC-96.100
Autologous Tumor Vaccines
Muragaki et al conducted a randomized phase IIb trial in 2023 enrolling 57 patients in Japan.101 The cohort of patients (n=63) had newly diagnosed GB and were intervened with an autologous formalin-fixed tumor vaccine (AFTV). The median OS was 25.6 months in the AFTV group and the placebo group with a median OS of 31.5 months (P=0.64). Both groups had no differences in PFS (13.3 months) (P=0.98). While there could be possible benefits of vaccine-based approaches, the survival differences were insignificant.101
In 2014, Ishikawa and colleagues led a phase I/IIa trial in Japan aimed at evaluating the efficacy of AFTV combined with TMZ in 24 patients with newly diagnosed GB.102 These patients had received standard RT with concurrent TMZ, which was followed by AFTV and TMZ maintenance treatment. The median OS was 22.2 months. The 2- and 3-year survival rates were 47% and 38%, respectively. The median PFS was 8.2 months overall, and only 33% of patients had PFS beyond 24 months in the subgroup with a delayed-type hypersensitivity response. On the whole, AFTV was well-tolerated and requires further exploration in different settings.102
Schijns and colleagues led a phase 0/I trial in 2015 where they assessed the efficacy of Gliovac (ERC1671) among 9 patients with recurrent GB.103 Gliovac is a combination of autologous and allogeneic tumor antigens and is given with GM-CSF and low-dose cyclophosphamide to improve immune response. The median OS was 7 months and the survival rate at 6 months was 100% and 77% at 40 weeks, which significantly exceeded historical controls (10% survival rate at 40 weeks). Transient and mild adverse events were noted including local erythema and grade-2 headaches. On assessing for radiographic responses, tumor reduction and stabilization were noted. One patient also achieved complete tumor regression.103
Bota and colleagues conducted a phase II, double-blind, placebo-controlled trial in the US in 2018 (NCT01903330).104 Nine patients who had failed prior standard therapy and were bevacizumab-naive were administered with ERC1671 (Gliovac). The ERC1671 plus bevacizumab group had a median OS of 12.1 months compared to the 7.6 months seen in the placebo group. Median PFS was 7.3 months in the ERC1671 group whereas 5.4 months were seen in the placebo group.104 The ORR was 75% in the interventional group (3 out of 4 patients) compared to 25% for the placebo arm (1 out of 4 patients). More interventions are required to improve survival in treatment-resistant and recurrent GB.104
Angiogenesis-Targeting Vaccines
Tanaka and colleagues conducted a phase 1, single-arm trial in 2013 to assess the preliminary efficacy and safety of the human umbilical vein endothelial cell (HUVEC) vaccine in 17 patients with recurrent GB who had exhausted all prior treatments. Weekly intradermal vaccine injections were administered and sought to inhibit tumor growth by inducing an immune response against endothelial cells, potentially impacting angiogenesis. The median OS was 11.4 months (95% CI: 7.9–14.9) and the median PFS of 5.5 months (95% CI: 3.1–7.9). The ORR was 5.9%, with 1 patient achieving a partial response and 23.5% experiencing disease progression within 3 months. The clinical activity was modest, and delayed-type hypersensitivity skin reactions were common.105
Oncolytic Viral Therapy
Oncolytic viral therapy has emerged as a promising approach in GB treatment, with several clinical trials investigating its efficacy and safety (Table 5).
 |
Table 5 Summary of Clinical Trials Investigating Oncolytic Viral Therapy in Glioblastoma |
The CAPTIVE (KEYNOTE-192) trial, a multicenter phase I/II, open-label, single-arm study, was conducted in 2023 among 49 patients with recurrent GB.63 This trial investigated the effects of combining an oncolytic virus (intratumoral DNX-2401) and a PD-1 inhibitor (pembrolizumab) on OS. The median OS was calculated as 12.5 months (95% CI: 10.7–13.5). ORR was based on the mRANO criteria which was computed as 10.4% (95% CI: 4.2–20.7%). These findings point to modest responses only within this population.63 However, as a single-arm trial, this study lacks a control group, making it difficult to determine whether these modest responses stem from the treatment itself or other confounding factors.
A multi-institutional, open-label, dose-escalation phase I trial conducted by Chiocca et al in 2022 enrolled 21 patients with recurrent GB across 3 dose-escalating cohorts.64 The study investigated the combination of nivolumab, an anti-PD-1 antibody, with Ad-RTS-hIL-12 gene therapy and veledimex (VDX), an oral gene therapy activator. Neoadjuvant nivolumab was administered and continued postoperatively, while VDX was given 3 hours before and for 14 days following tumor resection. The median OS was 9.8 months (95% CI: 5.2–17.4) for all subjects, whereas patients receiving 10mg of VDX with nivolumab had a median OS of 16.9 months (95% CI: 3.8–24.0). Toxicities were dose-related, reversible, and comparable to IL-12 gene monotherapy. The findings suggest that gene therapy in combination with immune checkpoint blockade may be a feasible therapeutic approach for recurrent GB, though further investigation is warranted.64
In a 2004 pilot study, Steiner and colleagues enrolled 23 newly diagnosed GB patients who were treated with autologous tumor vaccines modified by Newcastle Disease Virus (ATV-NDV).106 The study was designed to activate immune targeting of residual tumor cells after standard treatment; patients could receive up to 8 doses after RT. The median OS was 23.0 months, ranging between 8.3 and 63.5 months. The median PFS was 9.2 months. The vaccine generated a strong immune response by increasing tumor-specific T-cell activity, tumor-infiltrating lymphocytes, and delayed-type hypersensitivity. Overall, ATV-NDV was well-tolerated.106
Oncolytic viral therapy shows promise as a novel approach for GB treatment, with DNX-2401 and pembrolizumab demonstrating modest responses, while Ad-RTS-hIL-12 gene therapy combined with nivolumab yielded encouraging survival outcomes. Additionally, the Newcastle Disease Virus-modified tumor vaccine (ATV-NDV) elicited strong immune activation and prolonged survival in newly diagnosed GB patients.
Emerging Therapies and Future Directions
Emerging immunotherapies tend to target alternate sites (ie, myeloid cells) to oppose tumor immunosuppression.107 TAMs enable tumors to grow and suppress patient immunity and are key targets as well.108 At present, preclinical studies reveal that blocking TAM infiltration can improve antitumoral immunity and slow progression of cancer cells.109 Recent trials, including a phase I/IIa TEM-GB study assessed genetically engineered myeloid cells called Temferon (macrophages-IFNα+/Tie2+) in newly diagnosed GB patients. The approach was well-tolerated and modified cells persisted up to 2 weeks post-treatment.110 Other approaches include engineering myeloid cells that release controlled IFNα and IL-12 levels via a dimerization-dependent mechanism to limit off-target toxicity.108 There are also Stimulators of Interferon Gene (STING) agonists that can be combined with ICIs and are being investigated for their potential role in improving GB immune responses. The goal of these approaches is to streamline more effective and less toxic immunotherapeutic approaches. In addition, multi-omics approaches including genomics, transcriptomics, proteomics, and metabolomics have uncovered key molecular targets in glioma-related conditions, such as BRAF V600E, IDH mutations, and PI3K/AKT/mTOR signaling. These technologies may offer valuable insights into GB disease mechanisms and may support the development of more precise, personalized therapies.111,112
Another approach is cytokine modulation, and early trials have been conducted to assess its efficacy for GB. The ATIM-15 trial used adenoviral vectors (Ad–RTS–hIL-12) to induce IL-12 expression in the host to boost immune responses in a controlled manner.113 This was a phase I study and supported the safety and feasibility of intervention in recurrent high-grade gliomas via increased T-cell infiltration. Early-phase trials are currently assessing monotherapy or combinations with ICIs such as pembrolizumab.113 Interventions like M032 are attempting to use modified Herpes simplex virus type 1 (HSV-1) viruses engineered to produce IL-12 to treat GB and even combined with ICIs in other trials.114 These approaches aim to primarily target cytokine levels, especially IL-12, that directly improve immune responses while also reducing systemic toxicity marking significant advances in potential curative regimens in GB care.
To realize the full potential of immunotherapy in GB, future efforts must move beyond proof-of-concept studies and toward integrated, clinically actionable strategies. One critical step will be the design of biomarker-stratified trials that incorporate genomic, transcriptomic, and immune profiling from the outset, enabling personalized patient selection. For example, leveraging IDH mutation status, MGMT promoter methylation, and markers such as tumor mutational burden or immune cell infiltration scores can guide immunotherapy decisions more effectively than one-size-fits-all approaches. Furthermore, sex-specific immune responses and age-related immune dysfunction remain underexplored but may play a significant role in treatment efficacy and tolerance, necessitating deliberate subgroup analyses in future studies.
Another need of the hour is the development of rational combination frameworks grounded in mechanistic synergy. Rather than empirically pairing treatments, future trials should be guided by models that predict additive or synergistic effects based on immune activation timing, tumor antigen load, and microenvironmental context. For instance, sequential delivery of oncolytic viruses followed by checkpoint blockade may be more effective than concurrent administration. Similarly, vaccine priming followed by CAR-T cell infusion could boost intratumoral T-cell infiltration and persistence. Finally, incorporating adaptive trial designs, such as basket trials and Bayesian platforms, will allow dynamic evaluation of multiple agents across molecular subtypes, accelerating clinical translation. GB immunotherapy requires orchestrated, patient-tailored regimens refined by deep biological insight and agile clinical experimentation.
Clinical Implications
The treatment landscape of GB continues to evolve even with limited improvements in survival. The data discussed within the scope of this review is summarized in Figure 1 to understand the current trends of ICI, CAR-T cell, and cancer vaccine therapies. ICIs, which largely comprised pembrolizumab and nivolumab, had a cumulative median OS of 11.15 months (IQR: 8.2–13.63) and PFS of 4.2 months (IQR: 3.9–5.03) across the reviewed GB studies. CAR-T cell therapies, which are primarily explored in hematologic malignancies, had a reported cumulative median OS of 8.18 months (IQR: 7.35–10.08) and PFS of 1.3 months. Regarding vaccines, the cumulative median OS was 18 months (IQR: 11.1–22.06) whereas the PFS was 6.4 months (IQR: 5.85–12.25). For oncolytic viral therapy, the median OS was 16.9 months (IQR: 14.7–19.95) and the PFS was 9.2 months (IQR: not available). While these non-exhaustive values provide a broad understanding of survival trends in different treatment modalities, they must be interpreted with caution.
Figure 1 Continued. Figure 1 Median OS and PFS Across Immunotherapy Trials for Glioblastoma. (A) ICIs, (B) CAR-T Cell Therapy, (C) Vaccines, (D) Oncolytic Viral Therapy.54–60,62–64,77–81,82–84,92–106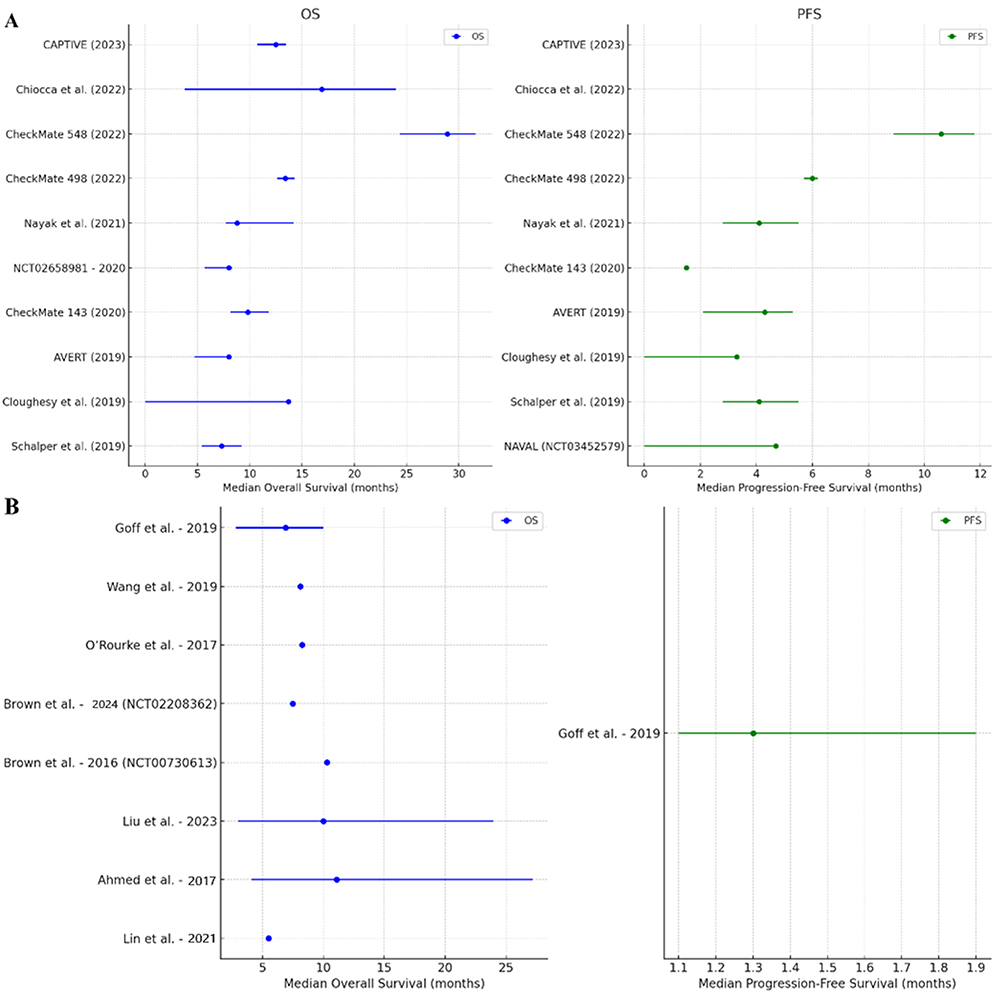
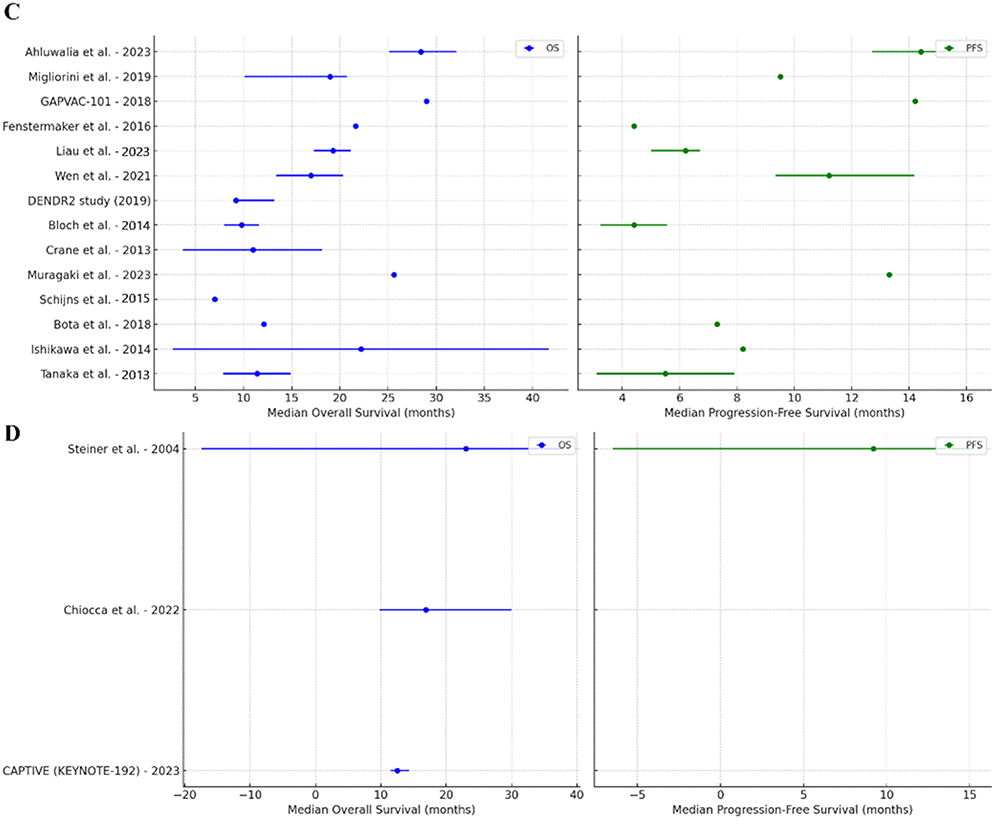
Each discussed modality faces certain challenges. CAR-T therapies have limitations concerning antigen heterogeneity, the BBB, and the immunosuppressive TME, which may collectively reduce their efficacy in solid tumors. On administering ICIs, improvement in clinical outcomes is often times barred from overcoming the immune evasion mechanisms commonly seen within GB. Vaccine-based approaches seek to use tumor-specific immune responses which, in theory, hold premise. As the immunotherapeutic landscape moves forward, combination therapies ought to be explored to overcome the innate anti-tumoral mechanisms of GB. ICIs, cancer vaccines, CAR-T cells, and other novel agents including cytokine modulators, STING agonists, and oncolytic viruses are still being tested to improve therapeutic effectiveness. Moreover, considering the development of biomarker-driven approaches and personalized therapies, there may be significant promise in achieving differential success against GB.
Conclusion
GB remains one of the most challenging malignancies to treat, owing to its aggressive biology, high recurrence rate, and immunosuppressive TME. While current immunotherapeutic strategies, including ICIs, CAR-T cell therapies, cancer vaccines, and oncolytic viruses, have yielded modest survival benefits to date, emerging data from clinical trials offer hope through targeted, multimodal approaches. This review highlights the growing importance of integrating immunotherapeutic strategies with patients’ genetic and immunologic profiles, including molecular markers such as IDH mutation status and MGMT promoter methylation. Additionally, sex-specific biological differences may be considered in future clinical designs, given their relevance in the BBB and immune landscape. Future GB treatment likely lies in rational combination therapies that apply multiple mechanisms of immune activation while overcoming tumor resistance. Continued research and innovation in immunotherapy are essential to not only extend survival but to improve the quality of life for those diagnosed with GB.
Abbreviations
GB, Glioblastoma, a highly aggressive type of brain cancer; ICI, Immune Checkpoint Inhibitor, a type of immunotherapy that blocks inhibitory immune pathways; CAR T-cell, Chimeric Antigen Receptor T-cell therapy, a type of immunotherapy where T-cells are engineered to target cancer; PD-1, Programmed cell death protein 1, a checkpoint receptor on T-cells; PD-L1, Programmed death-ligand 1, a ligand that binds PD-1 to suppress T-cell activity; CTLA-4, Cytotoxic T-Lymphocyte Antigen 4, an immune checkpoint receptor; DC, Dendritic Cell, an antigen-presenting cell important for initiating immune responses; TMZ, Temozolomide, a chemotherapy drug used for GB treatment; TME, Tumor Microenvironment, the environment around a tumor including immune and stromal cells; BBB, Blood-Brain Barrier, a protective barrier that restricts the entry of substances into the brain; Tregs, Regulatory T cells, a subset of T cells that suppress immune responses; MDSCs, Myeloid-Derived Suppressor Cells, cells that suppress T-cell responses in cancer; GSCs, Glioma Stem Cells, therapy-resistant tumor-initiating cells; EGFRvIII, Epidermal Growth Factor Receptor Variant III, a mutation common in GB; IL13Rα2, Interleukin 13 Receptor Subunit Alpha 2, a tumor-associated antigen; GD2, Disialoganglioside GD2, a surface molecule targeted in some cancers; HER2, Human Epidermal Growth Factor Receptor 2, often overexpressed in cancers; EphA2, Ephrin Type-A Receptor 2, a receptor involved in tumor growth and targeted in CAR T-cell therapy; DCVax-L, A dendritic cell vaccine made from patient tumor lysate; ORR, Objective Response Rate, the proportion of patients with tumor size reduction; PFS, Progression-Free Survival, the length of time during which a patient’s disease does not worsen; OS, Overall Survival, the duration a patient survives from the start of treatment; KPS, Karnofsky Performance Scale, a measure of a cancer patient’s functional status; MGMT, O6-Methylguanine-DNA Methyltransferase, a DNA repair enzyme and biomarker for GB; IDH, Isocitrate Dehydrogenase, a gene commonly mutated in GB; HSPPC-96, Heat Shock Protein Peptide Complex-96, a vaccine derived from heat shock proteins and tumor peptides; STING, Stimulator of Interferon Genes, involved in innate immune sensing of tumors; IFNα, Interferon alpha, a cytokine used to enhance immune response; IL-12, Interleukin-12, a cytokine promoting T-cell activity; RT, Radiation Therapy, a common cancer treatment; VEGF, Vascular Endothelial Growth Factor, promotes tumor blood vessel formation; rCBV, Relative Cerebral Blood Volume, an imaging metric used in assessing GB response; APVAC, Personalized peptide-based vaccine derived from tumor and patient HLA type; NAVAL, Nivolumab and Avastin for Recurrent Glioblastoma trial; AFTV, Autologous Formalin-Fixed Tumor Vaccine, a personalized vaccine from patient’s tumor tissue.
Disclosure
J.D.L.: Named as an inventor on cancer therapy patents held by the Cleveland Clinic but these are not directly related to the topic of this review
V.A.V: Reports grants from Mimivax, during the conduct of the study; grants from Mimivax, outside the submitted work;
Y.O.: Serves on the Data and Safety Monitoring Board (DSMB) for Actuate Therapeutics, Oncoceutics (unpaid), and GammaTile. Receives clinical trial support from Bristol Myers Squibb (BMS), Novocure, Cantex Pharmaceuticals, Carthera, Chimerix, CNS Pharmaceuticals, Exelixis, Karyopharm, MimVax, and VBI Vaccines. Consultant for PharPoint Research. No financial conflicts of interest.
M.S.A.: Received consulting fees from Bayer, Xoft, Apollomics, Viewray, Cairn Therapeutics, Anheart Therapeutics, Theraguix, Menarini Ricerche, Sumitomo Pharma Oncology, Autem Therapeutics, GT Medical Technologies, Allovir, EquilliumBio, QV Bioelectronics, Servier Pharmaceuticals, Incyte, and Recordati. He has served on a Data Safety Monitoring Committee (DSMC) for VBI Vaccines and on the Scientific Advisory Board for Modifi biosciences and Bugworks. M.S.A. is a shareholder in Mimivax, CytoDyn, MedInnovate Advisors LLC, Trisalus Life Sciences, and LiveAI. His research has been supported by grants 1R01CA277728-01A1 and 1R01CA264017-01A1.
The funders had no role in the study design, data collection, analysis, interpretation, manuscript writing, or the decision to publish the results.
The authors report no other conflicts of interest in this work.
References
1. Department of Neurosurgery, Jordan University Hospital and Medical School, University of Jordan, Amman, Jordan.AF T, Juweid M.Department of Radiology and Nuclear Medicine, Jordan University Hospital and Medical School, University of Jordan, Amman, Jordan. Epidemiology and Outcome of Glioblastoma.Department of Neurosurgery, University Hospitals Leuven.Leuven, Belgium, De;Vleeschouwer S;eds.;2017;143–153.doi:10.15586/codon.glioblastoma.2017.ch8
2. Grochans S, Cybulska AM, Simińska D, et al. Epidemiology of glioblastoma multiforme–literature review. Cancers. 2022;14(10):2412. doi:10.3390/cancers14102412
3. Ostrom QT, Price M, Neff C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2015–2019. Neuro-Oncology. 2022;24(Supplement_5):v1–v95. doi:10.1093/neuonc/noac202
4. Schaff LR, Mellinghoff IK. Glioblastoma and other primary brain malignancies in adults: a review. JAMA. 2023;329(7):574. doi:10.1001/jama.2023.0023
5. Lin D, Wang M, Chen Y, et al. Trends in intracranial glioma incidence and mortality in the United States, 1975-2018. Front Oncol. 2021;11:748061. doi:10.3389/fonc.2021.748061
6. McCutcheon IE, Preul MC. Historical perspective on surgery and survival with glioblastoma: how far have we come? World Neurosurg. 2021;149:148–168. doi:10.1016/j.wneu.2021.02.047
7. Gerges C, Elder T, Penuela M, et al. Comparative epidemiology of gliosarcoma and glioblastoma and the impact of Race on overall survival: a systematic literature review. Clinl Neurology Neurosurg. 2020;195:106054. doi:10.1016/j.clineuro.2020.106054
8. Tan AC, Ashley DM, López GY, Malinzak M, Friedman HS, Khasraw M. Management of glioblastoma: state of the art and future directions. Ca a Cancer J Clin. 2020;70(4):299–312. doi:10.3322/caac.21613
9. Cone EB, Marchese M, Paciotti M, et al. Assessment of time-to-treatment initiation and survival in a cohort of patients with common cancers. JAMA Netw Open. 2020;3(12):e2030072. doi:10.1001/jamanetworkopen.2020.30072
10. Madhugiri VS, Moiyadi AV, Shetty P, et al. Analysis of factors associated with long-term survival in patients with glioblastoma. World Neurosurg. 2021;149:e758–e765. doi:10.1016/j.wneu.2021.01.103
11. Stupp R, Mason WP, Van Den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi:10.1056/NEJMoa043330
12. Fisher JP, Adamson DC. Current FDA-approved therapies for high-grade malignant gliomas. Biomedicines. 2021;9(3):324. doi:10.3390/biomedicines9030324
13. Cruz JVR, Batista C, Afonso BDH, et al. Obstacles to glioblastoma treatment two decades after temozolomide. Cancers. 2022;14(13):3203. doi:10.3390/cancers14133203
14. Lang F, Liu Y, Chou FJ, Yang C. Genotoxic therapy and resistance mechanism in gliomas. Pharmacol Ther. 2021;228:107922. doi:10.1016/j.pharmthera.2021.107922
15. Seker-Polat F, Pinarbasi Degirmenci N, Solaroglu I, Bagci-Onder T. Tumor cell infiltration into the brain in glioblastoma: from mechanisms to clinical perspectives. Cancers. 2022;14(2):443. doi:10.3390/cancers14020443
16. Abbas MN, Kausar S, Cui H. Therapeutic potential of natural products in glioblastoma treatment: targeting key glioblastoma signaling pathways and epigenetic alterations. Clin Transl Oncol. 2020;22(7):963–977. doi:10.1007/s12094-019-02227-3
17. Datta N, Chakraborty S, Basu M, Ghosh MK. Tumor suppressors having oncogenic functions: the double agents. Cells. 2020;10(1):46. doi:10.3390/cells10010046
18. Filippone A, Lanza M, Mannino D, et al. PD1/PD-L1 immune checkpoint as a potential target for preventing brain tumor progression. Cancer Immunol Immunother. 2022;71(9):2067–2075. doi:10.1007/s00262-021-03130-z
19. Li XP, Guo ZQ, Wang BF, Zhao M. EGFR alterations in glioblastoma play a role in antitumor immunity regulation. Front Oncol. 2023;13:1236246. doi:10.3389/fonc.2023.1236246
20. Himes BT, Geiger PA, Ayasoufi K, Bhargav AG, Brown DA, Parney IF. Immunosuppression in glioblastoma: current understanding and therapeutic implications. Front Oncol. 2021;11:770561. doi:10.3389/fonc.2021.770561
21. Widodo SS, Dinevska M, Furst LM, Stylli SS, Mantamadiotis T. IL-10 in glioma. Br J Cancer. 2021;125(11):1466–1476. doi:10.1038/s41416-021-01515-6
22. Mirlekar B. Tumor promoting roles of IL-10, TGF-β, IL-4, and IL-35: its implications in cancer immunotherapy. SAGE Open Med. 2022;10:20503121211069012. doi:10.1177/20503121211069012
23. Goldmann T, Wieghofer P, Jordão MJC, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. 2016;17(7):797–805. doi:10.1038/ni.3423
24. Van Hove H, Martens L, Scheyltjens I, et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci. 2019;22(6):1021–1035. doi:10.1038/s41593-019-0393-4
25. Valtorta S, Salvatore D, Rainone P, Belloli S, Bertoli G, Moresco RM. Molecular and cellular complexity of glioma. focus on tumour microenvironment and the use of molecular and imaging biomarkers to overcome treatment resistance. IJMS. 2020;21(16):5631. doi:10.3390/ijms21165631
26. Liebelt BD, Shingu T, Zhou X, Ren J, Shin SA, Hu J. Glioma stem cells: signaling, microenvironment, and therapy. Stem Cells Int. 2016;2016(1):7849890. doi:10.1155/2016/7849890
27. Codrici E, Enciu AM, Popescu ID, Mihai S, Tanase C. Glioma stem cells and their microenvironments: providers of challenging therapeutic targets. Stem Cells Int. 2016;2016(1):5728438. doi:10.1155/2016/5728438
28. Colwell N, Larion M, Giles AJ, et al. Hypoxia in the glioblastoma microenvironment: shaping the phenotype of cancer stem-like cells. Neuro-Oncology. 2017;19(7):887–896. doi:10.1093/neuonc/now258
29. Peng Y, Zhao Y, Chen Y, et al. Dual-targeting for brain-specific liposomes drug delivery system: synthesis and preliminary evaluation. Bioorg Med Chem. 2018;26(16):4677–4686. doi:10.1016/j.bmc.2018.08.006
30. Rui Y, Green JJ. Overcoming delivery barriers in immunotherapy for glioblastoma. Drug Deliv Transl Res. 2021;11(6):2302–2316. doi:10.1007/s13346-021-01008-2
31. Prasad S, Pati A, Panda S, Saxena S. Integrative data analysis of MGMT methylation and IDH1 mutation in glioblastoma: a comprehensive review. Radiomics Radiogenomics Neuro-Oncology Elsevier. 2025:181–202. doi:10.1016/B978-0-443-18509-0.00003-7
32. Fares J, Wan Y, Mair R, Price SJ. Molecular diversity in isocitrate dehydrogenase-wild-type glioblastoma. Brain Comm. 2024;6(2):fcae108. doi:10.1093/braincomms/fcae108
33. Asioli S, Gatto L, Vardy U, et al. Immunophenotypic profile of adult glioblastoma IDH-wildtype microenvironment: a cohort study. Cancers. 2024;16(22):3859. doi:10.3390/cancers16223859
34. Donson AM, Addo‐Yobo SO, Handler MH, Gore L, Foreman NK. MGMT promoter methylation correlates with survival benefit and sensitivity to temozolomide in pediatric glioblastoma. Pediatr Blood Cancer. 2007;48(4):403–407. doi:10.1002/pbc.20803
35. Le VH, Minh TNT, Kha QH, Le NQK. A transfer learning approach on MRI-based radiomics signature for overall survival prediction of low-grade and high-grade gliomas. Med Biol Eng Comput. 2023;61(10):2699–2712. doi:10.1007/s11517-023-02875-2
36. Kha QH, Le VH, Hung TNK, Le NQK. Development and validation of an efficient mri radiomics signature for improving the predictive performance of 1p/19q Co-deletion in lower-grade gliomas. Cancers. 2021;13(21):5398. doi:10.3390/cancers13215398
37. De Souza N, Zhao S, Bodenmiller B. Multiplex protein imaging in tumour biology. Nat Rev Cancer. 2024;24(3):171–191. doi:10.1038/s41568-023-00657-4
38. Fougner V, Hasselbalch B, Lassen U, Weischenfeldt J, Poulsen HS, Urup T. Implementing targeted therapies in the treatment of glioblastoma: previous shortcomings, future promises, and a multimodal strategy recommendation. Neurooncol Adv. 2022;4(1):vdac157. doi:10.1093/noajnl/vdac157
39. Roda D, Veiga P, Melo JB, Carreira IM, Ribeiro IP. Principles in the Management of Glioblastoma. Genes. 2024;15(4):501. doi:10.3390/genes15040501
40. Rabin EE, Huang J, Kim M, et al. Age-stratified comorbid and pharmacologic analysis of patients with glioblastoma. Brain, Behavior, & Immunity - Health. 2024;38:100753. doi:10.1016/j.bbih.2024.100753
41. Ladomersky E, Zhai L, Lauing KL, et al. Advanced age increases immunosuppression in the brain and decreases immunotherapeutic efficacy in subjects with glioblastoma. Clin Cancer Res. 2020;26(19):5232–5245. doi:10.1158/1078-0432.CCR-19-3874
42. Johnson M, Bell A, Lauing KL, et al. Advanced age in humans and mouse models of glioblastoma show decreased survival from extratumoral influence. Clin Cancer Res. 2023;29(23):4973–4989. doi:10.1158/1078-0432.CCR-23-0834
43. Khaddour K, Johanns T, Ansstas G. The landscape of novel therapeutics and challenges in glioblastoma multiforme: contemporary state and future directions. Pharmaceuticals. 2020;13(11):389. doi:10.3390/ph13110389
44. Zhang H, Dai Z, Wu W, et al. Regulatory mechanisms of immune checkpoints PD-L1 and CTLA-4 in cancer. J Exp Clin Cancer Res. 2021;40(1):184. doi:10.1186/s13046-021-01987-7
45. Shen S, Hong Y, Huang J, et al. Targeting PD-1/PD-L1 in tumor immunotherapy: mechanisms and interactions with host growth regulatory pathways. Cytokine Growth Factor Rev. 2024;79:16–28. doi:10.1016/j.cytogfr.2024.08.001
46. Shi H, Li K, Ni Y, Liang X, Zhao X. Myeloid-derived suppressor cells: implications in the resistance of malignant tumors to T cell-based immunotherapy. Front Cell Dev Biol. 2021;9:707198. doi:10.3389/fcell.2021.707198
47. Liu J, Chen Z, Li Y, Zhao W, Wu J, Zhang Z. PD-1/PD-L1 checkpoint inhibitors in tumor immunotherapy. Front Pharmacol. 2021;12:731798. doi:10.3389/fphar.2021.731798
48. Fudaba H, Wakimoto H. Oncolytic virus therapy for malignant gliomas: entering the new era. Expert Opin Biol Ther. 2023;23(3):269–282. doi:10.1080/14712598.2023.2184256
49. Kreidieh FY, Tawbi HA. The introduction of LAG-3 checkpoint blockade in melanoma: immunotherapy landscape beyond PD-1 and CTLA-4 inhibition. Ther Adv Med Oncol. 2023;15:17588359231186027. doi:10.1177/17588359231186027
50. Balta E, Wabnitz GH, Samstag Y. Hijacked immune cells in the tumor microenvironment: molecular mechanisms of immunosuppression and cues to improve T cell-based immunotherapy of solid tumors. IJMS. 2021;22(11):5736. doi:10.3390/ijms22115736
51. Abedi Kiasari B, Abbasi A, Ghasemi Darestani N, et al. Combination therapy with nivolumab (anti-PD-1 monoclonal antibody): a new era in tumor immunotherapy. Int Immunopharmacol. 2022;113:109365. doi:10.1016/j.intimp.2022.109365
52. Cillo AR, Cardello C, Shan F, et al. Blockade of LAG-3 and PD-1 leads to co-expression of cytotoxic and exhaustion gene modules in CD8+ T cells to promote antitumor immunity. Cell. 2024;187(16):4373–4388.e15. doi:10.1016/j.cell.2024.06.036
53. Tobin JWD, Bednarska K, Campbell A, Keane C. PD-1 and LAG-3 checkpoint blockade: potential avenues for therapy in B-cell lymphoma. Cells. 2021;10(5):1152. doi:10.3390/cells10051152
54. Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med. 2019;25(3):470–476. doi:10.1038/s41591-018-0339-5
55. Omuro A, Brandes AA, Carpentier AF, et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: an international randomized phase III trial. Neuro-Oncology. 2023;25(1):123–134. doi:10.1093/neuonc/noac099
56. Lim M, Weller M, Idbaih A, et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro-Oncology. 2022;24(11):1935–1949. doi:10.1093/neuonc/noac116
57. Cloughesy TF, Mochizuki AY, Orpilla JR, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25(3):477–486. doi:10.1038/s41591-018-0337-7
58. Reardon DA, Brandes AA, Omuro A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the checkmate 143 phase 3 randomized clinical trial. JAMA Oncol. 2020;6(7):1003. doi:10.1001/jamaoncol.2020.1024
59. Lim M, Ye X, Piotrowski AF, et al. Updated safety phase I trial of anti-LAG-3 alone and in combination with anti-PD-1 in patients with recurrent GBM. JCO. 2020;38(15_suppl):2512. doi:10.1200/JCO.2020.38.15_suppl.2512
60. Nayak L, Molinaro AM, Peters K, et al. Randomized phase ii and biomarker study of pembrolizumab plus bevacizumab versus pembrolizumab alone for patients with recurrent glioblastoma. Clin Cancer Res. 2021;27(4):1048–1057. doi:10.1158/1078-0432.CCR-20-2500
61. Ahluwalia MS, Peereboom DM, Ozair A, et al. A randomized, controlled, phase 2 trial of nivolumab plus standard-dose or low-dose bevacizumab for recurrent glioblastoma (NAVAL). JCO. 2024;42(16_suppl):2072. doi:10.1200/JCO.2024.42.16_suppl.2072
62. Peters KB, Archer GE, Norberg P, et al. Safety of nivolumab in combination with dendritic cell vaccines in recurrent high-grade glioma. JCO. 2019;37(15_suppl):e13526–e13526. doi:10.1200/JCO.2019.37.15_suppl.e13526
63. Nassiri F, Patil V, Yefet LS, et al. Oncolytic DNX-2401 virotherapy plus pembrolizumab in recurrent glioblastoma: a phase 1/2 trial. Nat Med. 2023;29(6):1370–1378. doi:10.1038/s41591-023-02347-y
64. Chiocca EA, Gelb AB, Chen CC, et al. Combined immunotherapy with controlled interleukin-12 gene therapy and immune checkpoint blockade in recurrent glioblastoma: an open-label, multi-institutional phase I trial. Neuro-Oncology. 2022;24(6):951–963. doi:10.1093/neuonc/noab271
65. Kieliszek AM, Mobilio D, Upreti D, et al. Intratumoral delivery of chimeric antigen receptor t cells targeting CD133 effectively treats brain metastases. Clin Cancer Res. 2024;30(3):554–563. doi:10.1158/1078-0432.CCR-23-1735
66. Kringel R, Lamszus K, Mohme M. Chimeric antigen receptor t cells in glioblastoma—current concepts and promising future. Cells. 2023;12(13):1770. doi:10.3390/cells12131770
67. Ma K, Hu P. Chimeric antigen receptor t-cell therapy for glioblastoma. Cancers. 2023;15(23):5652. doi:10.3390/cancers15235652
68. Zhu G, Zhang Q, Zhang J, Liu F. Targeting tumor-associated antigen: a promising CAR-T therapeutic strategy for glioblastoma treatment. Front Pharmacol. 2021;12:661606. doi:10.3389/fphar.2021.661606
69. Chokshi CR, Shaikh MV, Brakel B, et al. Targeting axonal guidance dependencies in glioblastoma with ROBO1 CAR T cells. Nat Med. 2024;30(10):2936–2946. doi:10.1038/s41591-024-03138-9
70. Thaci B, Brown CE, Binello E, Werbaneth K, Sampath P, Sengupta S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro-Oncology. 2014;16(10):1304–1312. doi:10.1093/neuonc/nou045
71. Alsajjan R, Mason WP. Bispecific T-cell engagers and chimeric antigen receptor t-cell therapies in glioblastoma: an update. Current Oncol. 2023;30(9):8501–8549. doi:10.3390/curroncol30090619
72. Garima G, Thanvi S, Singh A, Verma V. Epidermal growth factor receptor variant iii mutation, an emerging molecular marker in glioblastoma multiforme patients: a single institution study on the Indian population. Cureus. doi:10.7759/cureus.26412
73. Nazha B, Inal C, Owonikoko TK. Disialoganglioside GD2 expression in solid tumors and role as a target for cancer therapy. Front Oncol. 2020;10:1000. doi:10.3389/fonc.2020.01000
74. Wang D, Starr R, Chang WC, et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci Transl Med. 2020;12(533):eaaw2672. doi:10.1126/scitranslmed.aaw2672
75. Mineo JF, Bordron A, Baroncini M, et al. Low HER2-expressing glioblastomas are more often secondary to anaplastic transformation of low-grade glioma. J Neurooncol. 2007;85(3):281–287. doi:10.1007/s11060-007-9424-1
76. Choi BD, Gerstner ER, Frigault MJ, et al. Intraventricular CARv3-TEAM-E T Cells in Recurrent Glioblastoma. N Engl J Med. 2024;390(14):1290–1298. doi:10.1056/NEJMoa2314390
77. Goff SL, Morgan RA, Yang JC, et al. Pilot Trial of adoptive transfer of chimeric antigen receptor–transduced t cells targeting EGFRvIII in patients with glioblastoma. J Immunother. 2019;42(4):126–135. doi:10.1097/CJI.0000000000000260
78. Wang S, O’Rourke DM, Chawla S, et al. Multiparametric magnetic resonance imaging in the assessment of anti-EGFRvIII chimeric antigen receptor T cell therapy in patients with recurrent glioblastoma. Br J Cancer. 2019;120(1):54–56. doi:10.1038/s41416-018-0342-0
79. O’Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399):eaaa0984. doi:10.1126/scitranslmed.aaa0984
80. Brown CE, Hibbard JC, Alizadeh D, et al. Locoregional delivery of IL-13Rα2-targeting CAR-T cells in recurrent high-grade glioma: a phase 1 trial. Nat Med. 2024;30(4):1001–1012. doi:10.1038/s41591-024-02875-1
81. Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor t-cell therapy. N Engl J Med. 2016;375(26):2561–2569. doi:10.1056/NEJMoa1610497
82. Liu Z, Zhou J, Yang X, et al. Safety and antitumor activity of GD2-Specific 4SCAR-T cells in patients with glioblastoma. Mol Cancer. 2023;22(1):3. doi:10.1186/s12943-022-01711-9
83. Ahmed N, Brawley V, Hegde M, et al. HER2-specific chimeric antigen receptor–modified virus-specific t cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 2017;3(8):1094. doi:10.1001/jamaoncol.2017.0184
84. Lin Q, Ba T, Ho J, et al. First-in-human trial of EphA2-Redirected CAR T-cells in patients with recurrent glioblastoma: a preliminary report of three cases at the starting dose. Front Oncol. 2021;11:694941. doi:10.3389/fonc.2021.694941
85. Brown CE, Badie B, Barish ME, et al. Bioactivity and Safety of IL13Rα2-redirected chimeric antigen receptor cd8+ t cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21(18):4062–4072. doi:10.1158/1078-0432.CCR-15-0428
86. Zhao Y, Baldin AV, Isayev O, Werner J, Zamyatnin AA, Bazhin AV. Cancer vaccines: antigen selection strategy. Vaccines. 2021;9(2):85. doi:10.3390/vaccines9020085
87. Buonaguro L, Tagliamonte M. Peptide-based vaccine for cancer therapies. Front Immunol. 2023;14:1210044. doi:10.3389/fimmu.2023.1210044
88. Baldin AV, Savvateeva LV, Bazhin AV, Zamyatnin AA. dendritic cells in anticancer vaccination: rationale for ex vivo loading or in vivo targeting. Cancers. 2020;12(3):590. doi:10.3390/cancers12030590
89. Haskell-Mendoza AP, Bloch O. Heat shock protein vaccines in glioblastoma. Immunotherapeutic Strategies Treatment Glioma Elsevier. 2022:39–53. doi:10.1016/B978-0-12-819755-4.00015-1
90. Yan Y, Zeng S, Gong Z, Xu Z. Clinical implication of cellular vaccine in glioma: current advances and future prospects. J Exp Clin Cancer Res. 2020;39(1):257. doi:10.1186/s13046-020-01778-6
91. Safety and Efficacy Study of SL-701, a glioma-associated antigen vaccine to treat recurrent glioblastoma multiforme. ClinicalTrials.Gov. 2024. https://www.clinicaltrials.gov/study/NCT02078648
92. Ahluwalia MS, Reardon DA, Abad AP, et al. Phase iia study of survaxm plus adjuvant temozolomide for newly diagnosed glioblastoma. JCO. 2023;41(7):1453–1465. doi:10.1200/JCO.22.00996
93. Fenstermaker RA, Ciesielski MJ, Qiu J, et al. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol Immunother. 2016;65(11):1339–1352. doi:10.1007/s00262-016-1890-x
94. Migliorini D, Dutoit V, Allard M, et al. Phase I/II trial testing safety and immunogenicity of the multipeptide IMA950/poly-ICLC vaccine in newly diagnosed adult malignant astrocytoma patients. Neuro-Oncology. 2019;21(7):923–933. doi:10.1093/neuonc/noz040
95. Wick W, Dietrich PY, Kuttruff S, et al. GAPVAC-101: first-in-human trial of a highly personalized peptide vaccination approach for patients with newly diagnosed glioblastoma. JCO. 2018;36(15_suppl). doi:10.1200/JCO.2018.36.15_suppl.2000
96. Liau LM, Ashkan K, Brem S, et al. Association of autologous tumor lysate-loaded dendritic cell vaccination with extension of survival among patients with newly diagnosed and recurrent glioblastoma: a phase 3 prospective externally controlled cohort trial. JAMA Oncol. 2023;9(1):112. doi:10.1001/jamaoncol.2022.5370
97. Wen PY, Reardon DA, Armstrong TS, et al. A randomized double-blind placebo-controlled phase ii trial of dendritic cell vaccine ICT-107 in newly diagnosed patients with glioblastoma. Clin Cancer Res. 2021;25(19):5799–5807. doi:10.1158/1078-0432.CCR-19-0261
98. Eoli M, Corbetta C, Anghileri E, et al. Expansion of effector and memory T cells is associated with increased survival in recurrent glioblastomas treated with dendritic cell immunotherapy. Neurooncol Adv. 2019;1(1):vdz022. doi:10.1093/noajnl/vdz022
99. Bloch O, Crane CA, Fuks Y, et al. Heat-shock protein peptide complex–96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro-Oncology. 2014;16(2):274–279. doi:10.1093/neuonc/not203
100. Crane CA, Han SJ, Ahn B, et al. Individual Patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin Cancer Res. 2013;19(1):205–214. doi:10.1158/1078-0432.CCR-11-3358
101. Muragaki Y, Ishikawa E, Maruyama T, et al. A multicenter, randomized, placebo-controlled phase IIb trial of an autologous formalin-fixed tumor vaccine for newly diagnosed glioblastomas. J Neurosurg. 2023;139(2):344–354. doi:10.3171/2022.12.JNS221221
102. Ishikawa E, Muragaki Y, Yamamoto T, et al. Phase I/IIa trial of fractionated radiotherapy, temozolomide, and autologous formalin-fixed tumor vaccine for newly diagnosed glioblastoma: clinical article. JNS. 2014;121(3):543–553. doi:10.3171/2014.5.JNS132392
103. Schijns VEJC, Pretto C, Devillers L, et al. First clinical results of a personalized immunotherapeutic vaccine against recurrent, incompletely resected, treatment-resistant glioblastoma multiforme (GBM) tumors, based on combined allo- and auto-immune tumor reactivity. Vaccine. 2015;33(23):2690–2696. doi:10.1016/j.vaccine.2015.03.095
104. Bota DA, Chung J, Dandekar M, et al. Phase II study of ERC1671 plus bevacizumab versus bevacizumab plus placebo in recurrent glioblastoma: interim results and correlations with CD4 + T-lymphocyte counts. CNS Oncol. 2018;7(3):CNS22. doi:10.2217/cns-2018-0009
105. Tanaka M, Tsuno NH, Fujii T, Todo T, Saito N, Takahashi K.
106. Steiner HH, Bonsanto MM, Beckhove P, et al. Antitumor vaccination of patients with glioblastoma multiforme: a pilot study to assess feasibility, safety, and clinical benefit. JCO. 2004;22(21):4272–4281. doi:10.1200/JCO.2004.09.038
107. Fekrirad Z, Barzegar Behrooz A, Ghaemi S, et al. Immunology meets bioengineering: improving the effectiveness of glioblastoma immunotherapy. Cancers. 2022;14(15):3698. doi:10.3390/cancers14153698
108. Canella A, Rajappa P. Therapeutic utility of engineered myeloid cells in the tumor microenvironment. Cancer Gene Ther. 2023;30(7):964–972. doi:10.1038/s41417-023-00600-7
109. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol. 2020;11:583084. doi:10.3389/fimmu.2020.583084
110. Finocchiaro G, Gentner B, Farina F, et al. A phase I-IIa study of genetically modified Tie-2 expressing monocytes in patients with glioblastoma multiforme (TEM-GBM Study). JCO. 2021;39(15_suppl):2532. doi:10.1200/JCO.2021.39.15_suppl.2532
111. Xu Y, Du Y, Zheng Q, et al. Identification of ferroptosis-related prognostic signature and subtypes related to the immune microenvironment for breast cancer patients receiving neoadjuvant chemotherapy. Front Immunol. 2022;13:895110. doi:10.3389/fimmu.2022.895110
112. Du Y, Li R, Fu D, et al. Multi‐omics technologies and molecular biomarkers in brain tumor‐related epilepsy. CNS Neurosci Ther. 2024;30(4):e14717. doi:10.1111/cns.14717
113. Yuan B, Wang G, Tang X, Tong A, Zhou L. Immunotherapy of glioblastoma: recent advances and future prospects. Hum Vaccines Immunother. 2022;18(5):2055417. doi:10.1080/21645515.2022.2055417
114. Chambers MR, Bentley RT, Crossman DK, et al. The One health consortium: design of a phase i clinical trial to evaluate m032, a genetically engineered hsv-1 expressing il-12, in combination with a checkpoint inhibitor in canine patients with sporadic high grade gliomas. Front Surg. 2020;7:59. doi:10.3389/fsurg.2020.00059
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.