Back to Journals » International Journal of Nanomedicine » Volume 21
Immunogenicity Enhancement via Self-Assembled mi3 Nanoparticles Displaying ESAT-6 Antigen
Authors Dong S, Wu S, Liang K, Zhang H, Guo F, Song Y, Hao Y, Qian Z, Wang X, Xu T, Wang H
Received 4 August 2025
Accepted for publication 11 December 2025
Published 8 January 2026 Volume 2026:21 558312
DOI https://doi.org/10.2147/IJN.S558312
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sachin Mali
Sihang Dong,1 Shuang Wu,1 Ke Liang,1 Hui Zhang,2 Fangzheng Guo,1 Yamin Song,1 Yanmei Hao,1 Zhongqing Qian,1 Xiaojing Wang,3 Tao Xu,1,2 Hongtao Wang1,2
1Anhui Province Key Laboratory of Immunology in Chronic Diseases, Medicine Experimental Center, Laboratory Medicine College, Bengbu Medical University, Bengbu, 233030, People’s Republic of China; 2Department of Neurosurgery, the First Afliated Hospital of Bengbu Medical University, Bengbu, 233000, People’s Republic of China; 3Anhui Province Key Laboratory of Respiratory Tumor and Infectious Disease, Molecular Diagnosis Center, First Affiliated Hospital of Bengbu Medical University, Bengbu, 233004, People’s Republic of China
Correspondence: Tao Xu, Email [email protected] Hongtao Wang, Email [email protected]
Purpose: The only World Health Organization (WHO)-approved vaccine for tuberculosis (TB), Bacillus Calmette–Guérin (BCG), shows limited efficacy, particularly against adult pulmonary TB. Moreover, traditional subunit vaccines often suffer from poor immunogenicity. To address these challenges, we aimed to enhance the immune activation potential of the early secreted antigenic target of 6 kDa (ESAT-6) by developing a novel nanoparticle-based vaccine platform.
Methods: We engineered a nanovaccine using mi3 nanoparticles to display the ESAT-6 antigen on their surface. These nanoparticles self-assembled into uniform dodecahedral structures, enabling enhanced antigen presentation. To evaluate its immunogenicity, mice (n= 6 per group) were primed with BCG and subsequently boosted with the ESAT-6–mi3 nanovaccine. Control groups included mice receiving BCG alone or unconjugated nanoparticles.
Results: Compared with mice that received BCG alone or unconjugated nanoparticles, those boosted with the ESAT-6–mi3 nanoparticle vaccine exhibited significantly stronger immune responses. Antigen-specific antibody titers increased by more than 10-fold compared to the BCG-only group. Additionally, elevated levels of pro-inflammatory cytokines such as TNF-α, IFN-γ, and IL-2 were observed, indicating a robust Th1/Th17-biased cellular immune response. Our findings indicate that the vaccine candidate can elicit effective immune responses in vitro. However, it should be noted that this study has limitations, primarily including the lack of an in vivo pathogen challenge to assess actual protective efficacy and the evaluation of immune responses against only a single antigen. Therefore, these results require further validation in future studies involving challenge experiments and multi-antigen strategies.
Conclusion: Our findings demonstrate that the ESAT-6–mi3 nanoparticle vaccine markedly enhances both humoral and cellular immune responses, outperforming traditional subunit vaccine approaches. This strategy shows strong potential as a next-generation booster vaccine to complement BCG and improve TB immunization outcomes.
Keywords: Mycobacterium tuberculosis, mi3, self-assembled, nanoparticle, vaccine
Introduction
Tuberculosis (TB) is a chronic infectious disease caused by Mycobacterium tuberculosis (Mtb), primarily affecting the lungs.1 Although combination chemotherapy for TB has advanced significantly over the past decade, challenges persist due to the spread of multidrug-resistant (MDR) and extensively drug-resistant (XDR) strains, rising rates of latent infections, and co-infection with HIV/AIDS.2–4 According to the World Health Organization’s Global Tuberculosis Report 2024, an estimated 8.2 million new TB cases were confirmed in 2023, which is the highest figure reported since global TB surveillance began in 1995. TB-related deaths reached 1.25 million, making TB the leading cause of death among all infectious diseases once again.1,2 The limited effectiveness of BCG in preventing adult TB, along with other contributing factors, further complicates TB control efforts and poses a serious challenge to the global goal of eliminating TB by 2035.5–7
Currently, BCG remains the only widely used vaccine for TB. While it offers some protection against primary infections in children, its efficacy in preventing TB in adults remains undetermined.5,6,8 Therefore, the adoption of effective booster vaccines will benefit the vast majority of the population who have been vaccinated with BCG, thereby reducing TB transmission and strengthening overall TB control.9 One key reason for BCG’s limited efficacy is the loss of region of difference (RD) genes from Mycobacterium bovis during attenuation, including the gene encoding early secreted antigenic target of 6 kDa (ESAT-6), a crucial virulence factor of Mtb. ESAT-6 is expressed only in pathogenic mycobacteria and is believed to contribute significantly to the suboptimal protective effect of BCG.10–13
In recent years, nanoparticle-based vaccines have emerged as a promising strategy to enhance antigen immunogenicity. Various platforms such as virus-like particles (VLPs), liposomes, and polymeric nanoparticles have been explored for TB subunit vaccines. However, many of these systems face limitations in structural uniformity, antigen loading capacity, or stability under physiological conditions. In contrast to these platforms, the mi3 nanoparticle used in this study offers a highly uniform dodecahedral geometry and exceptional thermodynamic stability, which are advantageous for consistent antigen presentation and in vivo performance.
mi3 is derived from 2-keto-3-deoxy-phosphogluconate (KDPG) aldolase from the superthermophilic anaerobic bacterium Thermotoga maritima. It originates from the i301 nanoparticle protein identified by Hsia et al. Brunn et al subsequently modified the i301 particles through computational design, after which the nanoparticles could spontaneously assemble into dodecahedrons with 60 subunits and a diameter of 25 nm, also known as mutant i301 (mi3).14–16 The mi3 nanoplatform is widely used in various studies in the biomedical field due to its high engineering and multi-functionality, which can provide enhanced bioavailability, high delivery efficiency of drugs and antigens, targeted delivery and high biological stability, unique antigen presentation methods, and self-assembly characteristics during the development process, including drug delivery, cancer immunotherapy, antimicrobial therapy, and antigen presentation.17–21
This study aims to utilize the self-assembling protein nanoparticle mi3 as a polymerized antigen delivery platform by displaying ESAT-6 on the mi3 dodecahedral cage, thereby constructing a novel chimeric nanoparticle vaccine for TB. The uniform architecture and high stability of mi3 distinguish it from previously reported nanocarriers such as polylactic-co-glycolic acid (PLGA) particles or alum-adsorbed formulations, potentially enabling improved codelivery of antigens and adjuvants and more potent T-cell activation. We systematically evaluate the assembly efficiency, structural integrity, and ability of the vaccine to induce antigen-specific T-cell responses.22–24
Materials and Methods
Cloning
Using complete sequences obtained from the NCBI gene library and relevant literature, gene sequences encoding ESAT-6 (GenBank: NP_178023), mi3 (GenBank: AXF54357.1), and the ESAT-6-mi3 fusion protein were synthesized in vitro. These sequences were constructed into expression vectors for Escherichia coli (E. coli), and the corresponding plasmids were generated. As shown in Figure 1A, expression plasmids for ESAT-6, mi3, and ESAT-6-mi3 were successfully constructed. To ensure structural integrity and flexibility of the fusion protein, ESAT-6 was linked to the N-terminal of mi3 via a flexible linker ((G2S)4), and a 6× His-tag was added to the N-terminal of ESAT-6 for purification purposes. After codon optimization, the target sequences were cloned into the pET-28a(+) vector. The recombinant plasmids were then sequenced to verify the correct gene and protein sequences for expression. All recombinant and fusion proteins were produced and purified using standard protocols.
 |
Figure 1 Characterization of recombinant proteins and nanoparticles. (A) Schematic diagram of the assembly of different plasmids in Escherichia coli. (B) SDS-PAGE analysis of the purified and identified proteins: 1: ESAT-6 (11.27kDa), 2: mi3 (22.87kDa), 3: ESAT-6-mi3 (34.07kDa). (C) Transmission electron microscopy (TEM) results. (D) Particle size analysis of mi3 and ESAT-6-mi3 by dynamic light scattering (DLS) characterization. |
Expression and Purification of Recombinant Protein and NPs
The correctly sequenced plasmids were transformed into E. coli BL21 (DE3) competent cells and plated onto LB agar containing 50 µg/mL kanamycin. Plates were incubated at 37°C for 16 hours. A single colony was inoculated into 50 mL of LB medium containing 50 µg/mL kanamycin and cultured overnight at 200 rpm and 37°C. The culture was then diluted 1:100 into 1 L of fresh LB medium with kanamycin and incubated under the same conditions until reaching an optical density of 0.6–0.8 at 600 nm (OD600, logarithmic growth phase). A small aliquot was collected as a pre-induction sample, and isopropyl-β-D-1-thiogalactopyranoside (IPTG, final concentration 0.2 mM) was added to induce expression. Cultures were incubated at 16°C for 12 hours. Cells were harvested and lysed by ultrasonication, and both the supernatant and pellet were analyzed by SDS-PAGE before and after IPTG induction.
For proteins in the supernatant, the lysate was loaded onto a pre-equilibrated nickel-nitrilotriacetic acid (Ni-NTA) Sepharose CL-6B affinity chromatography column. The column was washed with Ni-NTA binding/washing buffer (20 mM Tris-HCl, 30 mM imidazole, 0.15 M NaCl, pH 8.0) until the OD280 returned to baseline. Target proteins were eluted using Ni-NTA elution buffer (20 mM Tris-HCl, 250 mM imidazole, 0.15 M NaCl, pH 8.0), and eluates were dialyzed overnight against phosphate-buffered saline (PBS). For proteins present in inclusion bodies, cells were resuspended in lysis buffer (20 mM Tris-HCl, 1 mM PMSF, pH 8.0), lysed by ultrasonication, and centrifuged at 12,000 rpm for 20 minutes at 4°C. The pellet was washed three times with inclusion body washing buffer (20 mM Tris-HCl, 1 mM EDTA, 2 M urea, 1 M NaCl, 1% Triton X-100, pH 8.0), then resuspended in solubilization buffer (20 mM Tris-HCl, 5 mM DTT, 8 M urea, pH 8.0) and incubated at room temperature. After centrifugation (12,000 rpm, 20 minutes), the supernatant was dialyzed overnight in refolding buffer (20 mM Tris-HCl, 0.15 M NaCl, pH 8.0) to facilitate protein renaturation, followed by a final dialysis against PBS. Renatured proteins were analyzed by 12% SDS-PAGE.
Characterization and Identification of Nanoparticles
The self-assembly of mi3 and ESAT-6-mi3 nanoparticles was evaluated by transmission electron microscopy (TEM). A 20 µL aliquot of each sample was loaded onto carbon-coated copper grids, allowed to adsorb for 5 minutes, and then stained with 2% uranyl acetate for 20 seconds. Samples were air-dried and imaged using a JEOL JEM-F200 TEM (Tokyo, Japan) at an accelerating voltage of 200 kV.
Particle size and distribution were analyzed using a Malvern ZS90 instrument. Aggregates were removed from the protein samples, which were then diluted to 0.5 mg/mL in PBS and transferred into disposable solvent-resistant microcuvettes. Measurements were performed at a 173° scattering angle at 25°C. The hydrodynamic diameter and polydispersity index (PDI) were recorded and analyzed using the manufacturer’s software (Malvern PANalytical).
Mice
Twenty-four specific-pathogen-free (SPF) grade female C57BL/6J mice (6–8 weeks old) were obtained from Jiangsu Jicui Yaokang Co., Ltd. Upon arrival, the mice were randomly divided into four groups and allowed to acclimate for at least one week in the animal facility. Mice were maintained in individually ventilated cages (IVC, Fengqiao Purification Equipment Co., Ltd., Suzhou, China) at a temperature of 20–23°C, relative humidity of 45–65%, and a 12-hour light/dark cycle. Sterile water and irradiated feed (HFK Bioscience Co., Ltd., Beijing, China) were provided ad libitum.
Immunization Regimens
To evaluate the vaccine’s efficacy and safety in vivo, 6–7-week-old female C57BL/6J mice were randomly divided into four groups: BCG, BCG+ESAT-6 (BE), BCG+mi3 (BM), and BCG+ESAT-6-mi3 (BEM), with six mice per group. All immunizations were administered subcutaneously at the neck. Prior to immunization, all recombinant protein solutions were quality-checked to ensure antigen integrity. The commercial BCG freeze-dried powder was reconstituted in sterile saline. Mice were immunized three times at two-week intervals (weeks 0, 2, and 4). The initial immunization was designated as Day 0. Booster immunizations were administered at defined intervals on Days 14 (Week 2) and 28 (Week 4). All immunizations were delivered via subcutaneous injection in the cervical region, using a standardized dose of 20 μg protein formulation in a 200 μL volume. The endpoint was determined on the 42nd day (the 6th week), and biological samples (serum and primary cells) were systematically collected two weeks after the last immunization. During the first immunization, mice received 200 µL of BCG suspension containing 2 × 106 CFU via intramuscular injection in the dorsal and cervical regions. For the second and third immunizations, groups BE, BM, and BEM received 20 µg of their respective recombinant proteins (in 200 µL total volume), while the BCG group received no additional treatment. All mice were euthanized by cervical dislocation two weeks after the final immunization.
Determination of Specific Antibodies
The levels of IgG, IgG1, and IgG2c antibodies against the antigen ESAT-6 in mouse serum were determined by enzyme-linked immunosorbent assay (ELISA). ELISA was performed as follows: ESAT-6 antigen protein was coated onto a microplate using Na2CO3-NaHCO3 buffer (pH 9.6) and incubated overnight at 4 °C. The plate was subsequently blocked to prevent non-specific binding. Mouse antiserum was serially diluted, starting from 1:200. At least eight dilutions (1:200, 1:400, 1:800, 1:1600, 1:3200, 1:6400, 1:12,800, 1:25,600, 1:51,200, 1:102400) were tested. Negative serum and serum dilutions were used as controls. Each concentration gradient was tested in triplicate wells with 100 μL per well. Plates were labeled accordingly and incubated at 37 °C for 1 hour. HRP-conjugated goat anti-mouse IgG, IgG1, and IgG2c antibodies, diluted 1:5000, were added and incubated at 37 °C for 45 minutes. Color development was allowed to proceed for 5 to 10 minutes. Measurements were taken within 5 minutes after stopping the reaction. OD values were recorded at 450 nm using an ELISA reader.
Lymphocyte Proliferation (LP) Assay
Spleen lymphocytes were isolated from individual mice and adjusted to a density of3–5 3-5 × 106 cells/mL. In 96-well culture plates, three groups (experimental, negative control and blank group) were prepared. Each was stimulated with 5 μg/well ESAT-6 protein. A total of 100 μL of splenic lymphocyte suspension was added to each well, typically containing 1000–3000 cells. Each group included at least three replicate wells. To minimize evaporation, 150 μL of sterile pre-cooled PBS was added around the outer wells of the 96-well plate. The cell suspension was mixed thoroughly to avoid sedimentation. Plates were incubated at 37 °C with 5% CO2 for 42 hours. The experimental groups received medium containing drug, while the blank group received drug-free medium. After incubation, 10 μL of CCK-8 reagent was added to each well, taking care to avoid bubbles that could affect OD measurements. Plates were incubated in the dark at 37 °C for 4 hours, and absorbance at 450 nm was measured using an ELISA reader. The stimulation index (SI) was calculated as follows: SI = (OD of experimental/positive stimulation well − OD of blank control well) / (OD of negative stimulation well − OD of blank control well).
Intracellular Cytokine Staining (ICS) and Flow Cytometry Analysis
Spleen lymphocyte suspensions from individual mice were seeded into 24-well plates at 1.5 mL per well and incubated with recombinant ESAT-6 protein (15 μg/mL) at 37 °C in a 5% CO2 atmosphere for 16 hours. Monensin (1 μg/mL; BioLegend, USA) was added for the final 5 hours to inhibit cytokine secretion. Cells were harvested by centrifugation, washed with cold PBS, and treated with 1 μL of TruStain FcX™ PLUS anti-mouse CD16/32 monoclonal antibody (clone S17011E, BioLegend, USA) to block non-specific binding. Fluorescent antibodies (FITC anti-mouse CD3, PE anti-mouse CD4, APC/Fire 750 anti-mouse CD8) were added to sample tubes in the dark. Blank tubes received no antibodies, and single-stained tubes received one antibody each. After fixation with FluoroFix™ buffer (BioLegend, CA, USA) and permeabilization with Perm-Wash buffer (BioLegend, CA, USA), cells were stained with APC-IFN-γ, PE/Cy7-TNF-α, and BV421-IL-2. After washing with PBS, cells were resuspended in FACS buffer and analyzed using a DxP Athena™ flow cytometer (Cytek Biosciences, CA, USA). Data were analyzed using FlowJo software.
Enzyme-Linked Immunosorbent Assay (ELISA)
The levels of various TB-related cytokines in the supernatant of cultured splenic lymphocytes were measured by ELISA. A total of 3 × 106 cells were added per well, and 15 μg of ESAT-6 protein was used to stimulate cell suspensions in four experimental groups. Plates were incubated at 37 °C in a 5% CO2 incubator for 48 hours. Supernatants were collected by centrifugation. Cytokine concentrations were measured directly from the cell culture supernatant after 48 hours of incubation using ELISA kits and are presented as picograms per milliliter (pg/mL). For all experiments, cells were plated at a standardized initial density of 4×106 cells per group to ensure comparability across conditions. Following the manufacturer’s instructions, the levels of IFN-γ, TNF-α, IL-2, IL-4, and IL-12 in the supernatants were measured using a murine ELISA kit (Dakewei Technology Co., Ltd). OD450 values were measured within 15 minutes of color development using a SpectraMax Mini microplate reader (Molecular Devices, Shanghai, China).
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA (1 × 107 cells) was extracted from the cell suspension samples extracted from mice using TRIzol reagent (Ambion, Thermo Fisher Scientific, USA). RNA concentration and purity were assessed using OD260/280 and OD260/230 ratios measured with a NanoDrop UV spectrophotometer (Thermo Fisher Scientific, USA) to ensure integrity for downstream applications. cDNA was synthesized via reverse transcription using the EasyScript® One-Step gDNA Removal and cDNA Synthesis SuperMix Kit (Transgen, Beijing, China). qPCR was performed using the PerfectStart® Green qPCR SuperMix Kit (Transgen) to assess cytokine transcription levels. Primers were synthesized by Sangon Biotech Co., Ltd. (China). Target genes and primer sequences are listed in Table S1. mRNA expression levels were calculated using the 2−ΔΔCT method.
H&E Staining
After the final immunization, tissues (heart, liver, spleen, lung, and kidney) were aseptically collected from one randomly selected mouse per group and fixed in 4% paraformaldehyde overnight. Tissues were then dehydrated in graded ethanol, embedded in paraffin, and sectioned into 5-μm slices. Sections were dewaxed, pretreated, and stained with hematoxylin and eosin, followed by dehydration and sealing. Microscopic imaging and analysis were conducted to assess histopathological changes. Sections were photographed using a digital camera (NIKON ECLIPSE E100, NIKON DS-U3, Japan) at 400× magnification. Histological evaluation of heart, liver, spleen, lung, and kidney tissues from five mice included assessments of congestion, hemorrhage, and inflammatory infiltration severity.
Statistical Analysis
Results are presented as mean ± standard deviation (SD) and visualized using GraphPad Prism 9.0. All groups were compared pairwise. Statistical differences between groups were assessed using one-way ANOVA followed by multiple comparison tests. Statistical analysis was performed using Tukey’s multiple comparison test in SPSS Version 29.0. The significance level was set at p < 0.05 in the two-tailed test. Significance levels were indicated as follows:*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Results
Expression, Purification, and Analysis of Mycobacterial Fusion Proteins
The electrophoresis results of purified ESAT-6, mi3 NPs, and ESAT-6-mi3 (Figure 1B) showed single, specific bands for all proteins. Transmission electron microscopy (TEM) demonstrated that both the individual mi3 nanoparticles and the mi3 nanoparticles fused with ESAT-6 were uniformly dispersed and exhibited consistent morphology (Figure 1C). Dynamic light scattering (DLS) was used to assess the size distribution of the nanoparticles (Figure 1D), revealing that both nanoparticle types were of uniform size. The ESAT-6-mi3 fusion nanoparticles (41.15 ± 0.17 nm) were larger than the mi3 nanoparticles alone (30.61 ± 0.11 nm).
Nanovaccine Promotes Lymphocyte Proliferation
Lymphocyte proliferation is a reliable indicator for evaluating cellular immune capacity.25 Two weeks after the final immunization, splenic lymphocytes were isolated from mice and stimulated in vitro with the ESAT-6 antigen. The results showed that, compared with the BCG group, spleen lymphocytes in the BE group exhibited significant proliferation upon antigen stimulation (Table S2). Moreover, the stimulation index (SI) of the BEM group was significantly higher than that of the traditional subunit vaccine BE group (Figure 2). It is noteworthy that the high stimulation index observed in this study likely results from the potent stimulation protocol used combined with the very low background signal of the negative controls, collectively indicating a robust specific lymphocyte response. These findings suggest that fusing mi3 nanoparticles with ESAT-6 enhances the ability of ESAT-6 to induce splenic lymphocyte proliferation. The combined formulation effectively promotes T-cell responses and induces a robust cellular immune response. In contrast, there was no significant difference in lymphocyte proliferation between the BCG and BM groups, indicating that mi3 nanoparticles alone lack immunostimulatory capability.
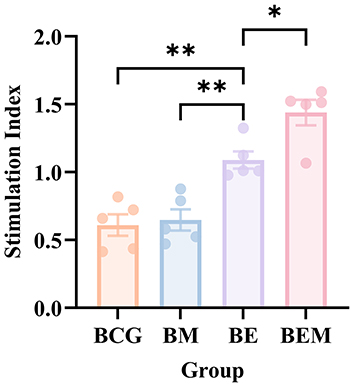 |
Figure 2 CCK-8 assay of splenocyte proliferation in different immunization groups. Splenocytes from immunized mice were stimulated with ESAT-6, and proliferation was assessed using the CCK-8 assay. The stimulation index (SI) was significantly increased in the BE and BEM groups compared to the BCG group (p < 0.01), with the BEM group showing the highest SI. Data are presented as mean ± SD, n = 5. *p < 0.05, **p < 0.01. |
Nanovaccine Induces Robust Antibody Responses in Mice
A key objective of TB vaccine development is to elicit a strong and sustained antibody response. Among antibody types, IgG, IgG1, and IgG2c serve as important indicators.26,27 The levels of these ESAT-6-specific antibodies in serum were measured by enzyme-linked immunosorbent assay (ELISA) 2 weeks after the final immunization. The geometric mean titers for the BE and BEM groups were 1:19,401 and 1:44,572, respectively. Compared with the BE group, the inclusion of mi3 nanoparticles in the BEM group significantly enhanced IgG production against ESAT-6, indicating a synergistic effect between mi3 and ESAT-6 in boosting the antibody response (Figure 3A).
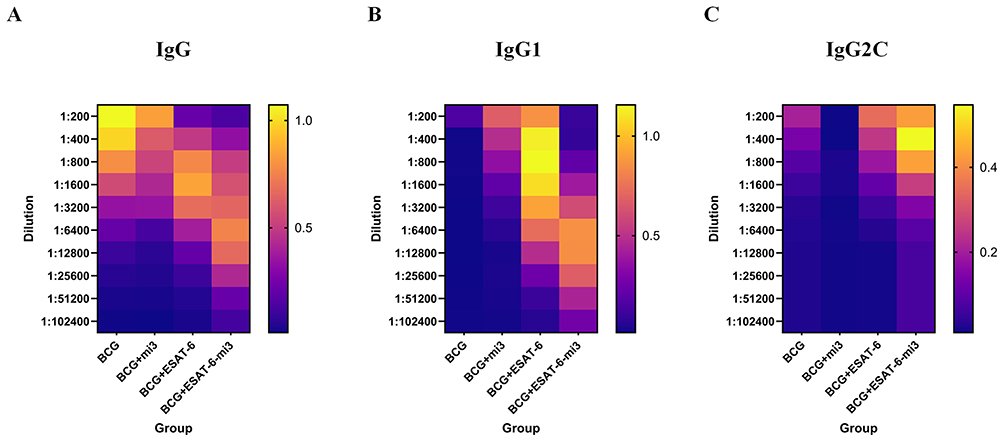 |
Figure 3 Detection of ESAT-6-specific antibodies by ELISA. Two weeks after last immunization, the IgG, IgG1, and IgG2c against ESAT-6 in serum were measured by ELISA. (A) The level of the ESAT-6-specific IgG. (B) The level of the ESAT-6-specific IgG1. (C) The level of the ESAT-6-specific IgG2c. Data were expressed as means ± SD. |
To further explore immune response types, IgG1 and IgG2c subtypes were analyzed. The BE group showed higher levels than the BCG and BM groups. The BEM group exhibited even higher IgG1 levels than the BE group (Figure 3B and C). Compared with the control, the combination of ESAT-6 and mi3 significantly increased IgG1-specific antibody titers, with the BEM group reaching the highest geometric mean titer (GMT = 1:31,945), far exceeding the BCG group (GMT = 1:280). The BEM group maintained high antibody titers across all dilutions, with IgG2c titers being the most elevated, indicating the strongest immune response. A heatmap was used to visually represent these results. Overall, BEM nanofusion vaccines elicited a significantly stronger IgG response than the traditional BE vaccine.
Analysis of CD4+ and CD8+ Specific Memory T Cell (TM)
To evaluate cellular immune responses across vaccine groups,28,29 flow cytometry was used to measure intracellular expression of TNF-α, IFN-γ, and interleukin (IL)-2 in CD4⁺ and CD8⁺ T cells following ESAT-6 stimulation. In CD4⁺ T cells, the BE group showed significantly higher cytokine expression than the BCG and BM groups but lower than the BEM group. The BEM group induced significantly higher proportions of TNF-α⁺, IFN-γ⁺, and IL-2⁺ cells than all the other groups (Figure 4A–F). A similar trend was observed in CD8⁺ T cells, with the BEM group showing the strongest cytokine responses, followed by the BE group, while the BCG and BM groups exhibited low and statistically insignificant responses (Figure 5A–F). These findings suggest that the BEM vaccine formulation more effectively induces moderate inflammation and promotes stronger cell-mediated immune responses than the ESAT-6 antigen alone.
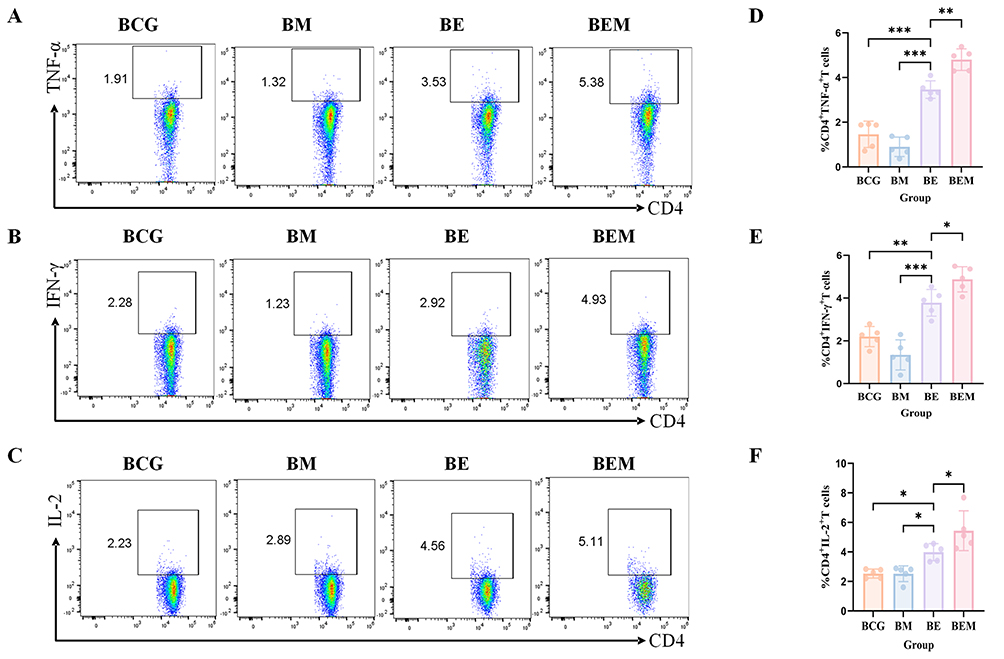 |
Figure 4 Frequencies of CD4+ T cells producing antigen-specific cytokines in mice. Two weeks after the last immunization, the mouse spleen lymphocytes were isolated and stimulated with antigen ESAT-6 in vitro. Intracellular production of cytokines was analyzed using flow cytometry. (A–C) The frequency of CD4+ T cells secreting TNF-α, IFN-γ and IL-2 was analyzed by flow cytometry. (D–F) Statistical analysis of the proportion of TNF-α, IFN-γ and IL-2-producing CD4+ T cells. At least three mice per group. Mean ± SD, *p <0.05; **p < 0.01; and ***p < 0.001. |
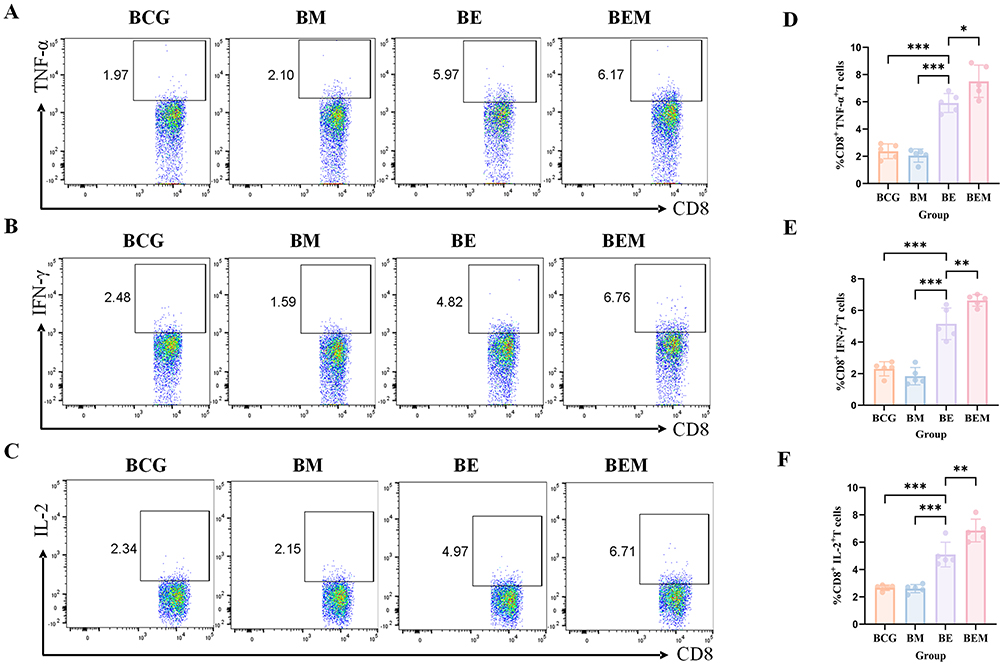 |
Figure 5 Frequencies of CD8+ T cells producing antigen-specific cytokines in mice. Two weeks after the last immunization, the mouse spleen lymphocytes were isolated and stimulated with antigen ESAT-6 in vitro. Intracellular production of cytokines was analyzed using flow cytometry. (A–C) The frequency of CD8+ T cells secreting TNF-α, IFN-γ and IL-2 was analyzed by flow cytometry. (D–F) Statistical analysis of the proportion of TNF-α, IFN-γ and IL-2-producing CD8+ T cells. At least three mice per group. Mean ± SD, *p <0.05; **p < 0.01; and ***p < 0.001. |
TB-Associated Cytokine Measurement
Cytokines play crucial roles in early infection defense and T-cell regulation.30–33 ELISA was used to quantify IFN-γ, TNF-α, IL-2, IL-12, and IL-17 in the culture supernatant. The BE group mice exhibited higher IFN-γ, TNF-α, and IL-2 secretion than the BCG and BM groups, suggesting that ESAT-6 enhances immune activation (Figure 6A–C). Notably, the BEM group showed significantly elevated levels of IFN-γ, TNF-α, and IL-2 compared with all other groups, particularly in TNF-α and IL-2, indicating potent activation of inflammation and T-cell proliferation. IL-12 secretion was also significantly elevated in the BEM group, further supporting enhanced cellular immunity (Figure 6D). Only the BEM group showed a significant increase in IL-17 secretion; the other three groups showed no such elevation (Figure 6E).
 |
Figure 6 Cytokine secretion levels in splenocyte cultures measured by ELISA after immunization. Splenocytes were isolated from mice immunized with BCG, BM, BE, or BEM. After in vitro stimulation, supernatants were collected and analyzed by ELISA for the secretion of TNF-α (A), IFN-γ (B), IL-2 (C), IL-12 (D), and IL-17 (E). Data are presented as mean ± SD. Statistical significance was determined by one-way ANOVA followed by Tukey′s post-hoc test. P < 0.05 (*), P < 0.01 (**), P < 0.001 (***). |
To validate cytokine expression at the transcriptional level, real-time quantitative PCR (qPCR) was conducted for TNF-α, IFN-γ, and IL-17 in splenic cells. The BEM group exhibited the highest mRNA expression levels for all three cytokines. TNF-α and IL-17 mRNA levels were higher in both the BEM and BE groups than in the BM and BCG groups, indicating that ESAT-6 activates Th17-associated gene expression, even though IL-17 secretion was not significantly increased in the BE group (Figure 7A–C). IFN-γ mRNA levels were significantly increased only in the BEM group, suggesting that ESAT-6 fusion with mi3 enhances transcription of this cytokine more effectively than ESAT-6 alone (Figure 7B).
 |
Figure 7 Relative mRNA expression levels of pro-inflammatory cytokines TNF-α, IFN-γ, and IL-17 in spleen cells after immunization. Spleens were harvested, and total RNA was extracted for quantitative RT-PCR analysis. The expression levels of TNF-α (A), IFN-γ (B), and IL-17 (C) mRNA were normalized to housekeeping genes and are presented as relative fold changes compared to the BCG group. Data are shown as mean ± SD. Statistical analysis was performed using one-way ANOVA with Tukey′s post-hoc test. P < 0.05 (*), P < 0.01 (**), P < 0.001 (***). |
These findings indicate that the BCG group alone induces IFN-γ, TNF-α, IL-2, IL-12, and IL-17 secretion. The BE group enhances cytokine secretion due to the ESAT-6 antigen, while the BEM group, via the synergistic effect of mi3 nanoparticles, further amplifies cellular immunity, particularly Th17-mediated responses.
In vivo Safety Evaluation
Two weeks after the final immunization, major immune and metabolic organs (heart, liver, spleen, lungs, kidneys) were collected from euthanized mice, stained with H&E, and analyzed histologically.34 All experimental mice remained healthy and gained weight, suggesting no long-term systemic toxicity in any group. These findings confirm the biological safety of the nanovaccine (Figure 8). No visible organ damage was observed in either the BCG or BEM groups. Cell nuclei appeared blue-purple, with morphology consistent with tissue function—for example, hepatocyte nuclei were large and round, and myocardial nuclei were centrally located. Cytoplasm was mainly eosinophilic (pink). All organs displayed intact structures with no necrosis, abnormal hyperplasia, or inflammatory infiltration (eg, no neutrophil or lymphocyte aggregation). Cardiac muscle fibers appeared short, columnar, and branched with centrally located oval nuclei. Cross-sections showed circular or polygonal outlines, and longitudinal sections revealed alternating light and dark striations. The interstitium contained minimal connective tissue and capillaries, without fibrosis or immune infiltration. Liver histology showed clearly defined hepatic lobules, central veins, and radially arranged hepatic cords. Hepatocytes were polygonal with abundant cytoplasm and central nuclei. In the portal areas, the portal vein, hepatic artery, and bile duct (forming the classic triad) were visible, with cuboidal epithelial cells lining the bile ducts. Spleen tissue displayed normal red pulp architecture with splenic cords rich in red blood cells and macrophages, and irregular splenic sinuses lined by elongated endothelial cells. Lung bronchi exhibited normal pseudostratified ciliated columnar epithelium with intact cilia, scattered goblet cells, and cartilage rings. Bronchial cytoplasm was lightly stained with visible cilia. Renal glomeruli appeared lobulated, composed of capillary plexuses. The histopathological evaluation demonstrated a favorable short-term safety profile for the nanoplatform, with no significant pathological changes observed in the heart or kidneys. However, findings of moderate-to-severe hepatic edema and mild inflammatory infiltration in the spleen and lungs indicate organ-specific responses, likely associated with nanoparticle accumulation and immune recognition. These results underscore the need for further investigation into the long-term biodistribution and chronic toxicity of the material (Table S3). These histological results further confirm the safety and tolerability of the nanoparticle vaccine.
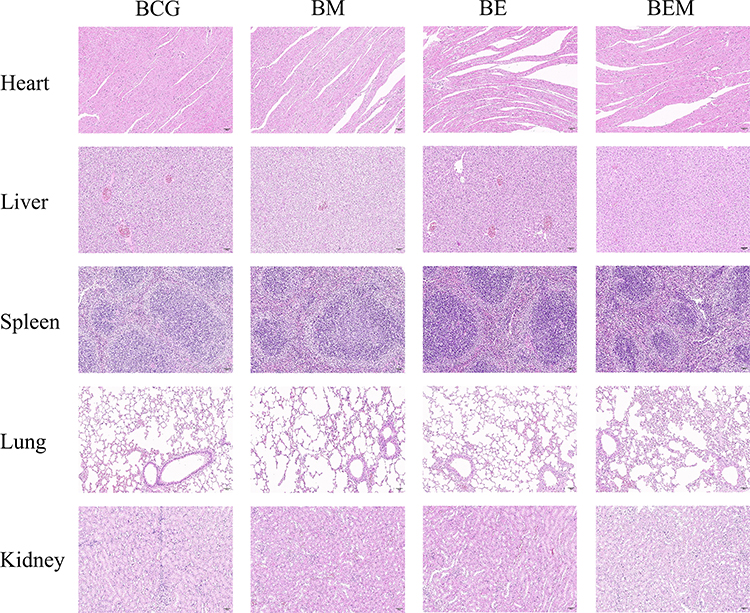 |
Figure 8 The safety analysis of the self-assembled nanoparticles vaccine after immunization. At the 6th week, the mice were sacrificed. The heart, liver, spleen, lung and kidney samples were collected and performed with H&E staining. The typical pathological changes in each group were shown (scar bar, 50 μm). |
Discussion
BCG is currently the only vaccine approved by the WHO for the prevention of TB. However, its protective efficacy remains controversial. In countries such as the United States, New Zealand, and Belgium, BCG has been excluded from routine immunization programs.5,6,35 Despite various interventions, the development of more effective TB vaccines remains a critical goal in global TB control efforts. As candidate vaccines advance through clinical trials, there is growing consensus on the need to diversify vaccine strategies to increase the likelihood of achieving broad and effective protection.36–40
In recent years, advancements in carrier system design have enabled the development of more effective vaccine platforms. Nanoparticle-based vaccines enhance immune responses by encapsulating antigens, adjuvants, or both within nanoscale carriers.41 Compared with traditional vaccines, nanoparticle vaccines offer several advantages, including improved stability, controlled release, increased immunogenicity, and targeted delivery, making them prominent candidates for next-generation vaccines.42,43 This versatile design allows for modulation of the immune response and offers the potential to restore immune protection where conventional vaccines fail. Among these, engineered mi3 nanoparticles have gained attention for their excellent immunogenicity and biocompatibility. These particles can self-assemble and present antigens repetitively and in an ordered fashion, thereby efficiently displaying epitopes and robustly inducing immune responses. Their applications span across bioimaging, drug delivery, vaccination, oncology, diagnostics, enzymology, and other biomedical fields.44–48
Specific antibodies, a key component of humoral immunity, are crucial for evaluating the immunogenicity of vaccines.49 Serum analysis in immunized mice revealed that the BEM group produced significantly higher levels of total and neutralizing antibodies than either BCG or ESAT-6 antigen alone. Further subtype analysis showed significantly increased IgG1 (associated with Th2 responses) and particularly strong IgG2c responses (indicative of Th1 immunity). Particularly, IgG2c maintained a relatively high OD value at high dilution concentrations, with the highest titer and strongest persistence, suggesting that this vaccine can induce a long-lasting and potent Th1-type humoral immune response. Meanwhile, the simultaneous upregulation of IgG1 and IgG2c and the stable trend in 10 consecutive dilution gradients further confirm that the vaccine with fused expression of ESAT-6-mi3 nanoparticles has the ability to rapidly induce high-level specific humoral immunity. Studies on the use of nanoparticles to encapsulate the Ag85B-ESAT-6 (H1) antigen also indicated that the fusion vaccine could enhance the expression of IgG2c while maintaining a certain level of IgG1.50 Similarly, vaccines using the fusion expression of Ag85B, Rv2608 and ESAT-6 also demonstrated the ability to simultaneously activate IgG1 and IgG2c.51 These findings all indicate that the nanoparticle vaccine with the fusion expression of ESAT-6-mi3 successfully induces a high level of humoral immune response in the body and shows potential in enhancing humoral and cellular immune responses.
The most effective vaccines typically stimulate both cellular and humoral immune responses in the body.52,53 In this study, we isolated spleens to analyze the proliferation of lymphocytes. Previously, Thomas et al immunized mice with the Ag85B-ESAT-6 fusion protein and reported significantly enhanced splenic lymphocyte proliferation levels.54 In addition, the HSP90-ESAT-6 fusion protein vaccine constructed by Kim et al also reported effectively enhanced proliferative activity of spleen cells.55 Similarly, in our results, compared with the BCG and BM groups, the BE group showed a more significant enhancement in inducing lymphocyte proliferation, which proved that the recombinant antigen protein in the RD region can improve the immunogenicity of BCG through an enhanced immune strategy. Compared with all the control groups, the BEM group had the highest intensity of lymphocyte proliferation induced in the splenic lymphocyte proliferation experiment. This indicates that the nanovaccine, by fusing and expressing immune enhancement antigens with the nanoparticle platform, not only improves antigen recognition by immune cells but also accelerates the development of adaptive immune responses in the spleen, emphasizing its advantages in activating cellular immunity.
The adaptive immune response includes both humoral and T-cell-mediated components, the latter of which is critical for eliminating infected or damaged cells and controlling intracellular pathogens.56,57 Prior studies using nanodelivery systems such as PLGA or liposomes to deliver ESAT-6 have shown upregulation of cytokines like TNF-α and IL-2.50,58 Similarly, the combination of S100A4 adjuvant with ESAT-6 elicited high expression of IFN-γ, TNF-α, and IL-17 in the lungs, indicating a Th1/Th17 synergistic immune response.59 In our study, we evaluated the cellular immunogenicity of the fusion vaccine by measuring cytokines including TNF-α, IFN-γ, IL-2, IL-12, and IL-17. Compared with BCG and other controls, the BEM group showed significantly elevated levels of these pro-inflammatory and Th1/Th17-associated cytokines, particularly TNF-α and IFN-γ, indicating a strong cellular immune response. Increased IL-2 and IL-12 levels further suggest effective T-cell proliferation and differentiation, supporting long-term immune memory. Our analysis of the mRNA and protein data revealed both concordant and discordant expression patterns among the cytokines examined. While TNF-α exhibited consistent upregulation at both transcriptional and translational levels, IFN-γ and IL-17 displayed a marked dissociation: IL-17 mRNA was significantly elevated without a proportional increase in protein secretion, whereas IFN-γ protein levels substantially exceeded its mRNA expression. These observations suggest distinct layers of post-transcriptional regulation. The attenuated translation of IL-17 may be attributable to microRNA-mediated suppression, reduced mRNA stability, or impaired secretion mechanisms.60 Conversely, the pronounced IFN-γ protein output likely reflects positive feedback amplification via the JAK-STAT pathway, alongside differential mRNA turnover and extracellular accumulation.61 These findings underscore the sophisticated, multi-tiered regulatory mechanisms shaping cytokine expression in the BEM-induced immune microenvironment.
Elevated IL-17 levels reflect the activation of the Th17 immune pathway, which may play a synergistic role in controlling intracellular pathogen infections and inflammatory responses.62,63 Collectively, these cytokine results demonstrate that this nanovaccine can synergistically stimulate the Th1/Th17 type immune pathway, significantly enhancing the cellular immune response. Not only is it superior to the traditional BCG vaccine in intensity but also it achieves a broader spectrum of immune regulation in response types, which is conducive to forming a more comprehensive cellular immune defense line, thereby providing an important basis for the continuous protective effect of the vaccine. In our study, the IL-17 level improvement suggests that the immune response we observed may encompass not only protective elements but also potential immunopathological risks. Future studies employing in vivo neutralization experiments or using genetically deficient models would be necessary to directly dissect the functional contribution of IL-17 in our specific model.
In this study, due to the fact that the nanoparticles carried only a single antigen, the induced immune response may be limited and insufficient to elicit broad protection against diverse TB strains.64 In contrast, multi-antigen nanoparticle vaccines can provide a richer array of epitopes, enhancing B- and T-cell cross-activation and eliciting stronger and more comprehensive immune responses. Although our data demonstrate strong short-term immunogenicity without acute toxicity, they cannot preclude long-term immunological risks. Such risks may arise from nanoparticle accumulation in RES organs, potentially leading to persistent low-level immune activation and disrupted homeostasis. Additionally, while our data demonstrate strong humoral and cellular immunogenicity, protective efficacy following pathogen challenge has not yet been evaluated.65 Further studies involving systematic challenge experiments in relevant animal models are required to assess real-world protective capacity and advance preclinical development. The true protective potential of this vaccine therefore remains to be fully clarified.
In summary, in this study, a novel TB vaccine based on the delivery of ESAT-6 antigen by mi3 nanoparticles was successfully constructed and evaluated. By fusing the immunodominant antigen ESAT-6 with the self-assembled mi3 nanoparticle platform and using it in combination with BCG immunization, the fusion vaccine significantly enhanced both humoral and cellular immune responses. It particularly promoted IgG2c-dominated Th1-type immunity and efficiently activated CD4⁺ and CD8⁺ T cells, with elevated production of IFN-γ, IL-2, and other key cytokines, reflecting a Th1/Th17-biased response. The vaccine demonstrated favorable stability and safety in animal models and outperformed BCG or traditional subunit vaccines in immunogenicity. The finding of this study that mi3 significantly enhances the immunogenicity of ESAT-6 provides a strong rationale for future evaluation of its protective efficacy in animal challenge models. These findings highlight its potential as a booster or alternative to BCG and lay a solid foundation for further preclinical development. Ultimately, this strategy could contribute to more effective TB prevention and enrich the technological arsenal of anti-infective nanocarriers.
Conclusions
In this study, a self-assembled nanoparticle vaccine was developed by fusing the ESAT-6 antigen with the mi3 nanoparticle platform, primarily aimed at enhancing the immune effects of the existing BCG vaccine. The results demonstrated significantly improved humoral and cellular immune responses, confirming strong immunogenicity and safety. These findings provide novel technical insights and an experimental foundation for the design of next-generation TB vaccines.
Ethics Approval
All animal procedures were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee at Bengbu Medical University (approval no. 2024502).
Acknowledgments
The authors thank all individuals who participated in this work.
Funding
This work was supported by the Key Projects of Natural Science Foundation of Bengbu Medical University.This work was supported by Natural Science Foundation of the Anhui Higher Education Institutions (2024AH051295), Bengbu City science and technology project (2023hm01), Key Projects of Natural Science Foundation of Bengbu Medical University (2023byzd020, 2024byzd045), Postgraduate Scientific Research Innovation Program of Bengbu Medical University (Byycxz24007).
Disclosure
The author(s) report no conflicts of interest in this work.
References
1. Organization WH. Global Tuberculosis Report 2024. Geneva: WHO. 2024. Available from: https://www.who.int/teams/global-tuberculosis-programme/tb-reports.
2. Organization WH. WHO consolidated guidelines on tuberculosis: module 4: treatment and care. 2025. Available from: https://www.who.int/publications/i/item/9789240107243.
3. Gandhi NR, Nunn P, Dheda K, et al. Multidrug-resistant and extensively drug-resistant tuberculosis: a threat to global control of tuberculosis. Lancet. 2010;375(9728):1830–16. doi:10.1016/s0140-6736(10)60410-2
4. Pawlowski A, Jansson M, Sköld M, Rottenberg ME, Källenius G. Tuberculosis and HIV co-infection. PLoS Pathog. 2012;8(2):e1002464. doi:10.1371/journal.ppat.1002464
5. Colditz GA, Brewer TF, Berkey CS, et al. Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of the published literature. JAMA. 1994;271(9):698–702. doi:10.1001/jama.1994.03510330076038
6. Mangtani P, Abubakar I, Ariti C, et al. Protection by BCG vaccine against tuberculosis: a systematic review of randomized controlled trials. Clin Infect Dis. 2014;58(4):470–480. doi:10.1093/cid/cit790
7. World Health Organization. The End TB Strategy. 2015. Available from: https://www.who.int/teams/global-programme-on-tuberculosis-and-lung-health/the-end-tb-strategy.
8. Zwerling A, Behr MA, Verma A, Brewer TF, Menzies D, Pai M. The BCG world atlas: a database of global BCG vaccination policies and practices. PLoS Med. 2011;8(3):e1001012. doi:10.1371/journal.pmed.1001012
9. World Health Organization. BCG vaccines: WHO position paper. 2018. Available from: https://www.who.int/publications/i/item/who-wer9308-73-96.
10. Brosch R, Gordon SV, Garnier T, et al. Genome plasticity of BCG and impact on vaccine efficacy. Proc Natl Acad Sci USA. 2007;104(13):5596–5601. doi:10.1073/pnas.0700869104
11. Pym AS, Brodin P, Brosch R, Huerre M, Cole ST. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol. 2002;46(3):709–717. doi:10.1046/j.1365-2958.2002.03237.x
12. Grode L, Seiler P, Baumann S, et al. Increased vaccine efficacy against tuberculosis of recombinant Mycobacterium bovis bacille Calmette-Guérin mutants that secrete listeriolysin. J Clin Invest. 2005;115(9):2472–2479. doi:10.1172/jci24617
13. Dijkman K, Sombroek CC, Vervenne RAW, et al. Prevention of tuberculosis infection and disease by local BCG in repeatedly exposed rhesus macaques. Nat Med. 2019;25(2):255–262. doi:10.1038/s41591-018-0319-9
14. Hsia Y, Bale JB, Gonen S, et al. Design of a hyperstable 60-subunit protein dodecahedron. [corrected]. Nature. 2016;535(7610):136–139. doi:10.1038/nature18010
15. Bruun TUJ, Andersson AC, Draper SJ, Howarth M. Engineering a rugged nanoscaffold to enhance plug-and-display vaccination. ACS Nano. 2018;12(9):8855–8866. doi:10.1021/acsnano.8b02805
16. Bale JB, Gonen S, Liu Y, et al. Accurate design of megadalton-scale two-component icosahedral protein complexes. Science. 2016;353(6297):389–394. doi:10.1126/science.aaf8818
17. Cohen AA, van Doremalen N, Greaney AJ, et al. Mosaic RBD nanoparticles protect against challenge by diverse sarbecoviruses in animal models. Science. 2022;377(6606):eabq0839. doi:10.1126/science.abq0839
18. Cohen AA, Yang Z, Gnanapragasam PNP, et al. Construction, characterization, and immunization of nanoparticles that display a diverse array of influenza HA trimers. PLoS One. 2021;16(3):e0247963. doi:10.1371/journal.pone.0247963
19. Eom S, Jun H, Kim E, Min D, Kim H, Kang S. Developing porous protein cage nanoparticles as cargo-loadable and ligand-displayable modular delivery nanoplatforms. ACS Appl Mater Interfaces. 2024;16(43):58464–58476. doi:10.1021/acsami.4c14505
20. Hartzell EJ, Lieser RM, Sullivan MO, Chen W. Modular hepatitis B virus-like particle platform for biosensing and drug delivery. ACS Nano. 2020;14(10):12642–12651. doi:10.1021/acsnano.9b08756
21. Kang YF, Sun C, Zhuang Z, et al. Rapid development of SARS-CoV-2 spike protein receptor-binding domain self-assembled nanoparticle vaccine candidates. ACS Nano. 2021;15(2):2738–2752. doi:10.1021/acsnano.0c08379
22. Ueda G, Antanasijevic A, Fallas JA, et al. Tailored design of protein nanoparticle scaffolds for multivalent presentation of viral glycoprotein antigens. Elife. 2020;9. doi:10.7554/eLife.57659
23. Kanekiyo M, Wei CJ, Yassine HM, et al. Self-assembling influenza nanoparticle vaccines elicit broadly neutralizing H1N1 antibodies. Nature. 2013;499(7456):102–106. doi:10.1038/nature12202
24. Tait DR, Hatherill M, Van Der Meeren O, et al. Final analysis of a trial of M72/AS01(E) vaccine to prevent tuberculosis. N Engl J Med. 2019;381(25):2429–2439. doi:10.1056/NEJMoa1909953
25. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. doi:10.1016/0022-1759(83)90303-4
26. Lu LL, Chung AW, Rosebrock TR, et al. A functional role for antibodies in tuberculosis. Cell. 2016;167(2):433–43.e14. doi:10.1016/j.cell.2016.08.072
27. Stavnezer J. Immunoglobulin class switching. Curr Opin Immunol. 1996;8(2):199–205. doi:10.1016/s0952-7915(96)80058-6
28. Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol. 2008;8(4):247–258. doi:10.1038/nri2274
29. Darrah PA, Patel DT, De Luca PM, et al. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med. 2007;13(7):843–850. doi:10.1038/nm1592
30. Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454(7203):428–435. doi:10.1038/nature07201
31. Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112(5):1557–1569. doi:10.1182/blood-2008-05-078154
32. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol. 2015;15(2):87–103. doi:10.1038/nri3787
33. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775–787. doi:10.1016/j.cell.2008.05.009
34. Dobrovolskaia MA, Aggarwal P, Hall JB, McNeil SE. Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol Pharm. 2008;5(4):487–495. doi:10.1021/mp800032f
35. Kaufmann SH. Novel tuberculosis vaccination strategies based on understanding the immune response. J Intern Med. 2010;267(4):337–353. doi:10.1111/j.1365-2796.2010.02216.x
36. Romanus V. Swedish experiences 12 years after the cessation of general BCG vaccination of newborns in 1975. Bull Int Union Tuberc Lung Dis. 1988;63(4):34–38.
37. Nemes E, Geldenhuys H, Rozot V, et al. Prevention of M. tuberculosis Infection with H4:IC31 Vaccine or BCG Revaccination. N Engl J Med. 2018;379(2):138–149. doi:10.1056/NEJMoa1714021
38. Nieuwenhuizen NE, Kulkarni PS, Shaligram U, et al. The recombinant bacille calmette-guérin vaccine VPM1002: ready for clinical efficacy testing. Front Immunol. 2017;8:1147. doi:10.3389/fimmu.2017.01147
39. Lacámara S, Martin C. MTBVAC: a tuberculosis vaccine candidate advancing towards clinical efficacy trials in TB prevention. Arch Bronconeumol. 2023;59(12):821–828. doi:10.1016/j.arbres.2023.09.009
40. Gonzalo-Asensio J, Marinova D, Martin C, Aguilo N. MTBVAC: attenuating the human pathogen of tuberculosis (TB) toward a promising vaccine against the TB epidemic. Front Immunol. 2017;8:1803. doi:10.3389/fimmu.2017.01803
41. Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol. 2010;10(11):787–796. doi:10.1038/nri2868
42. Reddy ST, van der Vlies AJ, Simeoni E, et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat Biotechnol. 2007;25(10):1159–1164. doi:10.1038/nbt1332
43. Pulendran B, Ahmed R. Immunological mechanisms of vaccination. Nat Immunol. 2011;12(6):509–517. doi:10.1038/ni.2039
44. Zhao L, Seth A, Wibowo N, et al. Nanoparticle vaccines. Vaccine. 2014;32(3):327–337. doi:10.1016/j.vaccine.2013.11.069
45. Zegeye ED, Diaz Y, Puntervoll P. Vaccine candidate double mutant variants of enterotoxigenic escherichia coli heat-stable toxin. Vaccines. 2022;10(2). doi:10.3390/vaccines10020241
46. Thavorasak T, Chulanetra M, Glab-Ampai K, et al. Novel neutralizing epitope of PEDV S1 protein identified by IgM monoclonal antibody. Viruses. 2022;14(1):125. doi:10.3390/v14010125
47. Tan TK, Rijal P, Rahikainen R, et al. A COVID-19 vaccine candidate using SpyCatcher multimerization of the SARS-CoV-2 spike protein receptor-binding domain induces potent neutralising antibody responses. Nat Commun. 2021;12(1):542. doi:10.1038/s41467-020-20654-7
48. Singha S, Shao K, Ellestad KK, Yang Y, Santamaria P. Nanoparticles for immune stimulation against infection, cancer, and autoimmunity. ACS Nano. 2018;12(11):10621–10635. doi:10.1021/acsnano.8b05950
49. Plotkin SA. Correlates of protection induced by vaccination. Clin Vaccine Immunol. 2010;17(7):1055–1065. doi:10.1128/cvi.00131-10
50. Malik A, Gupta M, Mani R, Bhatnagar R. Single-dose Ag85B-ESAT6-loaded poly(lactic-co-glycolic acid) nanoparticles confer protective immunity against tuberculosis. Int J Nanomed. 2019;14:3129–3143. doi:10.2147/ijn.S172391
51. Lu Y, Xu Y, Yang E, et al. Novel recombinant BCG coexpressing Ag85B, ESAT-6 and Rv2608 elicits significantly enhanced cellular immune and antibody responses in C57BL/6 mice. Scand J Immunol. 2012;76(3):271–277. doi:10.1111/j.1365-3083.2012.02726.x
52. Kaufmann SH. Protection against tuberculosis: cytokines, T cells, and macrophages. Ann Rheum Dis. 2002;61(Suppl 2):ii54–8. doi:10.1136/ard.61.suppl_2.ii54
53. Divangahi M, Aaby P, Khader SA, et al. Trained immunity, tolerance, priming and differentiation: distinct immunological processes. Nat Immunol. 2021;22(1):2–6. doi:10.1038/s41590-020-00845-6
54. Lindenstrøm T, Agger EM, Korsholm KS, et al. Tuberculosis subunit vaccination provides long-term protective immunity characterized by multifunctional CD4 memory T cells. J Immunol. 2009;182(12):8047–8055. doi:10.4049/jimmunol.0801592
55. Kwon KW, Choi HG, Kim KS, Park SA, Kim HJ, Shin SJ. BCG-booster vaccination with HSP90-ESAT-6-HspX-RipA multivalent subunit vaccine confers durable protection against hypervirulent Mtb in mice. NPJ Vaccines. 2024;9(1):55. doi:10.1038/s41541-024-00847-7
56. Chi H, Pepper M, Thomas PG. Principles and therapeutic applications of adaptive immunity. Cell. 2024;187(9):2052–2078. doi:10.1016/j.cell.2024.03.037
57. Sun L, Su Y, Jiao A, Wang X, Zhang B. T cells in health and disease. Signal Transduct Target Ther. 2023;8(1):235. doi:10.1038/s41392-023-01471-y
58. Diogo GR, Hart P, Copland A, et al. Immunization with Mycobacterium tuberculosis antigens encapsulated in phosphatidylserine liposomes improves protection afforded by BCG. Front Immunol. 2019;10:1349. doi:10.3389/fimmu.2019.01349
59. Abil OZ, Liu S, Yeh YW, et al. A mucosal vaccine formulation against tuberculosis by exploiting the adjuvant activity of S100A4-A damage-associated molecular pattern molecule. Vaccine. 2024;42(25):126151. doi:10.1016/j.vaccine.2024.07.052
60. Harding H P, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M and Ron D. (2000). Regulated Translation Initiation Controls Stress-Induced Gene Expression in Mammalian Cells. Molecular Cell, 6(5), 1099–1108. 10.1016/S1097-2765(00)00108-8
61. Stark G and Darnell J. (2012). The JAK-STAT Pathway at Twenty. Immunity, 36(4), 503–514. 10.1016/j.immuni.2012.03.013
62. Li Y, Wei C, Xu H, et al. The immunoregulation of Th17 in host against intracellular bacterial infection. Mediators Inflamm. 2018;2018:6587296. doi:10.1155/2018/6587296
63. Lin Y, Slight SR, Khader SA. Th17 cytokines and vaccine-induced immunity. Semin Immunopathol. 2010;32(1):79–90. doi:10.1007/s00281-009-0191-2
64. Song X, Li Y, Wu H, Qiu H, Sun Y. T-cell epitope-based vaccines: a promising strategy for prevention of infectious diseases. Vaccines. 2024;12(10). doi:10.3390/vaccines12101181
65. Chugh S, Bahal RK, Dhiman R, Singh R. Antigen identification strategies and preclinical evaluation models for advancing tuberculosis vaccine development. NPJ Vaccines. 2024;9(1):57. doi:10.1038/s41541-024-00834-y
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.