Back to Journals » Therapeutics and Clinical Risk Management » Volume 12
IgG4-related disease: current challenges and future prospects
Authors Lang D ![]() , Zwerina J, Pieringer H
, Zwerina J, Pieringer H
Received 6 November 2015
Accepted for publication 19 December 2015
Published 15 February 2016 Volume 2016:12 Pages 189—199
DOI https://doi.org/10.2147/TCRM.S99985
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Garry Walsh
David Lang,1 Jochen Zwerina,2 Herwig Pieringer1,3
1Second Department of Internal Medicine, Kepler University Clinic, Linz, 2First Department of Internal Medicine, Ludwig Boltzmann-Institute of Osteology, Hanusch Hospital, Vienna, 3Paracelsus Private Medical University, Salzburg, Austria
Abstract: Immunoglobulin G4-related disease (IgG4-RD) represents an immune-mediated fibroinflammatory condition with a characteristic histopathological appearance that can affect various organs. Although numerous single-organ manifestations have been described more than a century ago, its systemic nature and unique features were only discovered in the last 2 decades, when IgG4-RD emerged as a new entity of disease. IgG4-RD is usually considered a rare disease, but its true epidemiology has not yet been fully clarified. Also, despite recent advances in the identification of the underlying immunological processes, its pathophysiology is only incompletely understood till now. The diagnostic workup of IgG4-RD is complex and usually requires a combination of clinical examination, imaging, histological, and serological analyses. However, no finding alone is specific for IgG4-RD. Therefore, its diagnosis requires careful interpretation of examination results in context with the patient’s clinical appearance as well as the exclusion of a broad variety of differential diagnoses. The past years brought rapid advances concerning this novel disease entity: diagnostic criteria, further insights into the underlying immunological processes, new biomarkers, and novel therapeutic approaches were proposed and widened the knowledge in the field of IgG4-RD. Still, a greater number of questions remain unanswered, and many recent developments require further discussion and proof from clinical trials. This review should give an overview on current knowledge and future perspectives in epidemiology, pathophysiology, diagnosis, and therapy of IgG4-RD.
Keywords: PET/CT, autoimmune pancreatitis, plasmablast, rituximab
Introduction
Immunoglobulin G4-related disease (IgG4-RD) is a condition characterized by an immune-mediated fibroinflammatory process with a tendency to form tumefactive lesions in various organs, occurring in a synchronous or metachronous fashion.1,2 The most frequent localizations include the pancreas and salivary glands. Other common manifestations are tubulointerstitial nephritis, dacryoadenitis, and periaortitis.3
Various single-organ manifestations of IgG4-RD have already been established in the 19th century, when histopathological tissue examination emerged. Those eponymous syndromes, such as Mikulicz’s disease, Riedel’s thyroiditis, Morbus Ormond, or Küttner’s tumor, were believed to be rare, isolated disease entities.2 The first step toward the discovery of IgG4-RD was the description of an autoimmune mediated, steroid responsive form of pancreatitis, today known as IgG4-related disease or type 1 autoimmune pancreatitis (AIP) in 1995.4 Subsequently, in 2001, Hamano et al reported an elevation of serum IgG4 levels in patients with AIP5 and described also the characteristic histopathological pattern in concomitant retroperitoneal fibrosis (RPF),6 thus paving the way for the recognition of the truly multiorgan nature of IgG4-RD.7 In 2012, a unified nomenclature of IgG4-RD was published, abandoning all other synonymous names for IgG4-RD and its organ manifestations.8 In the same year, Japanese groups introduced comprehensive diagnostic criteria for IgG4-RD, and an international consensus on the pathology of IgG4-RD was reached.9,10
IgG4-RD represents a very young entity of disease, having emerged within only 1 decade. Although it is generally thought to be a rare condition, it is likely that with growing awareness and recent advances in research, the number of patients diagnosed with IgG4-RD will increase in the future. Despite rapid progress within the last years, there are still a greater number of challenges and open questions regarding epidemiology, pathophysiology, diagnosis, and treatment of IgG4-RD. Those including the recent developments and future perspectives are discussed in this article.
Epidemiology
Prevalence, incidence, age, and sex
Up to now, only little is known about the epidemiology of IgG4-RD and its organ manifestations. It is still unclear whether Asian patients are more susceptible to IgG4-RD than others; hence, the available epidemiologic data, mainly derived from Japanese cohorts, must be interpreted very carefully.11 Uchida et al estimated the annual incidence of IgG4-RD at 0.28–1.08/100,000. For the year 2009, a total of 8,000 patients were expected in Japan, which accounted for a prevalence of ~62 patients per million inhabitants.12
IgG4-RD usually affects individuals of middle to upper age, with an onset at 50–70 years,3,11,13 although rare pediatric cases have been described.14 Most studies report an overall predilection for the male sex,3,12,13 especially for IgG4-related pancreatitis with an M:F ratio of 3:7.15 However, IgG4-related sialadenitis and dacryoadenitis may occur more frequently among females.3
Organ distribution
Traditionally, IgG4-related pancreatitis is regarded as the most frequent manifestation of IgG4-RD,3,16 although organ distribution varies between the studies and is influenced by hospital and medical specialty.13 In a cohort of 235 Japanese patients with IgG4-RD, 60% presented with pancreatitis, further 34% with sialadenitis, 23% with tubulointerstitial nephritis, 23% with dacryoadenitis, and 20% with periaortitis. In total, 58% had multiple organs involved, whereas the most frequent solitary manifestation was pancreatitis.3 Importantly, it cannot be ruled out that European/American and Asian patients differ not only in the overall prevalence of IgG4-RD but also in the frequency of certain organ manifestations.13 Additionally, localized patterns of organ involvement may exist: AIP has been reported to be frequently associated with IgG4-related cholangitis and kidney disease, while sialadenitis often coexists with dacryoadenitis.16 Conversely, RPF may apparently only rarely coincide with IgG4-related kidney disease.13
IgG4-RD and allergy
Various studies from the past years have reported frequent atopic symptoms and concomitant laboratory findings among patients with IgG4-RD.1,3 Contrary to that, a recent study found no higher prevalence (31%) of atopy than in the matched general population. Elevated serum IgE and blood eosinophilia were not only predominately present in atopic patients with IgG4-RD but also occurred in nonatopic subjects with no significant difference between the two groups.17 Current knowledge suggests that these laboratory findings rather represent an immunological bystander phenomenon in IgG4-RD, than being actually attributable to real atopy.17,18
IgG4-RD and malignancies
Several publications have stated an increased incidence of malignancies in patients with IgG4-RD, especially of pancreatic cancer in IgG4-related pancreatitis.19,20 Conversely, however, for all IgG4-RD manifestations, more recent epidemiologic studies found no significantly different age- and sex-adjusted incidence compared to the general population.3,21 Diabetes and age >65 years were identified as risk factors for malignancy among patients with IgG4-RD, while factors directly associated with IgG4-RD or steroid treatment did not pose any difference between the groups.21
Still, in spite of a possible selection bias by extensive diagnostic workup among patients with IgG4-RD,21 IgG4-related pancreatitis probably cannot yet be exculpated from being linked with a higher risk of cancer. IgG4-related pancreatitis and pancreatic carcinoma allegedly could either coincide or be associated with each other in a causal way.20,22 Therefore, further research is definitely necessary to ultimately clarify this issue.
IgG4-RD, cerebral aneurysms, and all-cause mortality
One recent study reported a relatively frequent occurrence of cerebral aneurysms (11%) among patients with IgG4-RD having undergone magnetic resonance tomography angiography for various reasons,3 but the significance of this finding has to be clarified in future studies.
All-cause mortality among patients with IgG4-RD was not higher than expected for an age- and sex-matched general population.3
Pathophysiology
IgG4 antibodies
IgG4 itself normally represents the smallest fraction of IgG subclasses, accounting for only <5%. Owing to an amino acid variance in the hinge region, half IgG4 molecules can split and randomly combine with other half molecules (FAB-arm exchange), creating asymmetric bispecific antibodies with two different antigen-binding sites.18,23,24 Owing to this special feature, IgG4 hardly forms immune complexes and binds Fc-receptors and complements only with low affinity. Thus, its ability to initiate immune responses is limited.23–25 Therefore, IgG4 is regarded as an anti-inflammatory and particularly antiallergic molecule that may counteract IgE functions. IgG4 has been reported to be increased after prolonged antigen exposure like in hyposensibilization.23,24,26
It has been repeatedly suggested that the enhanced production of IgG4 in IgG4-RD rather represents a bystander phenomenon than a causative factor.18 Still, IgG4 antibodies play a role in certain vasculitides and thrombotic thrombocytopenic purpura.27,28 Thus, a direct pathogenic role of IgG4 antibodies cannot be fully precluded yet.
B-cells
The extensive presence of IgG4+ plasma cells in the affected tissue1,23 and the finding that B-cell depletion with rituximab (RTX) is effective in treatment29 suggest an important role of B-lymphocytes in the pathogenesis of IgG4-RD.
Recent studies have shown elevated levels of CD19+CD20–CD27+CD38+ plasmablasts in peripheral circulation, correlating with disease activity.30 As these cells display oligoclonal expansion and somatic hypermutation, a specific antigen-driven immune response seems to be present in IgG4-RD.18,31 Relapses of IgG4-RD after RTX therapy are paralleled by an increase in new distinct clonally expanded plasmablast lines, possibly indicating a recruitment of naïve B-cells from the bone marrow under the stimulatory influences by T-cells.32
T-cells and cytokines
Since the discovery of IgG4-RD, a Type 2 helper T-cell (Th2)-driven immunological mechanism has been proposed in its pathogenesis.33 Th2 cytokines, such as interleukin (IL)-4, IL-5, and IL-13, next to T-regulatory-associated cytokines, such as IL-10, and transforming growth factor beta (TGF-β) are present in IgG4-RD.18,23,34,35 IL-13 and TGF-β may be accountable for fibrosis by activating fibroblasts, while IL-4 and IL-10 are thought to cause the IgG4-specific class-switch recombination in B-lymphocytes.18,34 However, this model approach to IgG4-RD by far cannot explain all of its features: an abundance of T-cells in IgG4-RD lesions has been described,36 and the latest research suggests that CD4+ T-cells are crucial in the initiation of IgG4-RD by activating B-cells.32 Nevertheless, it is still unclear whether T-cells themselves produce these cytokines or rather act at a different level.
Recent advances in research also highlight other cytokines, such as IL-21 and IL-27, produced by macrophages and other antigen-presenting cells. Both seem to be involved in the differentiation of T-helper cells, ultimately augmenting IL-10 production.18,23,37
Innate immunity and complement
Latest studies have cast doubt over the model of IgG4-RD as a primarily T-cell-driven disease. In immunostaining studies, mast cells rather than T-cells seemed to produce IL-4, IL-10, and TGF-β. Additionally, these mast cells were highly positive for IgE compared to controls, suggesting nonspecific IgE as a further important factor in IgG4-RD.36 Additionally, M2 macrophages, activated by Th2- and T-regulatory cytokines such as IL-4 or IL-13,18 probably take part in various fibrosing processes.18,23,38 Toll- and nucleotide-binding oligomerization domain-like receptors on macrophages convey class-switch recombination via B-cell activating factor and may lead to further activation and proliferation of IgG4-positive B-cells.39 Increasing interest is also taken in the role of basophil and eosinophil granulocytes in IgG4-RD.18,40 Importantly, the seemingly extensive activation of innate immunity could suggest the presence of an immune response against a yet unknown microbial trigger antigen as a causative factor in IgG4-RD.39
Hypocomplementemia is frequently reported in IgG4-RD, especially among patients with renal involvement.13 However, research suggests that low complement levels may be rather attributable to concomitant elevations in IgG subclasses other than IgG4.13 Further knowledge about complement activation in IgG4-RD will possibly arise from the ongoing research on antihinge antibodies, which was recently reported in patients with rheumatoid arthritis.41
Future prospects
Summing up, it appears that the role of innate immunity in IgG4-RD has been underestimated in the past and needs to be further clarified by future research, just like the question, whether IgG4 antibodies represent a causative factor or just a bystander in the disease. Other essential research targets are the elucidation of possible antigenic triggers initiating IgG4-RD and B- and T-cell interactions, including the mechanisms ultimately leading to fibrosis.18
Diagnostics
Signs and symptoms
In a Japanese cohort, reportedly 70% of patients later diagnosed with IgG4-RD presented with symptoms, while 30% were found incidentally. Most frequently, symptoms were related to tumefaction, such as obstructive jaundice and exophthalmos (41%), followed by general abdominal symptoms (18%) and malaise (4%). Renal (4%) and pulmonary symptoms (3%) were only present in a minority of patients.3 Another frequently reported symptom preceding the diagnosis of IgG4-RD is weight loss.42
For IgG4-related pancreatitis, abdominal pain (65%), jaundice (62%), and weight loss (42%) were the most commonly reported symptoms.43
Differential diagnosis
All those mentioned signs and symptoms are highly unspecific. Therefore, the list of possible differential diagnoses consists of a large number of neoplastic and inflammatory conditions, which need to be excluded before one can arrive at the diagnosis of IgG4-RD.
In the case of tumefactive lesions or infiltrating processes, it is self-evident that malignancy is usually the main differential diagnosis, especially for IgG4-related pancreatitis, as already described previously in this article. Lymphoproliferative disorders such as multicentric Castleman’s disease or antineutrophil cytoplasmic antibody-associated vasculitides such as granulomatosis with polyangiitis may mimic IgG4-RD in certain cases and can also present with an increased number of IgG4+ plasma cells in lesions.10
Other common differential diagnoses depend on the organ systems or regions where IgG4-RD is suspected. For example, in the case of pancreatic lesions, also type 2 AIP should be thought of. Concerning liver lesions, primary sclerosing cholangitis has to be kept in mind, while Sjögren’s syndrome or sialolithiasis has to be distinguished from IgG4-related sialadenitis and dacryoadenitis. Diagnosis of pulmonary manifestations of IgG4-RD can be especially challenging as signs may be very unspecific and subtle. Besides malignancy, a very broad spectrum of interstitial lung diseases may mimic pulmonary IgG4-RD.2
Standard laboratory testing
Besides renal, pancreatic, and liver manifestations, which may result in organ-specific laboratory findings, there is no standard laboratory parameter that would explicitly mark the presence of IgG4-RD. C-reactive protein may be elevated, and low complement levels, especially in patients with IgG4-related kidney disease, have been frequently reported.11,13,42 Also, peripheral eosinophilia and elevated serum IgE are often present among patients with IgG4-RD.17 Altogether, the diagnostic value of these findings is only limited and does not allow to draw any conclusions on the localization and extent of IgG4-RD.
Antinuclear antibodies (ANA) and rheumatoid factor have been found in a relatively large percentage of patients3,44 but usually at low serum titers.42 Reportedly, the ANA found in IgG4-RD are not IgG4-, but usually IgG2-type ANA, and it remains unclear whether and how they contribute to IgG4-RD.44 Still, screening for ANA and other autoantibodies is certainly important in differential diagnosis toward rheumatologic conditions, possibly mimicking IgG4-RD.45
Serum IgG4 concentration
Serum IgG4, eponymous for the disease, was initially thought to be the major diagnostic criterion for IgG4-RD.5 However, it soon became clear that elevated IgG4 neither was present in all patients with histologically proven IgG4-RD nor was specific for it. Only a small minority of patients with elevated IgG4 levels actually have definite IgG4-RD but a broad variety of respiratory, biliary, liver, or pancreatic diseases as well as rheumatic disorders.46 Importantly, also patients with pancreatic cancer, being the major differential diagnosis to IgG4-related pancreatitis, were reported to display increased serum IgG4 levels in up to 10% of cases.47
The diagnostic value of IgG4 levels strongly depends on the underlying population: studies using databases of patients with any IgG4 elevation usually report a sensitivity for IgG4-RD at ~90% and a specificity of 80% or less.3,11,42,48 On the other hand, among biopsy-proven patients, the retrospective diagnostic value of IgG4 levels seems to be even worse.42 Recently, elevated IgG4 levels were reported in only 51% in such a cohort.13
Another possible pitfall in measuring IgG4 levels is the so-called “Prozone effect”: this phenomenon happens due to excess antigen in the test system, which can inhibit agglutination, leading to artificially low serum levels of IgG4. It can be prevented by proper sample dilution.42,49
An interesting finding in recent years was that serum IgG4 levels seem to be associated with disease activity, marking more “inflammatory” types of IgG4-RD. Patients with elevated IgG4 tend to have more extended organ involvement, higher inflammatory marker alterations, and lower complement levels.3,13,30,42 Also, the location of IgG4-RD may have an influence on serum IgG4 levels, which were higher in patients with pancreatic manifestations and lower in most patients with IgG4-related RPF,13 probably reflecting the scarcity of IgG4+ plasma cells in the usually highly fibrotic tissue in RPF.10
Summing up, it appears that the initially highly esteemed measurement of serum IgG4 for the diagnosis of IgG4-RD has been surpassed by reality. IgG4 is certainly not a screening tool, and “blind” measurement of IgG4 levels in unclear situations may either cause over- or underdiagnosis, depending on the pretest probability. Still, despite the ongoing discussion and its substantial limitations, measurement of serum IgG4 should generally be performed in the diagnostic workup of IgG4-RD as it is easy to obtain and inexpensive. In combination with a solid clinical suspicion and especially in situations when a biopsy is not possible or inconclusive, measuring IgG4 is likely to reveal relevant additional information.
Histopathology
According to most experts, biopsy and histological proof is the gold standard in the diagnosis of IgG4-RD and should be acquired if anyhow possible.50 An international consensus statement on the pathology of IgG4-RD was published in 2012, according to which the diagnosis should be primarily based on general morphological criteria in routine staining, which is usually performed with hematoxylin and eosin, periodic acid–Schiff, and periodic acid methenamine silver. Immunohistochemical staining for IgG4 and IgG using monoclonal antibodies can help to support the diagnosis of IgG4-RD.10
The three major findings, of which two should be present upon diagnosis, are: 1) a dense lyphoplasmacytic infiltrate; 2) storiform fibrosis; and 3) obliterative phlebitis. Other, less characteristic features may be phlebitis without obliteration, tissue eosinophilia, or nonnecrotizing arteritis. Findings such as epitheloid cell granulomas and neutrophilic infiltration argue against the presence of IgG4-RD.10
Concerning immunostaining, >50 IgG4+ plasma cells per high-power field are generally regarded highly specific to IgG4-RD, whereas various organ-specific thresholds have been suggested. However, counting absolute numbers of IgG4+ plasma cells can be substantially misleading in highly inflammatory or fibrotic lesions with an absolute abundance or lack of plasma cells, respectively.10 Therefore, a ratio of IgG4+ to IgG+ plasma cells with a cutoff value of >40% has been established,10,42 for which a sensitivity of 94.4% and a specificity of 85.7% have been reported.48
Just like serum IgG4, the presence of IgG4+ plasma cells is not only strictly confined to IgG4-RD but may also occur in other inflammatory or neoplastic diseases.10,22,51 Thus, the diagnosis of IgG4-RD must never be based on the presence of IgG4+ cells alone but also on the concomitant typical histological appearance of IgG4-RD, as well as on a close clinicopathological correlation.10,42
Flow cytometry
CD19lowCD38+CD20−CD27+ plasmablasts represent a stage between B-cells and plasma cells. Under normal conditions, they are only rarely found in peripheral blood but have been known to be increased in certain autoimmune disorders like systemic lupus erythematosus.52
Recently, plasmablast counts were reported to be significantly increased in patients with active, untreated IgG4-RD compared to patients with other inflammatory conditions and healthy controls.30 Besides, plasmablast levels have been shown to be significantly correlated with IgG4 in serum as well as with the IgG4-responder index (RI) – a recently introduced score for objective assessment of disease activity in IgG4-RD.13,53 For a plasmablast count of 900/mL, a sensitivity of 95%, a specificity of 82%, a positive predictive value of 86%, and a negative predictive value of 97% have been calculated.30
Additionally, IgG4+ plasmablasts were counted in a subset of patients. These amounted to 61% of total plasmablast count and tended to be higher in patients with serum IgG4 elevations.30 Also, the correlation between IgG4+ plasmablast level and IgG4-RI was stronger than with total plasmablast count.13
Importantly, plasmablast levels seem to reflect disease activity, and elevations may precede a disease flare. Therefore, serial measurement of plasmablast levels has been suggested as a longitudinal marker for the monitoring of disease activity.13,30,32,42
The recognition of plasmablasts as relevant players in the pathophysiology of IgG4-RD is certainly one of the most promising recent developments in this field. However, measurement of plasmablasts is yet not widely available and adds further complexity to the diagnostic approach to IgG4-RD. Importantly, just like IgG4 concentrations in serum, plasmablasts are also not exclusive for IgG4-RD.30,52 In any case, further studies with larger cohorts are needed in order to establish plasmablasts in the diagnosis and follow-up of patients with IgG4-RD; especially, the measuring of IgG4+ plasmablasts requires further investigation.
Imaging techniques
As IgG4-RD frequently presents itself with organ enlargement and tumefactive lesions, imaging is obviously essential as malignancy is usually the main differential diagnosis. As a result, a majority of patients will have computed tomography (CT) or magnetic resonance imaging (MRI) scans probably before IgG4-RD is even thought of. Generally, imaging techniques are certainly useful differential diagnosis and useful in judging the extent of disease, but they do not provide signs specific of IgG4-RD.42
18F-fluorodeoxyglucose positron emission tomography (FDG PET)/CT may be an effective diagnostic utility in IgG4-RD as it can highlight active inflammatory lesions and thus enables estimating the extent of disease. Furthermore, FDG PET/CT is a useful tool for staging and monitoring of disease activity, for assessing response to treatment, and also for guiding biopsies.54–57 In one study, PET/CT revealed a larger extent of organ involvement than assumed before imaging in >70% of patients with IgG4-RD.55 Still, its value is limited, especially in the case of small lesions and in brain or kidney manifestations.54
Experience from cases in our center implies that PET/CT is very helpful in the diagnostic workup of patients with IgG4-RD. However, findings can also be substantially misleading, especially in differentiating IgG4-RD and malignant lesions, as demonstrated in the following imaging studies (Figures 1 and 2).
 | Figure 1 PET/CT scan of a patient later histologically diagnosed with a retroperitoneal diffuse large B-cell lymphoma, whereas upon biopsy, the enhancement in the submandibular glands turned out to be IgG4-related sialadenitis. |
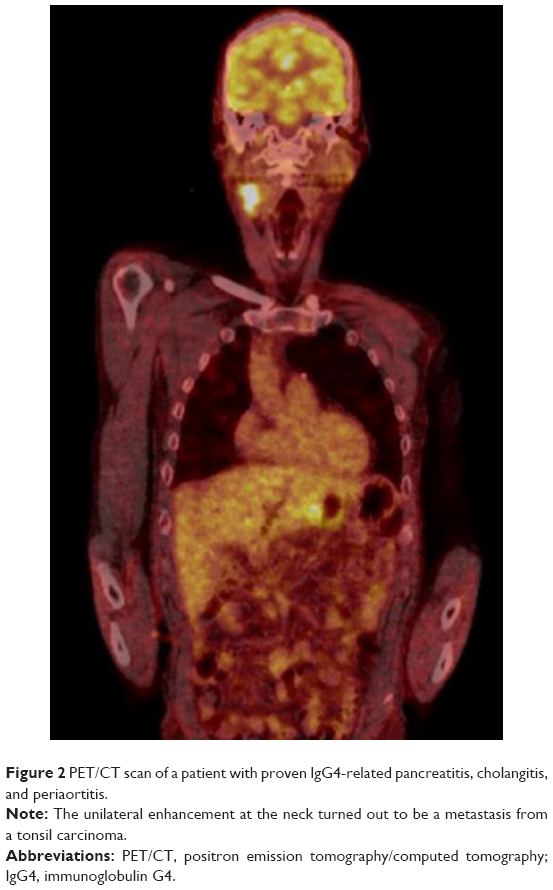 | Figure 2 PET/CT scan of a patient with proven IgG4-related pancreatitis, cholangitis, and periaortitis. |
Summing up, the utility of PET/CT seems to be promising not only for diagnosis of IgG4-RD but also for monitoring of disease activity. However, at the moment, its availability is limited and its broad application in clinical practice requires further investigation.
Diagnostic criteria
In 2012, Japanese investigators proposed comprehensive diagnostic criteria for IgG4-RD, according to which diagnosis should be based on three pillars (Table 1).9,58
 | Table 1 Comprehensive diagnostic criteria for IgG4-RD |
Next to these comprehensive diagnostic criteria, several organ-specific criteria have been suggested, like for IgG4-related pancreatitis or for IgG4-related kidney disease.59,60
Regardless of which criteria used, reliance on IgG4 levels poses a potential limitation, as those may be within the normal range in a considerable fraction of histologically proven patients with IgG4-RD.13 Just like IgG4 levels, also clinical, laboratory, imaging, and histopathological findings are never totally exclusive for IgG4-RD, which makes valid diagnostic criteria hard to establish. New biomarkers such as plasmablasts will probably improve sensitivity and specificity of such diagnostic tools. Nevertheless, until having proven evidence in larger clinical studies, scores and criteria can only support diagnosis but cannot replace expert experience in the assessment of clinical history and cautious interpretation of examination results.
Therapy
Indication for treatment
Untreated IgG4-RD in vital organs such as pancreas and kidney can – even if asymptomatic – lead to irreversible organ damage within only months and therefore requires immediate treatment.50 Still, spontaneous remissions have repeatedly been reported,61,62 and disease progression in untreated patients can be absent for a long period of time.3
Thus, watchful waiting with close follow-up examinations may be an option to be considered in certain patients with no or only mild symptoms, without signs of organ dysfunction and with IgG4-RD in locations not likely to cause major complications.50 Nonetheless, missing the right point for the initiation of treatment worsens organ function, reduces the chance of remission, and increases the incidence of complications.50,63–65 “Burnt-out”, inactive IgG4-RD lesions may be less responsive to drug therapy. However, even highly fibrotic disease can respond to steroids. In some cases, not only surgical debulking but also endoluminal or endovascular interventions should be considered when indicated and are in some cases required as urgent surgical interventions,50,55 especially when organ function is at risk.
Initial treatment and maintenance therapy
Glucocorticoids (GCs) have been considered the first-line therapy in IgG4-RD ever since the first description of AIP in 1995, when prompt response to GC treatment was reported as a characteristic finding.4
According to the recently published consensus statement on the treatment of IgG4-RD, the vast majority of IgG4-RD experts still regard GC as the first-line therapy for active, untreated disease.50 Remission induction is commonly initialized with 30–40 mg/day prednisone or a weight-adjusted dose at 0.6 mg/kg of body weight per day, which can be varied according to disease activity and urgency.50,66 Response to GC therapy is usually seen within days or weeks, and remission can be achieved within months in the majority of patients.3,50 Slow tapering of the GC dose should begin 2–4 weeks after induction of therapy and last for 3–6 months, whereas various regimes have been suggested.43,50,65 Whether long-term maintenance therapy with low-dose GC (2.5–5 mg/d prednisolone) for up to 3 years is indicated, as suggested by Japanese guidelines for AIP,66 is still object to discussion.50
At least for IgG4-related pancreatitis, maintenance therapy seems to decrease the risk of a relapse.65 Patients with multiorgan disease, especially with localization in vital organs and with a history of relapses, will probably benefit from long-term maintenance therapy.50 Expert opinions are divided on the question, if steroid-sparing agents such as azathioprine, methotrexate, or mycophenolate-mofetil should be used in induction and maintenance therapy besides GC.43,50,64 The choice, if and which of these substances should be administered strongly, depends on the region and medical specialty treating IgG4-RD.50 Further research is definitely needed in this field as evidence concerning steroid-sparing drugs is limited to small case series. At the moment, most experts recommend an add-on therapy with steroid-sparing agents after relapses under GC treatment alone.50 Recently, the introduction of B-cell depletion with RTX has brought a new impetus into the treatment of IgG4-RD. RTX therapy leads to specific IgG4 reductions together with apparently very effective disease control, even in steroid refractory cases.13,30,50,64,67,68 RTX seems to be a very specific agent interfering with the immunological processes underlying IgG4-RD, though it has not yet been fully elucidated.18 Still, although recently reported results are promising,67,68 relapses also happen under RTX.69
At the moment, evidence concerning RTX in IgG4-RD is still limited due to low patient numbers and the lack of larger prospective and especially randomized trials. Most experience with RTX is confined to European and North American centers, while most guidelines have been fitted to Asian patients. Besides, there is still no consensus regarding dosage and frequency of RTX application, so that the efficacy of RTX in IgG4-RD cannot be ultimately judged yet.
Disease activity
IgG4-RD is a chronic, frequently relapsing disease, which often requires long-term maintenance therapy.50 Thus, it is essential to reevaluate patients regularly in order to adapt therapy to disease activity and prevent flares during remission.
For this reason, an IgG4-RD RI based on the Birmingham Vasculitis Activity Score for Wegener’s Granulomatosis has been proposed in 2012 and has been applied in several studies up to now.13,30,32,53 The IgG4-RI has been reported to be associated with IgG4 concentration in serum30 as well as with total and IgG4+ plasmablasts.13
Importantly, plasmablasts recently emerged as an apparently good indicator for disease activity, although their application as a longitudinal monitoring parameter still requires further research.13,30,32,42
Furthermore, serum IgG4, with all its limitations, as extensively described in this article, may characterize a more inflammatory and therapy-resistant type of IgG4-RD and thus also help estimate the disease activity.13,30,42
Future prospects
Regardless of the therapy modality ultimately chosen, it seems that relapses happen frequently in IgG4-RD. Up to now, there is no striking evidence of superiority for GC, alone or together with steroid-sparing drugs or for RTX in either remission induction or maintenance therapy. Also, the question, whether timely GC discontinuation or long-term maintenance therapy with or without steroid-sparing agents is advisable after remission remains unanswered due to the lack of evidence from randomized trials. Anti-CD20 therapy approaches seem to be a promising new therapeutic option on the horizon. However, there is no consensus about dosage, frequency, and indication for RTX treatment, and especially, trials comparing RTX versus GC with or without steroid-sparing drugs are still lacking.
From the present point of view, GC are still regarded as the first-line therapy for induction and maintenance, while steroid-sparing drugs and RTX should be considered in relapsing or steroid-resistant cases as well as in patients with intolerance to GC treatment. Long-term maintenance therapy seems to be advisable at least in patients with a history of relapses and disease localizations in vital organs.
Conclusion
IgG4-RD is a complex condition with a highly variable clinical appearance that can mimic various inflammatory and neoplastic diseases. Its pathophysiology has still not been fully understood, though recent research suggests the existence of an antigenic trigger, initiating and perpetuating the disease.
Having emerged within only 1 decade, its epidemiology is still widely unknown. IgG4-RD is probably underdiagnosed, especially in Europe and North America, where the awareness of its existence may be lower than in Asian countries, where the disease was originally discovered.
Diagnosis of IgG4-RD is challenging as there is no laboratory, imaging, or histopathological finding that would be exclusive for IgG-RD. Especially, the serum IgG4 levels, originally considered highly significant, have certainly lost importance in the diagnosis of IgG4-RD. Histopathological proof is still regarded a gold standard and should be obtained if possible. Measurement of plasmablast counts in flow cytometry has recently emerged as a promising new diagnostic modality; however, more evidence in this field is certainly needed. In any case, all diagnostic findings need to be put into context with the clinical presentation of the patient carefully. Differential diagnoses, especially malignancies, are numerous and have to be excluded before the treatment is initiated.
GCs are effective in initial and relapse treatment and are widely used in maintenance therapy. From the present point of view, neither RTX nor GC treatment has been shown to be superior in therapy and the application of steroid-sparing drugs is still object to discussion. Anyway, the true challenge in IgG4-RD is probably its long-term management, as relapses happen frequently, regardless of the therapy chosen. Thus, further advances in the treatment of IgG4-RD will most likely rather come with new parameters such as plasmablasts, which could allow monitoring of disease activity and exact timing of retreatment in order to prevent relapses before they actually happen.
Disclosure
JZ received an unrestricted research grant for clinical research on IgG4-RD from Roche. JZ received honoraria for educational activities and advisory board participation from Roche. The authors report no other conflicts of interest in this work.
References
Umehara H, Okazaki K, Masaki Y, et al; Research Program for Intractable Disease by Ministry of Health, Labor and Welfare (MHLW) Japan G4 team. A novel clinical entity, IgG4-related disease (IgG4RD): general concept and details. Mod Rheumatol. 2012;22(1):1–14. | ||
Pieringer H, Parzer I, Wöhrer A, Reis P, Oppl B, Zwerina J. IgG4-related disease: an orphan disease with many faces. Orphanet J Rare Dis. 2014;9:110. | ||
Inoue D, Yoshida K, Yoneda N, et al. IgG4-related disease: dataset of 235 consecutive patients. Medicine (Baltimore). 2015;94(15):e680. | ||
Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci. 1995;40(7):1561–1568. | ||
Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344(10):732–738. | ||
Hamano H, Kawa S, Ochi Y, et al. Hydronephrosis associated with retroperitoneal fibrosis and sclerosing pancreatitis. Lancet. 2002;359(9315):1403–1404. | ||
Kamisawa T, Nakajima H, Egawa N, Funata N, Tsuruta K, Okamoto A. IgG4-related sclerosing disease incorporating sclerosing pancreatitis, cholangitis, sialadenitis and retroperitoneal fibrosis with lymphadenopathy. Pancreatology. 2006;6(1–2):132–137. | ||
Stone JH, Khosroshahi A, Deshpande V, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum. 2012;64(10):3061–3067. | ||
Okazaki K, Umehara H. Are classification criteria for IgG4-RD now possible? The concept of IgG4-related disease and proposal of comprehensive diagnostic criteria in Japan. Int J Rheumatol. 2012;2012:357071. | ||
Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25(9):1181–1192. | ||
Brito-Zerón P, Ramos-Casals M, Bosch X, Stone JH. The clinical spectrum of IgG4-related disease. Autoimmun Rev. 2014;13(12):1203–1210. | ||
Uchida K, Masamune A, Shimosegawa T, Okazaki K. Prevalence of IgG4-related disease in Japan based on Nationwide Survey in 2009. Int J Rheumatol. 2012;2012:358371. | ||
Wallace ZS, Deshpande V, Mattoo H, et al. IgG4-related disease: clinical and laboratory features in one hundred twenty-five patients. Arthritis Rheumatol. 2015;67(9):2466–2475. | ||
Griepentrog GJ, Vickers RW, Karesh JW, Azari AA, Albert DM, Bukat CN. A clinicopathologic case study of two patients with pediatric orbital IgG4-related disease. Orbit. 2013;32(6):389–391. | ||
Kanno A, Nishimori I, Masamune A, et al; Research Committee on Intractable Diseases of Pancreas. Nationwide epidemiological survey of autoimmune pancreatitis in Japan. Pancreas. 2012;41(6):835–839. | ||
Koizumi S, Kamisawa T, Kuruma S, et al. Organ correlation in IgG4-related diseases. J Korean Med Sci. 2015;30(6):743–748. | ||
Della Torre E, Mattoo H, Mahajan VS, Carruthers M, Pillai S, Stone JH. Prevalence of atopy, eosinophilia, and IgE elevation in IgG4-related disease. Allergy. 2014;69(2):269–272. | ||
Della-Torre E, Lanzillotta M, Doglioni C. Immunology of IgG4-related disease. Clin Exp Immunol. 2015;181(2):191–206. | ||
Yamamoto M, Takahashi H, Tabeya T, et al. Risk of malignancies in IgG4-related disease. Mod Rheumatol. 2012;22(3):414–418. | ||
Shiokawa M, Kodama Y, Yoshimura K, et al. Risk of cancer in patients with autoimmune pancreatitis. Am J Gastroenterol. 2013;108(4):610–617. | ||
Hirano K, Tada M, Sasahira N, et al. Incidence of malignancies in patients with IgG4-related disease. Intern Med. 2014;53(3):171–176. | ||
Witkiewicz AK, Kennedy EP, Kennyon L, Yeo CJ, Hruban RH. Synchronous autoimmune pancreatitis and infiltrating pancreatic ductal adenocarcinoma: case report and review of the literature. Hum Pathol. 2008;39(10):1548–1551. | ||
Umehara H, Nakajima A, Nakamura T, et al. IgG4-related disease and its pathogenesis-cross-talk between innate and acquired immunity. Int Immunol. 2014;26(11):585–595. | ||
Rispens T, Ooijevaar-de Heer P, Bende O, Aalberse RC. Mechanism of immunoglobulin G4 Fab-arm exchange. J Am Chem Soc. 2011;133(26):10302–10311. | ||
Bindon CI, Hale G, Brüggemann M, Waldmann H. Human monoclonal IgG isotypes differ in complement activating function at the level of C4 as well as C1q. J Exp Med. 1988;168(1):127–142. | ||
Aalberse RC, Stapel SO, Schuurman J, Rispens T. Immunoglobulin G4: an odd antibody. Clin Exp Allergy. 2009;39(4):469–477. | ||
Holland M, Hewins P, Goodall M, Adu D, Jefferis R, Savage CO. Anti-neutrophil cytoplasm antibody IgG subclasses in Wegener’s granulomatosis: a possible pathogenic role for the IgG4 subclass. Clin Exp Immunol. 2004;138(1):183–192. | ||
Ferrari S, Mudde GC, Rieger M, Veyradier A, Kremer Hovinga JA, Scheiflinger F. IgG subclass distribution of anti-ADAMTS13 antibodies in patients with acquired thrombotic thrombocytopenic purpura. J Thromb Haemost. 2009;7(10):1703–1710. | ||
Khosroshahi A, Bloch DB, Deshpande V, Stone JH. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum. 2010;62(6):1755–1762. | ||
Wallace ZS, Mattoo H, Carruthers M, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis. 2015;74(1):190–195. | ||
Maillette de Buy Wenniger LJ, Doorenspleet ME, Klarenbeek PL, et al. Immunoglobulin G4+ clones identified by next-generation sequencing dominate the B cell receptor repertoire in immunoglobulin G4 associated cholangitis. Hepatology. 2013;57(6):2390–2398. | ||
Mattoo H, Mahajan VS, Della-Torre E, et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol. 2014;134(3):679–687. | ||
Lighaam LC, Aalberse RC, Rispens T. IgG4-related fibrotic diseases from an immunological perspective: regulators out of control? Int J Rheumatol. 2012;2012:789164. | ||
Tsuboi H, Matsuo N, Iizuka M, et al. Analysis of IgG4 class switch-related molecules in IgG4-related disease. Arthritis Res Ther. 2012;14(4):R171. | ||
Nakashima H, Miyake K, Moriyama M, et al. An amplification of IL-10 and TGF-beta in patients with IgG4-related tubulointerstitial nephritis. Clin Nephrol. 2010;73(5):385–391. | ||
Takeuchi M, Sato Y, Ohno K, et al. T helper 2 and regulatory T-cell cytokine production by mast cells: a key factor in the pathogenesis of IgG4-related disease. Mod Pathol. 2014;27(8):1126–1136. | ||
Maehara T, Moriyama M, Nakashima H, et al. Interleukin-21 contributes to germinal centre formation and immunoglobulin G4 production in IgG4-related dacryoadenitis and sialoadenitis, so-called Mikulicz’s disease. Ann Rheum Dis. 2012;71(12):2011–2019. | ||
Furukawa S, Moriyama M, Tanaka A, et al. Preferential M2 macrophages contribute to fibrosis in IgG4-related dacryoadenitis and sialoadenitis, so-called Mikulicz’s disease. Clin Immunol. 2015;156(1):9–18. | ||
Watanabe T, Yamashita K, Fujikawa S, et al. Involvement of activation of toll-like receptors and nucleotide-binding oligomerization domain-like receptors in enhanced IgG4 responses in autoimmune pancreatitis. Arthritis Rheum. 2012;64(3):914–924. | ||
Watanabe T, Yamashita K, Sakurai T, et al. Toll-like receptor activation in basophils contributes to the development of IgG4-related disease. J Gastroenterol. 2013;48(2):247–253. | ||
van de Stadt LA, de Vrieze H, Derksen NI, et al. Antibodies to IgG4 hinge can be found in rheumatoid arthritis patients during all stages of disease and may exacerbate chronic antibody-mediated inflammation. Arthritis Rheumatol. 2014;66(5):1133–1140. | ||
Stone JH, Brito-Zerón P, Bosch X, Ramos-Casals M. Diagnostic approach to the complexity of IgG4-related disease. Mayo Clin Proc. 2015;90(7):927–939. | ||
Raina A, Yadav D, Krasinskas AM, et al. Evaluation and management of autoimmune pancreatitis: experience at a large US center. Am J Gastroenterol. 2009;104(9):2295–2306. | ||
Kiyama K, Yoshifuji H, Kandou T, et al. Screening for IgG4-type anti-nuclear antibodies in IgG4-related disease. BMC Musculoskelet Disord. 2015;16:129. | ||
Chang SY, Keogh K, Lewis JE, Ryu JH, Yi ES. Increased IgG4-positive plasma cells in granulomatosis with polyangiitis: a diagnostic pitfall of IgG4-related disease. Int J Rheumatol. 2012;2012:121702. | ||
Ryu JH, Horie R, Sekiguchi H, Peikert T, Yi ES. Spectrum of disorders associated with elevated serum IgG4 levels encountered in clinical practice. Int J Rheumatol. 2012;2012:232960. | ||
Ghazale A, Chari ST, Smyrk TC, et al. Value of serum IgG4 in the diagnosis of autoimmune pancreatitis and in distinguishing it from pancreatic cancer. Am J Gastroenterol. 2007;102(8):1646–1653. | ||
Masaki Y, Kurose N, Yamamoto M, et al. Cutoff values of serum IgG4 and histopathological IgG4+ plasma cells for diagnosis of patients with IgG4-related disease. Int J Rheumatol. 2012;2012:580814. | ||
Khosroshahi A, Cheryk LA, Carruthers MN, Edwards JA, Bloch DB, Stone JH. Brief report: spuriously low serum IgG4 concentrations caused by the prozone phenomenon in patients with IgG4-related disease. Arthritis Rheumatol. 2014;66(1):213–217. | ||
Khosroshahi A, Wallace ZS, Crowe JL, et al; Second International Symposium on IgG4-Related Disease. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheumatol. 2015;67(7):1688–1699. | ||
Strehl JD, Hartmann A, Agaimy A. Numerous IgG4-positive plasma cells are ubiquitous in diverse localised non-specific chronic inflammatory conditions and need to be distinguished from IgG4-related systemic disorders. J Clin Pathol. 2011;64(3):237–243. | ||
Jacobi AM, Odendahl M, Reiter K, et al. Correlation between circulating CD27high plasma cells and disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2003;48(5):1332–1342. | ||
Carruthers MN, Stone JH, Deshpande V, Khosroshahi A. Development of an IgG4-RD Responder Index. Int J Rheumatol. 2012;2012:259408. | ||
Ebbo M, Grados A, Guedj E, et al. Usefulness of 2-[18F]-fluoro-2-deoxy-D-glucose-positron emission tomography/computed tomography for staging and evaluation of treatment response in IgG4-related disease: a retrospective multicenter study. Arthritis Care Res (Hoboken). 2014;66(1):86–96. | ||
Zhang J, Chen H, Ma Y, et al. Characterizing IgG4-related disease with 18F-FDG PET/CT: a prospective cohort study. Eur J Nucl Med Mol Imaging. 2014;41(8):1624–1634. | ||
Vasaitis L. IgG4-related disease: a relatively new concept for clinicians. Eur J Intern Med. Epub 2015 Oct 16. | ||
Nakatani K, Nakamoto Y, Togashi K. Utility of FDG PET/CT in IgG4-related systemic disease. Clin Radiol. 2012;67(4):297–305. | ||
Umehara H, Okazaki K, Masaki Y, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol. 2012;22(1):21–30. | ||
Shimosegawa T, Chari ST, Frulloni L, et al; International Association of Pancreatology. International consensus diagnostic criteria for autoimmune pancreatitis: guidelines of the International Association of Pancreatology. Pancreas. 2011;40(3):352–358. | ||
Kawano M, Saeki T, Nakashima H, et al. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol. 2011;15(5):615–626. | ||
Seki N, Yamazaki N, Kondo A, Nomura K, Himi T. Spontaneous regression of lung lesions after excision of the submandibular gland in a patient with chronic sclerosing sialadenitis. Auris Nasus Larynx. 2012;39(2):212–215. | ||
Ohshima K, Sato Y, Yoshino T. A case of IgG4-related dacryoadenitis that regressed without systemic steroid administration. J Clin Exp Hematop. 2013;53(1):53–56. | ||
Shimizu Y, Yamamoto M, Naishiro Y, et al. Necessity of early intervention for IgG4-related disease – delayed treatment induces fibrosis progression. Rheumatology (Oxford). 2013;52(4):679–683. | ||
Hart PA, Topazian MD, Witzig TE, et al. Treatment of relapsing autoimmune pancreatitis with immunomodulators and rituximab: the Mayo Clinic experience. Gut. 2013;62(11):1607–1615. | ||
Kamisawa T, Shimosegawa T, Okazaki K, et al. Standard steroid treatment for autoimmune pancreatitis. Gut. 2009;58(11):1504–1507. | ||
Kamisawa T, Okazaki K, Kawa S, et al; Working Committee of the Japan Pancreas Society and the Research Committee for Intractable Pancreatic Disease supported by the Ministry of Health, Labour and Welfare of Japan. Amendment of the Japanese consensus guidelines for autoimmune pancreatitis, 2013 III. Treatment and prognosis of autoimmune pancreatitis. J Gastroenterol. 2014;49(6):961–970. | ||
Khosroshahi A, Carruthers MN, Deshpande V, Unizony S, Bloch DB, Stone JH. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine (Baltimore). 2012;91(1):57–66. | ||
Carruthers MN, Topazian MD, Khosroshahi A, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis. 2015;74(6):1171–1177. | ||
Murakami J, Matsui S, Ishizawa S, et al. Recurrence of IgG4-related disease following treatment with rituximab. Mod Rheumatol. 2013;23(6):1226–1230. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.