Back to Journals » Journal of Asthma and Allergy » Volume 12
IgE-Mediated Systemic Anaphylaxis And Its Association With Gene Polymorphisms Of ACE, Angiotensinogen And Chymase
Authors Varney VA ![]() , Nicholas A, Warner A, Sumar N
, Nicholas A, Warner A, Sumar N
Received 21 April 2019
Accepted for publication 5 September 2019
Published 8 October 2019 Volume 2019:12 Pages 343—361
DOI https://doi.org/10.2147/JAA.S213016
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Amrita Dosanjh
VA Varney,1,2 A Nicholas,2 A Warner,2 N Sumar2
1Department of Medicine, St Helier Hospital, Carshalton, Surrey SM5 1AA, UK; 2Department of Allergy and Immunology, St Helier Hospital, Carshalton, Surrey SM5 1AA, UK
Correspondence: VA Varney
Department of Medicine, St Helier Hospital, Wrythe Lane, Carshalton, Surrey SM5 1AA, UK
Tel +44 20 8296 2401
Email [email protected]
Background: The renin-angiotensin system (RAS) protects the circulation against sudden falls in systemic blood pressure via generation of angiotensin II (AII). Previously, we demonstrated that patients with anaphylaxis involving airway angioedema and cardiovascular collapse (AACVS) had significantly increased “I” gene polymorphisms of the angiotensin-converting-enzymes (ACE). This is associated with lower serum ACE and AII levels and was not seen in anaphylaxis without collapse nor atopics and healthy controls.
Objectives: To examine the angiotensinogen (AGT-M235T) and chymase gene (CMA-1 A1903G) polymorphisms in these original subjects.
Method: 122 patients with IgE-mediated anaphylaxis, 119 healthy controls and 52 atopics had polymorphisms of the AGT gene and chymase gene examined by polymerase chain reactions and gel electrophoresis. Their previous ACE genotypes were included for the analysis.
Results: AGT-MM genes (associated with low AGT levels) were significantly increased in anaphylaxis (Terr’s classification). When combined with ACE, anaphylaxis showed increased MM/II gene pairing (p<0.0013) consistent with lower RAS activity. For chymase, there was increased pairing of MM/AG (p<0.005) and AG/II and AG/ID (p<0.0073) for anaphylaxis consistent with lower RAS activity. A tri-allelic ensemble of the 6 commonest gene combinations for the healthy controls and anaphylaxis confirmed this difference (p=0.0001); for anaphylaxis, genes were predominately MM/AG/II or ID, while healthy controls were DD/MT/AG or GG patterns.
Conclusion: Our gene polymorphisms show lower RAS activity for anaphylaxis especially AACVS. Animal models of anaphylaxis are focused on endothelial nitric oxide (eNO) which is shown to be the mediator of fatal shock and prevented by eNO-blockade. The interaction of AII and eNO controls the microcirculation in man. High serum AII levels reduce eNO activity, so higher RAS-activity could protect against shock. Our data shows low RAS activity in anaphylaxis especially AACVS, suggesting the influence of these genes on shock are via AII levels and its effects on eNO.
Keywords: IgE-mediated anaphylaxis, angiotensinogen M235T, chymase CMA1-1903, angioedema, ACE genotype, endothelial nitric oxide
Introduction
IgE-mediated anaphylaxis can result in clinical symptoms ranging from mild cutaneous effects (Grade I) to cardiac arrest from profound hypotension and circulatory collapse (Grade 4) as described by Terr’s classification.1 To date, the severity of these reactions cannot be predicted from the IgE levels alone nor the allergen involved. The most severe anaphylaxes involve angioedema of the airway and severe hypotension with cardiovascular collapse.2–5 The effects of released histamine are central to this, and produce falls in systolic blood pressure and venous return along with bronchoconstriction and angioedema resulting from its effects on the microcirculation and bradykinin generation.6–8
There are unanswered questions as to which host factors may be influencing the effects of the released histamine.9–14 Anaphylaxis is increasing on a global scale, with a 9-year Australian study showing an increase of 150% in anaphylaxis admissions and a 300% increase in fatalities, especially in children.15
Epidemiological factors have suggested a high incidence of anaphylaxis in young children <5yrs due to food allergy and also in pre-menopausal women.16–19
In the UK 1-2% of the adult population carry adrenaline.4 A 15-year UK study also confirmed a 7-fold increase in hospital admission for anaphylaxis with a disproportionate increase in younger children <4yrs, without a clear explanation for this increase.13,18
Post-mortem findings in fatal anaphylaxis show 80% of cases to have upper airway oedema with acute pulmonary hyperinflation and circulatory collapse frequently as the only features.20,21
Prior cardiovascular and respiratory comorbidities in adults increase the risk of death, while prior asthma in a young child increases the risk of a severe or fatal outcome.12,16,17,19,22,23 Small children have reduced lung capacity allowing histamine-induced bronchoconstriction to give greater respiratory difficulty than adults.11,19,21
From a cardiovascular aspect, children have lower blood pressure (both systolic and diastolic) which starts to increase steadily after the age of 4yrs with adult values reached after 15yrs of age.24,25 Childhood blood pressure (BP) is related to age, height, BMI and subscapular skin fold. Children >8yrs have significantly higher systolic and diastolic BP than younger children.26–28 In infancy and childhood, increased activity of the renin-angiotensin-system (RAS) is well recognised.27,29–31 Plasma renin and aldosterone levels are 9-times higher in infants and young children showing an inverse relationship to their diastolic BP, age and body surface area noted up to the age of 12yrs. After 12yrs, both hormones decrease slowly down to adult values.27,32–34 These raised levels appear related to immaturity of the renal tubules and relative resistance to aldosterone giving high sodium and water loss in the kidney that stimulates renin to support both blood pressure and the autonomic nervous system.27,32,33,35
Young children <4yrs also have lower serum ACE levels that start to increase over the age of 4 years with stable adult levels by 18yrs of age but they remain lower in females due to oestrogen’s inhibitory effects on ACE biosynthesis.31,36,37 Studies show ACE is important for growth and differentiation of the kidney in young animals.38
Angiotensin-2 (AII) levels follow serum ACE and increase between 4 and 12yrs in order to elevate both BP and correct the low serum sodium levels of childhood.27 Systolic blood pressure in all children is correlated with AII and serum ACE levels.39 The renin substrate “angiotensinogen” (AGT) that generates angiotensin-1 (AI) via renin is a rate-limiting step if deficient.34 AGT levels reach adult values between the ages of 6–9 years, and are significantly related to BMI and race; with 20% higher levels in children with T alleles, found most commonly in African-Americans.34,40,41 Testosterone levels have an inverse relationship to AGT, while oestrogen has a significant positive relationship, possibly related to AGT production by fat cells under the influence of oestrogen.41
All these complex and evolving hormonal changes could give one explanation for increased hospitalization with anaphylaxis in children <4yrs of age, that later decreases.11,17,41,42 This may mask the influence of gene polymorphisms in youth due to the confounding effects of hormonal, autonomic and renal immaturity at that stage in their development.43
Hermann and Ring (1993) were the first to examine the renin-angiotensin system in adults with hymenoptera venom-induced anaphylaxis and healthy controls.44 Using Terr’s classification, they showed a reduction in renin, AI and AII plasma levels in anaphylaxis grades 1–3.1 This gave the first laboratory-measured parameter linked to the severity of clinical symptoms.45 The findings fitted well with Pumphrey’s observations that serum ACE levels <37mmol/L in patients with tree/nut allergy were a risk factor for pharyngeal oedema but not asthma.46 He suggested that since ACE levels are stable in adult life they reflected genetic inheritance.
Niedoszytko examined ACE gene polymorphisms (I/D) in insect venom allergy of grades 3–4 on the Mueller scale, showing 80% of subjects to have ID or II genotypes.47,48 Our previous study also showed that IgE reactions to food, venom or drugs involving angioedema and cardiovascular collapse were significantly associated with II or ID gene polymorphisms of ACE (odds ratio=44) not seen in atopics and healthy controls subjects.49
“I” (insertion) gene polymorphisms are associated with lower serum ACE activity giving reduced AII generation and increased bradykinin (BK) levels from reduced catabolism of BK by ACE. This pattern would be consistent with hypotension and angioedema reported in these cases of anaphylaxis. Both II and ID genotypes share lower serum ACE levels and activity, with lower AI and AII plasma levels under both exercise and rest.50–56
Oestrogen has inhibitory effects upon mRNA synthesis of ACE giving cardio-protection for women and contributes to the hypotension of pregnancy (as oestrogen levels climb) that is lost at menopause.37,57–59 This renders pre-menopausal women more ACE deficient than men for the same genotype, and could offer one explanation for the increased rates of anaphylaxis in pre-menopausal women that decrease in menopause. There is also evidence that oestrogen may augment mast cell histamine release in rodent studies.58 The role of serum ACE in the generation of AII-induced vasoconstriction is well documented, but Chymase from mast cell “leak” is now recognised to be an important Non-ACE source of AII in the circulation and linked with circulatory disease and hypertension. Chymase levels have been measured in anaphylaxis, but chymase genes not been directly examined in anaphylaxis cases unlike the genes of the renin-angiotensin system. We have therefore included the Chymase promotor gene CMA-1903 to our analysis as the GG polymorphism is strongly associated with hypertension.
We have examined further our original study subjects for Angiotensinogen (AGT-polymorphism M235T) and Chymase (CMA1-1903-polymorphisms A/G) genes. We have linked these findings to their prior ACE genotype, to see if there is evidence for bi- or tri-allelic ensemble of genes that could be a host factor for angioedema and cardiovascular collapse in allergic reactions compared with atopics and healthy controls.60
Methods
Objectives
To establish the M235T and A1903G genotypes of all 293 subjects and combine that with their prior determined ACE (I/D) genotype. To examine these gene polymorphisms for bi- or tri-allelic patterns that may differ between healthy control, atopics and anaphylaxis especially those with angioedema and cardiovascular collapse compared with cutaneous and respiratory symptoms only.
Subjects
A parallel-group study of 293 subjects, 119 healthy (non-atopic) controls without medication, 52 atopics without anaphylaxis (minor pollen or oral allergy symptoms) and 122 subjects with IgE-mediated anaphylaxis on two or more occasions to food, venom or drugs.49 No subjects were taking ACE-inhibitors or angiotensin-receptor antagonists. The anaphylaxis patients had no personal history of hypertension, although 5% of venom patients and 3% of food/drug confirmed a family history of hypertension. Serum ACE levels were lower in the anaphylaxis group. Any subjects <18yrs old had informed consent from the parent to participate in the study.
Study Ethics
The study was performed at St. Helier Hospital Allergy Clinic in Surrey, with permission granted by our London Surrey Borders regional Ethics Committee initially in 1997 (REC approval No. 14/97) and re-submitted and expanded in 2005 (Rec. No. 14/97) to examine further aspects of the RAS system and genotypes. The study conformed to the declaration of Helsinki. Each patient gave written informed consent and enrolment of cases and controls were completed by 2009. The statistically calculated sample size was a minimum of 50 subjects in each of the 3 main groups. This was comparable at the time with other studies linking genotype to disease states. ISRCTN registry number: 10,465,389. Funding was from a local R&D dept. grant initially and also by funds in our departmental charity account.
Clinical Assessment
All subjects including healthy controls (HC) were skin prick tested to airborne allergens, venom and food to confirm their exact atopic status. Detailed records of allergic history, past medical history, drug medication and family history were recorded in all subjects.
Total Serum IgE And Allergen-Specific IgE
This was measured in all cases of hymenoptera venom allergy and drug-induced anaphylaxis with intradermal testing as appropriate. The food-induced anaphylaxis subjects had IgE and specific IgE tests only if strong confirmation by skin prick test was judged unsatisfactory.
Most measurements were performed “in house” by our Immunology Laboratory Department at St. Helier Hospital by Phadia 250 Unicap analysis.
Skin Prick Tests
A range of products were used, Alk-Abello for airborne allergens and Hymenoptera venom, Hollister–Stier for food allergens with “fresh food” tests in some subjects. Skin prick tests were read at 15 mins with appropriate positive and negative controls.
Assessment Of Anaphylaxis
All reactions were recorded in detail including (route of allergen exposure, time to onset, duration and treatment) including a proforma of Terr’s classification for grading of the anaphylaxis (Figure 1).1,49
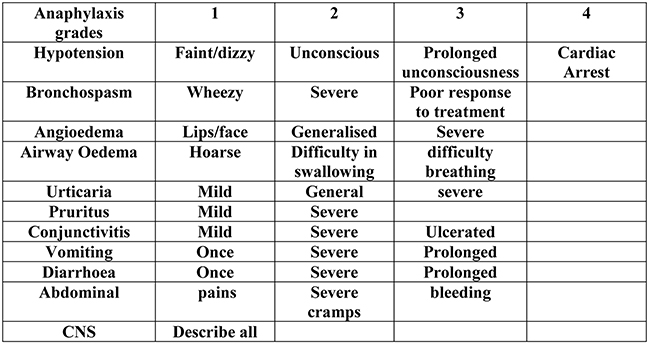 |
Figure 1 Terr’s classification of anaphylaxis grades 1–4. Note: Data from Terr.1 |
The anaphylaxis group was further subdivided into:49
- Those with Airway Angioedema & CardioVascular System collapse (AACVS) suggestive of reduced renin-angiotensin function n=95 subjects.
- Those with Cutaneous and Respiratory Allergy (CRA) symptoms only and suggestive of histamine effects including skin itch with or without rash, eye and nasal symptoms with a mild cough or chest tightness or mild wheeze n=27 subjects.
Angiotensinogen And Chymase Gene Polymorphisms
EDTA samples previously collected (and stored at −20°C) and had their buffy coats used for DNA extraction.
Isolation Of Genomic DNA
Genomic DNA was prepared from the buffy coats (300 µL) using the Promega Maxwell 16 semi-automated extraction system. The extracted DNA samples were quantified (Eppendorf Biophotometer) and stored at −20°C until ready for PCR.
Angiotensinogen M235T Gene And Polymerase Chain Reaction Method
PCR: The PCR reaction was set up in a total volume of 20 µL containing 15 µL of PCR Master Mix (ABgene Ltd., UK, Thermo Scientific) containing 1.5 mM MgCl2, 20mM ammonium sulphate in 75 mM Tris-HCl buffer and 50pmol of the forward and reverse oligonucleotide primer (MWG Biotech, Germany) and 5µL (50–100ng) of genomic DNA template.61
The sense and antisense primers had the following sequences:
Downstream 5ʹ-CAG GGT GCT GTC CAC ACT GGA CCC C-3ʹ
Upstream 5’-CCG TTT GTG CAG GGC CTG GCT CTC T-3ʹ
PCR cycling conditions used were initial denaturation at 90°C for 3 mins followed by amplification for 10 cycles; at 94°C for 1 mins, 68°C for 1 min, 72°C for 1 min. This was followed by 30 cycles at 90°C for 30 s, 68°C for 1 min, 72°C for 30s, and final extension at 72°C for 10 mins.
Enzyme digest
10 µL of PCR product was incubated for 30mins at 65°C with 0.2µL of enzyme (Tth III-I), 2 µL buffer (X10 concentration), and 8 µL H2O followed by analysis by gel electrophoresis.
Gel Electrophoresis
The PCR products (10μL) were separated on 2% agarose gel with ethidium bromide using Triboreate EDTA buffer solution at 110V for 60 mins. A 100bp (basepair) DNA ladder size marker (8μL) was used. The amplified PCR products were visualised as bands under UV light. M and T alleles were identified at 165 bp and 141 bp, respectively.
Chymase Gene (CMA1-A1903G) And Polymerase Chain Reaction Method
PCR: The PCR reaction was set up in a total volume of 25 µL with 12.5 µL of master mix containing 1.5 mM MgCl2, 20mM ammonium sulphate in 75 mM Tris-HCl buffer (ABgene Ltd, UK, Thermo Scientific) 1.25 µL of primer-1 and 1.25 µL of primer-2 (50pmol of the forward and reverse oligonucleotide primer, MWG Biotech, Germany), 3.5 µL of genomic DNA (100–300ng) and 6.5 µL of water.62,63
The sense and antisense primers had the following sequences:
5ʹ-GGA AAT GTG AGC AGA TAG TGC AGT C-3ʹ
5ʹ-AAT CCG GAG CTG GAG AAC TCT TGT C-3ʹ
The PCR cycling conditions used were heating at 95°C for 5 mins followed by amplification for 39 cycles; each cycle consisted of denaturation at 94°C for 30s, annealing at 51°C for 15s and 72°C for 30s extension.
Enzyme digests
10 µL of PCR product was incubated for 30mins at 37°C in an enzyme mix containing 2 µL of fast digest buffer (X10), 1 µL restriction enzyme (BST X1) and 17µL of nuclease-free water followed by analysis by gel electrophoresis.
Gel Electrophoresis
The PCR products (10μL) were separated on 2% agarose gel with ethidium bromide using Tri-boreate EDTA buffer solution for 1 hr at 110V. A 100 bp DNA ladder size marker (8μL) was used. The amplified PCR products were visualised as bands under UV light. “A” genotypes were identified at 190+90 bp and “G” at 280 bp, respectively.
Statistical Analysis
Statistical analysis was carried out using Sigma Stats 3.1 and Prism 3.03 statistical package.
Genotype profiles for all the groups and subgroups were compared by Pearson’s Chi-Square test for categorical data. Chi-Square test was used for assessment of the Hardy–Weinberg equilibrium for the distribution of genotypes.
The analysis was performed for all patients as a group. The genotype frequencies for atopics and anaphylaxis groups were firstly compared with healthy controls using a chi-square test (2df) for both AGT and Chymase genes. Genotype subgroups with AACVS were compared with CRA and Terr’s traditional anaphylaxis grades 1–4. The level of significance was adjusted by the Bonferroni correction for multiple comparisons to avoid false-positive findings. For example, 10 comparisons for the paired gene analysis gave a statistical significance p valve of 0.05/10=0.005.
Bi-Allelic Pairing Of Genotype Patterns For The Groups
The pattern of the shared genotypes (AGT/CMA, AGT/ACE and CMA/ACE) for the groups was examined by Chi-Square analysis with significance determined by the Bonferroni correction and indicated in tables.
Tri-Allelic Ensembles For The 3 Commonest Gene Patterns (AGT, Chymase And ACE)
The 2 main groups HC and anaphylaxis (with AACVS) were examined for tri-allelic patterns.60 The 27 gene combinations were generated, limiting reliable statistical analysis for all combinations together, Figure 2 shows the tri-allelic pattern for HC and anaphylaxis. By selecting the 6 commonest tri-allelic gene patterns seen in HC and anaphylaxis (that accounted for 47–58% of all subjects), analysis by Pearson’s Chi-square testing could be used as shown in Table 7.
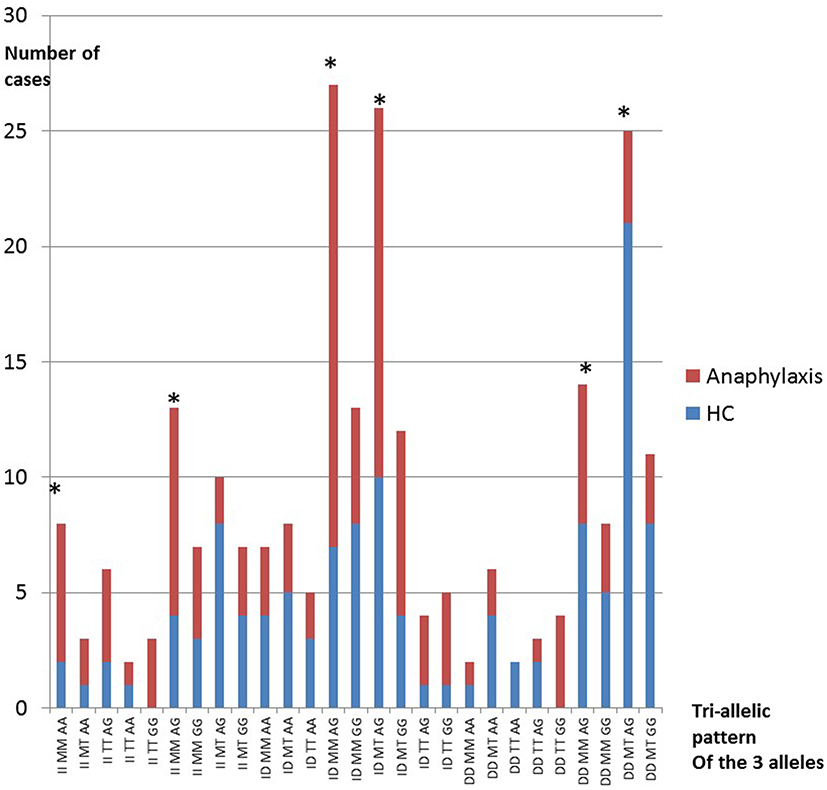 |
Figure 2 Tri-allelic ensemble of the 27 genes combinations of AGT, CMA1 and ACE for healthy controls (HC) and anaphylaxis.Note: *Genes in the analysis. |
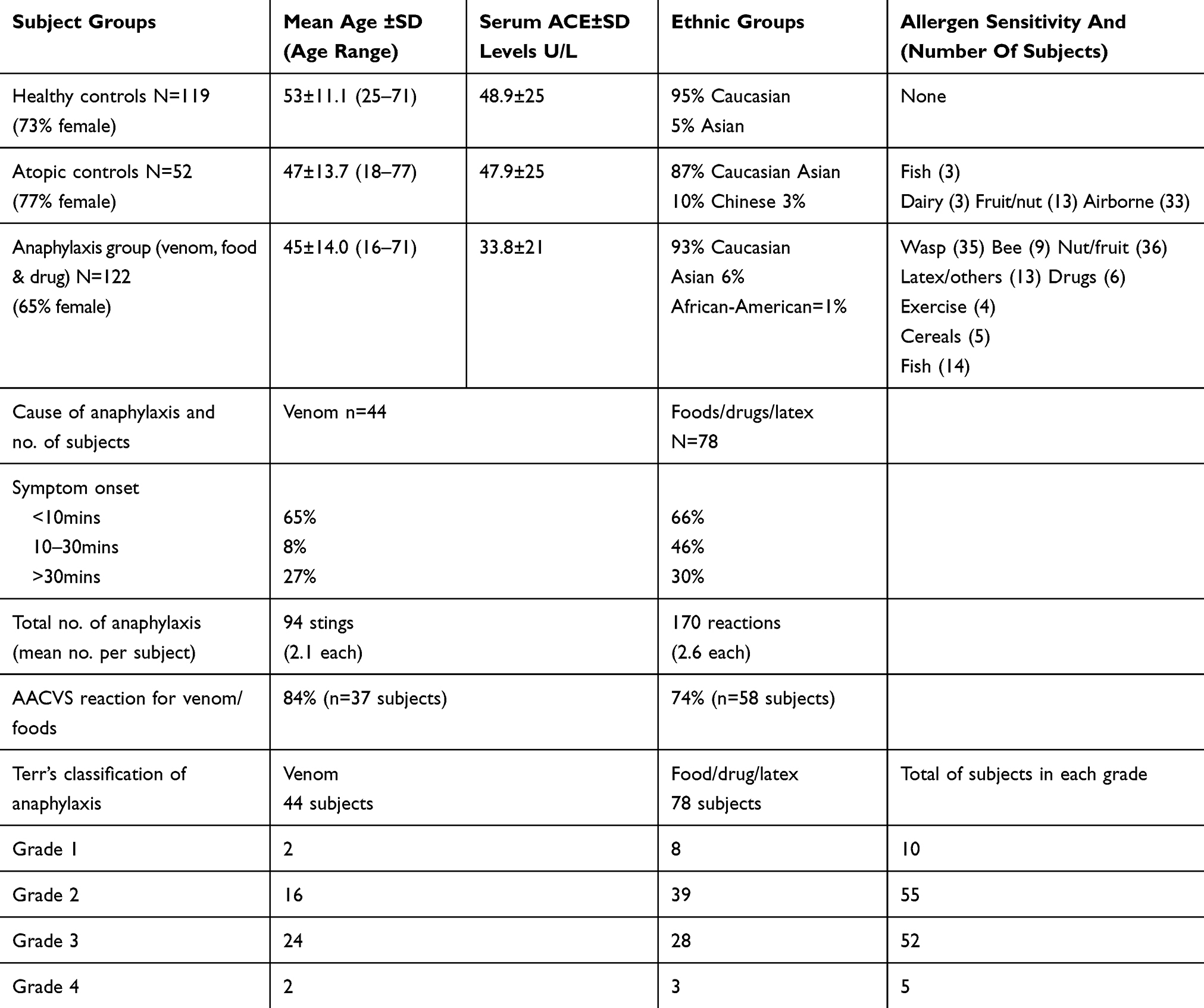 |
Table 1 Demographics Of Subjects |
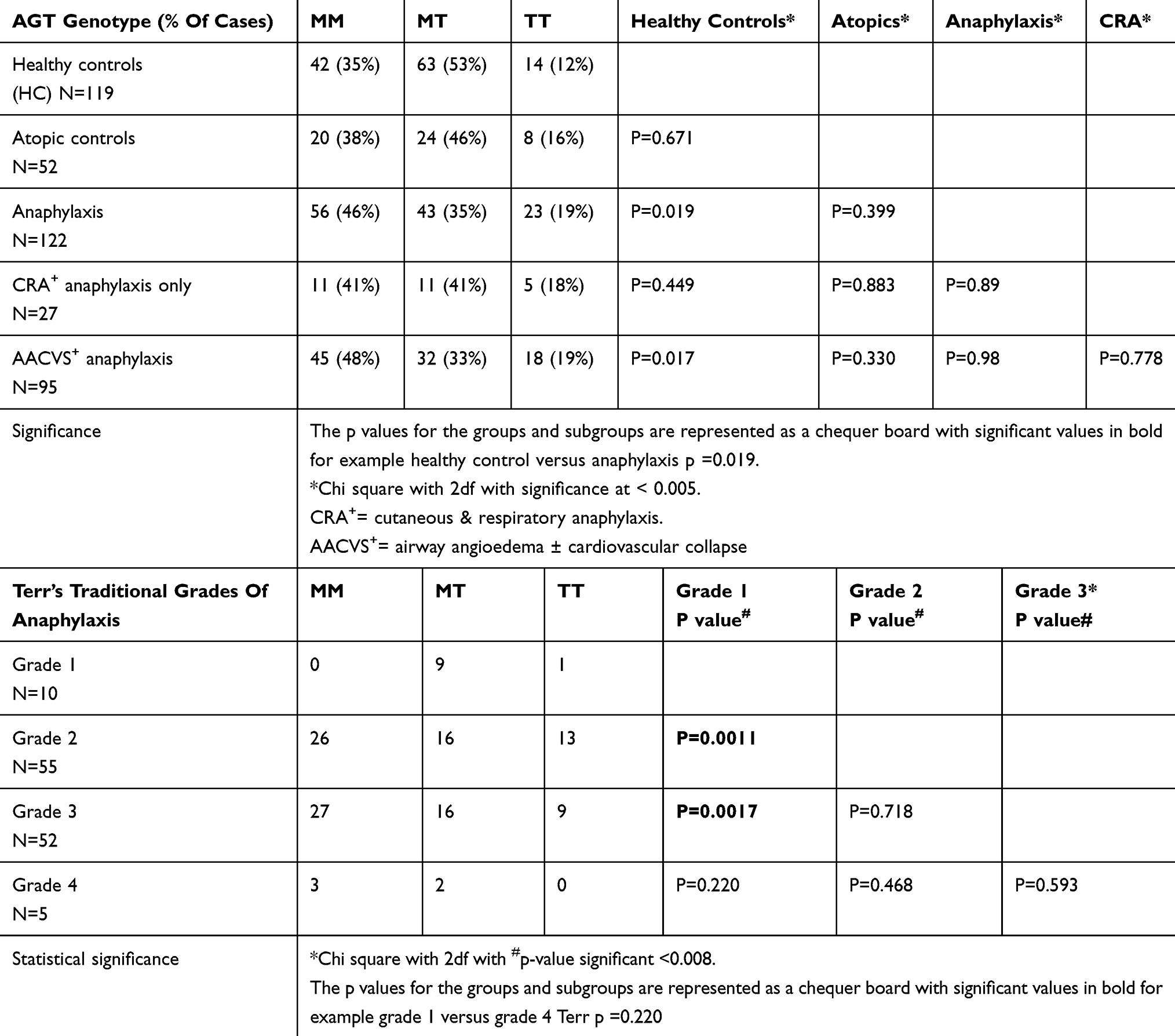 |
Table 2 Angiotensinogen Genes For The Groups And Terr’s Classification |
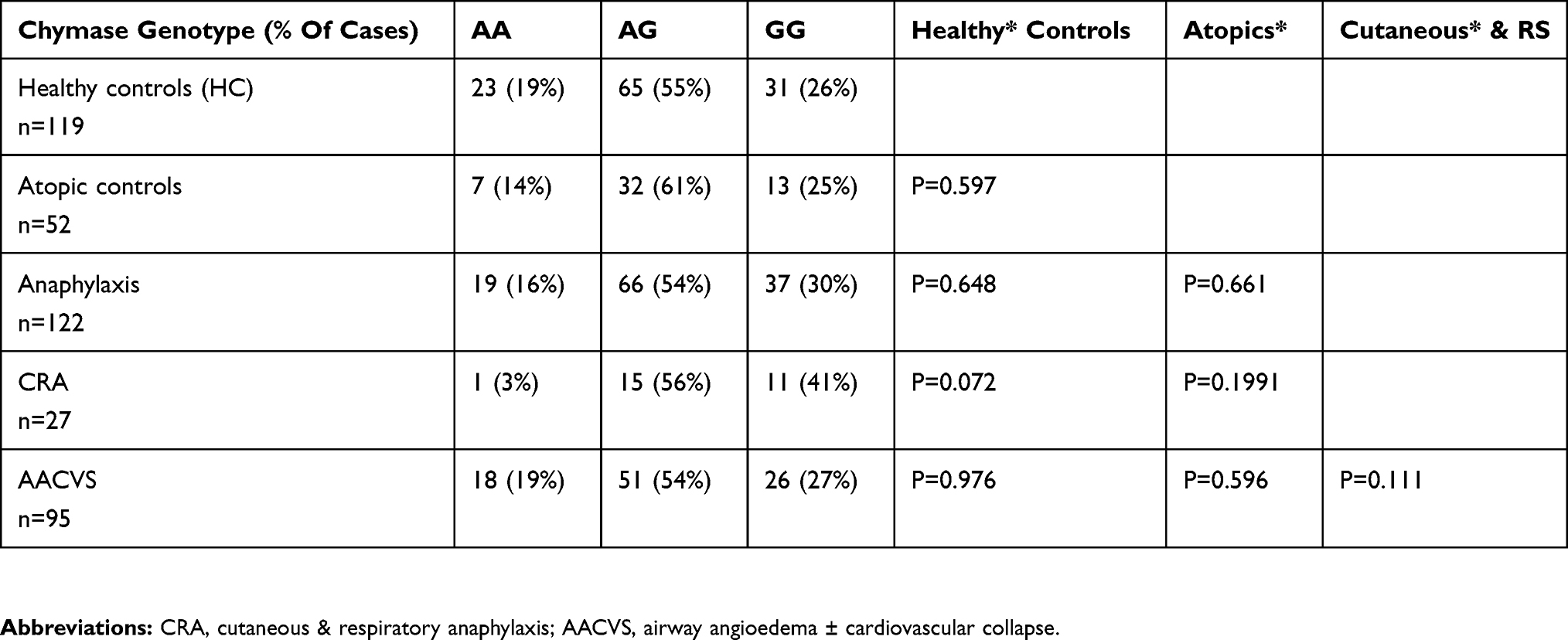 |
Table 3 Chymase CMA1-1903 Genotype Frequency For The Groups |
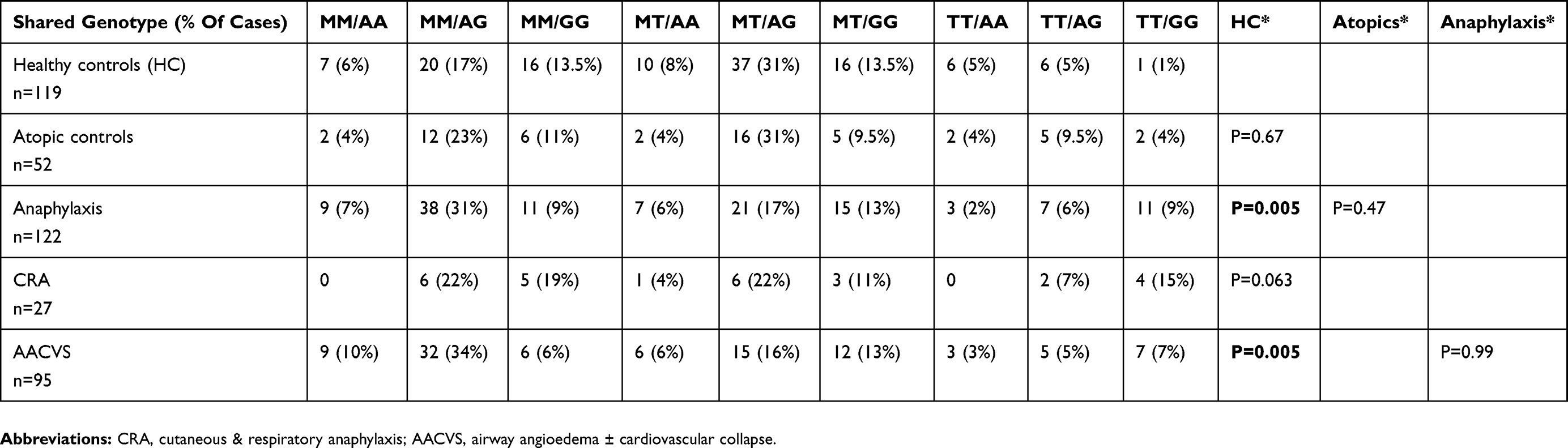 |
Table 4 Chymase CMA-1 A1903G And AGT M235T Bi-Allelic Pairing For The Groups |
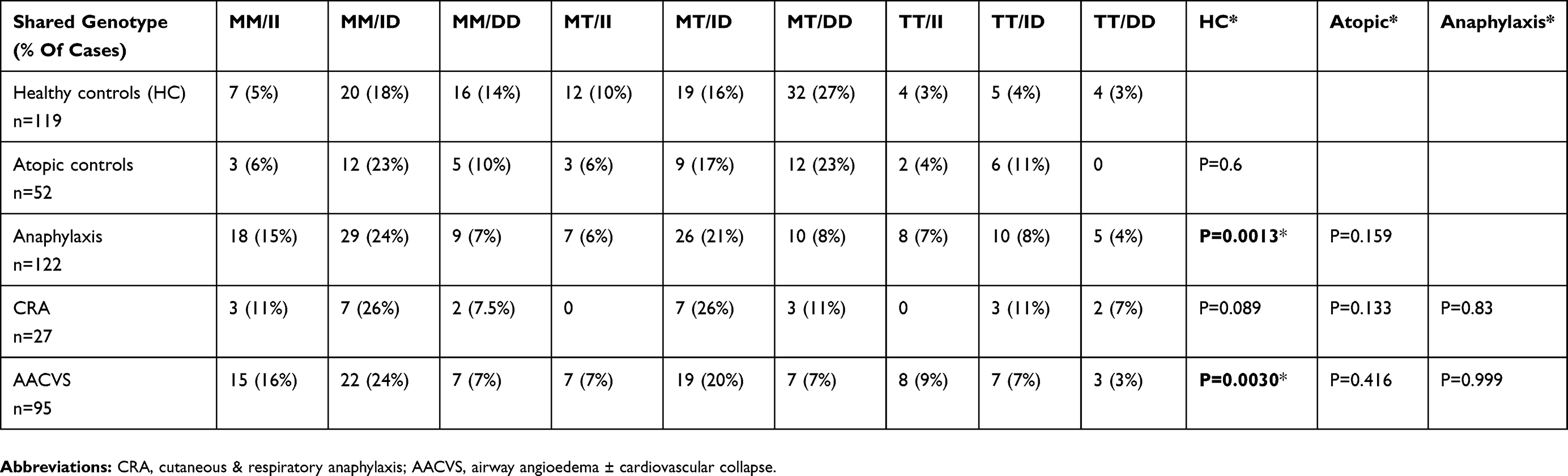 |
Table 5 AGT M235T And ACE I/D Gene Bi-Allelic Pairing For The Groups |
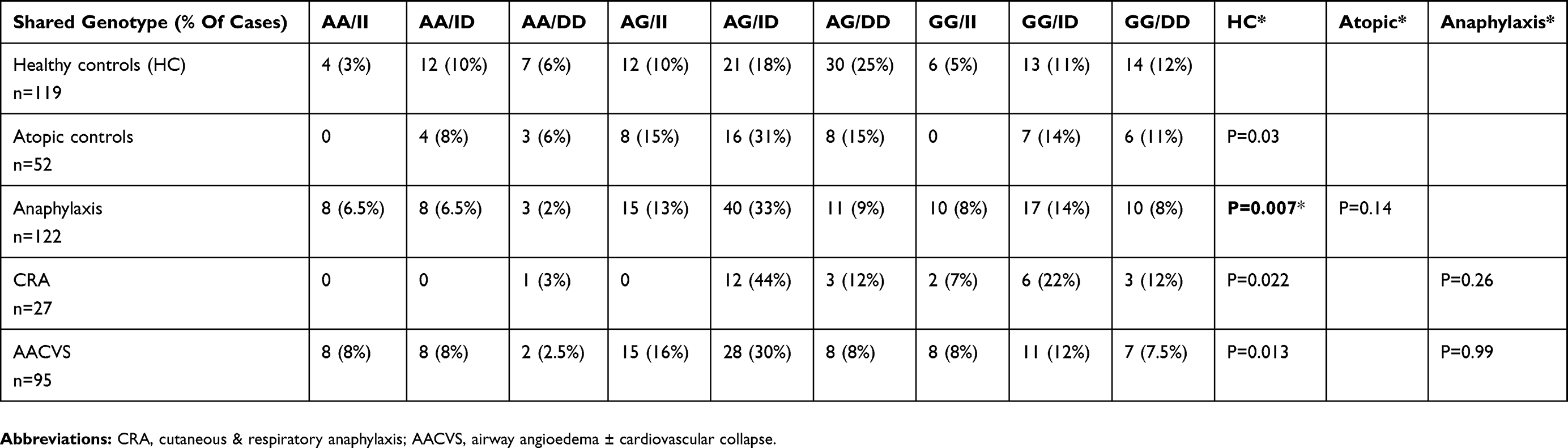 |
Table 6 Bi-Allelic Pairing Of Chymase CMA-1 A1903G And ACE I/D Gene Combinations For The Different Groups |
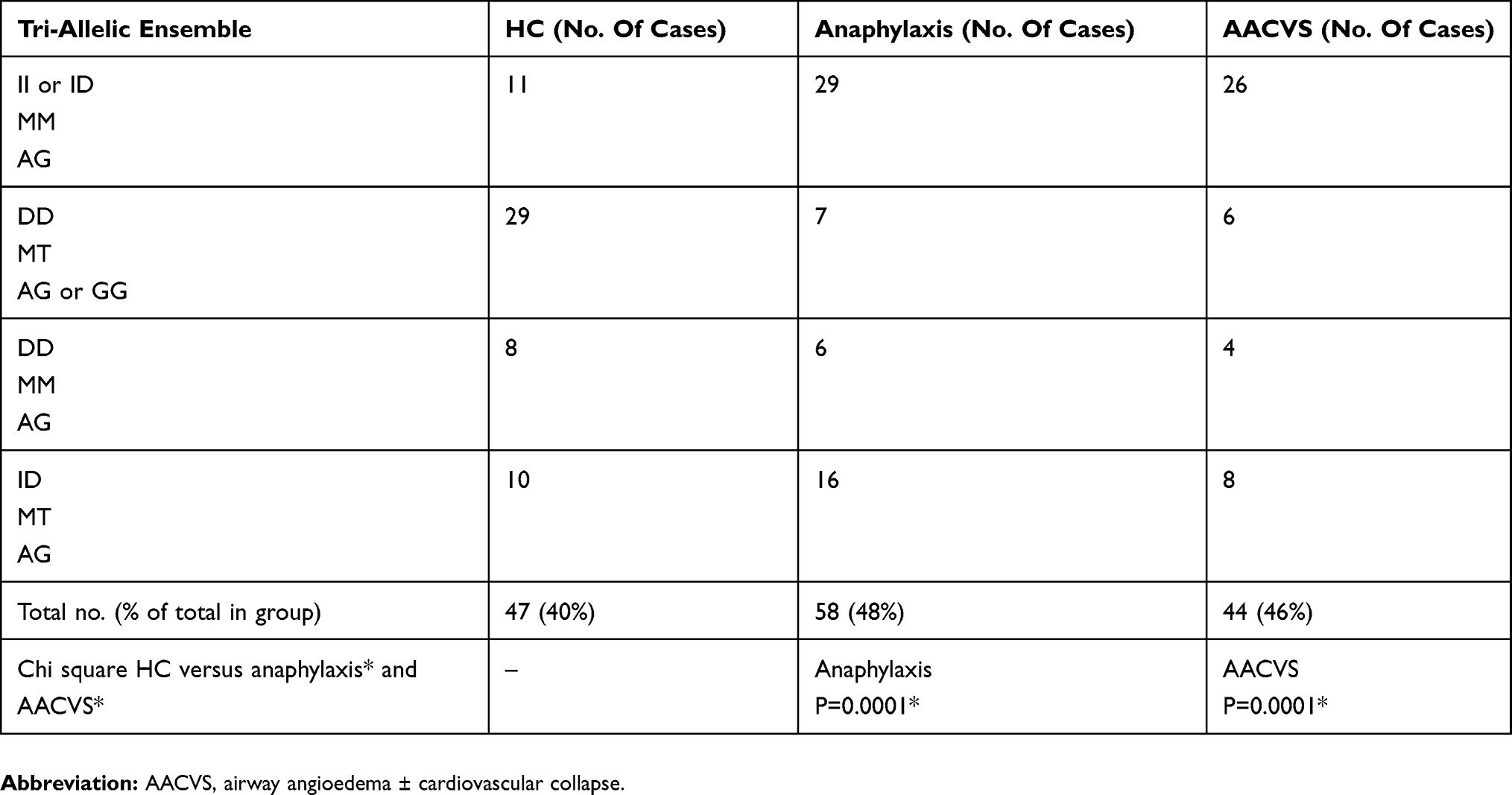 |
Table 7 Ensemble Of 6 Common Gene Combinations |
Results
Table 1 shows the demographics of the 3 main groups: includes ethnic groups, allergen sensitivity, total number of anaphylaxis events and time to onset (Table 1). Terr’s grading 1–4 for anaphylaxis (see Figure 1) and the percentage of cases that involved AACVS (airway angioedema and cardiovascular collapse) is shown. The female predominance of the group reflects that reported in the UK.4
Table 2 shows angiotensinogen (AGT) gene frequency for the 3 groups. In our healthy controls, the AGT gene frequency is entirely consistent with European data (MM 30%, MT 49%, TT 21%).64
There were no statistical differences in AGT gene patterns between HC and atopics (Table 2). Anaphylaxis patients, however, showed a predominance of the MM genotype in 46%, of subjects (p=0.019) with a reduce MT gene frequency compared to HC. This difference was most marked in the AACVS group where the MM frequency was 48% (p=0.017 but did not reach significance). The MT/TT gene pattern predominated in our HC and atopic subjects with 50–65% having 1 or 2 T-alleles compared with only 36% in anaphylaxis.65
MM genes are associated with 20% lower plasma AGT levels, which is the precursor for AI and AII generation and results in lower BP through lower activity of the angiotensin system. It is therefore more likely to produce hypotension than MT or TT. T-alleles are strongly linked to diastolic hypertension, left ventricular hypertrophy and myocardial infarction especially in European Caucasians.45,66–70
For Terr’s traditional grading, MM genes increased significantly from grade 1 (0%), grade 2 (47%), grade 3 (52%) and grade 4 (60%) but 5 subjects only. Chi-squared analysis showed a significant increase in MM frequency between Terr grade 1 versus 2 (p=0.0011) and Terr grades 1 versus 3 (p=0.0017) with significance at p=0.008 as shown in Table 2.
Table 3 shows the chymase gene frequency (CMA1-1903) for the 3 groups.
The HC group was consistent with the European population data for the Chymase gene A1903G (AA=17%, AG=52%, GG=29%).65 There were no statistical differences between HC, atopics and anaphylaxis including subgroups (Table 3). For Terr’s classification, the AG genotype was commonest and increased through the Terr’s grades 2–4 (44%, 61%, and 80%, respectively) data not shown but did not reach significance. The AG frequency was equal across the 2 main groups (HC and anaphylaxis) at 53% and 54% although higher at 62% of all atopics. The data does not differ from the expected frequency of this gene at 52% in the European population.
G alleles are very strongly associated with hypertension and higher IgE blood levels, and hypertensive risk. There is increasing evidence that alpha-2 macroglobulin captures released mast cell chymase, and enters the circulation where chymase contributes to 80% of AII generation within the circulation. No data is available from the literature on AII levels for the Chymase CMA1-1903, despite its strong association with hypertension. In our previous study, we had measured serum ACE and renin as described.49 Renin levels did not vary between the Chymase polymorphism, but in anaphylaxis serum ACE levels ± S.D. (U/L) were lower in the AG genotype (39± 19) compared with AG of HC (47± 28), but similar for GG in anaphylaxis and HC (47± 21 and 48± 41, respectively). This difference did not reach significance but may suggest a yet unrecognised cross-regulation between ACE and Non-ACE activity in those prone to anaphylaxis.
Table 4 shows the paired (bi-allelic) gene frequency for AGT and CMA I-1903 genes.
When the AGT (M/T) and chymase genes (A/G) were paired for each individual within the 3 main groups (+ anaphylaxis sub-groups); a statistically significant difference in gene sharing between HC and anaphylaxis occurred (Table 4). This showed statistically significant increased pairing of AG/MM in anaphylaxis. This was not seen for Chymase alone, although the Terr’s grading had suggested increased AG genotypes within grades 2–4. MM genotypes alone had been increased in anaphylaxis (46%), but were not significantly different from HC (35%).
The MM/AG gene frequency in anaphylaxis subjects (including AACVS) was doubled (17% in HC to 31% in anaphylaxis; p<0.005). This was significantly different from HC, atopics and CRA where MT/AG was 31%, 31%, 22% respectively compared with 17% in anaphylaxis. The combination of AGT with chymase had increased the statistical significance between the HC and anaphylaxis groups that had not been reached for either gene polymorphism alone.
The MM pattern would reduce circulating angiotensinogen levels by 20% for the anaphylaxis group as discussed previously, while the presence of T alleles is associated with higher BP responses and AGT levels. Blood pressure elevation is also associated with the number of G alleles.71,72
Table 5 shows the paired (bi-allelic) gene frequency for AGT and ACE genes.
When AGT (M/T) and ACE (I/D) genes were paired for each individual subject and their group (Table 5), anaphylaxis differed significantly from the HC, atopics and CRA groups due to an increased MM/II and MM/ID gene sharing (p=0.0013). These are gene polymorphisms that give lower AGT levels and lower ACE activity, respectively, with a tendency to lower BP and potentially angioedema from reduced BK catabolism. This gene pattern also included AACVS (p=0.003 with significance <0.005).
HC, atopics and CRA showed increased MT/DD and MM/DD gene sharing consistent with higher activity of the renin-angiotensin system. Higher Serum ACE is very strongly associated with the DD genotype giving higher BP levels with increased BK catabolism that would reduce a tendency to angioedema. Equally, the presence of a T-allele is also associated with higher BP through genetically determined higher circulating levels of AGT.
Table 6 shows the paired (bi-allelic) gene frequency for CMA1-1903 and ACE gene.
Chymase (A/G) and ACE (I/D) gene pairing, showed significant difference between anaphylaxis and HC (p<0.0073). There was an increased frequency of II/AG and ID/AG sharing in the anaphylaxis group (Table 6), which would be associated with lower ACE, AI and AII circulating levels and reduced BK catabolism.
Conversely, HC and atopics showed increased DD/AG pattern with DD strongly associated with higher activity of serum ACE and higher blood pressure along with increased BK catabolism. For CRA, the pattern was mixed with 56% showing genes for higher BP (DD or GG), with 44% having the same II/AG pattern seen for anaphylaxis.
Studies clearly show that II and ID genotypes have lower ACE levels and activity with higher basal BK levels.53,54 Interestingly, the potency of intravenous AI is 5× greater in people with the DD genotype. An observation not explained by AII levels nor angiotensin-2 receptor density. DD genes have the highest serum ACE and AII levels with shortest half-life for BK catabolism which may protect against a tendency to hypotension and angioedema.54,55
Figure 2 shows the 3 genotype combinations (Tri-allelic ensembles) for HC and anaphylaxis.60
Twenty-seven possible gene combinations were generated from the 3 alleles. These genes or “tri-allelic patterns or ensembles” are shown in Figure 2 for the HC and anaphylaxis groups only. The 6 commonest tri-allelic gene ensembles from Figure 2 (marked by *) were analysed as shown in Table 7. This confirmed statistically significant differences between HC and anaphylaxis for the 6 commonest combinations (including AACVS). Comparison tri-allelic graphs (data not shown) for HC and atopics and also HC, atopics and CRA showed a similar distribution of gene combinations without any evident differences. Tri-allelic graphs of AACVS versus CRA and atopics showed 4 clear peaks of difference again II/MM/AG and ID/MM/AG for AACVS and while DD/MT/AG and ID/MT/AG for CRAand atopics. The findings support the bi-allelic gene combinations already demonstrated.
In anaphylaxis, the tri-allelic genes patterns were predominately II or ID/MM/AG.
While in HC (including atopics and CRA) they were predominantly DD/MT/AG or GG.
These gene polymorphisms would support differences in BP responses under cardiovascular stress between HC and Anaphylaxis. The atopics and CRA groups despite their allergic disease followed the HC group’s tendency to higher angiotensin activity and higher BK catabolism through their gene pattern.
Discussion
There remains limited understanding of host factors in anaphylaxis despite 2% of the UK adult population carrying adrenaline.2 Many genes for atopy, bronchial hyper-responsiveness and asthma are recognised and form a polygenetic pattern of inheritance, but so far there are no links between individuals with anaphylaxis and any specific “anaphylaxis inducing genes.” Current research is examining food allergy and conformational and sequential antibody-antigen binding sites that may ultimately confirm a difference in IgE binding that could influence levels of histamine and other mediators in these reactions.
Our study demonstrates a potential genetic link between ACE, AGT and CMA gene polymorphisms with IgE-mediated anaphylaxis to food, venom and drugs, particularly for reactions that include cardiovascular collapse and severe angioedema.
The subdivision of our anaphylaxis patients into AACVS and CRA fits more closely with the Australian Classification of Anaphylaxis (life threatening) with cardio-respiratory effects, laryngeal oedema and hypotension or generalised allergic reactions (non-life threatening) that involve mainly cutaneous and abdominal symptom.15 AACVS reactions could be consistent with lower activity of the renin-angiotensin system through effects on ACE, AI, AII, BK and angiotensinogen levels that may encourage hypotension with angioedema in severe allergic reactions involving endothelial nitric oxide (eNO) and its known interactions with AII.12
The classical view of the RAS is that of an endocrine system in which AII is generated within the circulation and responsible for salt and water balance and blood pressure regulation via multiple target receptors (Figure 3). Multiple gene polymorphisms of RAS have been extensively examined for associations with hypertension in a large sample of sibling pairs.60 Tri-allelic gene ensembles were examined for predisposition to hypertension, with the D alleles of ACE and G of Chymase clearly linked to idiopathic hypertension with normal plasma renin and aldosterone levels. M235T was not examined but the T genes are clearly linked to hypertension in many other studies.60,63
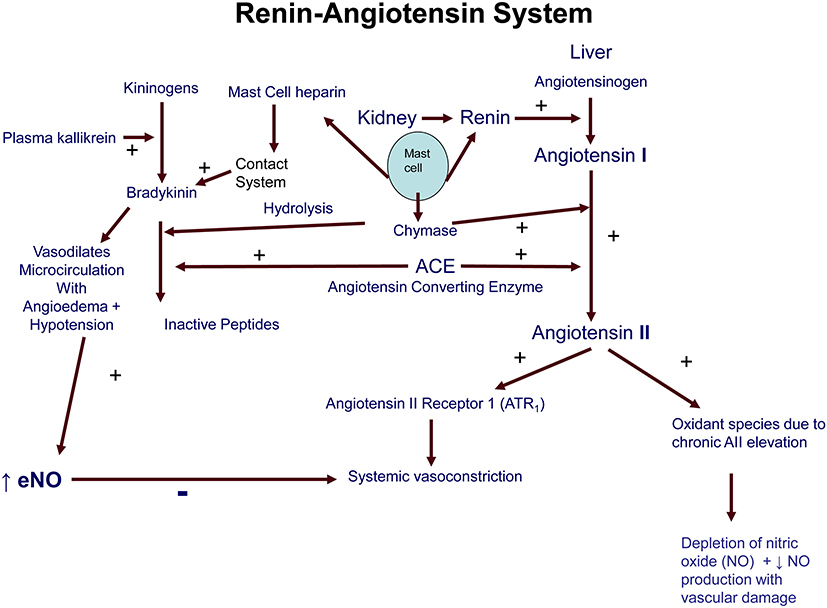 |
Figure 3 Diagram of the renin-angiotensin-aldosterone system. |
The human genome project has shown that 30–50% of BP variance is attributable to genetic heritability with 50% affected by environment. Although multiple genes contribute a 1–2mm increases in systolic and diastolic pressure, genes for the renin-angiotensin system, kallikrein, nitric oxide and natriuretic peptide B feature strongly in the analysis of relevant genes.73 Previous work in anaphylaxis and the RAS has focused on venom-induced anaphylaxis and its association with the ACE and AGT genes. Niedoszytko examined ACE gene polymorphisms in 30 patients with insect venom allergy of Grade III or IV severity on the Mueller scale. They found ID or II genotypes in 80% of subjects.47 Studies clearly show that II and ID genotypes have 50% lower ACE levels and activity with higher basal BK levels that could explain their findings.53,54 A recent study of exercise-induced anaphylaxis to wheat also confirmed increased II or ID gene polymorphisms of ACE in subjects with the condition compared with a very large control group.74 Likewise, our prior study of ACE genes found II/ID genes linked to anaphylaxis involving hypotension and angioedema.49
Our findings for AGT show the MM genotype to be increased in anaphylaxis (46%), but significant only for Terr’s grades 2+3 relative to grade 1. Prior data for venom anaphylaxis also showed an MM− gene frequency of 29–39% in grade 3 and 4 reactions, with 17% in healthy controls.47
M− alleles are reported to be more frequent in asthma, allergic rhinitis and atopic dermatitis.65 M235T genes predict AGT levels from which AI and AII are generated. Since the MM genotype has 20% lower angiotensinogen levels this is likely to produce more hypotension in cardiovascular stress. In contrast, the T alleles have the highest blood levels of AGT, AI and AII levels; with the number of T-alleles linked to diastolic hypertension, left ventricular hypertrophy and myocardial infarction especially in European Caucasians.45,66–69 The MT/TT gene pattern predominated in our HC and atopic subjects with 62–65% having 1 or 2 T-alleles.
When AGT was paired with ACE, a significant increase in bi-allelic sharing of MM/II or MM/ID for anaphylaxis especially AACVS occurred again, supporting the lowered activity of RAS as already reported for venom anaphylaxis.44,46,47,49 HC favoured the DD/MT combinations consistent with greater RAAS activity and hypertension. As mentioned previously, the effects of intravenous AI is 5 times more potent in people with the DD genotype, which is not explained by AII levels or ATR1-receptor density alone, suggesting additional unrecognised factors. DD genes have the highest serum ACE and AII levels, with shortest half-life for BK that could protect against a tendency to hypotension, nitric oxide activation and angioedema in anaphylaxis.54,55,75,76
Chymase (CMA1-1903) alone showed no statistical differences between the 3 groups and subgroups. However, its pairing with AGT showed significantly increased MM/AG in anaphylaxis and AACVS, while HC showed increased MT/AG or GG pairing.
Likewise, the pairing of Chymase and ACE also showed increased AG/II or AG/ID for anaphylaxis compared with AG/DD genes in HC.
The AG pattern of the Chymase gene was consistently observed in our analysis, but unlike ACE and AGT, no data are available in the literature that links CMA1-1903 with measured AII levels despite the G-allele link to hypertension and GG-homozygosity being strongly associated with cardiovascular disease. As outlined in “Results“ section, serum ACE levels were lower in the AG anaphylaxis group compared with AG healthy controls, and may suggest a yet unrecognised cross-regulation between ACE and Non-ACE activity in those prone to anaphylaxis.
When all 3 genes were examined for tri-allelic patterns, the same findings were confirmed with the generation of greater p values for statistical significance. The genes for low AGT and ACE activity (MM/II/AG or MM/ID/AG) were seen in anaphylaxis and AACVS and associated with the Chymase AG polymorphism; While higher RAS activity was seen in HC, CRA and atopics (MT/DD/AG or GG) and associated with AG or GG.60 This suggests a polygenetic contribution from genes of RAS that could influence hypotension and angioedema in allergic reactions. The AG frequency for the Chymase gene in the general population is 52% from European data, and this was unchanged between HC (53%) and anaphylaxis (54%) but increased in atopics (62%). This AG polymorphism of chymase was therefore equally associated with anaphylaxis and HC and may reflect its gene frequency although it had a significant influence on the tri-allelic pattern and its differences between HC and anaphylaxis. The role of the serine protease Chymase in BP control and anaphylaxis is not well understood and warrants a brief review in the light of these findings.
Chymase is generated by mast cell degranulation in anaphylaxis. The non-ACE generating system of Chymase involves its secretion from mast cells and its capture by alpha-2 macroglobulin (alpha-2M) which facilitates entry to the circulation.77,78 Data show Chymase to be responsible for 80% of our circulating AII levels and unaffected by ACE-inhibitors and angiotensin-receptor blockers.79,80 The mast cells of the skin, heart and blood vessels are the main source of Chymase, with only 7% from bronchial mast cells.80–83
Chymase is normally stored in secretory granules bound to its inhibitor heparin and was thought to mediate only local AII generation in allergic reactions due to rapid inhibition by other serine proteases.77–79 When captured by alpha-2M was recognised, this theory changed.78,84,85
Upon capture, access to small molecules like AI continues, giving an important role in systemic BP control.77,81,86 Standard chymase assays do not detect captured chymase due to shielding of the antibody site.78,87 Newer assays do, and suggests a steady leak of chymase from mast cells as responsible for tonic AII effects on vascular smooth muscle.78,87,88 Homozygosity for G alleles is assumed to give higher AII production accounting for its strong association with hypertension, atherosclerosis, atrial fibrillation and reduced left ventricular function.89–91
In anaphylaxis, blood Chymase levels increase after 1hr and remain raised for 8–24hrs.87,92–94 An autopsy study showed mean Chymase blood levels of 89.8ng/mL in anaphylaxis compared with <3ng/mL in cases without anaphylaxis.95–97
After mast cell release, chymase can inactivate peptides such as BK, kallikrein and substance P, while its co-released heparin activates the “contact system” with auto-activation of factor XII giving Factor X-mediated further BK formation94,98 (Figure 3). Heparin is recognised in human anaphylaxis and animal models, where it is linked to both the circulating BK levels and the severity of anaphylaxis.99–101
Mice deficient in Factor X and the BK-receptor do not develop hypotension in systemic anaphylaxis.102
Raised IgE levels are associated with CMA 1-1903-GG (297 KU/L) and also AG (144KU/L) polymorphisms that may influence the level of mast cell activation with secondary effects on the contact system and BK generation.103 The reason for the significant association of G with raised IgE levels is not understood but thought to involve regulation of IgE. The G-allele is also linked with bronchial asthma and chronic dermatitis particularly for GG homozygosity in Caucasians.104–106
The chymase AA genotype has the lowest IgE levels (48.4KU/L) and no association with hypertension and did not feature significantly in our data.89,104,106 Greater understanding of the Chymase CMA1-1903 promotor gene polymorphism is required along with its effects on non-ACE generated AII production and IgE levels. Since chymase potentiates histamine and IgE levels, mast cell activation could give increased BK levels in those with a G-allele.93,96,105–108
Human mast cells contain renin so local AI production can occur; anaphylaxis is associated with the release renin and histamine from mast cells in guinea pig models.109,110 The drug Aliskiren is an orally active non-peptide direct-inhibitor of the enzyme renin used in hypertension. It was noted on post-marketing surveillance to show the occurrence of anaphylaxis and angioedema in some patients with histories of hypersensitivity.111 This effect was deemed similar to other medicinal products acting on the renin-angiotensin system, including the severe anaphylaxis in patients on ACE-inhibitor drugs seen especially with hymenoptera venom field stings.112
Anaphylaxis research is predominately focused upon animal models of IgE-mediated reactions due to the difficulty of such studies in man. In man, IgE levels alone do not explain a patients susceptibility to anaphylaxis, as some subjects experience near-fatal anaphylaxis with low levels of circulating allergen-specific IgE, while others with higher levels have no symptoms on exposure, suggesting other pathways play a part that are yet to be understood.113
Studies in rodents suggest that platelet-activating factor (PAF) is involved in anaphylaxis-induced cardiovascular collapse and can be prevented by a PAF antagonist.114,115 In PAF-knockout mice, no hypotension or death in anaphylaxis occurs. PAF levels have been measured in man, and are reduced immediately following anaphylaxis, but are normal if measured away from the event.116,117 Anaphylaxis in wild-type mice can be blocked by anti-histamines and the PAF antagonists, suggesting a synergistic effect between histamine and PAF. Animal models examining the depletion of monocytes/macrophage in anaphylaxis suggests that they may also be the source of PAF.113,118,119
PAF, like BK maybe acting through endothelial nitric oxide (eNO) when mediating its hypotensive effect in anaphylaxis, as the NO-inhibitor L-NAME (L-Nitro Arginine Methyl Ester) can block severe hypotension induced by PAF and prevent shock.114–117 Endothelial NO is now of interest in murine models of anaphylaxis, and chronic blockade of eNO increases the expression of mRNA for renin, ACE and ATR1- receptors in the aorta with the risk of hypertension.120,121 In man, eNO is involved in the regulation of the micro-circulation.122 Activation of RAS enhances eNO, producing a vasodilator effect to protect against AII vasoconstriction.50 Local administration of an NO-synthase inhibitor into human brachial arteries causes a dose-dependant fall in forearm blood flow, confirming that it participates in vascular tone. A functional feedback exists between AII and eNO under normal conditions. AII and eNO interact at the level of the endothelium, where ACE converts AI to AII. eNO has been shown to downregulate the synthesis of ACE in the endothelium, as well as ATR1- receptors in vascular smooth muscle, thus decreasing AII production and its action. Endothelial NO can be activated by BK, substance P and physical shear forces on the blood vessel walls. BK exerts a powerful arterial vasodilatation through NO synthase; while ACE degrades BK. ATR2- receptor stimulation by AII can enhance eNO release.
Vascular production of superoxide radicals is enhanced by AII and accompanied by impairment of eNO vasodilatation. AII causes oxidant damage to the vascular system via activation of NADPH oxidase. The oxidant species leads to depletion of eNO and blood vessel injury with vascular remodelling and cardiovascular disease. AII generated oxidants also inactivate extracellular NO. Higher basal AII levels are shown to reduce eNO levels.9 An imbalance in AII-NO, rather than the absolute concentration of either is what determines cardiovascular physiology and pathophysiology. Oestrogens have been shown to increase eNO levels probably through effects on eNO synthase and so protect the circulation in women leading to the better renal function and resistance to renal injury.121,123,124 Individuals with the DD genotype of ACE and T-alleles of AGT and G-alleles of Chymase may be more protected from the risk of cardiovascular collapse through chronically depleted eNO secondary to chronically raised AII levels.113,121,125–127. The converse may be true for the “I” genotypes of ACE, where eNO levels are shown to be higher giving benefit at high altitude from eNO- maintained vasodilatation of the microcirculation that is not seen with DD genotype.124 Likewise, MM would have lower AII levels and higher eNO activity.
Our data may suggest that individuals with the DD-ACE genes and T-alleles of AGT and the GG-alleles of Chymase would be more protected from cardiovascular collapse from chronically higher AII levels suppressing the eNO system.121,125 This may not be true for the ID/II and MM genotypes where AII levels are lower.113,124–130
Assessment of other gene polymorphisms of nitric oxide synthase and renin in our patients could be interesting.
Conclusion
Clearly, there are complex changes in the RAS between birth and adulthood, some of which could explain the increased hypotension and angioedema to allergic mediators manifesting more in childhood and pre-menopausal women. The influence of the RAS genes may be disguised in childhood by the systems immaturity and changing hormone levels that once stabilized in adulthood offers easier identification of gene polymorphisms and their influence in anaphylaxis. This data in adults suggest an influence from several genes involved in the generation of AII and the catabolism of BK as relevant; With the gene patterns for lower ACE, AI, AII and BK seen in AACVS reactions possibly reflecting higher eNO activity. In animal studies, eNO appears to be the main mediator of shock, and AII-NO balance controls the microcirculation. As with all gene studies, confirmation requires larger case numbers and controls while environmental and epidemiological studies can address other reason for the increasing incidence of anaphylaxis in adults and children.
Abbreviations
ACE, angiotensin-converting enzyme; HC, healthy controls; AACVS, airway angioedema & cardiovascular collapse; CRA, cutaneous & mild respiratory anaphylaxis; BK, bradykinin; AI, angiotensin-1; AII, angiotensin-2; RAS, renin-angiotensin system; AGT, angiotensinogen; CMA-1 1903, Chymase gene polymorphisms; alpha-2M, alpha-2-macroglobulin; IgE, immunoglobulin E; BP, blood pressure; BMI, body mass index; DNA, deoxyribonucleic acid; ATR1- receptor, angiotensin-II receptor-1; ACEI, angiotensin-converting enzyme inhibitor; eNO, endothelial nitric oxide.
Acknowledgments
The authors thank Irina Chisster for statistical analysis and advice. This study received funding from the R&D department at St Helier Hospital. A Warner is currently affiliated with Allergy UK Charity and N Sumar is currently affiliated with Centre for Clinical Education, Institute of Medical & Biomedical Education, St Georges University of London.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Terr AI. Anaphylaxis. Clin Rev Allergy. 1985;3:3–23.
2. Mullins RJ, Wainstein BK, Barnes EH, Liew WK, Campbell DE. Increases in anaphylaxis fatalities in Australia from 1997–2013. Clin Exp Allergy. 2016;46:1099–1110. doi:10.1111/cea.12748
3. Tejeclor A, Lonson MA, Moromoro M, Mugica Garcia MV. Epidemiology of anaphylaxis. Clin Exp Allergy. 2014;45:1027–1039.
4. Sheikh A, Alves B. Age, sex, geographical and socio-economic variations in admissions for anaphylaxis: analysis of four years of English hospital data. Clin Exp All. 2001;31:1571–1576. doi:10.1046/j.1365-2222.2001.01203.x
5. Bjornsson I, Graffeo LS. Improving diagnostic accuracy of anaphylaxis in acute care setting. West J Emerg Med. 2010;11(5):456–461.
6. Lee J, Vadas P. Anaphylaxis: mechanisms and management. Clin Exp Allergy. 2011;41:923–938. doi:10.1111/j.1365-2222.2011.03779.x
7. Pumphrey R, Stanworth SJ. The clinical spectrum of anaphylaxis in north-west England. Clin Exp All. 1996;26(12):1364–1370. doi:10.1111/j.1365-2222.1996.tb00537.x
8. Pumphrey RS. Lessons for management of anaphylaxis from a study of fatal reactions. Clin Exp Allergy. 2000;30:1144–1150. doi:10.1046/j.1365-2222.2000.00864.x
9. Kajiwara N, Sasaki T, Bradding P. Activation of human mast cells through the platelet-activating factor receptor. J Allergy Clin Immunol. 2010;125:1136–1137. doi:10.1016/j.jaci.2010.01.056
10. Finkleman FD, Rothenburg ME, Brandt EB, Morris SC, Strait RT. Molecular mechanisms of anaphylaxis; lessons from studies with murine models. J Allergy Clin Immunol. 2005;115:449–457. doi:10.1016/j.jaci.2004.12.1125
11. Umsasunthar T, Leonardi-Bel J, Hodes M, et al. Incidence of fatal food anaphylaxis in people with food allergy: a systematic review and meta-analysis. Clin Exp Allergy. 2013;43:1333–1341. doi:10.1111/cea.12211
12. Lieberman P, Simons FER. Anaphylaxis and cardiovascular disease: therapeutic dilemmas. Clin Exp Allergy. 2015;45:1288–1295. doi:10.1111/cea.12520
13. Rutkowski K, Dua S, Nasser S. Anaphylaxis: current state of knowledge for the modern physician. Postgrad Med J. 2012;88:458–464. doi:10.1136/postgradmedj-2011-130634
14. Berlin MC. Pathogenesis of IgE mediated food allergy. Clin Exp Allergy. 2015;45:1483–1496. doi:10.1111/cea.12598
15. Braganza SC, Acworth JP, McKinnon DRL, Peake JE, Brown AFT. Paediatric emergency department anaphylaxis: different patterns from Adults. Arch Dis Child. 2006;91:159–163. doi:10.1136/adc.2004.069914
16. Macdougall CF, Cart AJ, Clover AF. How dangerous is food allergy in childhood? The incidence of severe and fatal allergic reactions across UK and Ireland. Arch Dis Child. 2002;86:236–237. doi:10.1136/adc.86.4.236
17. Smith PK, Hourihane JO, Lieberman P. Risk multipliers for severe food anaphylaxis. World Allergy Organ J. 2005;8:30. doi:10.1186/s40413-015-0081-0
18. Gupta R, Sheikh A, Strachan DP. Time trends in allergic disorders in the UK. Thorax. 2007;62:91–96. doi:10.1136/thx.2004.038844
19. Vetander M, Protudjer JLP, Liija G, et al. Anaphylaxis to foods in a population of adolescents: incidence, characteristics and associated risks. Clin Exp Allergy. 2016;46:1575–1587. doi:10.1111/cea.12842
20. Delage C, Irey NS. Anaphylactic deaths: a Clinicopathologic study of 43 cases. J Forensic Sci. 1972;17:525–540. doi:10.1520/JFS10141J
21. Greenberger PA, Rotskoff BD, Lifschultz B. Fatal anaphylaxis: post-mortem findings and associated comorbid diseases. Ann Allergy Asthma Immunol. 2007;98:252–257. doi:10.1016/S1081-1206(10)60714-4
22. Stumpf JL, Shehab N, Patel AC. Safety of ACE-inhibitors in patients with insect venom allergies. Am Pharmacother. 2006;40:699–703. doi:10.1345/aph.1G295
23. Slade CA, Douglass JA. Changing practice: no need to stop ACE inhibitors for venom immunotherapy. Clin Exp Allergy. 2014;44:617–619. doi:10.1111/cea.12295
24. Londe S. Blood pressure in children under office conditions. Clin Paediatr. 1966;5(2):71–78. doi:10.1177/000992286600500204
25. Strufald MW, Silva EMK, France MC, Puccini RF. Blood pressure levels in childhood:-probing the relative importance of birthweight and current size. Eur J Pediatr. 2009;168:619–625. doi:10.1007/s00431-008-0813-z
26. Strufaldi MW, Silva EM, Franco MC, Puccini RF. Blood pressure in childhood: probing the relative importance of birth weight and current size. Eur J Pediatr. 2009;168:619–625. doi:10.1007/s00431-008-0813-z
27. Pipkin FB, Smales OR, O’Callaghan MJ. Renin and angiotensin levels in children. Arch Dis Child. 1981;56:298–302. doi:10.1136/adc.56.4.298
28. Vlajinac H, Miljus D, Adanja B, Marinkovic J, Sipetic S, Kovec N. Blood pressure levels in 7 to 14 year old Belgrade children. J Human Hypertens. 2003;17:761–765. doi:10.1038/sj.jhh.1001618
29. VanAcker KJ, Scharpe SL, Depreltere JR, Neels HM. Renin-angiotensin-aldosterone system in the healthy infant and child. Kidney Int. 1979;16:196–203. doi:10.1038/ki.1979.121
30. Silva AC, Diniz JS, Filho AR, Santos RAS. The renin angiotensin system in childhood hypertension: selective increase of angiotensin (1-7) in essential hypertension. J Pediatr. 2004;145:93–98. doi:10.1016/j.jpeds.2004.03.055
31. Fiselier TJW, Lunen P, Monnens L, Van Munster JM, Peer P. Levels of renin, angiotensin-I and 2, ACE and aldosterone in infancy and childhood. Eur J Pediat. 1983;141:3–7. doi:10.1007/BF00445660
32. Sassard A, Incent M, Francois R, Cier JF. Plasma renin activity in normal subjects from infancy to puberty. J Clin Endocrinol Metab. 1975;40:524–525. doi:10.1210/jcem-40-3-524
33. Dillon MJ, Ryness JM. Plasma renin activity and aldosterone concentration in children. Bmj. 1975;4:316–319. doi:10.1136/bmj.4.5992.316
34. Slalker HP, Holland NH, Kotchen JM, Kotchen TA. Plasma renin activity in healthy children.The. J Pediatr. 1976;89:256–258. doi:10.1016/S0022-3476(76)80460-X
35. Kruger C, Rauh M, Dorr HG. Immunoreactive renin concentrations in healthy children from birth to adolescence. Clin Chimica Acta. 1998;274:15–27. doi:10.1016/S0009-8981(98)00044-8
36. Beneteau-Burnat B, Baudin B, Morgant G, Baumann FCH, Gilboudeau J. Serum angiotensin-converting enzyme in healthy and sarcoidotic children: comparison with the reference intervals for adults. Clin Chem. 1990;36:344–348.
37. Gallagher PE, Li P, Lenhart MC, Brosnihan KB. Estrogen regulation of angiotensin-converting enzyme mRNA. Hypertension. 1999;33:323–328. doi:10.1161/01.hyp.33.1.323
38. Fogo A, Yoshido Y, Yared A, Ichikawa I. Important of angiogenic action of angiotensin II in the glomerular growth of maturing kidneys. Kidney Int. 1990;38:1068–1074. doi:10.1038/ki.1990.314
39. Franco MC, Casarini DE, Carneiro-Ramos MS, Sawaya AL, Barreto-Chaves MLM, Sesso R. Circulating renin-angiotensin system and catecholamine’s in childhood: is there a role for birthweight? Clin Sci. 2008;114:375–380. doi:10.1042/CS20070284
40. Sippell WG, Dorr HG, Bidlingmaier F, Knorr D. Plasma levels of aldosterone, cortisone, 11-deoxycorticosterone, progesterone, 17-hydroxyprogerone, cortisol during infancy and childhood. Pediatr Res. 1980;14:39–46. doi:10.1203/00006450-198001000-00010
41. Howard PJ, Ambrosuis WT, Tewksbury DA, Wagner MA, Zhou L, Hanna MP. Serum Angiotensinogen concentration in relation to gonadal hormones, body size and genotype in growing young people. Hypertension. 1998;32:875–879. doi:10.1161/01.hyp.32.5.875
42. Giacchetti G, Faloia E, Mariniello B, et al. Over expression of the renin angiotensin system in human visceral adipose tissue in normal and overweight subjects. Am J Hypertension. 2002;15(5):381–388. doi:10.1016/S0895-7061(02)02257-4
43. Staessen JA, Ginocchio B, Wang JG, et al. Genetic variability in the renin-angiotensin system: prevalence of alleles and genotypes. J Cardiovasc Risk. 1997;4:401–422.
44. Herman K, Ring J. The renin angiotensin system and hymenoptera venom anaphylaxis. Clin Exp Allergy. 1993;23(9):762–769. doi:10.1111/j.1365-2222.1993.tb00364.x
45. Herman K, Ring J. Hymenoptera venom anaphylaxis: may decrease levels of angiotensin peptides play a role? Clin Exp All. 1990;20(5):569–570. doi:10.1111/j.1365-2222.1990.tb03151.x
46. Summers CW, Pumphrey RS, Woods CN, Mcdowell G, Pemberton PW, Arkwright PD. Factors predicting anaphylaxis to peanuts and tree nuts in patients referred to a specialist centre. J Allergy Clin Immunol. 2008;121(3):632–638. doi:10.1016/j.jaci.2007.12.003
47. Niedoszytko M, Ratajska M, Jassem E. AGT(M235T), ACE(I/D,I/I,D/D) polymorphism in patients with insect venom allergy preliminary results. Allergy. 2007;62(suppl 83):111. doi:10.1111/j.1398-9995.2007.01456.x
48. Mueller UR. Clinical Presentation and Pathogenesis in Venom Allergy. Stuggart: Gustav Fischer; 1990:35–60.
49. Varney VA, Warner A, Ghosh A, Nicholas A, Sumar N. IgE-mediated anaphylaxis to foods, venom, and drugs; influence of serum angiotensin converting enzyme levels and genotype. 2012. J Allergy. 9. article ID 258145.
50. Dimitropoulou C, Chatterjee A, McCloud L, Yetik-Anacak G, Catravas JD. Angiotensin, Bradykinin and the Endothelium. Hypertension. 2006;176:255–294.
51. Persson K, Safholm ACE, Andersson RGG, Ahlner J. GTN-induced angiotensin-converting enzyme (ACE) inhibition in healthy volunteers is dependent on ACE genotype. J Physiol Pharmacol. 2005;83:1117–1122. doi:10.1139/y05-118
52. Reyes-Engel A, Morcillo L, Aranda FJ, et al. Influence of Gender and Genetic Variability on Plasma Angiotensin Peptides. J Renin Aldosterone Angiotensin Sys. 2006;7(2):92–97. doi:10.3317/jraas.2006.015
53. Jan Danser AH, Deinum J, Osterop APRM, Admiral PJJ, Schalekamp M. Angiotensin I to angiotensin II conversion in the human forearm and leg: effects of angiotensin converting enzyme gene insertion/deletion polymorphisms. Hypertension. 1999;17:1867–1872. doi:10.1097/00004872-199917121-00014
54. Van Dijik MA, Kroon K, Kamper AM, Boomsma F, Danser AHJ, Chang PC. The angiotensin-converting enzyme gene polymorphism and responses to angiotensin and bradykinin in the human foreman. J Cardiovascular Pharmacol. 2000;35(30):484–490. doi:10.1097/00005344-200003000-00020
55. Woods D, Sanders J, Jones A, et al. The serum angiotensin-converting enzyme and angiotensin II response to altered posture and acute exercise, and the influence of ACE genotype. Eur J Appl Physiol. 2004;91:342–348. doi:10.1007/s00421-003-0993-1
56. Zee RYL, Schrader AP, Morris BJ. Effect of angiotensin-converting enzyme genotype on the renin-angiotensin components in hypertensives. Clin Chimica Acta. 1996;252:33–39. doi:10.1016/0009-8981(96)06310-3
57. Sumino H, Ichikawa S, Kanda T, et al. Hormone replacement therapy in postmenopausal women with essential hypertension increases circulating plasma levels of bradykinin. Ajh. 1999;12:1044–1047. doi:10.1016/s0895-7061(99)00094-1
58. Vliagoftis H, Dimitriadou V, Boucher W, et al. Estradiol augments while tamoxifen inhibits rat mast cell secretion. Int Arch Allergy Immunol. 1992;98:398–409. doi:10.1159/000236217
59. Tsuda M, Iwai M, Li J-M, et al. Inhibitory effects of AT1 receptor blocker, olmesartan and estrogen on atherosclerosis via anti-oxidative stress. Hypertension. 2005;45:545–551. doi:10.1161/01.HYP.0000157409.88971.fc
60. Chikhladze NM, Samedova KF, Sudomoina MA, et al. Comparative genetic analysis of different forms of low-renin arterial hypertension. Mol Biol. 2008;42:521–530. doi:10.1134/S0026893308040067
61. Stein U, Grob W. Rapid detection of the hypertension associated Met234 toThr allele of the human angiotensinogen gene. Hum Mol Genet. 1993;2(5):609–610. doi:10.1093/hmg/2.5.609
62. Nalogowska K, Glosnicka BI, Lacka MJ, et al. Angiotensin-2 type-1 receptor A1166c polymorphism is associated with increased risk of pregnancy associated hypertension. Med Sci Monit. 2000;6(3):523–529.
63. Pfeufer A, Osterziel KJ, Urata H, et al. Angiotensin-converting enzyme and heart Chymase gene polymorphisms in hypertrophic cardiomyopathy. Am J Cardiol. 1996;78:362–364. doi:10.1016/S0002-9149(96)00296-2
64. Ortlepp JR, Metmkat J, Mevissen V, et al. Relation between the angiotensinogen (AGT) M235T gene polymorphism and blood pressure in a large homogenous study population. J Human Hypertens. 2003;17:555–559. doi:10.1038/sj.jhh.1001587
65. Holla L, Vasku A, Znojil V, Siskova L, Vache J. Association of 3 gene polymorphisms with atopic diseases. J Allergy Clin Immunol. 1999;103(4):702–708. doi:10.1016/s0091-6749(99)70246-0
66. Joong J, Hyun-ju K, Lee I-K, Chung HT, Lee JH. Association between polymorphisms of the angiotensin-converting enzyme and angiotensinogen genes and allergic rhinitis in a Korean population. Ann Otol Rhinol Laryngol. 2004;113:297–302. doi:10.1177/000348940411300408
67. Li H, Du Z, Zhang L, et al. The relationship between angiotensinogen gene polymorphisms and essential hypertension in a northern Han Chinese population. Hypertension. 2014;65(5):614–619.
68. Al-Hazzan A, Daoud MS, Ataya FS, Fouad D, Al-Jafari AA. Renin-angiotensin system gene polymorphisms among Saudi patients with coronary artery disease. J Biol Res. 2014;21(8):1–9.
69. Ortlepp JR, Metrikat J, Mevissen V, et al. Relation between the angiotensinogen (AGT) M235T gene polymorphism and blood pressure in a large, homogenous study population. J Human Hypertens. 2003;17:555–559. doi:10.1038/sj.jhh.1001587
70. Glavnik N, Petrovic D. M235T polymorphisms of the angiotensinogen gene and insertion/deletion polymorphisms of the angiotensin-1 converting enzyme gene in essential arterial hypertension in Caucasians. Folia Biol. 2007;53(2):69–73.
71. Buraczynska M, Pijanowski Z, Spasiewicz D, et al. Renin-angiotensin system gene polymorphisms: assessment of the risk of coronary heart disease. Kardiol Pol. 2003;58(1):1–9.
72. Fernandez-Arcas N, Dieguez-lucena JL, Munoz-Moran E, et al. Both alleles of the M235T polymorphism of angiotensinogen gene can be a risk factor for myocardial infarction. Clin Genet. 2001;60:52–57. doi:10.1034/j.1399-0004.2001.600108.x
73. Murugan M, Ramalingam K, Nazzuredin M, Rashed HA, Punamalai G. SNP’s and its correltion with hypertension: a comprehensive review. Dent Med Res. 2013;1:3–6.
74. Sugiyama A, Kishikawa R, Honjo S, et al. Angiotensin-converting enzyme genotype is a risk for wheat-dependant exercise-induced anaphylaxis sensitized with hydrolysed wheat protein. Allergol Int. 2016;65:115–116. doi:10.1016/j.alit.2015.09.003
75. Sethi AA, Nordesgaard BG, Agerholm-larsen B. Angiotensinogen polymorphisms and elevated blood pressure in the general population. The copenhagen city heart study. Hypertension. 2001;37:875–881. doi:10.1161/01.hyp.37.3.875
76. Jimenez PM, Conde C, Casanegra A, Romero C, Tabares AH, Orias M. Association of ACE genotype and predominantly diastolic hypertension: a preliminary study. J RAA Sys. 2007;8(1):42–44.
77. Miyazaki M, Takai S, Jin D, Muramatsu M. Pathological roles of angiotensin II produced by mast cell chymase and the effects of Chymase inhibition in animal models. Pharmacol Ther. 2006;112:668–676. doi:10.1016/j.pharmthera.2006.05.008
78. Raymond WW, Su S, Makarova A, et al. α2-Macroglobulin capture allows detection of mast cell Chymase in serum and creates a reservoir of angiotensin II-generating activity. J Immunol. 2009;182:5770–5777. doi:10.4049/jimmunol.0900127
79. Fukani H, Okunishi H, Miyazaki M. Chymase: its pathophysiological roles and inhibitors. Curr Pharm Drugs. 1998;4:439–453.
80. Dell’talia LJ, Husain A. Dissecting the role of Chymase in angiotensin II formation and heart and blood vessels disease. Curr Opin Cardiol. 2002;17:374–379.
81. Dell’italia LJ, Husain A. Dissecting the role of Chymase in angiotensin II formation and heart and blood vessel diseases. Curr Opin Cardiol. 2002;17:374–379.
82. Mao XQ, Shirakawa T, Yoshikawa T, et al. Association between genetic variants of mast-cell chymase and eczema. Lancet. 1996;348:581–583. doi:10.1016/s0140-6736(95)10244-2
83. Welle M. Development, significance and heterogeneity of mast cells with particular regard to the mast cell specific proteases Chymase and tryptase. J Leucocyte Biol. 1997;61:233–245. doi:10.1002/jlb.61.3.233
84. Bacani C, Frishman WH. Chymase: a new pharmacological target in cardiovascular disease. Cardiol Rev. 2006;14(4):187–193. doi:10.1097/01.crd.0000195220.62533.c5
85. Takai S, Jin D, Miyazaki M. New approaches to blockade of the renin-angiotensin-aldosterone system: chymase as an important target to prevent organ damage. J Pharmacol Sci. 2010;113:301–309. doi:10.1254/jphs.10r05fm
86. Heuston S, Hyland NP. Chymase inhibition as a pharmacological target: a role in inflammatory and functional gastrointestinal disorders. Brit J Pharmacol. 2012;167:732–740. doi:10.1111/j.1476-5381.2012.02055.x
87. Zhou X, Whitworth HS, E-Khedr M, et al. Mast cell chymase: a useful marker in anaphylaxis. J Allergy Clin Immunol. 2011;127(2):abstracts AB143 number 539. doi:10.1016/j.jaci.2011.01.057
88. He S, Walls AF. The induction of a prolonged increase in microvascular permeability by human mast cell chymase. Eur J Pharmacol. 1998;352:91–98. doi:10.1016/S0014-2999(98)00343-4
89. Ortlepp JR, Janssens U, Bleckmann F, et al. A chymase gene variant is associated with atherosclerosis in venous coronary bypass grafts. Coron Artery Dis. 2001;12:493–497.
90. Ortlepp JR, Janssens U, Bleckmann F, et al. A chymase gene variant is associated with atherosclerosis in venous coronary artery bypass grafts. J Coronary Artery Dis. 2001;12:493–497. doi:10.1097/00019501-200109000-00008
91. Ahmad S, Varagic J, Groban L, et al. Angiotensin (1-12); A chymase mediated cellular angiotensin II substrate. Curr Hypertens Res. 2014;16:429–437. doi:10.1007/s11906-014-0429-9
92. Schwartz LB, Yunginger JW, Miller J, Bokhari R, Dull D. Time course of appearance and disappearance of human mast cell tryptase in the circulation after anaphylaxis. J Clin Invest. 1989;83:1551–1555. doi:10.1172/JCI114051
93. Rubinstein I, Nadel JA, Graf PD, Caughey GH. Mast cell chymase potentiates histamine-induced wheal formation in the skin of ragweed-allergic dogs. J Clin Invest. 1990;86:555–559. doi:10.1172/JCI114744
94. Saarinen JV, Harvima RJ, Naukkarinen A, Horsmanheimo M, Harvima IT. The release of histamine is associated with the inactivation of mast cell chymase during immediate allergic wheal reactions in the skin. Clin Exp Allergy. 2001;31:593–601. doi:10.1046/j.1365-2222.2001.01030.x
95. Nisho H, Takai S, Miyazaki M, et al. Usefulness of serum mast cell-specific chymase levels for postmortem diagnosis of anaphylaxis. Int J Legal Med. 2005;119:331–334. doi:10.1007/s00414-005-0524-1
96. Wong CK, Ng SSM, Lun SWM, Cao J, Lam CWK. Signaling mechanisms regulating the activation of human eosinophils by mast-cell-derived Chymase; implications for mast-cell-eosinophil interaction in allergic inflammation. Immunology. 2009;126:579–587. doi:10.1111/j.1365-2567.2008.02916.x
97. Caughey GH. Mast cell tryptases and chymases in inflammation and host defense. Immunol Rev. 2007;217:141–154. doi:10.1111/j.1600-065X.2007.00509.x
98. Guilarte M, Sala-Cunill A, Luengo O, Labrador-Horrillo M, Cardona V. The mast cell contact and coagulation system connection in anaphylaxis. Front Immunol. 2017;8:846.
99. Proud D, Togias A, Nacleiro RM, Crush SA, Norman PS, Lichtenstein LM. Kinins are generated in vivo following airway challenge of allergic individuals with allergen. J Clin Invest. 1983;72(5):1678–1685. doi:10.1172/JCI111127
100. Stone SF, Brown SG. Mediators released during human anaphylaxis. Curr Allergy Asthma Rev. 2012;12(1):33–41. doi:10.1007/s11882-011-0231-6
101. Caughey GH. Mast cell proteases as pharmacological targets. Eur J Pharmacol. 2016;778:44–55. doi:10.1016/j.ejphar.2015.04.045
102. Imamura T, Dubin A, Moore W, Tanaka R, Travis J. Induction of vascular permeability enhancement by human tryptase : dependence on activation of prekallikrein and direct release of bradykinin from kininogens. Lab Invest. 1996;74(5):861–870.
103. Weidinger S, Rummier L, Klopp N, et al. Association study of mast cell Chymase polymorphisms with atopy. Allergy. 2005;60(10):1256–1261. doi:10.1111/j.1398-9995.2005.00879.x
104. Sharma S, Rajan UM, Kumar A, Soni A, Ghosh B. A novel (TG) n (GA) m repeat polymorphism 254bp downstream of the mast cell Chymase (CMA1) gene is associated with atopic asthma and total serum IgE levels. J Hum Genet. 2005;50(6):276–282. doi:10.1007/s10038-005-0252-x
105. Hossny EM, Amr NH, Elsayed SB, Nasr RA, Ibraheim EM. Association of polymorphisms in the mast cell chymase gene promoter region (−1903 G/A) and (TG) n (GA) m repeat downstream of the gene with bronchial asthma in children. J Investing Allergol Clin Immunol. 2008;18(5):376–381.
106. Iwanaga T, McEuen A, Walls AF, et al. Polymorphisms of the mast cell Chymase(CMA1) promotor region: lack of association with asthma but association with serum total Immunoglobulin E levels in aduld atopic dermatitis. Clin Exp Allergy. 2004;34(7):1037–1042. doi:10.1111/j.1365-2222.2004.02000.x
107. He S, Walls AF. The prolonged increase in microvascular permeability by human mast cells Chymase. Eur J Pharmacol. 1998;352:91–98. doi:10.1016/S0014-2999(98)00343-4
108. Orlowska-Baranowska E, Gora J, Baranowski R, et al. Association of the commeon genetic polymorphisms and haplotypes of the Chymase gene with left ventricular mass in male patients with symptomatic aortic stenosis. PLoS ONE. 2014;9(5):1–10.
109. Veerappan A, Reid AC, Estephan R, et al. Mast cell renin and a local renin-angiotensin system in the airway: role in bronchoconstriction. PNAS. 2008;105(4):1315–1320. doi:10.1073/pnas.0709739105
110. Renne T. The vascular side of plasma kallikrein. Blood. 2015;125(4):589–590. doi:10.1182/blood-2014-11-609016
111. European Marketing Authorization website Date 24/8/2012 EU/1/07/405/001-010.
112. Przybilla RF, Bilo B, Muller U, Aberer W. Predictors of severe systemic anaphylaxic reactions in patients with hymenoptera venom allergy. J Allergy Clin Immunol. 2009;124:1047–1054. doi:10.1016/j.jaci.2009.08.027
113. Reber LL, Hernandez JD, Galli SJ. The pathophysiology of anaphylaxis. J Allergy Clin Immunol. 2007;140:335–348. doi:10.1016/j.jaci.2017.06.003
114. Fukuda Y, Kawashima H, Saito K, Inomata N, Matsui M, Nakanishi T. Effect of human plasma-type plateletactivating factor acetylhydrolase in two anaphylactic shock models. Eur J Pharmacol. 2000;390(1–2):203–207. doi:10.1016/S0014-2999(99)00920-6
115. Arimura A, Harada M. Differential effect of a PAF antagonist CV-3988 on active and passive anaphylactic shock in various mouse strains. Lipids. 1991;26(12):1386–1390. doi:10.1007/BF02536572
116. Vadas P, Gold M, Perelman B, et al. Platelet-activating factor, PAF acetylhydrolase, and severe anaphylaxis. N Engl J Med. 2008;358(1):28–35. doi:10.1056/NEJMoa070030
117. Campbell DJ, Alexiou T, Xiao HD, et al. Effect of reduced angiotensin-converting enzyme gene expression and angiotensin-converting enzyme inhibition on angiotensin and bradykinin peptide levels in mice. Hypertension. 2004;43(4):854–859. doi:10.1161/01.HYP.0000119190.06968.f1
118. Gill P, Jindal NL, Jagdis A, Vadas P. Platelets in the immune response; Revisiting platelet activating factor in anaphylaxis. J Allergy Clin Immunol. 2015;135(6):1424–1432. doi:10.1016/j.jaci.2015.04.019
119. Kajiwara N, Sasaki T, Braddind P, et al. 2010. Activation of human mast cells through the platelet-activating factor receptor. J Allergy Clin Immunology. 125(5):1137–1145.
120. Pretorius M, Luther JM, Murphey LJ, Vaughan DE, Brown NJ. Angiotensin-converting enzyme inhibition increases basal vascular tissue plasminogen activator release in women but not in men. Arterioscler Thromb Vasc Biol. 2005;25(11):2435–2440. doi:10.1161/01.ATV.0000186185.13977.94
121. Ortega T, Paul M, Ackermann A, Fern´andez-Alfonso MS, De Rojas RS, Gonz´alez C. Modulation of angiotensinconverting enzyme by nitric oxide. Br J Pharmacol. 1998;124(2):291–298. doi:10.1038/sj.bjp.0701836
122. Finkelman FD, Rothenberg ME, Brandt EB, Morris SC, Strait RT. Molecular mechanisms of anaphylaxis: lessons from studies with murine models. J Allergy Clin Imunol. 2005;115(3):449–457. doi:10.1016/j.jaci.2004.10.029
123. Hox V, Desai A, Bandara G, Gilfillan AM, Metcalfe DD, Olivera A. Estrogen increases the severity of anaphylaxis in female mice through enhanced endothelial nitric oxide synthase expression and nitric oxide production.J. Allergy Clin Immunol. 2015;135(3):729–736. doi:10.1016/j.jaci.2014.11.003
124. Dimitropoulou C, Chatterjee A, McCloud L, Yetik-Anacak G, Catravas JD. Angiotensin, bradykinin and the endothelium. Handb Exp Pharmacol. 2006;(176):255–294.
125. Mombouli JV, Vanhoutte PM. Heterogeneity of endothelium-dependent vasodilator effects of angiotensinconverting enzyme inhibitors: role of bradykinin generation during ACE inhibition. J Cardiovasc Pharmacol. 1992;20(9):S74–S82. doi:10.1097/00005344-199200209-00014
126. Lowenstein CJ, Michel T. What’s in a name? eNOS and anaphylactic shock. Jci. 2006. doi:10.1172/JCI29406
127. Castells M. Diagnosis and management of anaphylaxis in precision medicine. J Allergy Clin Immunol. 2017;140(2):321–333. doi:10.1016/j.jaci.2017.06.012
128. Rajj L. Hypertension and cardiovascular risk factors: role of the angiotensin II-nitric oxide interaction. Hypertension. 2001;37(2):767–773. doi:10.1161/01.hyp.37.2.767
129. Husain K, Hernandez W, Ansari RA, Ferder L. Inflammation, oxidative stress and renin angiotensin system in atherosclerosis. World J Boil Chem. 2015;6(3):209–217. doi:10.4331/wjbc.v6.i3.209
130. Schmermund A, Lerman LO, Ritman EL, Rumberger JA. Cardiac production of Angiotensin II and its pharmacologic inhibition: effects on the coronary circulation. Mayo Clin Proc. 1999;74:503–513. doi:10.4065/74.5.503
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.