Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 8
IgA-dominant post-infectious glomerulonephritis presenting as a fatal pulmonary-renal syndrome
Authors Saad M, Daoud M, Nasr P, Syed R, El-Sayegh S
Received 5 March 2015
Accepted for publication 8 April 2015
Published 5 August 2015 Volume 2015:8 Pages 77—81
DOI https://doi.org/10.2147/IJNRD.S84061
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pravin Singhal
Marc Saad,1 Magda Daoud,1 Patricia Nasr,1 Rafeel Syed,2 Suzanne El-Sayegh2
1Department of Medicine, 2Department of Nephrology, Staten Island University Hospital, Staten Island, NY, USA
Abstract: Over the last decades, post-infectious glomerulonephritis underwent major changes in its epidemiology, pathophysiology, and outcomes. We are reporting a case of IgA-dominant post-infectious glomerulonephritis (IgA-PIGN) presenting as a fatal pulmonary-renal syndrome. An 86-year-old Filipino man presented with worsening dyspnea, hemoptysis, and decreased urine output over 2 weeks. Past medical history is significant for hypertension, chronic kidney disease stage III, and pneumonia 3 weeks prior treated with intravenous cefazolin for methicillin-sensitive Staphylococcus aureus bacteremia. Physical examination was remarkable for heart rate of 109/min and respiratory rate of 25/min saturating 99% on 3 liters via nasal cannula. There were bibasilar rales in the lungs and bilateral ankle edema. A chest radiograph showed bibasilar opacifications. Blood work was significant for hemoglobin of 8.3 g/dL and creatinine of 9.2 mg/dL (baseline of 1.67). TTE showed EF 55%. Urinalysis revealed large blood and red blood cell casts. Kidney ultrasound showed bilateral echogenicity compatible with renal disease. Pulse methylprednisolone therapy and hemodialysis were initiated with patient's condition precluding kidney biopsy. Serology workup for rapidly progressive glomerulonephritis was negative. On day 7, the patient required mechanical ventilation; bronchoscopy showed alveolar hemorrhage and plasmapheresis was initiated. Renal biopsy revealed IgA-PIGN with endocapillary and focal extracapillary proliferative and exudative features. IgA-PIGN occurs in diabetic elderly (mean age of 60 years), 0–16 weeks after an infection mainly by Staphylococcus. However, this nondiabetic patient had normal complement IgA-PIGN with fatal pulmonary-renal syndrome. Understanding the pathogenesis and identifying the nephrotoxic bacteria species and the aberrant IgA molecule will open new insights toward prevention and treatment.
Keywords: acute kidney injury, alveolar hemorrhage, subepithelial deposits, starry-sky
Case presentation
An 86-year-old Filipino man presented to the emergency room for dyspnea at rest. His shortness of breath had worsened over the previous 2 days, and was associated with substernal dull chest pain and hemoptysis. Patient also reported a decrease in urine output and bilateral ankle swelling that had been worsening over the past 2 weeks. He had no history of recent travel, sick contacts or blood transfusion. Past medical history is significant for hypertension, chronic kidney disease stage III, gout, and COPD. He had no allergies. He had quit smoking 9 years ago, did not drink alcohol or use illicit drugs.
The patient was hospitalized for pneumonia 3 weeks before presentation, and was treated with intravenous cefazolin for 2 weeks for methicillin-sensitive Staphylococcus aureus bacteremia. On examination, the patient was tachypneic (respiratory rate 25/min), temperature 97.6°F, blood pressure 155/71, pulse 109 bpm, with a saturation of 99% on 3 liters via nasal cannula. The skin and conjunctiva were pale. There were bibasilar rales in the lungs and bilateral lower extremity pitting edema 2+; the remainder of the examination was normal.
Electrocardiogram showed sinus tachycardia and nonspecific ST segment and T wave abnormalities. Chest radiograph showed bibasilar opacifications and bilateral pleural effusions. Furosemide was administered intravenously, sublingual nitroglycerin, and oral aspirin were given. Blood work was significant for hemoglobin of 8.3 g/dL (baseline 10), creatinine of 9.2 mg/dL (baseline of 1.67 GFR 39 mL/min/1.73m2), troponin of 0.6 and albumin 2.6 g/dL. Lower extremity duplex study showed right peroneal deep vein thrombosis and heparin infusion was initiated for possible pulmonary embolism and non-ST elevation myocardial infarction (NSTEMI). In addition, broad-spectrum antibiotics (vancomycin and meropenem) were initiated empirically. Urinalysis showed large blood, and red blood cell (RBC) casts were seen on light microscopy. Sputum staining showed epithelial cells and few white blood cells.
Transthoracic echocardiogram revealed a left ventricular ejection fraction of 55%, with moderate mitral regurgitation and a small pericardial effusion. Ultrasonography of the abdomen revealed bilateral renal echogenicity, compatible with renal disease. The right kidney was measured to be 10 cm in length by 4.4 cm in width by 4.4 cm anteroposterior diameter. The left kidney measured 9.3 cm by 4.6 cm by 4.9 cm. Vascular flow was demonstrated within each kidney. Serology workup for rapidly progressive glomerulonephritis (RPGN) was sent.
On day 2 of the hospitalization, the patient became more tachypneic and fatigued necessitating oxygen supplementation with bi-level positive airway pressure. He was anuric (urine output 100 cc/24 hours) and was started on hemodialysis for hypervolemia; blood and urine cultures were negative. The patient had acute drop in hemoglobin (7 g/dL); heparin was stopped and he remained on acetylsalicylic acid and clopidogrel. On day 4, the patient was started on pulse steroids Solu-Medrol 1 g daily for 3 days. Patient’s condition and thrombocytopenia had precluded kidney biopsy up to this point. On day 6, sputum culture showed heavy growth of Klebsiella oxytoca.
Rapidly progressive GN workup: hepatitis viral panel was negative. Hematological and immunological workup was normal including antinuclear antibody, anti-DNA, anti-glomerular basement membrane antibody, anticardiolipin IgG and IgM, cryoglobulin, anti-neutrophil cytoplasmic antibody (proteinase-3 AB <1.0, myeloperox AB <1.0), C3 and C4. Serum protein electrophoresis showed polyclonal gammopathy (gamma fraction 2.2 g). Rheumatoid factor titers were elevated 45 H (<14); elevated free kappa and lambda serum with normal ratio (1) (free kappa serum 205.8 mg/L [3.3–19.4], free lambda serum 205.0 mg/L [5.7–26.3]).
On day 7, the patient was intubated for impending respiratory failure and put on mechanical ventilation. Bronchoscopic examination revealed blood in both bronchial trees and alveolar hemorrhage and the patient was started on total plasma exchange with fresh frozen plasma on a daily basis (total of seven sessions); bronchoalveolar lavage cultures grew Streptococcus bovis and Aspergillus fumigatus and antifungal were added to meropenem.
On day 13, renal biopsy and staining with hematoxylin and eosin stain, Periodic acid Schiff stain, and Jones methionine silver stain revealed core of renal cortex contains 19 glomeruli, 18 of which are globally sclerotic and one of which has ischemic tuft retraction associated with tubular atrophy, interstitial fibrosis and a moderate chronic inflammatory infiltrate. There is moderate narrowing of small arteries and arterioles by intimal and medial sclerosis. The glomeruli appear enlarged with diffuse and global mild to moderate mesangial sclerosis forming segmental nodules. The glomerular basement membranes appear diffusely thickened with focal duplications. (Figure 1A) Superimposed on this nodular sclerosis, there is mild to moderate mesangial hypercellularity and segmental to global endocapillary proliferation (Figure 1B) including infiltrating neutrophils. (Figure 1C) There is endocapillary and focal extracapillary proliferative and exudative features: cellular/fibrocellular crescents; arteries and arterioles are narrowed by moderately severe intimal sclerosis; (Figure 1D) RBC casts (Figure 1E) and focal acute tubular injury. Figure 1F; the immunofluorescence (IF) findings of granular global mesangial and “starry-sky” pattern of glomerular capillary wall positivity for IgA and C3 (Figure 1G and H) support IgA-dominant post-infectious glomerulonephritis (IgA-PIGN) with endocapillary and focal extracapillary proliferative and exudative features associated with staphylococcal infection. Smoking-related moderate nodular glomerulosclerosis, (associated with longstanding smoking and COPD), focal subcapsular cortical scar with ischemic glomerulosclerosis, consistent with hypertensive arterionephrosclerosis, and moderately severe arteriosclerosis were also noted. On electronic microscopy, there was evidence of “hump-shaped” subepithelial deposits.
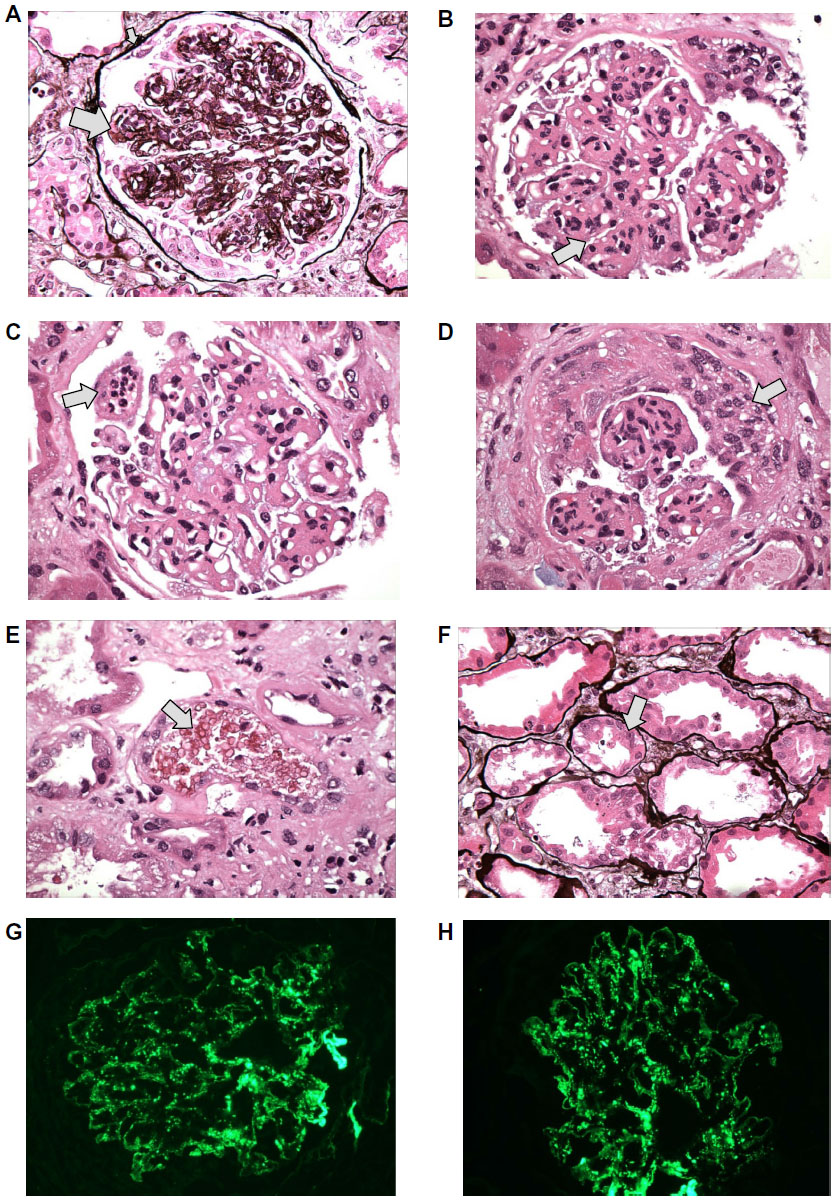 | Figure 1 Microscopic finding of kidney biopsy with hematoxylin and eosin stain Periodic acid Schiff (PAS) stain, and Jones methionine silver (JMS) stain. |
The hospital course was complicated by pneumonia, pulmonary edema, and hemorrhage. The patient remained anuric requiring hemodialysis. The patient expired on day 18 secondary to respiratory failure, hypoxia, and cardiac arrest.
Discussion
PIGN is an entity well known to occur in childhood following streptococcal upper respiratory tract or skin infections. During the past 3 decades although, a major shift has occurred in the epidemiology, characteristics, and outcomes of this disease. It is recognized increasingly more in the adult population, and can occur after streptococcal and more commonly staphylococcal infections from various sites.1–7
Both post-streptococcal and staphylococcal infectious RPGN share identical histopathological appearance, with diffuse endocapillary proliferative and exudative RPGN on light microscopy, glomerular immune deposits that stain for either IgG and C3 or C3 only on IF, and hump-shaped subepithelial deposits on electron microscopy. More recently, several cases of acute post-infectious glomerulonephritis (APIGN) have been identified with intense deposits of IgA as the sole or dominant Ig on IF, and mesangial and subepithelial deposits on EM, characterizing a new entity named IgA-dominant APIGN.1–6 To signify the ongoing infection at the time of diagnosis, the term “IgA dominant infection-related glomerulonephritis (IRGN)” is employed.4
Codominance of IgA and IgG staining can lead to a diagnostic dilemma in terms of distinguishing IgA dominant IRGN from IgA nephropathy. Classically, IgA nephropathy is associated with a stronger C3 staining in a “starry-sky” appearance. In addition, staphylococcal associated APIGN is associated with hypocomplementemia in 69% of the cases.1,3,5,6
On average, it takes 4 weeks from the time of infection to kidney injury. Epidemiologically, the mean age of diagnosis is 60 years of age, with a male to female predominance of nearly 4:1. It has never been reported in children, and is most commonly associated with diabetes mellitus (55%). Fifty-one percentage of the cases (in which an infection site was identified) were due to cellulitis, and Staphylococcus is the most commonly associated pathogen, but there are also reports indicating occurrence in patients with urinary tract infections.1–6,8,9
The clinical manifestations include acute nephritic syndrome with severe renal failure (acute or rapidly progressive), new onset of gross hematuria with or without RBC casts (25% of the cases), nephrotic and non-nephrotic range proteinuria, edema, and hypertension (~75% at presentation). Extrarenal manifestations rarely occur in adults with IRGN.
Prognosis of IRGN is guarded, despite therapy against the infectious pathogen. Eighty-four percentage of the cases result in chronic kidney disease, nearly half of which progress to end stage renal disease requiring renal replacement therapy. Interestingly, and perhaps not surprisingly, two-thirds of the patients progressing to end stage renal disease have underlying diabetic glomerulosclerosis.1,3,5,6
Histopathology of IgA-dominant APIGN
IgA dominant APIGN has a broad spectrum of glomerular injury, which cannot be precisely specified. It mainly presents as diffuse (53%) or focal (28%) endocapillary proliferative and exudative, but also as pure mesangial proliferative RPGN (13%), membranoproliferative RPGN, or crescentic RPGN.2 It is differentiated by IF and EM findings.4 On IF, glomerular mesangial and glomerular capillary injury resembling the “starry-sky pattern” is seen in 69% of the cases. On EM, electron dense deposits were present in 92% of the patients and exhibited a “hump-shaped” appearance, which is a key histopathological finding in terms of diagnosing IgA dominant IRGN. Interestingly, in cases of resolving IRGN, the subepithelial deposits were preferentially located at the glomerular basement membrane as opposed to the mesangium. Subendothelial deposits tend to be small and infrequent, but are seen in up to 66% of the cases. IF for C3 tends to be the strongest, with a “starry-sky” pattern. However, unlike usual PIGN, IgA is prominent and IgG is either weak or absent.2,3
Pathogenesis of IgA-dominant APIGN
The pathogenesis of the IgA predominant deposition is not well known, and is likely to involve specific host-pathogen interactions.1,5 Increased serum IgA levels in some reported cases suggest activation of selective IgA immune responses. Koyama et al10,11 suggested that post-methicillin-resistant S. aureus (MRSA) RPGN may be induced by a staphylococcal enterotoxin acting as a superantigen.3 This leads to massive T-cell activation and production of high amounts of proinflammatory cytokines and IgA–IgG immune complex formation.1,5
Recently, a S. aureus cell envelope antigen designated as “probable adhesion” was identified in IgA nephropathy, Henoch-Schonlein Purpura, and post-MRSA RPGN, co-localizing with glomerular IgA deposits. It appears that S. aureus, a common colonizing bacterium in nasal and oral flora, contributes to the pathogenesis of IgA nephropathy, and can result in PIGN in the setting of disease causing infections. Although “probable adhesion” may be the antigen trigger for IgA-dominant PIGN, it is still unknown if the IgA secreted by the host is abnormal as is the case in IgA nephropathy.1,4,12 Interestingly, diabetic patients develop IgA-rich immune responses in circulation when compared to the normal population, especially in the presence of diabetic complications and end organ damage. High serum IgA levels in diabetic patients likely represent an immune response to advanced glycosylation end products. The IgA-rich environment in diabetics coupled with IgA-specific responses to the proposed staphylococcal superantigen may precipitate a unique variant of PIGN with glomerular IgA deposition and explain the predilection for more severe IgA dominant IRGN in diabetic patients.3,5,8,9
Diagnosis
The consensus for diagnosis requires three out of the following five criteria: 1) clinical or laboratory evidence of infection preceding or at the onset of RPGN; 2) depressed serum complement; 3) endocapillary proliferative and exudative RPGN; 4) C3-dominant or codominant glomerular IF staining; and 5) hump-shaped subepithelial deposits shown on electron microscopy.1,13
Kidney biopsy is recommended in most adults suspected of having IRGN to confirm the diagnosis and rule out other glomerulonephritides that have similar clinical presentations and may require prompt aggressive immunosuppressive therapy, which has not yet been shown to be of utility in IRGN.
This case represents the first reported case to our knowledge of IgA-dominant IRGN presenting with pulmonary-renal syndrome. Our patient eventually succumbed to his illness due to severe sepsis and respiratory failure. Our aim is to advance knowledge of this phenomenon, and hopefully spur a better understanding of the pathogenesis involved, which may lead to new treatment paradigms.
Acknowledgment
Dr Vivette D’Agati, Department of pathology, Columbia University Medical Center, NY, USA.
Disclosure
The authors report no conflicts of interest in this work.
References
Worawichawong S, Girard L, Trpkov K, Gough JC, Gregson DB, Benediktsson H. Immunoglobulin A-dominant postinfectious glomerulonephritis: frequent occurrence in nondiabetic patients with Staphylococcus aureus infection. Hum Pathol. 2011;42(2):279–284. | |
Nasr SH, Fidler ME, Valeri AM, et al. Postinfectious glomerulonephritis in the elderly. J Am Soc Nephrol. 2011;22(1):187–195. | |
Kambham N. Postinfectious glomerulonephritis. Adv Anat Pathol. 2012;19(5):338–347. | |
Nasr SH, D’Agati VD. IgA-dominant postinfectious glomerulonephritis: a new twist on an old disease. Nephron Clin Pract. 2011;119(1):c18–c25. | |
Nast CC. Infection-related glomerulonephritis: changing demographics and outcomes. Adv Chronic Kidney Dis. 2012;19(2):68–75. | |
Koo TY, Kim GH, Park H. Clinicopathologic Features of IgA-Dominant Postinfectious Glomerulonephritis. Korean J Pathol. 2012;46(2):105–114. | |
Nasr SH, Radhakrishnan J, D’Agati VD. Bacterial infection-related glomerulonephritis in adults. Kidney Int. 2013;83(5):792–803. | |
Erqou S, Kamat N, Moschos S, Im A, Bastacky S. A 61 year diabetic man with methicillin-sensitive Staphylococcus aureus septic arthritis and acute renal failure: a case of IgA-dominant postinfectious glomerulonephritis. Nephrology (Carlton). 2012;17(8):780–781. | |
Arakawa Y, Shimizu Y, Sakurai H, et al. Polyclonal activation of an IgA subclass against Staphylococcus aureus cell membrane antigen in post-methicillin-resistant S. aureus infection glomerulonephritis. Nephrol Dial Transplant. 2006;21(5):1448–1449. | |
Koyama A, Kobayashi M, Yamaguchi N, et al. Glomerulonephritis associated with MRSA infection: a possible role of bacterial superantigen. Kidney Int. 1995;47:207–216. | |
Koyama A, Sharmin S, Sakurai H, et al. Staphylococcus aureus cell envelope antigen is a new candidate for the induction of IgA nephropathy. Kidney Int. 2004;66:121–132. | |
Nasr SH, Share DS, Vargas MT, D’Agati VD, Markowitz GS. Acute poststaphylococcal glomerulonephritis superimposed on diabetic glomerulosclerosis. Kidney Int. 2007;71(12):1317–1321. | |
Stratta P, Musetti C, Barreca A, Mazzucco G. New trends of an old disease: the acute post infectious glomerulonephritis at the beginning of the new millenium. J Nephrol. 2014;27(3):229–239. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.