Back to Journals » Journal of Asthma and Allergy » Volume 13
Idiopathic Angioedema: Current Challenges
Authors Belbézier A, Bocquet A, Bouillet L
Received 6 August 2019
Accepted for publication 13 March 2020
Published 17 April 2020 Volume 2020:13 Pages 137—144
DOI https://doi.org/10.2147/JAA.S205709
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Luis Garcia-Marcos
Aude Belbézier,* Alexis Bocquet,* Laurence Bouillet
National Reference Center for Angioedema (CREAK), Department of Internal Medicine/Clinical Immunology, Grenoble Alpes University Hospital, Grenoble, France
*These authors contributed equally to this work
Correspondence: Laurence Bouillet
Centre de référence des angioedèmes (CREAK), Servicede médecine interne/immunologie clinique, Centre Hospitalier Universitaire de Grenoble Alpes, CS10217, Grenoble cedex 09 38043, France
Tel +33 476765513
Fax +33 76765816
Email [email protected]
Abstract: The etiological diagnosis of isolated recurrent angioedema poses problems because it must often be done urgently. Angioedema secondary to nonspecific mast cell activation (MC-AE) is the most frequent form and is usually mild. Bradykinin mediated angioedema (BK-AE) is rarer but potentially fatal in the absence of the correct treatment. Few biological markers exist. The C1-inhibitor (C1-inh) functional assay can exclude AE due to C1-inh deficiency. Genetic diagnoses of hereditary AE due to abnormal C1-inh AE have progressed with four currently known mutations. However, determining the physiopathological mechanism leading to some isolated AE cases is sometimes very difficult. In such cases, therapeutic tests are then the only solution: antihistamines at high doses and omalizumab for suspected MC-AE, icatibant for suspected AE-BK. Identifying new markers would be a great help.
Keywords: angioedema, mast cell, bradykinin, C1 inhibitor, histamine
Angioedema
Angioedema (AE) is defined as transitory recurrent episodes of subcutaneous and/or submucosal swelling, which mainly affect the skin, the gastrointestinal tract and the upper airways. It is due to a sudden and localized increase in vascular permeability and vasodilation.1,2 In the United-States, it accounts for over 100,000 emergency department visits per year.3 AE can be mediated by either bradykinin (BK) or mast cell (MC) mediators secondary to specific (IgE-dependent) or nonspecific activation (non-IgE dependent) (Figures 1 and 2). An appropriate clinical approach and sound diagnosis are essential before concluding on one or the other because the prognostics and treatment of these two forms differ. The risk of mortality is 45‐fold higher for BK-AE than for MC‐AE.4 In the case of isolated AE (without hives), C1-Inhibitor (C1Inh) tests are mandatory so as to exclude AE due to C1-inh deficiency. If C1 inhibition is normal, it is not easy to distinguish between the two possible etiologies and diagnosis is problematic.
 |
Figure 1 Schematic diagram of biochemical pathways responsible for angioedema. Part I: Mast cell angioedema. (A) Mast cell activation by different modes (allergic or non-allergic). (B) Release of various mediators responsible for angioedema. Part II: Bradykinin angioedema (kallikrein-kinin system). Abbreviations: C1-inh, C1inhibitor; ACE, angiotensin-converting enzyme. |
 |
Figure 2 Etiologies of angioedema without wheals (hives). Abbreviations: AE, angioedema; HAE, hereditary angioedema; AAE, acquired angioedema; MGUS, monoclonal gammopathy of unknown signification; ACEi, angiotensin-converting enzyme inhibitors; MC-AE, mast cell-induced angioedema. FXII, Hageman factor gene; PLG, plasminogen gene; ANGPT1, angiopoietin 1 gene; KNG1, kininogen1 gene. |
Mast-Cell Induced AE
Mast-cell induced AE (MC‐AE) is the most frequent form, seen in 96% of consultations.1 MC‐AE can last from only a few hours up to several days and is often associated with hives, concomitant with or at some distance in time from the AE episode. So, searching for episodes of hives in the patient’s medical history is the first step in this challenging diagnosis. A history of hives, or hives concomitant with the AE, points to MC-AE, but the absence of hives poses a dilemma. The next step is to try high dose antihistamine treatment as for the chronic spontaneous urticaria (CSU). If there is an improvement, then the AE can be assumed to be histaminergic (Hist-AE). Hist-AE is secondary to either the specific activation of mast cells (allergy) or to a nonspecific activation (e.g. spontaneous or nonsteroid anti-inflammatory drug induced) (Figures 1 and 2). Recurrent spontaneous MC-AE forms part of the CSU nosological framework.5 CSU/CIU (chronic inducible urticaria) is a mast cell-driven disease with possibly an auto-immune mechanism (Figure 1).6 In a series of patients with CSU/CIU, around 10% of patients showed exclusively angioedema without wheals or hives.7 Some patients with CSU/CIU do not respond to antihistamines;8 for example, Staevska et al reported that some patients in their study were non-responders to a fourfold dose of antihistamine.9 Consequently, such patients presenting with isolated CSU/CIU and not responding to high doses of antihistamines could be diagnosed as having non-Histaminergic-AE.10 Nevertheless, non-Hist-AE does not exclude a mast cell origin. Histamine is probably not the only mediator of these AEs with tryptase, leukotrienes, and other cytokines undoubtedly being involved. Indeed, Oschatz et al reported that mast cells could increase vascular permeability by the in vivo formation of heparin-initiated bradykinin.11 Using a mast-cell-targeted treatment in place of antihistamine or concomitant with antihistamine could be a proof of this concept. As such, several case series have reported patients with partial or complete response to omalizumab12–18 Omalizumab is a humanized monoclonal anti-Immunoglobulin E (IgE) antibody approved for use in CSU with or without angioedema as second-line therapy in patients with failure of a fourfold dose of antihistamine.6 The mechanism of action of omalizumab in MC-driven diseases like CSU/CIU is only partially understood. It could lower IgE levels, downregulate IgE receptors (FcεRI) on basophils/MC and decrease MC susceptibility.19 Interestingly, Bucher et al reported one patient case with non-Hist-AE who responded to omalizumab and who presented a low density of FcεRI on basophils.17 Clinically, patients with MC-AE who responded to omalizumab were more often males presenting a higher prevalence of upper airway involvement than patients with CSU/CIU in the main case series.15–17 Autologous serum skin tests that evaluate the presence of serum histamine-releasing factor are sometimes used to screen for CSU/CIU. However, the usefulness of this test has not been proven in cases of non-Hist-AE.6 Diagnostic biomarkers that could help to diagnose these AE might be needed. Compared to healthy controls, patients with CSU/CIU had higher levels of D-Dimer, C-reactive protein, tumor necrosis factor, factor VIIa, and prothrombin fragment 1+2, but the specificity of these biomarkers had never been investigated.20 Lara-Marquez et al reported that stimulated plasma kallikrein activity was increased only in patients with non-Hist-AE and normal C1-Inh-HAE compared to patients with Hist-AE.21 However, in that study omalizumab appears not to have been tested, making it difficult to conclude.
A therapeutic test with omalizumab is essential in the diagnostic approach to non-Hist-AE. Only in the case of failure to response does a mast cell origin seem unlikely, and we should then consider BK-AE. The development of new treatments such as ligelizumab will probably allow us to improve the diagnosis.22
Bradykinin Induced AE (BK-AE)
BK-AE are isolated attacks with no associated wheal(s), although erythema Marginatum could appear in the form of a non-pitting, non-pruritic rash that manifests hours or days before AE attacks.23 Swelling attacks always last over 24 h (2–7 days) and can affect all parts of the body including two particularly problematic sites: the upper airways and the gastrointestinal tract. Patients with gastrointestinal tract BK-AE present with severe pain, nausea/vomiting and diarrhea;24 looking like a sub-occlusive syndrome with ascites. These abdominal attacks can mimic surgical emergencies, such as pancreatitis.25 There is often a high degree of diagnostic uncertainty and patients sometimes undergo laparotomy unnecessarily. When the upper airways are affected, the risk of asphyxiation is high.26
BK-AE is often associated with C1-Inhibitor (C1-inh) deficiency (Figure 2), although this is not always the case. It occurs following activation of the kallikrein-kinin pathway by activated factor XII or directly by kallikrein. C1-inh is the main inhibitor of the kallikrein-kinin pathway. BK has a very short half-life because it is quickly degraded by different kininases including angiotensin-converting enzyme (ACE) to 65% (Figure 1).27
BK-AE Associated with C1-inh Deficiency
The differential diagnosis of the two types of BK-AE is simple and is based on assays of C1-inh protein concentration and activity.10 C1Inh deficiency can be assumed if C1-inh activity is below 50% of normal values. It is recommended to assay blood C1-Inh activity, and C1-inh and C4 protein concentrations in all suspected cases (Table 1).28 If C1-inh assays are not available, a C4 is useful because C4 is almost always decreased in C1-inh deficit.29 Tests should be repeated to confirm the diagnosis.
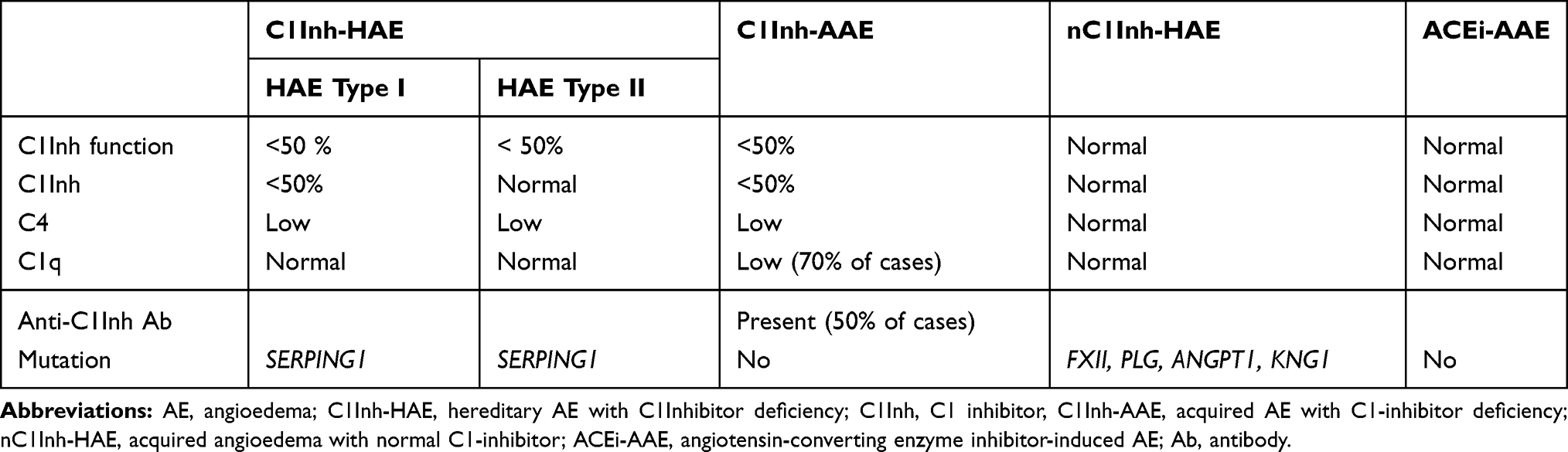 |
Table 1 Biological Characteristics and Profiles of Bradykinin Angioedema |
Hereditary AE with C1-inh Deficiency (C1-inh-HAE)
C1-inh-HAE is a rare genetic disease affecting around 1 in 50 000 people,30,31 due either to decreased production of C1-inh (type I) or production of a non-functional C1-inh (type II). It is secondary to the SERPING1 mutation (over 250 mutations reported).32 The disease follows an autosomal dominant pattern of inheritance with childhood onset. However, in 30% of cases, a mutation appears de novo and in 15% patients can be asymptomatic.27
Acquired AE with C1-inh Deficiency (C1-inh-AAE)
This is a very rare disease.33 C1-inh-AAE patients have no family history of angioedema and usually have late-onset symptoms; the median age of the first attack is around 50. The phenotype does not differ from that of C1-inh-HAE being localized to the face, tongue, ENT, extremities and abdomen.33,34 Low levels of C1q are highly specific to C1-inh-AAE and seen in 7°% of cases. However, some genetically proven C1-inh-HAE can also show low C1q levels s.35,36 Anti-C1-inh antibodies are present in 60% of cases.37 Hemopathies and AAE seem to be linked, with, 40% of C1-inh-AAE associated with a monoclonal gammopathy of undetermined significance, in which monoclonal and anti-C1-inh antibodies share the same isotype.33 While angioedema can precede the appearance of a hemopathy by several months or years, a search for the underlying hemopathy is essential.34 Sometimes, acquired C1-inh deficiency is associated with an autoimmune disease such as systemic lupus erythematosus.38
BK-AE with Normal C1-inh
Normal C1-inh activity excludes C1-inh deficiency.
Hereditary Angioedema with Normal C1-inh (nC1-inh-HAE)39
The diagnosis of nC1-inh-HAE is extremely difficult because very few patients have the corresponding genetic signature: Factor XII (FXII), plasminogen (PLG), or Angiopoietin 1 (ANGPT1) gene mutations.40–42
HAE with FXII gene mutation (FXII-HAE) is principally symptomatic in women and is dependent on high estrogen exposure.39,43,44 The first symptoms often appear on commencing oral contraception or during pregnancy. For men carrying an FXII mutation, half are symptomatic. The diagnosis is based on FXII gene mutation assessment, with four mutations having been recently described.44,45 Knowledge of these mutations is important because of the high risk of complications during pregnancy necessitating closer monitoring.46 Tranexamic acid (TA) and icatibant seem to be more effective than other therapies for this type of HAE.47
HAE with PLG mutation (PLG-HAE) has been recently described 41 and has been identified in more than 80 patients.41,48-51 The median age of the first angioedema attack was around 20. The PLG-HAE phenotype seems to have some particularities with patients developing face and tongue swelling. Angiotensin-converting-enzyme inhibitor (ACEi) and Angiotensin II receptor blocker (ARA) seem to be triggering factors.48 In this type of HAE, tranexamic acid (TA) as long-term prophylaxis could be very efficient.
HAE with ANGPT1 mutation (ANGPT1-HAE) has been described only once by Bafunno et al42. They noted that these patients did not respond to antihistamines and steroids for either acute attacks or as prophylactics, but responded to tranexamic acid.42
HAE with unknown mutations (U-HAE): Sometimes the clinical suspicion of nC1-inh-HAE is very strong particularly if the patient is female with AE at the extremities (as well as having typical abdominal attacks), is particularly symptomatic during pregnancy, identical crises have been described in her family, and the patient improved considerably under prophylactic treatment with tranexamic acid. In such cases, HAE is likely, even if the search for a mutation is negative. New mutations are regularly discovered. Recently, a new mutation that concerns the kininogen 1 gene (KNG 1) has been added to those already known.52
Acquired Angioedema with Normal C1-inh: Angiotensin-Converting Enzyme Inhibitor Induced AE (ACEi-AAE)
ACEi-AAE appear in 1.6 per 1000 person-years of ACE inhibitor use.53 A small number (0.7-1%) of patients taking ACEi may develop AE. This risk seems to be higher in the black – than in the white – American population, and when gliptin or mTOR inhibitor treatments are concomitantly prescribed.54,55 The risk also increases for patients reporting a cough when on ACEi. This AE risk is higher at the initiation of ACEi use (during the first 30 days) than during long-term use,53 but ACEi-AAE could nevertheless occur some years later. ACEi-AAE is commonly localized to the tongue, lips and upper airway tract.56,57 In some cases, ACEi use can reveal a hereditary disease such as FXII-HAE.58 The diagnosis of ACEi-AAE should be made with caution and familial cases should be systematically sought. In all cases it is essential to check for the absence of C1-inh deficiency, and if in doubt to also check for FXII and PLG mutations.
The diagnosis of ACEi-AAE is very challenging. One must be certain that the patient has not experienced AE before starting ACEi and continue to monitor for AE after discontinuing ACEi. A recurrence of AE after 3 months argues against an ACEi-AAE, especially if accompanied by hives. In our experience, more than 50% of cases eventually turn out to be MC-AE. If the diagnosis of ACEi-AAE is confirmed, then ACEi must be contraindicated for life.59
Challenging Idiopathic Non-MC-AE (INMC-AE)
Sometimes, after having ruled out all the different AE diagnoses, the patient has a recurrence of AE despite continuous administration of a 4-fold antihistamine dose. Such patients are considered to have idiopathic non-histaminergic AE. However, this does not automatically mean that they have BK-AE; it could still be AE secondary to nonspecific MC activation. It is then necessary to propose omalizumab treatment. In our experience, more than 90% of AE that are resistant to antihistamines improve with omalizumab.16 Omalizumab, an anti-IgE monoclonal antibody, can nowadays be considered to be a second-line treatment of MC-AE that is poorly controlled by antihistamine therapy, as for chronic spontaneous urticaria (CSU). For this reason, we recommend to treat the patient for 6 months with omalizumab before concluding an idiopathic non-mast cell angioedema (INMC-AE). During this time, one cannot affirm that the INMC-AE is synonymous with BK-AE. However, one can test a prophylactic treatment of tranexamic acid (TA) (1 g 3 times a day).47,60 An improvement would argue in favor of BK-AE (Figure 3). The most specific test is probably improvement with on icatibant treatment. At this point, if mutations associated with nC1-inh-HAE have not been sought, this needs to be done.
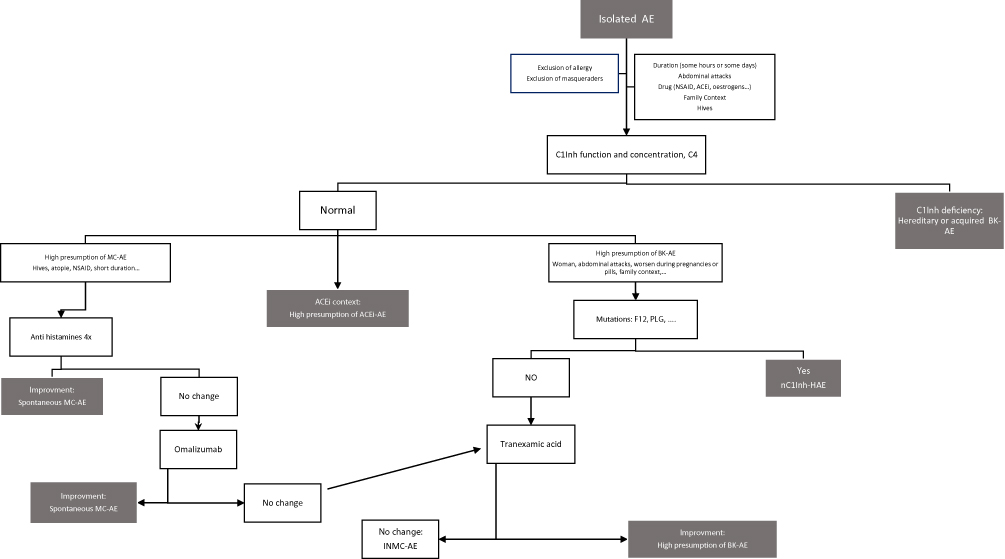 |
Figure 3 AE diagnostic process. Abbreviations: AE, angioedema; BK-AE, bradykinin mediated angioedema; MC-AE, mast cell angioedema; nC1INH-HAE, hereditary angioedema with normal C1-inhibitor; ACEi, angiotensin-converting enzyme inhibitors; INMC-AE, idiopathic non-mast cell angioedema; NSAID, nonsteroid anti-inflammatory drugs; FXII, Hageman factor gene; PLG, plasminogen gene; C1Inh, C1Inhibitor. |
The situation is highly problematic if the patient does not improve with icatibant. Firstly, the whole diagnostic process needs to be reassessed by checking: That it is true AE and not a look-alike.61
- The patient has adhered to the various treatments (antihistamines, TA, omalizumab)
- The absence of aggravating factors (regular nonsteroid anti-inflammatory drugs for MC-AE, estrogens or ACEi for BK-AE).
These difficult cases must be discussed in a multidisciplinary consultation meeting. Sometimes an outside view is useful.
The diagnostic impasses described above highlight the absence of biological markers to identify the key mediator of AE. Different diagnostic strategies have been proposed such as:21,62-65
- For MC-AE: Tryptase, D-Dimers, urinary histamine and methylhistamine excretion;
- For BK-AE: VE-Cadherin, HWK cleavage, plasma kallikrein activity, C4a.
In conclusion, isolated recurrent AE is a diagnostic challenge. One key test is the C1-inh activity assay. In case of normal C1-inh activity, therapeutic tests must be done to identify the main pathway responsible for the angioedema: mast cell induction or bradykinin mediation (Figure 3). At present, there remain some therapeutic impasses. Patient management is very challenging because there are two opposing diagnoses: one that is often benign, the other that can cause death by asphyxiation in 25% of cases without immediate specific treatment.
Acknowledgment
We thank Dr. Alison Foote (Grenoble Alpes University Hospital, France) for editing the manuscript.
Disclosures
Aude Belbézier and Alexis Bocquet have no conflict of interest. Laurence Bouillet has received honoraria from BioCryst, CSL Behring, Novartis, Pharming, and Shire, her institute has received research funding from CSL Behring, GlaxoSmithKline, Novartis, Roche, and Shire, and she reports grants and personal fees from Takeda during the conduct of the study, and grants and personal fees from Novartis outside the submitted work.
References
1. Bouillet L, Boccon-Gibod I, Berard F, Nicolas JF Recurrent angioedema: diagnosis strategy and biological aspects. Eur J Dermatol. 2014; 24:293–296. doi:10.1684/ejd.2014.2276
2. Radonjic-Hoesli S, Hofmeier KS, Micaletto S, Schmid-Grendelmeier P, Bircher A, Simon D. Urticaria and angioedema: an update on classification and pathogenesis. Clin Rev Allergy Immunol. 2018; 54:88–101. doi:10.1007/s12016-017-8628-1.
3. Kelly M, Donnelly JP, McAnnally J-R, Wang HE National estimates of emergency department visits for angioedema and allergic reactions in the United States. Allergy Asthma Proc. 2013; 34:
4. Crochet J, Lepelley M, Yahiaoui N, et al. Bradykinin mechanism is the main responsible for death by isolated asphyxiating angioedema in France. Clin Exp Allergy. 2019; 49:252–254. doi:10.1111/cea.13297.
5. Faisant C, Boccon-Gibod I, Mansard C, et al. Idiopathic histaminergic angioedema without wheals: a case series of 31 patients. Clin Exp Immunol. 2016; 185:81–85. doi:10.1111/cei.12789.
6. Zuberbier T, Aberer W, Asero R, et al. The EAACI/GA2LEN/EDF/WAO guideline for the definition, classification, diagnosis and management of urticaria. Allergy. 2018; 73:1393–1414. doi:10.1111/all.13397.
7. Kanani A, Betschel SD, Warrington R Urticaria and angioedema. Allergy Asthma Clin Immunol. 2018; 14:59. doi:10.1186/s13223-018-0288-z.
8. Maurer M, Weller K, Bindslev-Jensen C, et al. Unmet clinical needs in chronic spontaneous urticaria A GA2LEN task force report. Allergy. 2011; 66:317–330. doi:10.1111/all.2011.66.issue-3.
9. Staevska M, Popov TA, Kralimarkova T, et al. The effectiveness of levocetirizine and desloratadine in up to 4 times conventional doses in difficult-to-treat urticaria. J Allergy Clin Immunol. 2010; 125:676–682. doi:10.1016/j.jaci.2009.11.047.
10. Cicardi M, Aberer W, Banerji A, et al. Classification, diagnosis, and approach to treatment for angioedema: consensus report from the Hereditary Angioedema International Working Group. Allergy. 2014; 69:602–616. doi:10.1111/all.12380.
11. Oschatz C, Maas C, Lecher B, et al. Mast cells increase vascular permeability by heparin-initiated bradykinin formation in vivo. Immunity. 2011;34:258–268. doi:10.1016/j.immuni.2011.02.008.
12. Sands MF, Blume JW, Schwartz SA Successful treatment of 3 patients with recurrent idiopathic angioedema with omalizumab. J Allergy Clin Immunol. 2007;120:979–981. doi:10.1016/j.jaci.2007.07.041.
13. von Websky A, Reich K, Steinkraus V, Breuer K Complete remission of severe chronic recurrent angioedema of unknown cause with omalizumab. J Dtsch Dermatol Ges. 2013;11:677–678. doi:10.1111/ddg.2013.11.issue-7.
14. Ozturk AB, Kocaturk E Omalizumab in recurring larynx angioedema: a case report. Asia Pac Allergy. 2014;4:129–130. doi:10.5415/apallergy.2014.4.2.129.
15. Azofra J, Díaz C, Antépara I, Jaúregui I, Soriano A, Ferrer M Positive response to omalizumab in patients with acquired idiopathic nonhistaminergic angioedema. Ann Allergy Asthma Immunol. 2015;114:418–419. doi:10.1016/j.anai.2015.02.007.
16. Faisant C, Du Thanh A, Mansard C, Deroux A, Boccon-Gibod I, Bouillet L. Idiopathic non-histaminergic angioedema: successful treatment with omalizumab in five patients. J Clin Immunol. 2017;37:80–84. doi:10.1007/s10875-016-0345-7.
17. Bucher MC, Petkovic T, Helbling A, Steiner UC Idiopathic non-histaminergic acquired angioedema: a case series and discussion of published clinical trials. Clin Transl Allergy. 2017;7:27. doi:10.1186/s13601-017-0164-9.
18. Brunetta E, Shiffer D, Folci M, et al. Omalizumab for idiopathic nonhistaminergic angioedema: evidence for efficacy in 2 patients. Case Rep Immunol. 2018: 8067610.
19. Kaplan AP, Giménez-Arnau AM, Saini SS Mechanisms of action that contribute to efficacy of omalizumab in chronic spontaneous urticaria. Allergy. 2017;72:519–533.
20. Kolkhir P, André F, Church MK, Maurer M, Metz M Potential blood biomarkers in chronic spontaneous urticaria. Clin Exp Allergy. 2017;47:19–36. doi:10.1111/cea.2017.47.issue-1.
21. Lara-Marquez ML, Christiansen SC, Riedl MA, Herschbach J, Zuraw BL Threshold-stimulated kallikrein activity distinguishes bradykinin- from histamine-mediated angioedema. Clin Exp Allergy. 2018;48:1429–1438. doi:10.1111/cea.2018.48.issue-11.
22. Arm JP, Bottoli I, Skerjanec A, et al. Pharmacokinetics, pharmacodynamics and safety of QGE031 (ligelizumab), a novel high-affinity anti-IgE antibody, in atopic subjects. Clin Exp Allergy. 2014;44:1371–1385. doi:10.1111/cea.12400.
23. Martinez-Saguer I, Farkas H Erythema marginatum as an early symptom of hereditary angioedema: case report of 2 newborns. Pediatrics. 2016;137:e20152411. doi:10.1542/peds.2015-2411.
24. Bork K, Staubach P, Eckardt AJ, Hardt J Symptoms, course, and complications of abdominal attacks in hereditary angioedema due to C1 inhibitor deficiency. Am J Gastroenterol. 2006;101:1–9. doi:10.1111/ajg.2006.101.issue-3.
25. Veronez CL, Campos RA, Constantino-Silva RN, Nicolicht P, Pesquero JB, Grumach AS Hereditary angioedema-associated acute pancreatitis in C1-inhibitor deficient and normal C1-inhibitor patients: case reports and literature review. Front Med. 2019;6:80. doi:10.3389/fmed.2019.00080.
26. Bork K, Bernstein JA, Machnig T, Craig TJ Efficacy of different medical therapies for the treatment of acute laryngeal attacks of hereditary angioedema due to C1-esterase inhibitor deficiency. J Emerg Med. 2016;50:567e1–580e1. doi:10.1016/j.jemermed.2015.11.008.
27. Longhurst H, Cicardi M Hereditary angioedema. Lancet. 2012;379:
28. Maurer M, Magerl M, Ansotegui I, et al. The international WAO/EAACI guideline for the management of hereditary angioedema-The 2017 revision and update. Allergy. 2018;73:1575–1596. doi:10.1111/all.2018.73.issue-8.
29. Farkas H, Veszeli N, Kajdácsi E, Cervenak L, Varga L Nuts and Bolts” of laboratory evaluation of angioedema. Clin Rev Allergy Immunol. 2016;51:140–151. doi:10.1007/s12016-016-8539-6.
30. Zanichelli A, Arcoleo F, Barca MP, et al. A nationwide survey of hereditary angioedema due to C1Inhibitor deficiency in Italy. Orphanet J Rare Dis. 2015; 10:11. doi:10.1186/s13023-015-0233-x.
31. Banerji A Hereditary angioedema: classification, pathogenesis, and diagnosis. Allergy Asthma Proc. 2011; 32:403–407. doi:10.2500/aap.2011.32.3492.
32. Germenis AE, Speletas M Genetics of hereditary angioedema revisited. Clin Rev Allergy Immunol. 2016;51:170–182. doi:10.1007/s12016-016-8543-x.
33. Gobert D, Paule R, Ponard D, et al. A nationwide study of acquired C1-inhibitor deficiency in France: characteristics and treatment responses in 92 patients. Medicine (Baltimore). 2016;95:e4363. doi:10.1097/MD.0000000000004363.
34. Castelli R, Zanichelli A, Cicardi M, Cugno M Acquired C1-inhibitor deficiency and lymphoproliferative disorders: a tight relationship. Crit Rev Oncol Hematol. 2013;87:323–332. doi:10.1016/j.critrevonc.2013.02.004.
35. Calvo-Ferrer MA, Martín-Rodríguez S, Ortega-Unanue N, ÁJ B-S, Bernardo-González I A novel variant in SERPING1 gene causing angioedema in a patient with C1q low levels. Ann Allergy Asthma Immunol. 2019; 27.
36. Mete Gökmen N, Gülbahar O, Onay H, et al. Deletions in SERPING1 lead to lower C1 inhibitor function: lower C1 inhibitor function can predict disease severity. Int Arch Allergy Immunol. 2019;178:50–59. doi:10.1159/000492583.
37. Cicardi M, Zingale LC, Pappalardo E, Folcioni A, Agostoni A Autoantibodies and lymphoproliferative diseases in acquired C1-inhibitor deficiencies. Medicine (Baltimore). 2003;82:274–281. doi:10.1097/01.md.0000085055.63483.09.
38. Cacoub P, Frémeaux-Bacchi V, De Lacroix I, et al. A new type of acquired C1 inhibitor deficiency associated with systemic lupus erythematosus. Arthritis Rheum. 2001;44:1836–1840. doi:10.1002/1529-0131(200108)44:8<1836::AID-ART321>3.0.CO;2-Y.
39. Binkley KE, Davis A Clinical, biochemical, and genetic characterization of a novel estrogen- dependent inherited form of angioedema. J Allergy Clin Immunol. 2000;106:
40. Dewald G, Bork K Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun. 2006;343:1286–1289. doi:10.1016/j.bbrc.2006.03.092.
41. Bork K, Wulff K, Steinmüller-Magin L, et al. Hereditary angioedema with a mutation in the plasminogen gene. Allergy. 2017.
42. Bafunno V, Firinu D, D’Apolito M, et al. Mutation of the angiopoietin-1 gene (ANGPT1) associates with a new type of hereditary angioedema. J Allergy Clin Immunol. 2018;141(3):
43. Deroux A, Boccon-Gibod I, Fain O, et al. Hereditary angioedema with normal C1 inhibitor and factor XII mutation: a series of 57 patients from the French National Center of Reference for Angioedema. Clin Exp Immunol. 2016;185(3):
44. Veronez CL, Moreno AS, Constantino-Silva RN, et al. Hereditary angioedema with normal C1 inhibitor and F12 mutations in 42 Brazilian families. J Allergy Clin Immunol Pract. 2018;6(4):1209–1216.e8. doi:10.1016/j.jaip.2017.09.025.
45. Zhang H, Liu S, Lin C, et al.Compound heterozygous mutations Glu502Lys and Met527Thr of the FXII gene in a patient with factor XII deficiency. Hematol Amst Neth. 2019;24(1):
46. Picone O, Donnadieu AC, Brivet FG, Boyer-Neumann C, Frémeaux-Bacchi V, Frydman R Obstetrical complications and outcome in two families with hereditary angioedema due to mutation in the F12 gene. Obstet Gynecol Int. 2010; 2010: 957507. doi:10.1155/2010/957507.
47. Wintenberger C, Boccon-Gibod I, Launay D, et al.Tranexamic acid as maintenance treatment for non-histaminergic angioedema: analysis of efficacy and safety in 37 patients. Clin Exp Immunol. 2014;178:112–117. doi:10.1111/cei.2014.178.issue-1.
48. Belbézier A, Hardy G, Marlu R, et al.Plasminogen gene mutation with normal C1 inhibitor hereditary angioedema: three additional French families. Allergy. 2018;73:2237–2239. doi:10.1111/all.2018.73.issue-11.
49. Germenis AE, Loules G, Zamanakou M, et al.On the pathogenicity of the plasminogen K330E mutation for hereditary angioedema. Allergy. 2018;73:1751–1753. doi:10.1111/all.2018.73.issue-8.
50. Recke A, Massalme EG, Jappe U, et al.Identification of the recently described plasminogen gene mutation p.Lys330Glu in a Family from Northern Germany with Hereditary Angioedema. Clin Transl Allergy. 2019;9:9.
51. Yakushiji H, Hashimura C, Fukuoka K, et al. A missense mutation of the plasminogen gene in hereditary angioedema with normal C1 inhibitor in Japan. Allergy. 2018;73(11):
52. Bork K, Wulff K, Rossmann H, et al. Hereditary angioedema cosegregating with a novel kininogen 1 gene mutation changing the N-terminal cleavage site of bradykinin. Allergy. 2019. doi:10.1111/all.v74.12.
53. Brown NJ, Ray WA, Snowden M, Griffin MR Black Americans have an increased rate of angiotensin converting enzyme inhibitor-associated angioedema. Clin Pharmacol Ther. 1996;60:8–13. doi:10.1016/S0009-9236(96)90161-7.
54. Brown NJ, Byiers S, Carr D, Maldonado M, Warner BA Dipeptidyl peptidase-IV inhibitor use associated with increased risk of ACE inhibitor-associated angioedema. Hypertension. 2009;54:516–523. doi:10.1161/HYPERTENSIONAHA.109.134197.
55. Duerr M, Glander P, Diekmann F, Dragun D, Neumayer -H-H, Budde K. Increased incidence of angioedema with ACE inhibitors in combination with mTOR inhibitors in kidney transplant recipients. Clin J Am Soc Nephrol. 2010;5:703–708. doi:10.2215/CJN.07371009.
56. Baş M, Greve J, Stelter K, et al. A randomized trial of icatibant in ACE-inhibitor-induced angioedema. N Engl J Med. 2015;372:418–425. doi:10.1056/NEJMoa1312524.
57. Roberts JR, Lee JJ, Marthers DA Angiotensin-converting enzyme (ACE) inhibitor angioedema: the silent epidemic. Am J Cardiol. 2012;109:774–775. doi:10.1016/j.amjcard.2011.11.014.
58. Veronez CL, Serpa FS, Pesquero JB A rare mutation in the F12 gene in a patient with ACE inhibitor-induced angioedema. Ann Allergy Asthma Immunol. 2017;118:
59. Faisant C, Armengol G, Bouillet L, et al. Angioedema triggered by medication blocking the renin/angiotensin system: retrospective study using the french national pharmacovigilance database. J Clin Immunol. 2016;36:95–102. doi:10.1007/s10875-015-0228-3.
60. Du-Thanh A, Raison-Peyron N, Drouet C, Guillot B Efficacy of tranexamic acid in sporadic idiopathic bradykinin angioedema. Allergy. 2010;65:
61. Fok JS, Katelaris CH Angioedema masqueraders. Clin Exp Allergy. 2019;49:1274–1282. doi:10.1111/cea.v49.10.
62. Deroux A, Dumestre-Perard C, Khalil-Mgharbel A, et al. BIOBRAD study: the search for biomarkers of bradykinin-mediated angio-oedema attacks. Int Arch Allergy Immunol. 2016;170:108–114. doi:10.1159/000446959.
63. Kaplan AP, Maas C The search for biomarkers in hereditary angioedema. Front Med. 2017;4:206. doi:10.3389/fmed.2017.00206.
64. Veszeli N, Kőhalmi KV, Kajdácsi E, et al. Complete kinetic follow-up of symptoms and complement parameters during a hereditary angioedema attack. Allergy. 2018;73:516–520. doi:10.1111/all.13327.
65. Bouillet L, Vilgrain IV E-cadherin, a potential marker for endothelial cell activation during hereditary angioedema attacks. J Allergy Clin Immunol. 2014;134:241. doi:10.1016/j.jaci.2014.04.016.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.