Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
Identification of Independent and Communal Differentially Expressed Genes as Well as Potential Therapeutic Targets in Ischemic Heart Failure and Non-Ischemic Heart Failure
Authors Wang Z, Zhang M, Xu Y ![]() , Gu Y, Song Y, Jiang T
, Gu Y, Song Y, Jiang T ![]()
Received 1 April 2021
Accepted for publication 25 May 2021
Published 14 June 2021 Volume 2021:14 Pages 683—693
DOI https://doi.org/10.2147/PGPM.S313621
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Zuoxiang Wang,1,2 Mingyang Zhang,1,2 Yinan Xu,1,2 Yiyu Gu,1,2 Yumeng Song,1,2 Tingbo Jiang1
1Department of Cardiology, The First Affiliated Hospital of Soochow University, Suzhou, Jiangsu, People’s Republic of China; 2Department of Medicine, Soochow University, Suzhou, Jiangsu, People’s Republic of China
Correspondence: Tingbo Jiang
Department of Cardiology, The First Affiliated Hospital of Soochow University, 188 Shizi Road, Suzhou, 215006, People’s Republic of China
Tel +86 18906201122
Email [email protected]
Background: Heart failure (HF) is a rapidly growing public health problem, and its two main etiological types are non-ischemic heart failure (NIHF) and ischemic heart failure (IHF). However, the independent and common mechanisms of NIHF and IHF have not been fully elucidated. Here, bioinformatic analysis was used to characterize the difference and independent pathways for IHF and NIHF, and more importantly, to unearth the common potential markers and therapeutic targets in IHF and NIHF.
Methods: Two data sets with accession numbers GSE26887 and GSE84796 were downloaded from the Gene Expression Omnibus (GEO) database. After identifying the independent and communal DEGs of NIHF and IHF, a functional annotation, protein–protein interaction (PPI) network analysis, co-expression and drug–gene interaction prediction analysis, and mRNA-miRNA regulatory network analysis were performed for DEGs.
Results: We found 1146 independent DEGs (DEGs2) of NIHF mainly enriched in transcription-related and 2595 independent DEGs (DEGs3) of IHF mainly enriched in immune-related. Moreover, 185 communal DEGs (DEGs1) were found between NIHF and IHF, including 93 upregulated genes and 92 downregulated genes. Pathway enrichment analysis results showed that GPCR pathways and biological processes are closely related to the occurrence of HF. In addition, three hub genes were identified from PPI network, including CCL5, C5 and TLR3.
Conclusion: The identification of DEGs and hub genes in this study contributes to a novel perception for potential functional mechanisms and biomarkers or therapeutic targets in NIHF and IHF.
Keywords: bioinformatical analysis, differentially expressed genes, hub genes, heart failure, ischemic heart failure, non-ischemic heart failure
Introduction
Heart failure (HF) is the inability of the heart to deliver sufficient blood to meet the demands of our body under normal filling pressure, which ultimately leads to a complex and severe disease syndrome.1,2 HF is a rapidly growing public health issue with an estimated prevalence of 37.7 million individuals globally, which confers a substantial burden to the health-care system.2,3 Although the classification system for HF causes remain debated, it’s undeniable that non-ischemic heart failure (NIHF) and ischemic heart failure (IHF) are the two main etiological categories of HF.4 IHF, the most common type, is defined as left ventricular (LV) systolic dysfunction due to the coronary artery disease (CAD).5 NIHF is the other heart failure that excludes CAD factors, including all remaining heterogeneous HF etiologies, like valvular diseases, toxic damage, metabolic conditions and genetic cardiomyopathies.6,7 Recent studies have shown that the expression levels of cytokines, relating to HF, are significantly different between IHF and NIHF.7 Meanwhile, large-scale clinical trials have reported that IHF and NIHF respond differently to interventional drug therapies.4,8 However, the exact biological mechanism of above is still unclear. Interestingly, although the etiology is different between IHF and NIHF, the pathology is very similar, especially in patients with end stage of HF.4 So, there could be a common biological mechanism in the development of IHF and NIHF.
Therefore, the purpose of this study is to use Bioinformatic analysis to characterize distinct and independent pathways for IHF and NIHF, and more importantly, to identify the common biological mechanisms of IHF and NIHF, so as to reveal potential biomarkers and therapeutic directions of HF. In this study, two mRNA microarray data sets were downloaded from the Gene Expression Omnibus (GEO) for screening the differentially expressed genes (DEGs) associated with IHF and NIHF. Then, gene ontology and pathway enrichment analysis and protein–protein interaction (PPI) network analysis were performed to help us understand the independent and shared molecular mechanisms of DM and HF. In conclusion, a total of 185 DEGs and 4 hub genes, which might play an important role in the common biological mechanisms between NIHF and IHF.
Materials and Methods
Data Source
GEO (https://www.ncbi.nlm.nih.gov/geo/) is a public gene expression database created by NCBI, which contains high throughput sequencing and microarray data sets.9 Among the inclusion criteria were (1) a. NIHF databases: HF patients was diagnosed dilated cardiomyopathy or no obvious evidence of ischemic heart disease; b. IHF database: HF patient was diagnosed with ischemic cardiomyopathy; (2) detection of gene level in left ventricular free wall heart tissue samples; (3) Datasets that included patients and healthy controls. Exclusion criteria included: Patients had participated in a clinical trial for drugs or other treatments. Finally, two datasets were selected from GEO: accession numbers GSE84796 (8 NIHF patients and 10 controls) and GSE26887 (12 IHF patients and 5 controls).
Identification of DEGs
GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/) is used to identify differentially expressed genes (DEGs).10 GEO2R is based on limma package that comes with the GEO databases. Differentially expressed genes (DEGs) were determined by |logFC| > 0.5 and adj.P-value < 0.05. Next, the online Venn software (http://bioinformatics.psb.ugent.be/webtools/Venn/) was applied to detect the overlap DEGs among three datasets.
Enrichment Analyses of DEGs
DAVID 6.8 (https://david.ncifcrf.gov/) was used for enrichment analyses to elucidate the biological functions of the overlapping DEGs.11 DAVID is a comprehensive bioinformatics analysis tool, providing a set of functional annotation tools for researchers to analyze the biological functions of massive genes. Further evaluation of the pathway enrichment analyses of DEGs was implemented by KOBAS 3.0 (http://kobas.cbi.pku.edu.cn), which annotates the input gene set with putative pathways by mapping to genes with known annotations from 5 pathway databases (KEGG PATHWAY, PID, BioCyc, Reactome and Panther).12 P-value < 0.05 was considered significant. The genes in modules were also analyzed in the same way.
Protein–Protein Interaction Network Construction and Module Analysis
Protein–protein interaction (PPI) network reveals the specific and unspecific interactions of proteins, and identifies the core protein genes. STRING (http://string-db.org, version 11.0) database was used to predict the PPI network of DEGs and analyze the interactions between proteins.13 An interaction with a combined score > 0.4 was recognized as statistical significance. The molecular interaction networks were visualized using the Cytoscape (version 3.7.0).14 Subsequently, we used the MCODE plugin to identify densely connected modules from the PPI network with the criteria of K-core = 2, degree cutoff = 2, max depth = 100, and node score cutoff = 0.2.15
Hub Genes Selection and Analyses
The hub genes were selected using the cytoHubba plugin, a Cytoscape plugin, was used to determine the hub proteins or genes in the PPI network.16 We randomly select 5 of the 12 algorithms in cytoHubba plugin, and take the intersection of the 5 algorithms results to determine hub gene. Subsequently, a network of genes and their co-expression genes was analyzed via GeneMANIA (http://www.genemania.org/), which is a convenient web portal for analyzing gene lists and predicting gene function.17 Finally, Drug-Gene Interaction database (DGIdb) 3.0 (http://www.dgidb.org/), which helps to predict drug–gene interaction networks, was adopted here to predict drugs based on the module genes.18 After the prediction of drug-gene pairs associated with the module genes, the network map was then formed by Cytoscape.
Construction of mRNA-miRNA Regulatory Network
We used Mirwalk to predict corresponding miRNA of hub gene. Mirwalk is a publicly available database that mainly focuses on miRNA-target interactions.19 In order to construct further accurate regulatory network, the screening condition was that the Predicted miRNA could be verified by experiments or other databases. After the prediction of mRNA-miRNA by Mirwalk, the regulatory network were visualized by Cytoscape.
Results
Identification of DEGs
After standardization and identification of the microarray results, DEGs were selected (Figure 1). There were 2780 DEGs in the GSE84796 dataset, including 1597 upregulated genes and 1183 downregulated genes (Figure 2A) (Supplementary Table 1). There were 1331 DEGs in the GSE26887 dataset, including 705 upregulated and 626 downregulated genes (Figure 2B) (Supplementary Table 2). A Venn diagram was generated to show the overlap between GSE26887 and GSE84796 datasets; these include 93 upregulated and 92 downregulated genes (DEGs1) (Figure 2C and D). Furthermore, 1146 DEGs (DEGs2) and 2595 DEGs (DEGs3) were identified independently from the DEGs in GSE26687 and GSE84796, respectively.
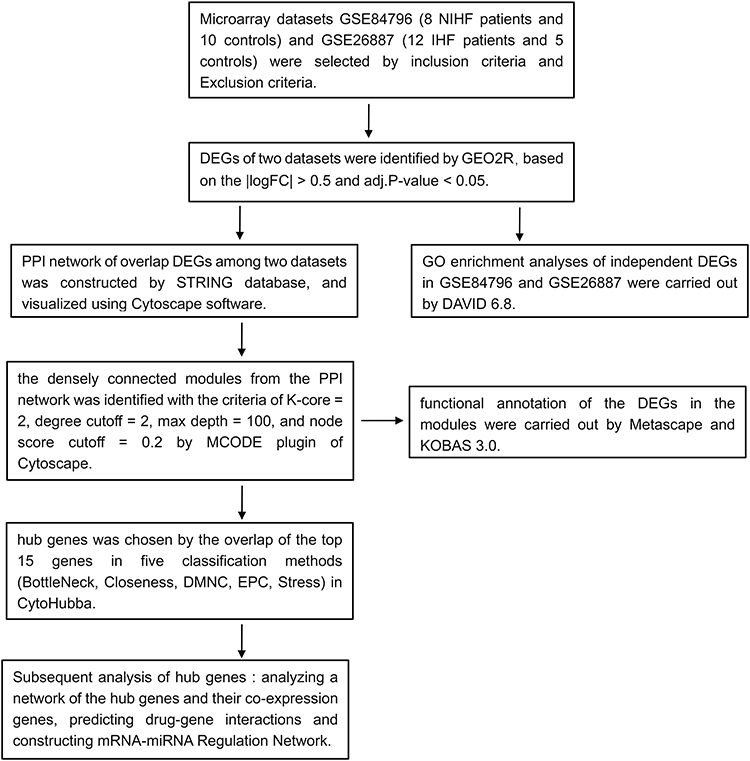 |
Figure 1 Flow diagram of the study design. |
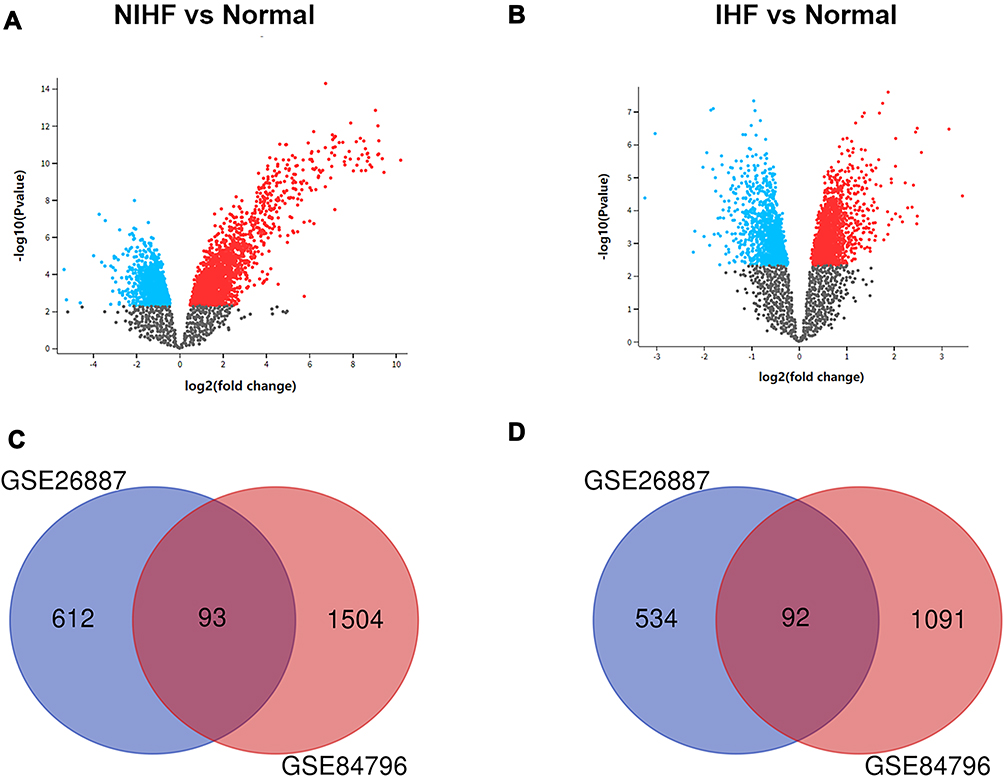 |
Figure 2 Identification of gene expression profiles in the two datasets. (A) Volcano plot of NIHF microarray data. (B) Volcano plot of IHF microarray data. (C) Venn diagram of the 93 communal upregulated DEGs between NIHF and IHF. (D) Venn diagram of the 92 communal downregulated DEGs between NIHF and IHF. |
GO Enrichment Analyses of Independent DEGs in IHF and NIHF
To determine the biological functions of DEGs2 and DEGs3, GO enrichment analysis was performed (Table 1). Results were divided into three functional categories, including biological processes (BP), cell component (CC), and molecular function (MF). GO analysis of DEGs2 indicated that changes in BP were significantly enriched in the positive regulation of pri-miRNA transcription from RNA polymerase II promoter (GO:1902895), amino acid transport (GO:0006865), regulation of cell shape (GO:0008360) and leukocyte migration (GO:0050900). As for CC, DEGs2 were particularly enriched in extracellular exosome (GO:0070062), extracellular matrix (GO:0031012), focal adhesion (GO:0005925) and cytosol (GO:0005829). Changes in MF were mostly enriched in low-density lipoprotein particle binding (GO:0030169), amino acid transmembrane transporter activity (GO:0015171), RAGE receptor binding (GO:0050786) and glycoprotein binding (GO:0001948). The GO analysis of DEGs3 returned that the terms response to immune response (GO:0006955), adaptive immune response (GO:0002250), regulation of immune response (GO:0050776) and innate immune response (GO:0045087) under BP were mainly enriched. The terms enriched under CC were external side of plasma membrane (GO:0009897), T cell receptor complex (GO:0042101), integral component of plasma membrane (GO:0005887) and plasma membrane (GO:0005886). Moreover, the terms enriched under MF were receptor activity (GO:0004872), non-membrane spanning protein tyrosine kinase activity (GO:0004715), protein binding (GO:0005515) and receptor binding (GO:0005102).
 |
Table 1 The GO Enrichment Analysis of DEGs2 and DEGs3 (Top 4 Terms According to p.adjust) |
Protein–Protein Interaction Network Construction and Module Analysis
The PPI network of DEGs with combined scores greater than 0.4 was generated by Cytoscape, which contained 109 nodes and 119 edges (Figure 3) (Supplementary Table 3). The MCODE plugin identified three densely connected modules in which 12 DEGs were among DEGs1 (Figure 4A). Then, we use online database DAVID 6.8 and KOBAS 3.0 to analyze the enrichment analyses of 12 genes. GO analysis of 12 genes were completed by DAVID 6.8, genes were significantly enriched in ribosomal large subunit biogenesis (GO:0042273) and G-protein coupled purinergic nucleotide receptor signaling pathway (GO:0035589) in BP. As for CC, genes were particularly enriched in nucleolus (GO:0005730) and integral component of plasma membrane (GO:0005887). Changes in MF were mostly enriched in poly(A) RNA binding and G-protein coupled purinergic nucleotide receptor activity (GO:0045028) (Figure 4B and C). Pathway analysis which was completed by KOBAS 3.0 revealed genes to be mainly involved in Class A/1 (Rhodopsin-like receptors) (R-HSA-373076), G alpha (i) signalling events (R-HSA-418594), P2Y receptors (R-HSA-417957) and GPCR ligand binding (R-HSA-500792) (Figure 4D).
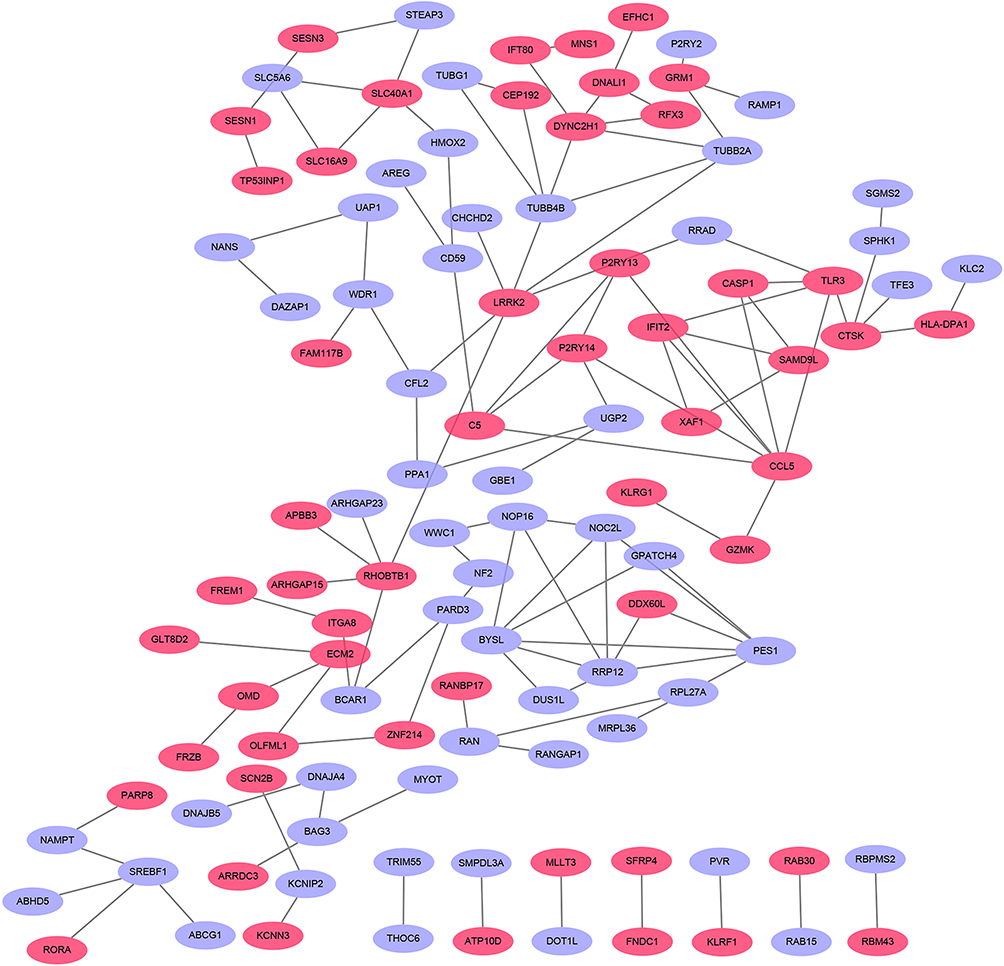 |
Figure 3 Based on database STRING and Cytoscape software, PPI networks of the DEGs were constructed. The pink point represents upregulated genes, and purple point represents downregulated genes. |
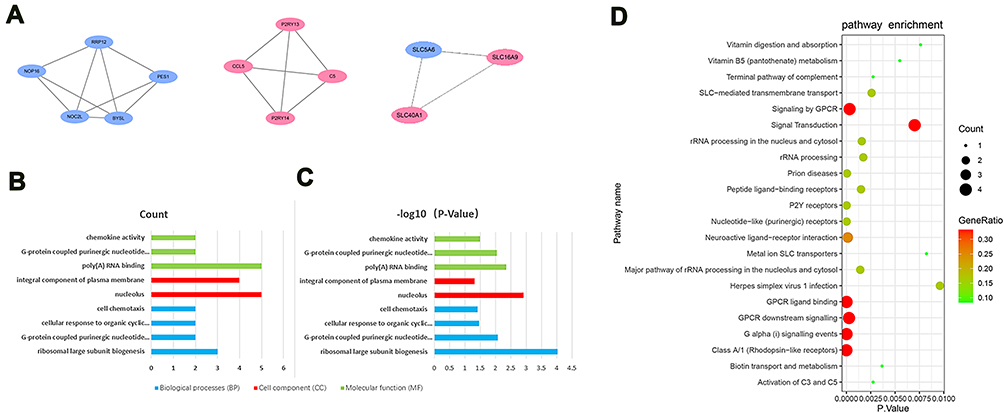 |
Figure 4 (A) Top modules from the protein–protein interaction network. (B) The biological process in functional enrichment of the DEGs in Modules was performed using the online biological tool DAVID between HF and T2DM with P-value and (C) gene count. (D) The pathway analysis of the DEGs in Modules by KOBAS 3.0. The abscissa represents the P-value, and the ordinate represents the terms. The size of the circle represents the number of genes involved, and the color represents the frequency of the genes involved in the term total genes. |
Hub Gene Selection and Analysis
In the present study, we used cytoHubba to choose hub genes. According to the five classification methods (BottleNeck, Closeness, DMNC, EPC, Stress) in cytoHubba, the top 15 hub genes selected by these ranked methods in cytoHubba are shown in Table 2. Finally, three central genes were identified by overlapping the first 15 genes (Figure 5). CCL5, C5, TLR3 were the three central genes, and three central genes were all upregulated genes. A network of the hub genes and their co-expression genes was analyzed by GeneMANIA online platform (Figure 6). The three genes showed the complex PPI network with the Physical interactions of 67.64%, Co-expression of 13.50%, Co-localization of 6.17%, Predicted of 6.35%, Pathway of 4.35%, Genetic Interactions of 1.40%, and Shared protein domains of 0.59%. Finally, based on the DGIdb predictions of the hub genes, we obtained 14 drug–gene interaction pairs, including two hub genes (C5, TLR3) and 14 drugs (FDA-listed + antitumor drugs) (Figure 7). These results may reveal the therapeutic targets related to HF.
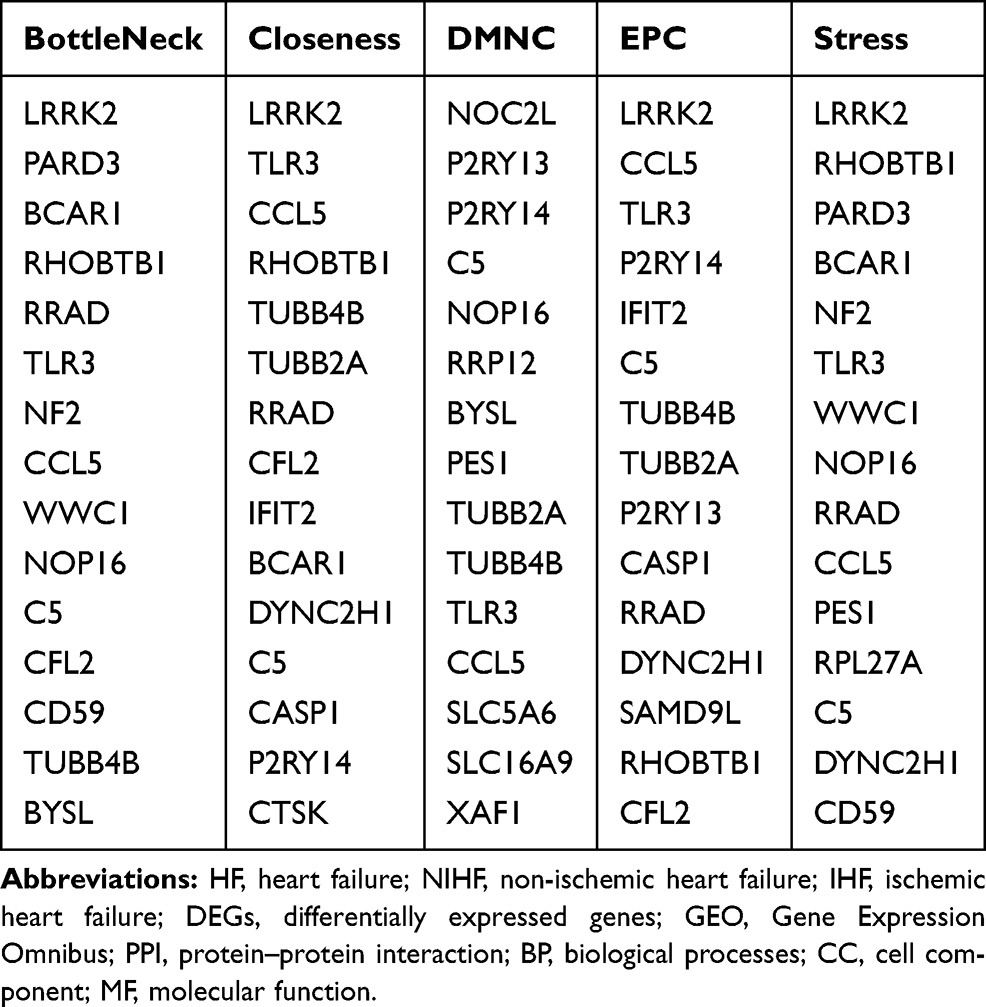 |
Table 2 The Top 15 Hub Genes Rank in cytoHubba |
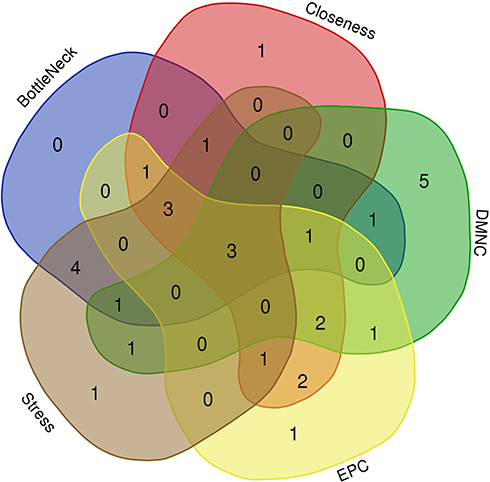 |
Figure 5 Three hub genes were identified by overlapping the first 15 genes in the five classification methods of cytoHubba. |
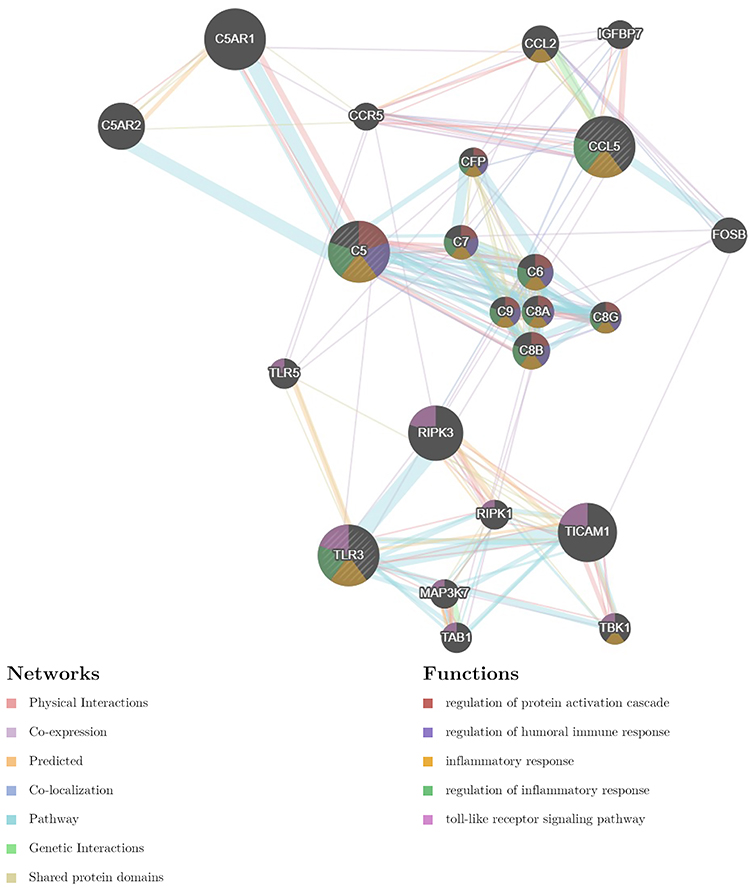 |
Figure 6 Hub genes and their co-expression genes were analyzed using GeneMANIA. |
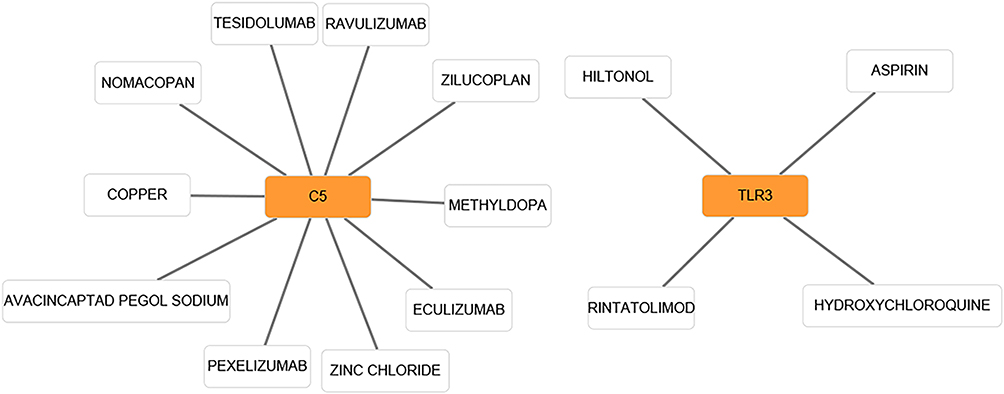 |
Figure 7 Based on the DGIdb predictions of the module genes, we obtained 14 drug–gene interaction pairs, including two hub genes (C5, TLR3) and 14 drugs. Yellow circle indicates the differentially expressed gene and blank square indicates the drug. |
mRNA-miRNA Regulation Network Construction
We used MiRwalk databases to predict the miRNAs of three hub genes and found a total of 45 miRNAs through screening condition that the predicted miRNA could be verified by experiments or other databases. The data of these hub genes and their miRNAs were integrated into a regulatory network, and visualized using Cytoscape software (Figure 8).
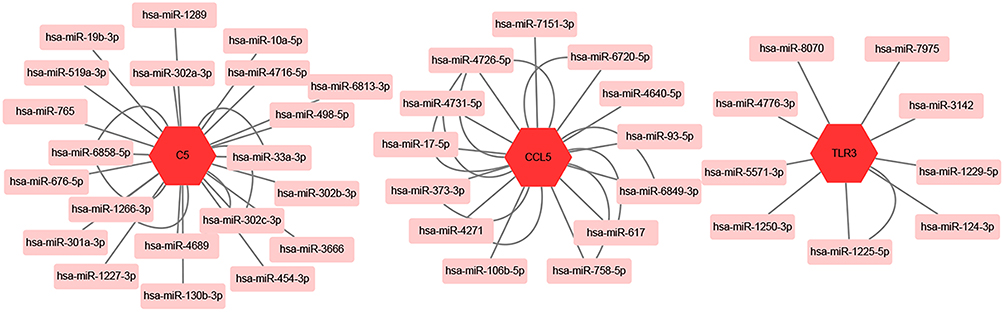 |
Figure 8 mRNA-miRNA regulation network of hub genes was constructed by MiRwalk. |
Discussion
In this study, we identified 185 overlapping DEGs (DEGs1) in both NIHF and IHF, of which 93 were upregulated and 92 were downregulated genes. Independent DEGs included 612 upregulated and 534 downregulated genes in NIHF (DEGs2), and 1504 upregulated and 1091 downregulated genes in IHF (DEGs3). GO analysis revealed that DEGs2 were mainly enriched in transcription-related, while DEGs3 were mainly enriched in immune-related. Enrichment analyses of the genes in the key modules of the constructed PPI network revealed that they were mainly enriched in some GPCR pathways and biological processes. Finally, three hub genes, CCL5, C5 and TLR3 were found in the PPI network. Then, we analyzed a network of the hub genes and their co-expression genes, predicted drug–gene interactions and construct mRNA-miRNA Regulation Network.
G protein-coupled receptors (GPCRs), as we know, are a major target of therapeutic intervention in most physiological processes including vision, smell, taste in addition to neurologic, cardiovascular, endocrine and reproductive function. GPCRs play a prominent role in the regulation of cardiovascular function.20 β-adrenergic receptors (Ars), as a member of a family of GPCRs, is widely confirmed closely related to the development of heart failure. Therefore, beta-blockers which can block Ars become one of the classic heart failure drugs.21,22 At present, most studies focused on the GRK, especially the GRK2 and GRK5.20 In the model of IHF, Phosphorylated GRK2 modified and desensitized AdipoR1 in failing cardiomyocytes, leading to post-MI remodeling and HF progression.23 Meanwhile, there is also some evidence indicate that GRK2 plays an important role in the development of NIHF. Rockman et al uncovered that upregulating level of GRK2 was closely related to the onset of dilated cardiomyopathy (DCM), and the effect could be reversed by GRK2-inhibitor peptide.21,24 Similarly, an experiment was also designed to use paroxetine to specifically inhibit GRK2 in mice and found an improvement in heart failure.25 Additional, GRK5 is a regulator of fibroblast activation and cardiac fibrosis.26 In conclusion, the GRCP-related field, especially CRK, remains a potential direction for various heart failure mechanisms and treatments.
CCL5 (C-C motif chemokine ligand 5) is one of several chemokine genes clustered on the q-arm of chromosome 17. Chemokines form a superfamily of secreted proteins involved in immunoregulatory and inflammatory processes.27,28 Moreover, CCL5 functions as one of the natural ligands for the chemokine receptor 5 (CCR5), which is a chemokine receptor belonging to the GPCR superfamily.28,29 CCL5 has been suggested to drive cell migration toward the heart tissue of patients with HF, which plays an important role in the occurrence and development of non-ischemic cardiomyopathy.30,31 The important role of CCL5 in IHF has also been revealed in numerous experiments. Stevenson et al found that CCL5 level increased significantly in the human hearts with ischemic cardiomyopathy compared to control nonfailing hearts.32 What’s more, Montecucco et al reported that the incidence of postinfarct heart failure was significantly reduced by treatment with anti-CCL5 mAb in chronic cardiac ischaemia mouse models.27 These findings show that CCL5 play an important role in the process of IHF and NIHF. So, we speculate that it could be a potential target for the common treatment of all types of HF.
Complement C5 (C5) encodes a component of the complement system, a part of the innate immune system that plays an important role in inflammation, host homeostasis, and host defense against pathogens.33 Moreover, C5 could be cleaved into the anaphylatoxin C5a and fragment C5b by convertases. During activation and amplification, C5a are constantly released and trigger proinflammatory signaling via their corresponding GPCR.34 Egerstedt et al observed that C5a and other complement levels were significantly elevated in the blood of patients with heart failure.33 Meanwhile, Lindsey et al reported that C5 gene levels were significantly higher in the extreme ventricular dilatation group than the moderate ventricular dilation group after MI.35 The strong association of C5a with HF and enrichment of complement activation pathways suggests activation of the complement cascade in subjects at risk of HF. It is exciting that recent experiment has pointed to a central role of C5a in myocardial repair and regeneration.36 Therefore, C5 is likely to be a potential marker and provide new therapeutic approaches.
TLR3 is a member of the Toll-like receptor (TLR) family which plays a fundamental role in pathogen recognition and activation of innate immunity. TLR3 primarily protects the heart against viral infection, also mediates inflammatory effects that may exacerbate heart damage.37–39 TLR3 has been widely confirmed as a potential target for the treatment of HF, Gao et al reported that germline knockout of TLR3 could attenuate HF and improve survival in mouse model of chronic myocardial infarction.40 However, the mechanisms and therapeutic direction is still unclear. Loniewski et al reported that the levels of GRK2 protein are markedly increased by TLR2, 3, 4 and 7, which uncover potential cross-talk mechanisms between TLRs and GPCRs.41 As previous researches mentioned, CRK2-related mechanisms have been confirmed to be closely related to the development of heart failure. So, we believe that potential cross-talk mechanisms between TLRs and GPCRs may be an important HF therapeutic direction.
Finally, based on the DGIdb predictions of the hub genes, we obtained 14 drugs (FDA-listed + antitumor drugs). These results may show potential treatment options to HF, but more experiments should be carried to verify and explore these possibilities. We used MiRwalk databases to predict a total of 45 miRNAs of three hub genes, which could reveal the potential mutual regulation between genes, allowing us to better understand the relationship and the potential regulation between hub genes.
Our study has some limitations.: The identified hub nodes need to be validated in future studies and the sample size in this study was relatively small. Hence, large sample size and further mechanism experiments are still needed to confirm our conclusion.
Conclusion
In conclusion, the independent DEGs provide us new perspectives on different mechanisms between NIHF and IHF, and communal DEGs identified in our study reveal potential common mechanism of NIHF and IHF. Besides, a total of three hub genes (CCL5, C5, TLR3) have been identified, which can be used as biomarkers for HF or as therapy targets.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Clinical trial ability improvement project of cardiovascular professional group in the First Affiliated Hospital of Soochow University (No:201900180019).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37(27):2129–2200. doi:10.1093/eurheartj/ehw128
2. Ziaeian B, Fonarow GC. Epidemiology and aetiology of heart failure. Nat Rev Cardiol. 2016;13(6):368–378. doi:10.1038/nrcardio.2016.25
3. Ponikowski P, Anker SD, AlHabib KF, et al. Heart failure: preventing disease and death worldwide. ESC Heart Fail. 2014;1(1):4–25. doi:10.1002/ehf2.12005
4. Park SY, Trinity JD, Gifford JR, et al. Mitochondrial function in heart failure: the impact of ischemic and non-ischemic etiology. Int J Cardiol. 2016;220:711–717. doi:10.1016/j.ijcard.2016.06.147
5. Xiao J, Xu F, Yang CL, et al. Preferred revascularization strategies in patients with ischemic heart failure: a meta-analysis. Curr Med Sci. 2018;38(5):776–784. doi:10.1007/s11596-018-1944-8
6. McNally EM, Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res. 2017;121(7):731–748. doi:10.1161/CIRCRESAHA.116.309396
7. Varricchi G, Loffredo S, Bencivenga L, et al. Angiopoietins, vascular endothelial growth factors and secretory phospholipase A2 in ischemic and non-ischemic heart failure. J Clin Med. 2020;9(6):1928. doi:10.3390/jcm9061928
8. Follath F, Cleland JGF, Klein W, Murphy R. Etiology and response to drug treatment in heart failure. J Am Coll Cardiol. 1998;32(5):1167–1172. doi:10.1016/S0735-1097(98)00400-8
9. Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi:10.1093/nar/30.1.207
10. Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41(D1):D991–995. doi:10.1093/nar/gks1193
11. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi:10.1038/nprot.2008.211
12. Wu J, Mao X, Cai T, Luo J, Wei L. KOBAS server: a web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006;34(Web Server):W720–724. doi:10.1093/nar/gkl167
13. Franceschini A, Szklarczyk D, Frankild S, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41(D1):D808–815. doi:10.1093/nar/gks1094
14. Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27(3):431–432. doi:10.1093/bioinformatics/btq675
15. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003;4:2. doi:10.1186/1471-2105-4-2
16. Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl4):S11. doi:10.1186/1752-0509-8-S4-S11
17. Warde-Farley D, Donaldson SL, Comes O, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38(Web Server issue):W214–220. doi:10.1093/nar/gkq537
18. Cotto KC, Wagner AH, Feng YY, et al. DGIdb 3.0: a redesign and expansion of the drug-gene interaction database. Nucleic Acids Res. 2018;46(D1):D1068–d1073. doi:10.1093/nar/gkx1143
19. Sticht C, De La Torre C, Parveen A, Gretz N, Campbell M. miRWalk: an online resource for prediction of microRNA binding sites. PLoS One. 2018;13(10):e0206239. doi:10.1371/journal.pone.0206239
20. Traynham CJ, Hullmann J, Koch WJ. Canonical and non-canonical actions of GRK5 in the heart. J Mol Cell Cardiol. 2016;92:196–202. doi:10.1016/j.yjmcc.2016.01.027
21. Cannavo A, Komici K, Bencivenga L, et al. GRK2 as a therapeutic target for heart failure. Expert Opin Ther Targets. 2018;22(1):75–83. doi:10.1080/14728222.2018.1406925
22. Ali DC, Naveed M, Gordon A, et al. beta-Adrenergic receptor, an essential target in cardiovascular diseases. Heart Fail Rev. 2020;25(2):343–354. doi:10.1007/s10741-019-09825-x
23. Wang Y, Gao E, Lau WB, et al. G-protein-coupled receptor kinase 2-mediated desensitization of adiponectin receptor 1 in failing heart. Circulation. 2015;131(16):1392–1404. doi:10.1161/CIRCULATIONAHA.114.015248
24. Rockman HA, Chien KR, Choi DJ, et al. Expression of a beta-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci U S A. 1998;95(12):7000–7005. doi:10.1073/pnas.95.12.7000
25. Schumacher SM, Gao E, Zhu W, et al. Paroxetine-mediated GRK2 inhibition reverses cardiac dysfunction and remodeling after myocardial infarction. Sci Transl Med. 2015;7(277):277ra231. doi:10.1126/scitranslmed.aaa0154
26. Eguchi A, Coleman R, Gresham K, et al. GRK5 is a regulator of fibroblast activation and cardiac fibrosis. Proc Natl Acad Sci U S A. 2021;118(5):e2012854118. doi:10.1073/pnas.2012854118
27. Montecucco F, Braunersreuther V, Lenglet S, et al. CC chemokine CCL5 plays a central role impacting infarct size and post-infarction heart failure in mice. Eur Heart J. 2012;33(15):1964–1974. doi:10.1093/eurheartj/ehr127
28. Wang X, Li W, Yue Q, et al. C-C chemokine receptor 5 signaling contributes to cardiac remodeling and dysfunction under pressure overload. Mol Med Rep. 2021;23(1):1.
29. Martins E, Brodier H, Rossitto-Borlat I, Ilgaz I, Villard M, Hartley O. Arrestin recruitment to C-C chemokine receptor 5: potent C-C chemokine ligand 5 analogs reveal differences in dependence on receptor phosphorylation and isoform-specific recruitment bias. Mol Pharmacol. 2020;98(5):599–611. doi:10.1124/molpharm.120.000036
30. Wang C, Yang H, Gao C. Potential biomarkers for heart failure. J Cell Physiol. 2019;234(6):9467–9474. doi:10.1002/jcp.27632
31. Batista AM, Alvarado-Arnez LE, Alves SM, et al. Genetic polymorphism at CCL5 is associated with protection in chagas’ heart disease: antagonistic participation of CCR1(+) and CCR5(+) cells in chronic chagasic cardiomyopathy. Front Immunol. 2018;9:615. doi:10.3389/fimmu.2018.00615
32. Stevenson MD, Canugovi C, Vendrov AE, et al. NADPH oxidase 4 regulates inflammation in ischemic heart failure: role of soluble epoxide hydrolase. Antioxid Redox Signal. 2019;31(1):39–58. doi:10.1089/ars.2018.7548
33. Egerstedt A, Berntsson J, Smith ML, et al. Profiling of the plasma proteome across different stages of human heart failure. Nat Commun. 2019;10(1):5830. doi:10.1038/s41467-019-13306-y
34. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11(9):785–797. doi:10.1038/ni.1923
35. Lindsey ML, Ma Y, Flynn ER, Winniford MD, Hall ME, DeLeon-Pennell KY. Identifying the molecular and cellular signature of cardiac dilation following myocardial infarction. Biochim Biophys Acta Mol Basis Dis. 2019;1865(7):1845–1852. doi:10.1016/j.bbadis.2018.09.023
36. Natarajan N, Abbas Y, Bryant DM, et al. Complement receptor C5aR1 plays an evolutionarily conserved role in successful cardiac regeneration. Circulation. 2018;137(20):2152–2165. doi:10.1161/CIRCULATIONAHA.117.030801
37. Becher PM, Hinrichs S, Fluschnik N, et al. Role of Toll-like receptors and interferon regulatory factors in different experimental heart failure models of diverse etiology: IRF7 as novel cardiovascular stress-inducible factor. PLoS One. 2018;13(3):e0193844. doi:10.1371/journal.pone.0193844
38. Chen C, Feng Y, Zou L, et al. Role of extracellular RNA and TLR3-Trif signaling in myocardial ischemia-reperfusion injury. J Am Heart Assoc. 2014;3(1):e000683. doi:10.1161/JAHA.113.000683
39. Yu L, Feng Z. The role of toll-like receptor signaling in the progression of heart failure. Mediators Inflamm. 2018;2018:9874109. doi:10.1155/2018/9874109
40. Gao T, Zhang SP, Wang JF, et al. TLR3 contributes to persistent autophagy and heart failure in mice after myocardial infarction. J Cell Mol Med. 2018;22(1):395–408. doi:10.1111/jcmm.13328
41. Loniewski K, Shi Y, Pestka J, Parameswaran N. Toll-like receptors differentially regulate GPCR kinases and arrestins in primary macrophages. Mol Immunol. 2008;45(8):2312–2322. doi:10.1016/j.molimm.2007.11.012
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.