Back to Journals » International Journal of Women's Health » Volume 18
Identification of a Novel MTM1 Mutation Associated with X-Linked Myotubular Myopathy: Clinical and Molecular Insights for Prenatal Diagnosis
Authors Chen S ![]() , Liang B
, Liang B ![]() , Lin N, Pan M
, Lin N, Pan M ![]() , Li L
, Li L
Received 2 December 2025
Accepted for publication 12 March 2026
Published 18 March 2026 Volume 2026:18 585909
DOI https://doi.org/10.2147/IJWH.S585909
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Matteo Frigerio
Shixin Chen,1,* Bin Liang,1,* Na Lin,1 Mian Pan,2 Li Li2
1Medical Genetic Diagnosis and Therapy Center, Fujian Key Laboratory for Prenatal Diagnosis and Birth Defects, Fujian Maternity and Child Health Hospital, College of Clinical Medicine for Obstetrics & Gynecology and Pediatrics, Fujian Medical University, Fuzhou, People’s Republic of China; 2Department of Obstetrics, Fujian Maternity and Child Health Hospital, College of Clinical Medicine for Obstetrics & Gynecology and Pediatrics, Fujian Medical University, Fuzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Li Li, Email [email protected]
Abstract: Given the clinical heterogeneity and generally poor prognosis of X-linked myotubular myopathy (XLMTM), the identification of prenatal indicators and relevant medical history is imperative. However, studies specifically addressing the prenatal manifestations of this condition remain limited. Therefore, we investigated a case of XLMTM characterized by persistent polyhydramnios and conducted a comprehensive literature review. A neonate diagnosed with XLMTM was identified at the Fujian Provincial Maternity and Children’s Hospital, Fuzhou, China. Genetic analysis, including whole exome sequencing (WES) and Sanger sequencing, was performed on the infant and his parents. Pathogenic variants were classified following American College of Medical Genetics and Genomics criteria. A literature review examined prenatal features of polyhydramnios in male infants with XLMTM. The mother experienced persistent idiopathic polyhydramnios and underwent amniotic fluid reduction and prenatal diagnosis. Despite normal fetal karyotyping and single nucleotide polymorphism-array findings, the neonate, born at 36 weeks, had severe respiratory failure and died 1.5 h post-resuscitation. WES identified a pathogenic hemizygous frameshift mutation within exon 10 of MTM1 (c.968_969delinsT, p.K323Mfs*2). A literature review revealed phenotypic variability in XLMTM, with single-nucleotide variations being the most common mutation type. The novel MTM1 mutation (c.968_969delinsT, p.K323Mfs*2) caused XLMTM in this case. The prenatal characteristics exhibited by the mother, including persistent amniotic fluid and reduced fetal movement, highlight the need for WES in high-risk pregnancies, offering critical insights for prenatal diagnosis and genetic counselling.
Keywords: X-linked myotubular myopathy, polyhydramnios, MTM1 gene, WES, prenatal diagnosis
Introduction
X-linked myotubular myopathy (XLMTM) is a hereditary myopathy caused by mutations in the MTM1 gene, which is essential for skeletal muscle function due to its role in encoding myotubularin.1 During early gestation, specifically between the 8th and 15th weeks, fetal muscle development is compromised because of halted myotube maturation, leading to the manifestation of XLMTM.2
XLMTM predominantly affects males, with an incidence of approximately one in every 40,000–50,000 male births.3 Approximately 80% of male infants diagnosed with XLMTM experience respiratory distress, often necessitating long-term ventilatory support, wheelchair assistance, and feeding tubes for nutrition.4 Conversely, 20% of affected males may present with milder symptoms, achieve independent ambulation, and survive into adulthood without requiring ventilatory assistance.5,6 However, approximately 50% of male infants with severe symptoms succumb before reaching 18 months of age.7,8 Given the clinical heterogeneity and generally poor prognosis of XLMTM, the identification of prenatal indicators and relevant medical history is imperative. Such information facilitates early genetic screening for MTM1 mutations using appropriate testing protocols. Although over a dozen cases of XLMTM have been documented in China, studies specifically addressing the prenatal manifestations of this condition remain limited.
Here, we aimed to explore the clinical implications and significance of prenatal features associated with XLMTM. The genetic attributes of XLMTM were analyzed in alongside a review of existing literature to improve understanding of this rare disorder and to support clinical diagnosis and genetic counselling. We identified a novel pathogenic variant of MTM1 in a neonate who exhibited polyhydramnios during prenatal assessment–a condition associated with XLMTM.
Object and Methods
Object
A neonate diagnosed with X-linked myotubular myopathy (XLMTM) at the Fujian Provincial Maternity and Children’s Hospital in 2024 was included in this investigation.
Methodology
Clinical Data Collection
Clinical data from the pregnant woman and her newborn were collected, encompassing routine blood examinations, liver and kidney function assessments, fasting venous glucose levels, cardiac enzyme profiles, and serological tests for herpes simplex virus, toxoplasmosis, cytomegalovirus, and rubella virus. Additional parameters included serum C-reactive protein (CRP), procalcitonin (PCT), and pathogenic tests. Prenatal evaluations involved nuchal translucency (NT) ultrasound and anomaly ultrasound scans.
Sample Collection
After providing informed consent, the pregnant woman underwent transabdominal amniocentesis to remove amniotic fluid. After discarding the initial 1–2 mL of fluid to minimize maternal cell contamination, 30 mL was collected. A 20-mL portion of the amniotic fluid was used for chromosome G-banding karyotype analysis, and 10 mL was used for chromosome microarray analysis.
Chromosome G-Banding Karyotype Analysis
The 20-mL sample of amniotic fluid was placed in two sterile centrifuge tubes (10 mL each), centrifuged, and then cultured in an aseptic environment. The samples were incubated at 37°C in two culture flasks with 5–6% CO2. After incubation, the fluid was switched, gathered, and prepared for G-banding. Five karyotypes and 20 metaphases were analyzed using an automated analysis system (model GS0120, Leica, Germany).
Chromosome Microarray Analysis
Genomic DNA was extracted from the uncultured 10-mL amniotic fluid sample using the Nucleic Acid Extraction Magnetic Bead Kit (Xiamen Zhisan Biotechnology, Xiamen, China). A genome-wide copy number assessment was conducted using the Affymetrix CytoScan 750 K high-density SNP chip, and results were analyzed with ChAS software (Thermo Fisher, Affymetrix, USA). The analysis protocol included quality control, enzymatic digestion, PCR amplification, purification, labeling, hybridization, and scanning in accordance with manufacturer’s guidelines. Variants were cross-referenced with databases such as the Database of Genomic Variants, ClinGen, ClinVar, Decipher, and local repositories.
Whole-Exome Sequencing (WES)
The QIAGEN extraction kit (No. 61104, QIAGEN, Hilden, Germany) was used to extract whole genomic DNA. The IDT ThexGen Exome Research Panel v2.0 Whole-Exome Capture Microarray (IDT, Coralville, IA, USA) was used at the Anji Kang’er Laboratory of Medical Laboratories, Shenzhen, China, to perform whole exome sequencing on samples from the newborn and its parents. Sequencing had > 99% coverage and an average depth of 100×.
Sanger Sequencing
Sanger sequencing of the MTM1 gene’s c.968_969delins region and T-variant site was performed using an ABI 3500XL Dx sequencer (Applied Biosystems, Waltham, MA, USA). The sequencing utilized forward primer 5′GCTTGCTTCATCTCTAGCTCTACCG3′ and reverse primer 5′CCTGCATGAAAGACTGAACGAC3′. Data were compared to normal population databases (gnomAD/Thousand Genomes/ExAC) to determine allele frequencies of variant loci. Pathogenicity was evaluated based on the American College of Medical Genetics and Genomics (ACMG) guidelines.
Literature Review
A systematic review of the PubMed, Web of Science, Scopus and Embase database identified cases of XLMTM linked to MTM1 gene mutations. Cases reporting polyhydramnios as a prenatal feature were screened and summarized in Table 1.
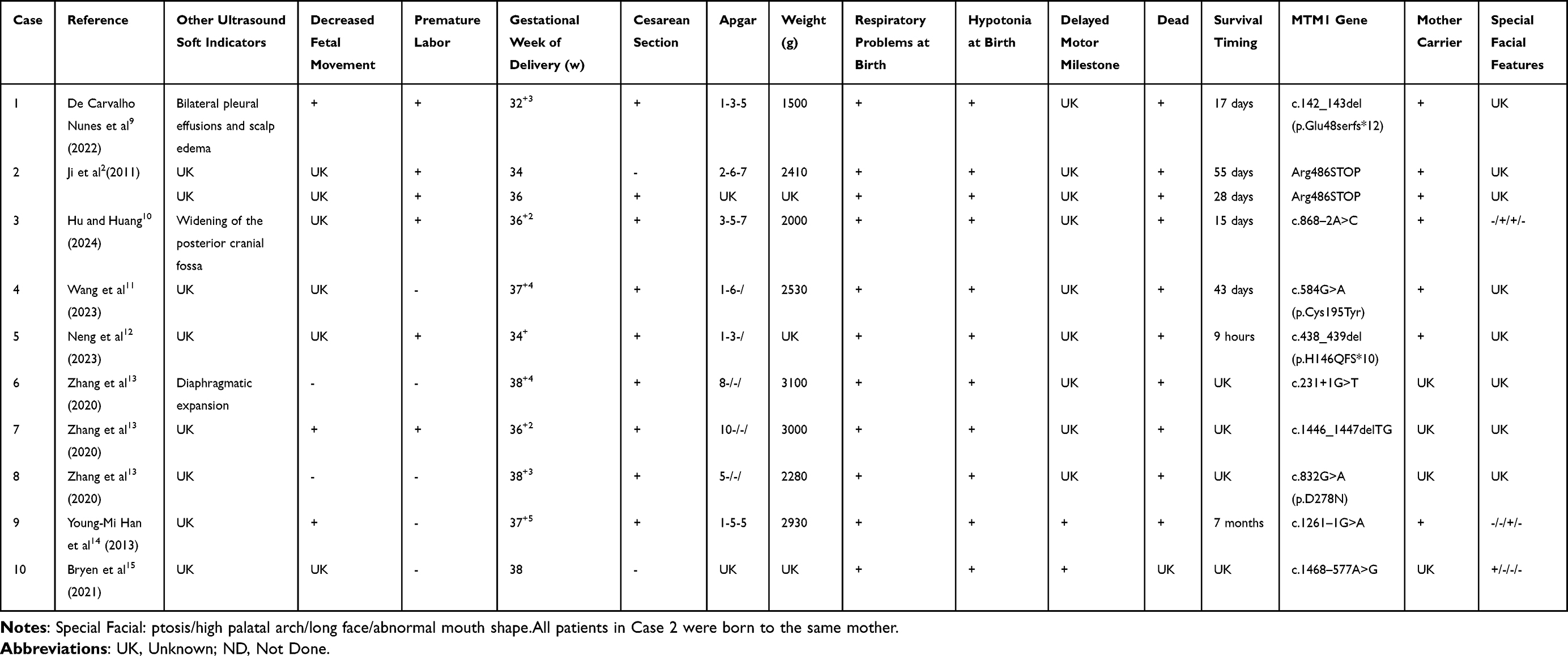 |
Table 1 Male XLMTM with Polyhydramnios as Prenatal Presentation |
Results
Analysis of Clinical Data
The expectant woman, age 33, had no notable medical or surgical history. No abnormalities were detected on prenatal anomaly ultrasound. However, polyhydramnios was first identified on ultrasound at 33 weeks and 3 days of gestation, with an amniotic fluid index (AFI) of 34.4 cm and slight inversion of the fetal feet. Two thousand milliliters of excess amniotic fluid were extracted via amniocentesis. Chromosome microarray studies and chromosome G-banding karyotyping were performed simultaneously on amniocytes. At 33 weeks and 5 days, the AFI measured 28 cm. However, a follow-up ultrasound at 35 weeks and 5 days showed that the AFI had grown to 37.4 cm, necessitating another amniocentesis, which involved the extraction of 1000 mL of amniotic fluid. At 36 weeks and 1 day, the AFI measured 33.9 cm.
A male baby was born at 36 weeks and 6 days (G2P1) as a result of the patient’s membranes rupturing prematurely at 36 weeks and 4 days of pregnancy. At delivery, the baby weighed 2765 g and was 47 cm long. At birth, the infant’s heart rate < 100 beats per minute.; he exhibited hypotonia, cyanosis, and no spontaneous breathing. The baby did not react. Initial resuscitation was initiated immediately, including airway suctioning, positive pressure oxygenation via mask T-piece resuscitator, and tactile stimulation.One minute after delivery, the newborn’s heart rate was 40 bpm and they were still cyanotic and hypotonic, resulting in an Apgar score of 1(0 for color, 0 for muscle tone, 0 for respiration, 0 for reflex irritability, 1 for heart rate).Advanced life support was initiated immediately, including endotracheal intubation, positive pressure ventilation, and chest compressions.Two milliliters of 1:10,000 adrenaline were administered via endotracheal instillation.Meanwhile, an umbilical venous access was established, followed by volume expansion with 13.8 mL of normal saline and correction of acidosis with 2.7 mL of 5% sodium bicarbonate.Despite the efforts, the Apgar score at the 5 and 10 min markers remained at 1. Thirty minutes after birth, the baby’s TcSaO2 levels were 40–50% and its heart rate was 20–30 beats per minute. The infant remained unresponsive, hypotonic, and apneic despite constant resuscitation. Forty-five minutes after delivery, the TcSaO2 levels vanished and the heart rate dropped to 10–15 beats per minute. The newborn’s limbs were flaccid and its skin was pallid. Whereas resuscitation efforts persisted, the heart rate did not improve significantly. An hour and thirty minutes after the baby’s birth, the family decided to end resuscitation efforts.
Genetic Analysis
Chromosomal Analysis
The prenatal chromosomal karyotype and chromosome microarray analysis returned normal results.
Whole Exome Sequencing (WES)
Whole Exome Sequencing (WES) of the affected child identified a de novo hemizygous frameshift mutation, c.968_969delinsT (p.K323Mfs*2), located in exon 10 of the MTM1 gene. This mutation had not been previously reported in normal reference population gene databases (allele frequency ExAC:; 1000 Genome:; gnomAD:). The variant was evaluated using the ACMG guidelines and classified as pathogenic. According to the criteria outlined in the ACMG guidelines (PMID: 25741868), this classification is supported by PVS1, PM6, and PM2.
Discussion
X-linked myotubular myopathy (XLMTM) is a rare and severe congenital myopathy caused by mutations in MTM1. The initial clinical manifestations of XLMTM were first documented in Korea in 1969.16 A hallmark of XLMTM is muscle weakness, which can range from mild to severe. Common symptoms observed at birth include pronounced hypotonia and muscular weakness, predominantly affecting the skeletal, medullary, and respiratory muscles. Additionally, affected children often present with distinct craniofacial features such as ptosis, high-arched palates, facial narrowing, and micrognathia.6 Nearly all patients experience respiratory failure, necessitating continuous ventilatory support.
This newborn died 1.5 hours after birth due to acute respiratory distress, extreme hypotonia, and difficulties during resuscitation. A de novo hemizygous variation, c.968_969delinsT (p.K323Mfs*2), was found in exon 10 of MTM1 of the boy and his parents by WES. The originality of this mutation is highlighted by the fact that it was not present in the parents and has not been documented in any genetic database of a typical reference group. To associate the mutant locus with clinical symptoms and find reliable diagnostic signs for obstetricians and geneticists, this work offers a comprehensive analysis of a neonatal XLMTM case using clinical and genetic studies, compared with previous research.
To date, over 400 distinct pathogenic mutations in MTM1 have been identified in XLMTM patients.17 The mutations are dispersed throughout the gene, with no specific hotspot regions. Most of the mutations result in severe classical manifestations of XLMTM. The MTM1 gene comprises 15 exons and has an open reading frame spanning 1.8 kb. Although mutations have been identified in all exons, the majority are concentrated in exons 3, 4, 8, 9, 11, and 12.2,14 Single nucleotide variants (SNVs) are the most prevalent mutation type in MTM1. Pathogenic variants include nonsense, missense, insertion/deletion mutations (resulting in frameshifts and premature stop codons), and splice site mutations that disrupt RNA processing.14,18 Additionally, intronic mutations can disrupt splice sites, facilitating pseudo-exon insertions15 or exon duplications.18 De novo mutations in MTM1 are reported in approximately 10–20% of cases.19
There is limited literature addressing the prenatal characteristics associated with XLMTM. The most frequently reported features are polyhydramnios and reduced fetal movement. Chromosomal anomalies in idiopathic polyhydramnios are primarily associated with trisomy 21 and trisomy 18.20 Polyhydramnios and other ultrasound-diagnosed fetal abnormalities are recognized as risk factors for chromosomal irregularities.21 A systematic review and meta-analysis of 20 studies involving 1,729 pregnant women with idiopathic polyhydramnios reported a mean incidence of chromosomal aberrations of 2.8 ± 3.7%.20 Given these findings, investigating potential genetic etiologies in pregnancies complicated by polyhydramnios is crucial for precise genetic counseling and prenatal diagnostics. To further elucidate the clinical and genetic characteristics of male XLMTM cases with polyhydramnios as a prenatal marker, this study analyzed 10 male XLMTM cases in addition to the neonate discussed (Table 1). In addition to polyhydramnios, diminished fetal movement emerges as the most prevalent prenatal features in XLMTM-diagnosed children. This reduction in fetal activity may be due to interference from excessive amniotic fluid during pregnancy. Accurate maternal assessment of fetal movement may indicate potential concerns regarding neuromuscular abnormalities in the fetus, warranting further prenatal diagnostic evaluations. Other prenatal features of XLMTM are generally nonspecific and depend on clinical manifestations associated with fetal hydrops, such as fetal pleural effusion, cephaloedema,9 widening of the posterior cranial fossa,10 and an elevated diaphragm.13
Notably, a cohort of 11 children diagnosed with XLMTM exhibited diverse mutation sites within the MTM1 gene, without any identified hotspot mutations. At our institution, we identified a hemizygous frameshift mutation in exon 10 of MTM1 (c.968_969delinsT, p.K323Mfs*2) in patients with XLMTM. This mutation substitutes lysine at position 323 in the protein sequence with methionine, leading to a frameshift and the emergence of a premature termination codon, which truncates protein translation.
Hyun et al2 documented a nonsense mutation (Arg486STOP) in MTM1 from the Department of Pediatrics, Yonsei University College of Medicine, South Korea. In their study, the mother was identified as a carrier. While the first female offspring was unaffected, her subsequent two male offspring inherited the mutation and developed the disease. Similarly, a family from China exhibited a truncating mutation (c.438_439del, p.H146Q fs*10) in exon 6 of MTM1, where the mother was also a carrier. In her subsequent pregnancies, she delivered two male offspring carrying the mutation, both of whom succumbed shortly after birth due to severe asphyxia and respiratory failure.12 These cases were associated with significant gestational polyhydramnios (AFI > 36.0 cm).12
A retrospective study at the Pediatrics Hospital of Fudan University in China investigated neonatal neuromuscular disorders and identified three children with XLMTM harboring mutations in MTM1. These mutations were located in exons 4, 9, and 13, characterized by specific mutation types: exon 4 (c.231+1G>T), exon 13 (c.1446_1447delTG), and exon 9 (c.832G>A, p.D278N), respectively.13 Another study by Young-Mi et al14 reported a mutation at the splice site between introns 10 and 11 (c.1261–1C>A). The affected neonate exhibited profound hypotonia, feeding difficulties, and absence of voluntary movements, requiring lifelong mechanical ventilation before succumbing to respiratory failure at seven months.
These findings emphasize that pregnant women with severe prenatal polyhydramnios should be monitored closely, particularly if ultrasonography reveals abnormalities or decreased fetal activity. Such circumstances necessitate a comprehensive genetic evaluation that includes chromosomal karyotyping and chromosomal microarray analysis. About 6% of instances had microdeletions or duplications, whereas 8–10% had karyotype abnormalities. However, in a significant proportion of cases, a definitive genetic diagnosis remains difficult.
WES has proven effective in identifying causative variants in 20–80% of cases with normal karyotype and CMA results.22 WES is a cost-effective approach for capturing protein-coding regions of the genome, as well as some untranslated regions (UTRs) and intron-exon boundaries. Despite technological advancements, WES coverage is inconsistent in areas such as the first exon, GC/AT-rich regions, and low-complexity segments, largely due to limitations in capture probe specificity. Whole-genome sequencing (WGS) offers an advantage by addressing these gaps.23 Samantha J. et al15 highlighted a case in which WES failed to identify anomalies in MTM1, despite the child displaying characteristic clinical symptoms of XLMTM. Using RNA sequencing, they discovered a variant (NG_008199.1(NM_000252.2):c. 1468–577A>G) in an intronic region that activated a pseudo-exon in intron 13, leading to a premature termination codon and the development of XLMTM.
In summary, the considerable phenotypic variability and lack of hotspot loci for MTM1 mutations complicates the intrauterine diagnosis of XLMTM. Pregnant women with recurrent polyhydramnios, diminished fetal movement, ultrasonography-detected abnormalities should be evaluated for neonatal neuromuscular disorders, including XLMTM. For cases with unremarkable karyotype and CMA results, WES or WGS should be considered to improve diagnostic precision.
Data Sharing Statement
Sequence data that support the findings of this study have been deposited in the ClinVAR with the primary accession code SUB15189952.
Ethical Approval
This study was approved by the Ethics Committee of Fujian Maternity and Child Health Hospital (approval number: 2024KY210). The publication of the case details has been approved by the Fujian Maternity and Child Health Hospital. The study was conducted in accordance with the Declaration of Helsinki.
Consent for Publication
Written informed consent for publication of the study is obtained from the parents of the neonate.
Acknowledgments
The authors would like to thank all participants involved in this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Supported by Foundation for Cultivated Young Talents of Fujian Province, China (2025350482).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Laporte J, Biancalana V, Tanner SM. et al. MTM1 mutations in X-linked myotubular myopathy. Hum Mutat. 2000;15(5):393–8. doi:10.1002/(SICI)1098-1004(200005)15:5<393::AID-HUMU1>3.0.CO;2-R
2. Jeon JH, Namgung R, Park MS, et al. X-linked myotubular myopathy in a family with two infant siblings: a case with MTM1 mutation. Yonsei Med J. 2011;52(3):547–550. doi:10.3349/ymj.2011.52.3.547
3. Vandersmissen I, Biancalana V, Servais L, et al. An integrated modelling methodology for estimating the prevalence of centronuclear myopathy. Neuromuscul Disord NMD. 2018;28(9):766–777. doi:10.1016/j.nmd.2018.06.012
4. Amburgey K, Tsuchiya E, de Chastonay S, et al. A natural history study of X-linked myotubular myopathy. Neurology. 2017;89(13):1355–1364. doi:10.1212/WNL.0000000000004415
5. Biancalana V, Caron O, Gallati S, et al. Characterisation of mutations in 77 patients with X-linked myotubular myopathy, including a family with a very mild phenotype. Hum Genet. 2003;112(2):135–142. doi:10.1007/s00439-002-0869-1
6. Annoussamy M, Lilien C, Gidaro T, et al. X-linked myotubular myopathy: a prospective international natural history study. Neurology. 2019;92(16):e1852–e1867. doi:10.1212/WNL.0000000000007319
7. McEntagart M, Parsons G, Buj-Bello A, et al. Genotype-phenotype correlations in X-linked myotubular myopathy. Neuromuscul Disord NMD. 2002;12(10):939–946. doi:10.1016/s0960-8966(02)00153-0
8. Beggs AH, Byrne BJ, De Chastonay S, et al. A multicenter, retrospective medical record review of X-linked myotubular myopathy: the recensus study. Muscle Nerve. 2018;57(4):550–560. doi:10.1002/mus.26018
9. de Carvalho Nunes G, Grenier K, Maedler Kron C, et al. Pulmonary lymphangiectasia in myotubular myopathy: a novel unrecognized association? Neuromuscul Disord NMD. 2022;32(6):512–515. doi:10.1016/j.nmd.2022.04.010
10. Hu Y, Huang X. Neonatal X-linked myotubular myopathy with a de novo mutation: a case report and literature review. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2024;49(3):491–496. doi:10.11817/j.issn.1672-7347.2024.230450
11. Tiantian W, Yu W, Huabin W. Clinical analysis of 2 cases of neonatal X-linked myotubular myopathy caused by MTM1 gene hemizygous mutation and RYR1 gene heterozygous mutation. J Chin Pract Diagn Ther. 2023;37(7):722–726. doi:10.13507/j.issn.1674-3474.2023.07.015
12. Jin N, Xv D, Xv YT, et al. Whole exome sequencing discloses a pathogenic MTM1 gene mutation in a continuous polyhydramnios family in China: case report and literature review. Eur J Obstet Gynecol Reprod Biol. 2023;291:34–38. doi:10.1016/j.ejogrb.2023.10.001
13. Peng Z, Lin Y, Lei Z, Bing-Bing W, Xi-Hua L, Guo-Qiang C. 25 cases of neonatal neuromuscular disease:A case series report. Chin J Evid Based Pediatr. 2020;15(3):177–181. doi:10.3969/j.issn.1673-5501.2020.03.003
14. Han YM, Kwon KA, Lee YJ, et al. X-linked recessive myotubular myopathy with MTM1 mutations. Korean J Pediatr. 2013;56(3):139–142. doi:10.3345/kjp.2013.56.3.139
15. Bryen SJ, Oates EC, Evesson FJ, et al. Pathogenic deep intronic MTM1 variant activates a pseudo-exon encoding a nonsense codon resulting in severe X-linked myotubular myopathy. Eur J Hum Genet EJHG. 2021;29(1):61–66. doi:10.1038/s41431-020-00715-7
16. Spiro AJ, Shy GM, Gonatas NK. Myotubular myopathy. Persistence of fetal muscle in an adolescent boy. Arch Neurol. 1966;14(1):1–14. doi:10.1001/archneur.1966.00470070005001
17. Biancalana V, Beggs AH, Das S, et al. Clinical utility gene card for: centronuclear and myotubular myopathies. Eur J Hum Genet EJHG. 2012;20(10). doi:10.1038/ejhg.2012.91
18. Oliveira J, Oliveira ME, Kress W, et al. Expanding the MTM1 mutational spectrum: novel variants including the first multi-exonic duplication and development of a locus-specific database. Eur J Hum Genet EJHG. 2013;21(5):540–549. doi:10.1038/ejhg.2012.201
19. Reumers SFI, Braun F, Spillane JE, et al. Spectrum of Clinical Features in X-Linked Myotubular Myopathy Carriers: an International Questionnaire Study. Neurology. 2021;97(5):e501–e512. doi:10.1212/WNL.0000000000012236
20. Sagi-Dain L, Sagi S. Chromosomal aberrations in idiopathic polyhydramnios: a systematic review and meta-analysis. Eur J Med Genet. 2015;58(8):409–415. doi:10.1016/j.ejmg.2015.06.010
21. Pri-Paz S, Khalek N, Fuchs KM, Simpson LL. Maximal amniotic fluid index as a prognostic factor in pregnancies complicated by polyhydramnios. Ultrasound Obstet Gynecol off J Int Soc Ultrasound Obstet Gynecol. 2012;39(6):648–653. doi:10.1002/uog.10093
22. Jelin AC, Vora N. Whole Exome Sequencing: applications in Prenatal Genetics. Obstet Gynecol Clin North Am. 2018;45(1):69–81. doi:10.1016/j.ogc.2017.10.003
23. Marwaha S, Knowles JW, Ashley EA. A guide for the diagnosis of rare and undiagnosed disease: beyond the exome. Genome Med. 2022;14(1):23. doi:10.1186/s13073-022-01026-w
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.