Back to Journals » Therapeutics and Clinical Risk Management » Volume 13
Identification of a new mutation in an Iranian family with hereditary multiple osteochondromas
Authors Akbaroghli S, Balali M, Kamalidehghan B, Saber S, Aryani O, Yong Meng G, Houshmand M
Received 30 April 2016
Accepted for publication 15 October 2016
Published 20 December 2016 Volume 2017:13 Pages 15—19
DOI https://doi.org/10.2147/TCRM.S111717
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Susan Akbaroghli,1,* Maryam Balali,2,* Behnam Kamalidehghan,3,4 Siamak Saber,4 Omid Aryani,5 Goh Yong Meng,6 Massoud Houshmand4
1Mofid Children’s Hospital, Shahid Beheshti University of Medical Sciences, 2ENT and Head & Neck Research Center and Department, Iran University of Medical Sciences (IUMS), 3Medical Genetics Department, School of Medicine, Shahid Beheshti University of Medical Sciences, 4Medical Genetics Department, National Institute for Genetic Engineering and Biotechnology, 5Department of Neuroscience, Iran Medical University, Tehran, Iran; 6Department of Veterinary Preclinical Sciences, Faculty of Veterinary Medicine, Universiti Putra Malaysia (UPM), Serdang, Malaysia
*These authors contributed equally to this work
Background: Hereditary multiple osteochondromas (HMO), previously named hereditary multiple exostoses (HME), is an autosomal dominant skeletal disorder characterized by the growth of multiple osteochondromas and is associated with bony deformity, skeletal growth reduction, nerve compression, restriction of joint motion, and premature osteoarthrosis. HMO is genetically heterogeneous, localized on at least three chromosomal loci including 8q24.1 (EXT1), 11p11-p13 (EXT2), and 19p (EXT3). The median age of diagnosis is 3 years; almost all affected individuals are diagnosed by age 12. The risk for malignant degeneration to osteochondrosarcoma increases with age, although the lifetime risk of malignant degeneration is low (~1%).
Methods and results: This study was performed on an Iranian family with nine affected individuals from three consecutive generations. Here, the proband was an affected woman who received genetic counseling prior to pregnancy. All exons of the three genes were examined in the proband using polymerase chain reaction and sequencing methods (the last member of this family is a male with severe deformities and lesions, especially around his large joints). Exon 4 of EXT1 (c.1235 G>A) was changed in affected individuals. This mutation alters tryptophan to a premature stop codon on amino acid position 412 (p.Trp412x).
Conclusion: The outcome of this study has extended the genotypic spectrum of Iranian patients with HMO, revealing a way for improving detection and genetic counseling in carriers.
Keywords: bony exostoses, exon 4 of EXT1, HMO, hereditary multiple exostoses, HME, c.1235 G>A, EXT1 and EXT2
Introduction
Hereditary multiple exostoses (HME) has also been known as hereditary multiple osteochondromas (HMO) because of the development of at least two or more benign outgrowths of the cartilage-capped bone tumors (osteochondromas) of the long bones that grow outward from the metaphyses of the long bones. HME is an autosomal dominant disorder characterized by the presence of the disorder in the consecutive generations.1,2 HME was first described by Boyer in 1814.3,4 The frequency of affected males is more than that of females, as females have incomplete penetrance for HME.5 Exostosin-1 (EXT1) has 746 amino acids and is responsible for heparan sulfate synthesis. However, exostosin-2 (EXT2) contains 14 exons that have two alternative exons. A previous study showed that mutations of these genes result in the synthesis of a truncated EXT protein with malfunctioned activity.6 Another study indicated that EXT proteins are essential enzymes for the synthesis of heparan sulfate, but the altered mechanism of heparan sulfate synthesis resulting in abnormal bone growth is still under debate. HME leads to distorted bone growth and may damage bony structures in the upper and lower limbs that can result in shortened stature.7 HME may also cause pain, limited range of joint motion, and pressure on the nerves, blood vessels, spinal cord, and tissues surrounding the exostoses.8 There are five homologous genes that are involved in HME, including two EXT-like genes (EXTL) in which no mutations have been reported earlier. The other genes are EXT1, EXT2, and EXT3, which have been located on the 8q24.1,9,10 11p13,11,12 and 19p chromosomes, respectively.13 It has been reported that most of the HME patients with EXT1 (60%–70%) and EXT2 (30%–40%) carry nonsense and frameshift mutations, respectively, resulting in the synthesis of truncated forms of the EXT proteins. The exact etiology of the exostoses is unclear, but it seems that exostoses occur most often in adults.14 Some authors suggest that both environmental and genetic factors are involved in HME disorder.15,16 Surgical interventions have been described for the treatment of patients with the disorder; however, there are many controversies.17 In this study, an Iranian male from a family with nine affected individuals for three consecutive generations was examined in order to identify a novel mutation related to this disorder. This index case is the last member of this family, who is a male with severe deformities and lesions, especially around his large joints, and with a mutation in exon 4 of EXT1 (c.1235 G>A).
Materials and methods
Case presentation
This study was performed on an Iranian family with nine affected individuals of three consecutive generations. Here, the proband is an affected woman who sought genetic counseling before pregnancy. The chief complaint of her family was pain, bony prominences, and deformities around large joints, including the shoulder, elbow, wrist, hip, knee, and ankle. The pedigree of this family was drawn, and the complaint was shown to be a typical autosomal dominant disorder. The affected woman had two affected sisters and two affected brothers (Figure 1). The index case is the last member of this family, who is a male with severe deformities and lesions, especially around his large joints. Typical bony lesions around his joints were found in the first visit (Figure 2). In this family, the father, grandfather, one aunt, and one uncle were previously affected. All these affected persons underwent multiple surgeries, but they experienced recurrence of the lesions. After clinical diagnosis, molecular study was carried out on the index case as well as on the involved mutant gene. Written informed consent, including consent to participate in the study for genetic analysis and publish the data was obtained from patients, and the Medical Ethics Committee of the Mofid Children’s Hospital, Tehran, Iran, specifically approved this study.
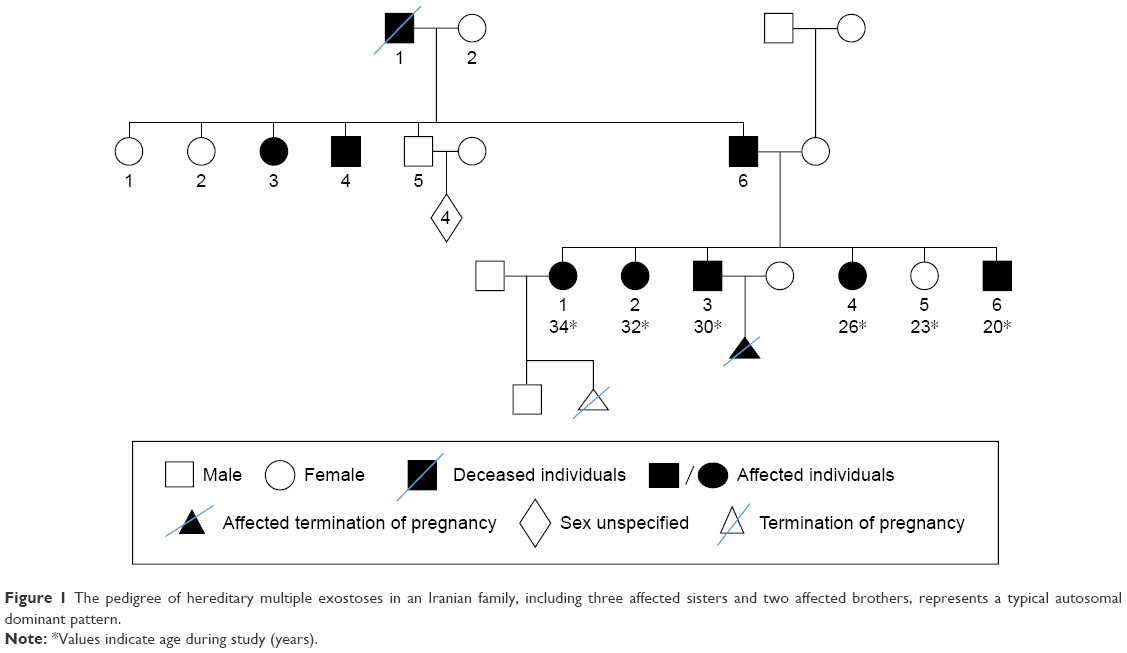 | Figure 1 The pedigree of hereditary multiple exostoses in an Iranian family, including three affected sisters and two affected brothers, represents a typical autosomal dominant pattern. |
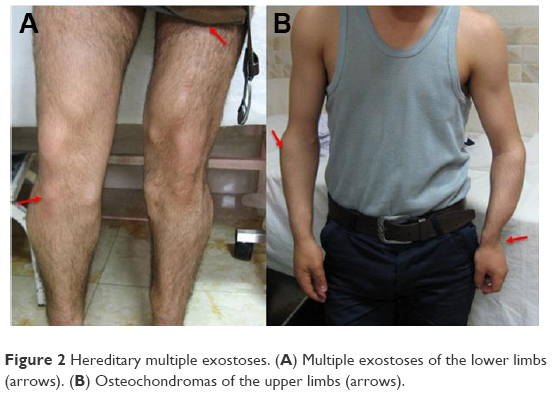 | Figure 2 Hereditary multiple exostoses. (A) Multiple exostoses of the lower limbs (arrows). (B) Osteochondromas of the upper limbs (arrows). |
DNA extraction and polymerase chain reaction (PCR)
Genomic DNA was extracted for the index case from his peripheral blood lymphocytes using salting-out method.18,19 PCRs were carried out in a 50 μL reaction, containing 80–110 ng of genomic DNA, 5 μL of 10× PCR buffer, 1.6 μL of MgCl2 (50 mM), 1 μL deoxynucleotide triphosphates (10 mM), primer pairs at 1.4 pmol/μL, and Super Taq (0.3–0.5 μL). PCR cycling consisted of 95°C for 5 min, followed by 35 cycles of 95°C for 1 min, annealing temperature of 60.4°C for 1 min and 72°C for 1 min, followed by a final elongation step at 72°C for 10 min. The sequences of the forward and reverse primers of EXT1 gene used in this experiment are 5′-TGGGTTATTTTGATCAAGTGC-3′ and 5′-TGGACCAATCACACATCCCTA-3′, respectively, whereas the primer sequences of the EXT2 gene are from previous study.20
In addition, molecular study of an affected sister of the index case who sought genetic counseling before pregnancy was performed. Similar tests were performed on the other affected family members.
Multiplex ligation-dependent probe amplification (MLPA) analysis
DNA was extracted from peripheral blood of individuals, using the SALSA MLPA KIT P215-B1 (MRC-Holland, Amsterdam, The Netherlands). The P215-B1 EXT probe mix contains 41 different MLPA probes with amplification products between 130 and 451 nt. In addition, it contains nine control fragments generating an amplification product <120 nt, four DNA quantity fragments (Q-fragments) at 64-70-76-82 nt, three DNA denaturation control fragments (D-fragments) at 88-92-96 nt, one X fragment at 100 nt, and one Y fragment at 105 nt. Q fragments do not require ligation due to short sizes. In other words, DNA fragments were exactly the same as the dependent probe and ligation fragment. This fragment appeared only when ligation was done and also the doses of DNA and denaturation were enough.
Results
Prior to genetic study, clinical pathology studies, including biopsy from bony lesions of the index case’s joints, radiography, and magnetic resonance imaging (MRI), were carried out in the affected family member with osteochondromas disorder (Figures 1–3). In this study, the pedigree indicated a classical autosomal dominant inheritance. After taking the history and studying the pedigree and clinical examination of three affected family members, variable expressivity was identified in this family with increasing severity in the consecutive generations and in the younger members in the same generation. The disease appeared at the end of the first decade of life for all of the patients, with the exception of the the youngest child for whom the disease appeared within the first decade. All the affected individuals underwent several orthopedic surgeries but experienced recurrence of the bony lesions. The growth of the exostoses was observed in these patients over the span of 4–5 months. For the genetic diagnosis, radiographic studies of the limbs were performed (Figure 3). X-rays revealed the prominent enlargement of the exostoses. For further imaging of bony lesions, MRI scans of the limbs were taken (Figure 4). No abnormal associated soft tissues were observed. Neuromuscular bundles were also normal. The removed bony lesions were studied pathologically for malignancy, but there was no evidence of malignancy. In this study, all exons of three genes, including EXT1, EXT2, and EXT3, were analyzed, and the results were compared with National Center for Biotechnology Information blast nucleotide. A heterozygous (c.1235 G>A) mutation was identified in Exon 4 of EXT1 in affected individuals. This mutation leads to a change in tryptophan to a premature stop codon on amino acid position 412 (p.Trp412x). Additionally, MLPA analysis did not reveal any duplications or deletions in the EXT1 gene (data not shown) (Figure 5).
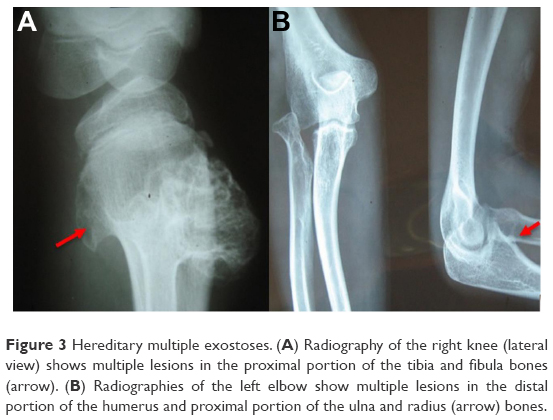 | Figure 3 Hereditary multiple exostoses. (A) Radiography of the right knee (lateral view) shows multiple lesions in the proximal portion of the tibia and fibula bones (arrow). (B) Radiographies of the left elbow show multiple lesions in the distal portion of the humerus and proximal portion of the ulna and radius (arrow) bones. |
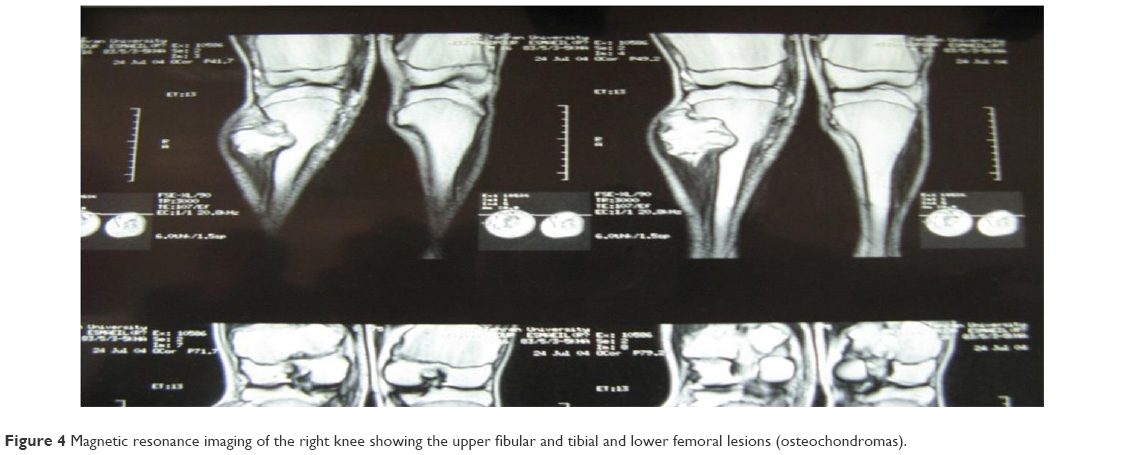 | Figure 4 Magnetic resonance imaging of the right knee showing the upper fibular and tibial and lower femoral lesions (osteochondromas). |
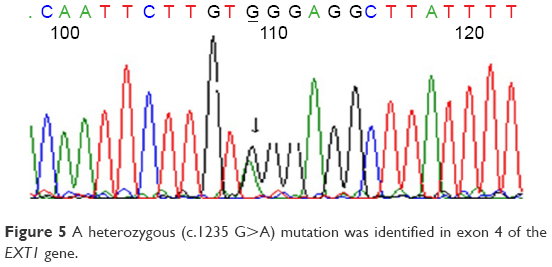 | Figure 5 A heterozygous (c.1235 G>A) mutation was identified in exon 4 of the EXT1 gene. |
Discussion
HMO was first described by Boyer in 181421 and then described by Guys in 1825. The prevalence of exostoses in the population is estimated to be 1 in 50,000.22 Exostoses are common in young people and are also the major type of benign bone tumors, called “osteochondromas.” Studies have shown that ~64%–76% of mutations appeared in the EXT1 gene and 21%–30% in the EXT2 gene. This mutant gene, for the first time, has been identified in an Iranian family. The clinical presentation in this family was a typical presentation for HME.23
Although the major function of the EXT protein is still under investigation, all EXT gene products exhibit glycosyltransferase activity associated with the proteoglycan heparan sulfate biosynthesis pathway.24 According to the literature, 60%–70% of HME patients revealed EXT1 gene mutations, belonging to either the nonsense or the frame shift mutations.12,25
Conclusion
A previous study showed that EXT1 and EXT2 consisted of inactivating frame shift, nonsense, and splice-site mutations.12 The most frequent EXT1 mutations are frameshift (44%), nonsense (24%), and splice-site mutations (11%). For EXT 2, frameshift (42%), nonsense (22%), and splice-site mutations (13%) are the most common mutations.26 Consequently, it is appropriate to say that inactivate mutations are responsible for 77%–80% of the multiple exostoses caused by EXT1/EXT2 mutations.27 A mutation in EXT1 exon 4 (c.1235 G>A) was reported previously,26 but this is the first study to confirm that the EXT1 gene has been found in an Iranian family with classic presentation of HME, and the molecular genetic testing for EXT1 should be done in HME patients in Iran. This mutation in the EXT1 gene is a frameshift mutation that could directly or indirectly lead to premature stop codons and then translation of truncated protein. In summary, the outcome of this study has extended the genotypic spectrum of Iranian patients with HMO, conferring a way for prediction of genetic risk in carriers.
Acknowledgment
The authors express their appreciation to all of the family members for donating their blood samples.
Disclosure
The authors report no conflicts of interest in this work.
References
Xiao CY, Wang J, Zhang SZ, et al. A novel deletion mutation of the EXT2 gene in a large Chinese pedigree with hereditary multiple exostosis. Br J Cancer. 2001;85(2):176–181. | ||
Trebicz-Geffen M, Robinson D, Evron Z, et al. The molecular and cellular basis of exostosis formation in hereditary multiple exostoses. Int J Exp Pathol. 2008;89(5):321–331. | ||
Solomon L. Hereditary multiple exostosis. J Bone Joint Surg Br. 1963;45(2):292–304. | ||
Peterson HA. Multiple hereditary osteochondromata. Clin Orthop Relat Res. 1989;239:222–230. | ||
Richardson RR. Variants of exostosis of the bone in children. Semin Roentgenol. 2005;40(4):380–390. | ||
Roentgenol S, Feta A, Presto J, et al. Contribution of EXT1, EXT2, and EXTL3 to heparan sulfate chain elongation. J Biol Chem. 2007;282(45):32802–32810. | ||
Philippe C, Porter DE, Emerton ME, Wells DE, Simpson AH, Monaco AP. Mutation screening of the EXT1 and EXT2 genes in patients with hereditary multiple exostoses. Am J Hum Genet. 1997;61:520–528. | ||
Kreutz RW, Sanders B. Bilateral coronoid hyperplasia resulting in severe limitation of mandibular movement: report of a case. Oral Surg Oral Med Oral Pathol. 1985;60(5):482–484. | ||
Cook A, Raskind W, Blanton SH, et al. Genetic heterogeneity in families with hereditary multiple exostoses. Am J Hum Genet. 1993;53(1):71. | ||
Hecht JT, Hogue D, Wang Y, et al. Hereditary multiple exostoses (EXT): mutational studies of familial EXT1 cases and EXT-associated malignancies. Am J Hum Genet. 1997;60(1):80. | ||
Potocki L, Shaffer LG. Interstitial deletion of 11 (p11. 2p12): a newly described contiguous gene deletion syndrome involving the gene for hereditary multiple exostoses (EXT2). Am J Med Genet. 1996;62(3):319–325. | ||
Wuyts W, Van Hul W. Molecular basis of multiple exostoses: mutations in the EXT1 and EXT2 genes. Hum Mutat. 2000;15(3):220–227. | ||
Le Merrer M, Legeai-Mallet L, Jeannin PM, et al. A gene for hereditary multiple exostoses maps to chromosome 19p. Hum Mol Genet. 1994;3(5):717–722. | ||
Mermer MJ, Gupta MC, Salamon PB, Benson DR. Thoracic vertebral body exostosis as a cause of myelopathy in a patient with hereditary multiple exostoses. J Spinal Disord Tech. 2002;15(2):144–148. | ||
Eggen S, Natvig B. Variation in torus palatinus prevalence in Norway: a statistical analysis using logistic regression. Community Dent Oral Epidemial. 1991;19:32–35. | ||
Raldi FV, Nascimento RD, Sá-Lima JR, Tsuda CA, Moraes MB. Excision of an atypical case of palatal bone exostosis: a case report. J Oral Sci. 2008;50(2):229–231. | ||
Kelly JP, James MA. Radiographic outcomes of hemiepiphyseal stapling for distal radius deformity due to multiple hereditary exostoses. J Pediatr Orthop. 2016;36(1):42–47. | ||
Rivero ER, Neves AC, Silva-Valenzuela MG, Sousa SO, Nunes FD. Simple salting-out method for DNA extraction from formalin-fixed, paraffin-embedded tissues. Pathol Res Pract. 2006;202(7):523–529. | ||
Laitinen J, Samarut J, Hölttä E. A nontoxic and versatile protein salting-out method for isolation of DNA. Biotechniques. 1994;17(2):316,318,320–312. | ||
Wuyts W, Van Hul W, De Boulle K, et al. Mutations in the EXT1 and EXT2 genes in hereditary multiple exostoses. Am J Hum Genet. 1998;62(2):346–354. | ||
Hennekam R. Hereditary multiple exostoses. J Med Genet. 1991;28(4):262–266. | ||
Kanik ZH, Gunaydin G, Sozlu U, Citaker S, Esen E. Eccentric training as an adjunct to rehabilitation program for hereditary multiple exostoses: a case report. J Clin Diagn Res. 2016;10(2):Yd03–Yd04. | ||
Foroughmand AM, Galehdari H, Rasouli M, Mohammadian G, Mohammadi M. Novel mutation in the EXT-1 gene in an iranian family affected with hereditary multiple exostoses. Pak J Biol Sci. 2008;11(7):1037–1041. | ||
Raskind WH, Conrad III EU, Matsushita M, et al. Evaluation of locus heterogeneity and EXT1 mutations in 34 families with hereditary multiple exostoses. Hum Mutat. 1998;11(3):231. | ||
Francannet C, Cohen-Tanugi A, Le Merrer M, Munnich A, Bonaventure J, Legeai-Mallet L. Genotype-phenotype correlation in hereditary multiple ehxostoses. J Med Genet. 2001;38(7):430–434. | ||
Jennes I, Pedrini E, Zuntini M, et al. Multiple osteochondromas: mutation update and description of the multiple osteochondromas mutation database (MOdb). Hum Mutat. 2009;30(12):1620–1627. | ||
Zhou Q, Yang C, Chen MJ, Li LZ. Detection of exostosin glycosyltransferase gene mutations in patients with non-hereditary osteochondromas of the mandibular condyle. Mol Clinical Oncol. 2016;5(3):295–299. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.