Back to Journals » International Journal of Nanomedicine » Volume 14
Idarubicin-loaded methoxy poly(ethylene glycol)-b-poly(L-lactide-co-glycolide) nanoparticles for enhancing cellular uptake and promoting antileukemia activity
Authors Liang B, Li N, Zhang S, Qi A, Feng J ![]() , Jing W, Shi C, Ma Z, Gao S
, Jing W, Shi C, Ma Z, Gao S
Received 7 October 2018
Accepted for publication 10 December 2018
Published 11 January 2019 Volume 2019:14 Pages 543—556
DOI https://doi.org/10.2147/IJN.S190027
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
Bin Liang,1,* Na Li,2,* Shuofei Zhang,3 Aihua Qi,4 Jianhua Feng,1 Weiwei Jing,5 Changcan Shi,2 Zhaipu Ma,6 Shenmeng Gao7
1Department of Hematology, The First Affiliated Hospital of Wenzhou Medical University, Nanbaixiang, Ouhai District, Wenzhou, Zhejiang 325000, China; 2Wenzhou Institute of Biomaterials and Engineering, CNITECH, CAS, Wenzhou, Zhejiang 325000, China; 3Department of Orthodontics, Stomatological Hospital, Wenzhou Medical University, Wenzhou, Zhejiang 325000, China; 4Department of Internal Medicine, Zaoqiang People’s Hospital, Zaoqiang, Hebei 053100, China; 5Department of Obstetrics and Gynecology, Wenzhou Hospital of Integrated Traditional Chinese and Western Medicine, Wenzhou, Zhejiang 325000, China; 6Department of Bioinformatics, College of Life Science, Hebei University, Baoding, Hebei 071002, China; 7Laboratory of Internal Medicine, The First Affiliated Hospital of Wenzhou Medical University, Nanbaixiang, Ouhai District, Wenzhou, Zhejiang 325000, China
*These authors contributed equally to this work
Purpose: Nanoparticle (NP)-based drug delivery approaches have tremendous potential for enhancing treatment efficacy and decreasing doses of chemotherapeutics. Idarubicin (IDA) is one of the most common chemotherapeutic drugs used in the treatment of acute myeloid leukemia (AML). However, severe side effects and drug resistance markedly limit the application of IDA.
Methods: In this study, we encapsulated IDA in polymeric NPs and validated their antileukemia activity in vitro and in vivo.
Results: NPs with an average diameter of 84 nm was assembled from a methoxy poly(ethylene glycol)-b-poly(l-lactide-co-glycolide) (mPEG-PLGA). After loading of IDA, IDA-loaded mPEG-PLGA NPs (IDA/mPEG-PLGA NPs) were formed. The in vitro release data showed that the IDA/mPEG-PLGA NPs have excellent sustained release property. IDA/mPEG-PLGA NPs had exhibited the lower IC50 than pure IDA. Moreover, IDA/mPEG-PLGA NPs in the same concentration substantially induced apoptosis than did pure IDA. Most importantly, IDA/MPEG-PLGA NPs significantly decreased the infiltration of leukemia blasts and improved the overall survival of MLL-AF9-induced murine leukemia compared with free IDA. However, the blank NPs were nontoxic to normal cultured cells in vitro, suggesting that NPs were the safe carrier.
Conclusion: Our data suggest that IDA/mPEG-PLGA NPs might be a suitable carrier to encapsulate IDA. Low dose of IDA/mPEG-PLGA NPs can be used as a conventional dosage for antileukemia therapy to reduce side effect and improve survival.
Keywords: idarubicin, mPEG-PLGA, acute myeloid leukemia, nanoparticles
Introduction
Acute myeloid leukemia (AML) is characterized by the blockage of differentiation or maturation of myeloid progenitor cells at different stages due to various genetic mutations and epigenetic dysregulation.1 Although current therapies involving intensive chemotherapy and stem cell transplantation substantially improve the overall survival (OS), AML is still fatal in about half of younger patients and approximately 80% of patients over the age of 60 years because of drug resistance, relapse, or treatment-related mortality.2 Conventional chemotherapy drugs are widely used in clinical practice and lead to rapid remission in AML patients. However, chemotherapy drugs are nonselective to kill leukemia blasts and normal cells, resulting in fatigue, severe immune suppression, cardiotoxicity, and gastrointestinal side effects.3 These severe side effects markedly limit the further usage of chemotherapy drugs in clinical applications.
Idarubicin (IDA, 4-demethoxydaunorubicin), a member of anthracycline antibiotics, was approved by the United States Food and Drug Administration (US FDA) in 1990 and is mainly used to treat AML and chronic lymphocytic leukemia (CLL).4 IDA is an analog of daunorubicin (DNR), but the absence of a methoxy group causes faster cellular uptake, superior DNA binding capacity, and consequently greater cytotoxicity than DNR.5 IDA is currently combined with cytosine arabinoside as a first-line treatment of AML.6 Although many AML patients achieve complete remission after initial treatment with IDA, about 70% of the patients eventually experience a relapse of leukemia. Furthermore, severe side effects finally lead to the discontinued treatment. As reported,7 low-dose IDA was well tolerated in elderly patients. Thus, overcoming dose-limiting side effects or reducing nonspecific toxicity might improve the OS in AML patients.
Biodegradable material-based nanoparticles (NPs) as effective carriers for conventional chemotherapeutic agents have been used to improve cancer treatment.8 Nanomaterial not only enhances drug delivery into tumor tissues but also minimizes systemic exposure and reduces nonspecific toxicity.9 Furthermore, nanomaterial-based carriers increase the circulation time of drugs in the blood, thereby enhancing the ability of drugs to reach their sites of action. Numerous cancer drugs including paclitaxel, tamoxifen, and anthracyclines have been reported to test the efficiency of biocompatibility material-based NPs as the effective carriers.
Because of the properties of biodegradable and biocompatible, poly(L-lactide-co-glycolide) (PLGA) is highly preferred for the preparation of NPs. For example, encapsulation of doxorubicin (Dox) in PLGA NPs improves the solubility and stability of Dox in K562 cells.10 Cytarabine was loaded in PLGA NPs by a modified nanoprecipitation method. The in vitro drug release from the PLGA-ara-c was sustained up to 24 hours, but the drug release from the pure drug was completed within 2 hours.11 Therefore, this sustained drug release of cytarabine would reduce the side effects associated with the conventional leukemia therapy by reducing dosing frequency. In the present study, we prepared amphiphilic diblock copolymer, namely, methoxy poly(ethylene glycol)-b-PLGA (mPEG-PLGA) and IDA-loaded mPEG-PLGA NPs (IDA/mPEG-PLGA NPs). The antileukemia activity of IDA/mPEG-PLGA NPs in leukemia cell lines and MLL-AF9-induced murine leukemia were investigated. IDA/mPEG-PLGA NPs have a lower IC50 in leukemia cell lines than does pure IDA. IDA/mPEG-PLGA NPs markedly induce apoptosis than pure IDA in the same concentration. Furthermore, IDA/mPEG-PLGA NPs reduce the infiltration of leukemia blasts in blood and prolong the OS in MLL-AF9-induced murine leukemia than pure IDA. Therefore, mPEG-PLGA presents sustained drug release and favorable antitumor properties, which can be used as a conventional carrier for antitumor therapy.
Materials and methods
Materials
L-Lactide (LLA) and glycolide (GA) were purchased from Huizhou Foryou Medical Devices Co., Ltd (Guangdong, China). Methoxy poly(ethylene glycol) (mPEG) (Mn =5,000) and Tin(II) 2-ethylhexanoate (Sn(Oct)2) were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Triethylamine (Et3N) and MTT reagents were supplied by Aladdin Biological Technology Co., Ltd (Shanghai, China). RPMI-1640 medium and PBS were purchased from Shanghai Chunsheng Biological Technology Co., Ltd (Shanghai, China). IDA was approved by Hanhui Pharmaceuticals Co, Ltd (Shanghai, China). Dialysis cellulose membrane (molecular weight cutoff, MWCO =3,500) was bought from Shanghai Baoman Biological Technology Co., Ltd. Ultra-purified water was supplied by a Milli-Q system (EMD Millipore, Billerica, MA, USA). Chemical reagents used in the present study were of analytical grade. All compounds were used without further purification.
Synthesis of amphiphilic diblock copolymer (mPEG-PLGA)
mPEG-PLGA diblock copolymers were synthesized using the ring-opening polymerization. In brief, LLA and GA (molar ratio 7:3) and mPEG (Mn =20,000, 30,000, and 40,000) were added into a dry ampoule bottle. Stannous octoate as a catalyst was dissolved in methylbenzene and introduced into the reaction mixture. The above mixture was degassed through vacuum/nitrogen cycles and melted at 135°C. The ampoule bottle was sealed under vacuum, and the polymerization reaction was carried out at 120°C for 24 hours. The obtained products were rinsed with chloroform for three times followed by precipitation in hexane thrice. The final precipitated copolymers (mPEG-PLGA1, mPEG-PLGA2, and mPEG-PLGA3) were dried under vacuum at 35°C for 24 hours.
Structural characterization of copolymers
To confirm the structure of prepared copolymer mPEG-PLGA, hydrogen nuclear magnetic resonance (1H NMR) (AMX500; BrukerOptik GmbH, Ettlingen, Germany), spectrum was recorded using CDCl3 as a solvent. Fourier transform infrared (FTIR) spectra were obtained using an FTIR spectrometer (Tensor II; BrukerOptik GmbH) in the frequency range from 4,000–400/cm by the method of attenuated total reflection.
IDA-loaded mPEG-PLGA NPs
Three kinds of IDA-loaded mPEG-PLGA NPs were prepared following a literature method with a slight modification.12 Briefly, 50 mg of mPEG-PLGA copolymer and 5 mg of IDA-HCl were dissolved in 5 mL of dimethyl formamide and stirred for 2 hours. Then, Et3N was introduced to the above solution to neutralize HCl and stirred for another 1 hour. The resulting solution was gradually added into 30 mL of PBS (10 mM, pH 7.4) to assemble to form NPs at the speed of 2.5 mL/h. After an additional 1 hour of stirring, the obtained NPs’ solution was dialyzed against PBS (10 mM, pH =7.4) for 48 hours using a dialysis bag (MWCO =3,500). The IDA/mPEG-PLGA NPs (IDA/mPEG-PLGA1 NPs, IDA/mPEG-PLGA2 NPs, and IDA/mPEG-PLGA3 NPs) were collected and stored at 4°C. The mPEG-PLGA NPs were also prepared without any IDA, respectively.
Size and zeta potential (ZP)
To measure the size and ZP of NPs, 100 μL of NPs was dispersed in double distilled water and subjected to size and ZP measurement using a Zetasizer (Nano ZS90; Malvern Instruments, Malvern, UK).
Morphology characterization of NPs
The morphology of NPs was determined by transmission electron microscopy (TEM, 2100; JEOL, Tokyo, Japan). For this, 3 μL of NPs’ solution was placed on a carbon-coated copper TEM grid. After drying completely, the images were visualized at an accelerating voltage of 200 kV.
Drug loading (DL) and encapsulation efficiency (EE)
The amount of IDA in IDA/mPEG-PLGA NPs was determined by the UV–vis spectrophotometer (CARY5000; Agilent Technologies, Santa Clara, CA, USA) at 485 nm. The standard curve was first built using different concentrations of IDA. Before the dialysis procedure, 0.5 mL of the formed NPs was pipetted into an ultrafiltration centrifuge tube (MWCO =3,000) and centrifuged for 20 minutes at the speed of 10,000 rpm. By this way, free IDA in the NPs’ solution could be separated and collected to measure the absorbance at 485 nm. The DL and EE were calculated following the formula13:
|
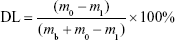
|
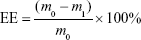
where the m0, m1, and mb represent the mass of total IDA, free IDA, and IDA in IDA/mPEG-PLGA NPs, respectively.
Research of release in vitro
The drug release behavior of IDA/mPEG-PLGA NPs was analyzed following the dialysis method. A total of 6 mL of fresh IDA/mPEG-PLGA NPs was put into a dialysis bag (MWCO =3,500). Then, the sealed bag was immersed into the release medium PBS and was subjected to shaking in water bath at 37°C at the speed of 120 rpm. At various time intervals, 2 mL of release medium was taken out and replaced with fresh PBS. Subsequently, the concentration of IDA was determined by UV–Vis. Three repeats were performed for each time point, and the cumulative release of IDA was calculated following the method of DL and EE measurement.
In vivo pharmacokinetics and biodistribution
For the biodistribution assay, 6-week-old wild-type C57BL/6J mice were intraperitoneally (IP) injected with IDA/mPEG-PLGA NPs (3 mg/kg). Then, liver, spleen, heart, lung, kidney, and intestine were dissected out after euthanizing the mice at 6 hours postinjection for subsequent ex vivo imaging analysis (IVIS Lumina X5; PerkinElmer Inc., Waltham, MA, USA) and analyzed using the Living Image software (PerkinElmer Inc.). For the pharmacokinetics assay, C57BL/6J mice were IP injected with IDA (3 mg/kg) or IDA/mPEG-PLGA NPs (3 mg/kg). Subsequently, liver and spleen were dissected out after euthanizing the mice at 2, 24, 48, and 96 hours postinjection for ex vivo imaging analysis (IVIS Lumina X5). The 540Ex/620Em was used to analyze DsRed fluorescence by IDA.
Flow cytometry analysis of intracellular IDA
To analyze intracellular IDA, leukemia cells were treated with IDA or IDA/mPEG-PLGA NPs and then washed three times with PBS and subjected to flow cytometry analysis system (Becton Dickinson, Mountain View, CA, USA). The PE fluorescence emitted by the particles bound or internalized by the leukemia cells was analyzed in the FL-2 channel, and the data were analyzed by FlowJo (TreeStar).
Cytotoxicity of IDA/mPEG-PLGA NPs in L929 cells in vitro
Murine L929 cells (Cell Bank of Typical Culture Collection of Chinese Academy of Sciences, Shanghai, China) were cultured in DMEM supplemented with 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA) and 1% penicillin–streptomycin in humidified 37°C incubator with 5% CO2. Normal human CD34+ hematological stem and progenitor cells (HSPCs) were isolated and enriched from umbilical cord blood (UCB) by positive selection (Stemcell Technologies, Vancouver, BC, Canada) and cultured in StemSpan™ SFEM (Stemcell Technologies) supplemented with human SCF, Flt3 ligand, erythropoietin, IL-3, and IL-6 at final concentrations 10 ng/mL. Murine L929 cells and normal human CD34+ HSPCs were used as normal cells to characterize the biocompatibility of NPs using the MTT-based colorimetric assay.14 Cells were seeded in a 96-well plate at the density of 1–2×104/well and incubated for 24 hours to obtain a monolayer of cells. Then, cells were treated with increasing concentrations (from 0.1 to 10 μM) of IDA. After incubation for 24 hours, 20 μL of MTT (5 mg/mL, in PBS) was added to corresponding wells and incubated for another 4 hours. The medium was discarded. The formed formazan crystals were dissolved in 150 μL of dimethyl sulfoxide. Then, the absorbance of each well was measured at 570 nm. Cells treated with DMEM and IDA were used as the negative and positive controls, respectively. Relative cell viability was calculated with the following equation:
|
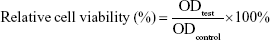
Antileukemia activity of IDA/mPEG-PLGA NPs in leukemia cells in vitro
Human leukemic cell lines including HL-60 and U937 (American Type Culture Collection [ATCC], Manassas, VA, USA) were purchased for the present study. Leukemic cell lines were cultured in RPMI-1640 supplemented with 10% FBS and 1% penicillin–streptomycin in humidified 37°C incubator with 5% CO2. Leukemic cells were plated at 2×105 cells/mL in six-well plate and then treated with PBS for vehicle, mPEG-PLGA, 0.01 μM IDA, and 0.01 μM IDA/mPEG-PLGA NPs. Apoptosis was detected by annexin V/PI staining (Thermo Fisher Scientific). Briefly, cells were collected and washed with binding buffer and were incubated in working solution (400 μL of binding buffer with 5.0 μL of Annexin V-FITC and 5.0 μL of PI) for 15 minutes. Cells were washed and resuspended with the binding buffer and analyzed by flow cytometry within 30 minutes after staining. Cell growth was measured by the MTT-based colorimetric assay.
Antileukemia activity of IDA and IDA/mPEG-PLGA NPs in vivo
Lineage-negative cells of mouse bone marrow (BM) were isolated from 6-week-old wild-type C57BL/6J at 6 days after 5-fluorouracil (5-FU) treatment. The Lin− cells were retrovirally transduced with MSCV-GFP-IRES-MLL-AF9 through two rounds of “spinoculation” as described previously.15,16 Retrovirally transduced donor cells (5×105) with whole BM cells (1×106, freshly isolated from B6 mice) were injected into lethally irradiated (8 Gy) 8-week-old B6 recipient mice within 24 hours after irradiation. After the mice developed full-blown leukemia, sorted GFP+ cells were injected into lethally irradiated recipient mice with freshly isolated BM cells from healthy mice. All the mice were divided into the following four groups: vehicle (untreated), mPEG-PLGA (3 mg/kg), IDA (3 mg/kg), and IDA/mPEG-PLGA NPs (3 mg/kg). Mice were started to inject by mPEG-PLGA, IDA, IDA/mPEG-PLGA NPs, or vehicle every other day after 2 weeks of transplantation. GFP+ cells were measured when the mice became moribund (lethargy, scruffy coat, or occasional limb paralysis) and then humanly sacrificed. Blast population of greater than 20% in the BM is confirmed as leukemia. Survival was evaluated from the first day of the experiment until death. Animals were housed under standard laboratory conditions (light–dark cycle conditions and controlled temperatures 22°C±2°C) with enough foods and fresh drinking water. All surgery was performed under sodium pentobarbital anesthesia with all efforts made to minimize suffering. The animal handling and procedures in this study were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory animals. The current study was reviewed and approved by the Institutional Animal Care and Use Committee of the First Affiliated Hospital of Wenzhou Medical University.
Statistical analyses
All the results were expressed as mean ± SD where applicable. For all the analyses, the P-values were two tailed, and a value of P<0.05 was considered statistically significant. OS probabilities were estimated by the Kaplan–Meier method, and differences in OS were compared using the log-rank test. OS was defined from the date of engraftment to death. All statistical analyses were performed using SPSS 22.0 (IBM Corporation, Armonk, NY, USA).
Results
Synthesis of mPEG-PLGA
The mPEG-PLGA copolymer was synthesized following the method of ring-opening polymerization. As shown in Figure 1A, mPEG was used as the initiator and expected to form a hydrophilic corona due to its high hydrophilicity and flexibility of the backbone. Herein, LLA and GA were used as monomers in the ring-opening polymerization, because PLGA copolymers are widely used in clinic as biodegradable material and in delivery systems for the controlled release for drug delivery. Therefore, mPEG-PLGA diblock copolymers with different molecular weights were synthesized.
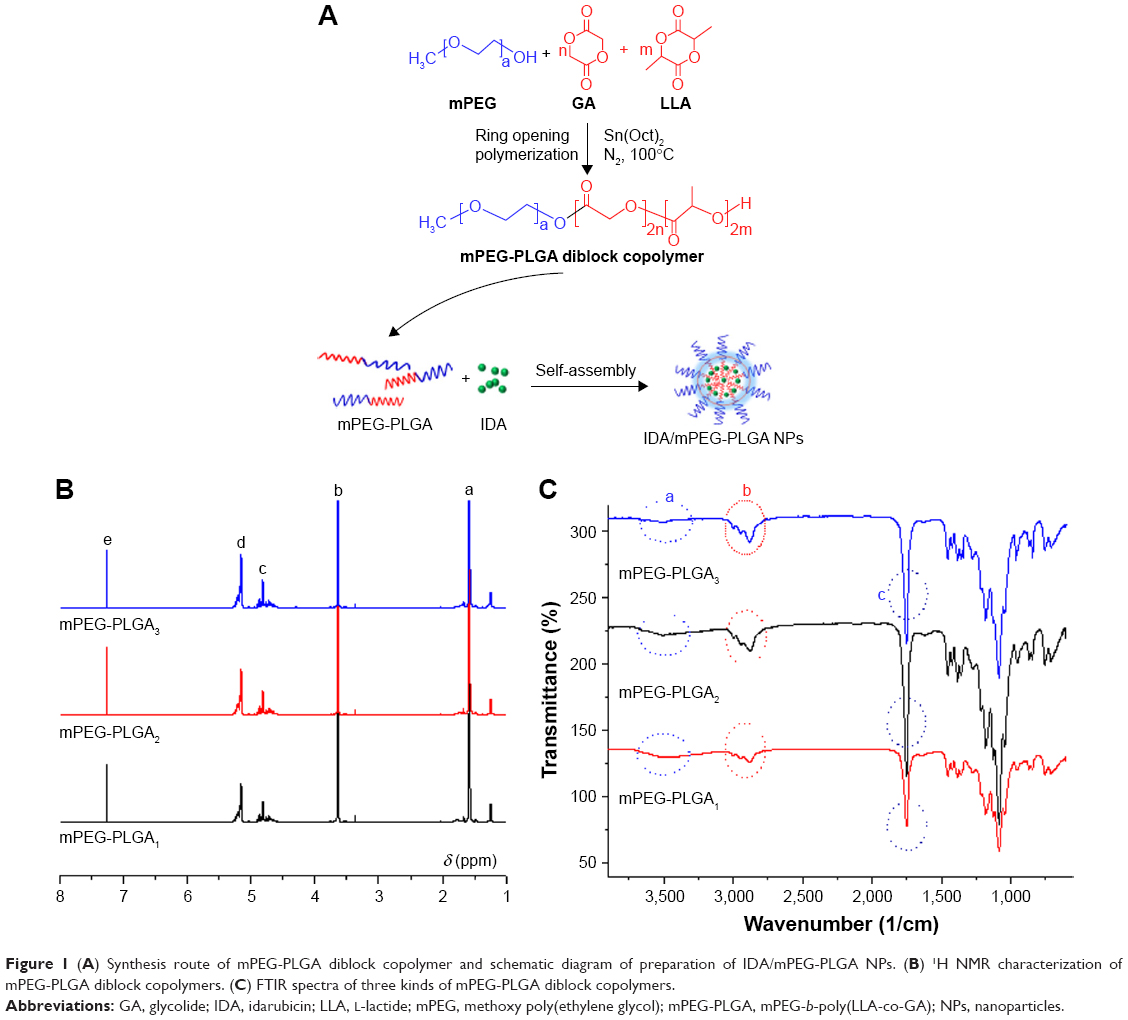 | Figure 1 (A) Synthesis route of mPEG-PLGA diblock copolymer and schematic diagram of preparation of IDA/mPEG-PLGA NPs. (B) 1H NMR characterization of mPEG-PLGA diblock copolymers. (C) FTIR spectra of three kinds of mPEG-PLGA diblock copolymers. |
To confirm the formation of diblock copolymers, the obtained materials were dissolved in CDCl3 for 1H NMR characterization. As shown in Figure 1B, with the following results: peak a (δ=1.57 ppm, LLA repeating unit: −O−CH(CH3)−CO−), b (δ=3.64 ppm, mPEG repeating unit: −O−CH2−CH2−), c (δ=4.68–4.91 ppm, GA repeating unit: −O−CH2−CO−), d (δ=5.13 ppm, LLA repeating unit: −O−CH(CH3)−CO−), and e (δ=7.26 ppm, CDCl3). These results demonstrated that mPEG-PLGA copolymers were synthesized successfully. Moreover, the molecular weights of mPEG-PLGA1, mPEG-PLGA2, and mPEG-PLGA3 were calculated using corresponding peaks. The molecular weight (Mn) of mPEG-PLGA1 was 15,000, that of mPEG-PLGA2 was 20,000, and that of mPEG-PLGA3 was 27,000.
To confirm that the mPEG-PLGA diblock copolymers had been successfully synthesized, FTIR spectra of mPEG-PLGA diblock copolymers were analyzed. As shown in Figure 1C, the width peak in 3,450/cm (a) was the typical peak of −OH at the end of the polymer. The absorption peak in 2,900–3,000/cm (b) was the contraction vibration peak of −CH3. The strong absorption peak in 1,760/cm (c) was the typical stretching vibration peak of −C=O, and the appearance of this peak revealed that LLA has reacted with GA and formed a straight chain ester bond. These results suggest that the mPEG-PLGA diblock copolymers have been successfully synthesized.
To further confirm the successful mPEG-PLGA synthesis, gel permeation chromatography (GPC) data are shown in Figure S1A–C. These data confirm the successful synthesis. Furthermore, the critical micelle concentrations of mPEG-PLGA1, mPEG-PLGA2, and mPEG-PLGA3 were 0.062, 0.053, and 0.072 μg/mL, respectively.
Size and ZP of NPs and IDA/mPEG-PLGA NPs
IDA/mPEG-PLGA NPs were prepared by a modified dialysis method,12 and the preparation schematic diagram is shown in Figure 1A. As mPEG-PLGA is an amphiphilic diblock copolymer. The PLGA segment is the hydrophobic part. IDA is a hydrophobic drug. Therefore, when they coexisted in water-soluble environment, hydrophobic part tended to far away from the environment. Then, IDA was encapsulated in the core, and the DL NPs were formed.
Surface properties and particle size of NPs play important roles in the drug release, cellular uptake, and cytotoxicity.17 The size and ZP distribution of NPs and IDA/mPEG-PLGA NPs are displayed in Table 1. The size of NPs and IDA/mPEG-PLGA NPs increased with the increased molecular weight of diblock copolymers. This phenomenon could be ascribed to the increase in hydrophobic segment (PLGA), which finally forms big hydrophobic cores. Furthermore, PDI of IDA/mPEG-PLGA NPs was rather narrow, which is benefit for cancer therapy.
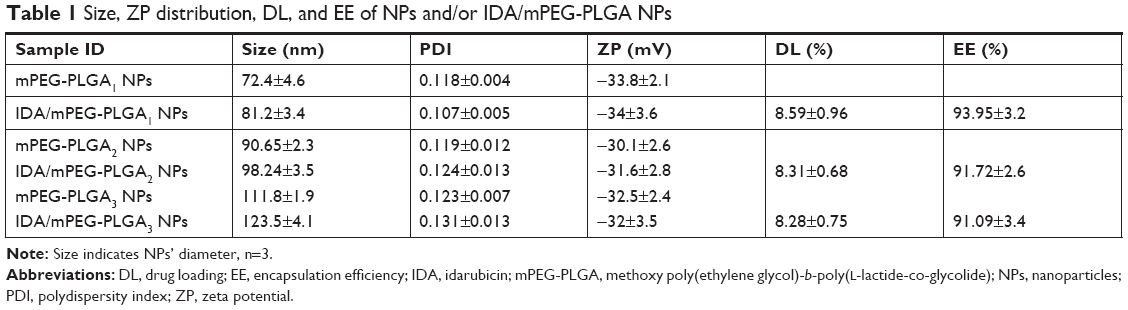 | Table 1 Size, ZP distribution, DL, and EE of NPs and/or IDA/mPEG-PLGA NPs |
The ZP of NPs and IDA/mPEG-PLGA NPs was both negative (−34±3.6 to −30.1±2.6 mV) due to the abundant −OH groups of mPEG segments. NPs with small diameter are easily swallowed by cells. These NPs with small diameter avoid elimination by reticuloendothelial system and promote accumulation in tumor vasculature18,19 Therefore, due to the suitable size and ZP, mPEG-PLGA1 NPs (72.4±4.6 nm) and IDA/mPEG-PLGA1 NPs (81.2±3.4 nm) were chosen for further experiments as the potential drug delivery system. Moreover, as shown in Table 1, the size of mPEG-PLGA1 NPs and IDA/mPEG-PLGA1 NPs was larger than that of TEM. The sizes measured by dynamic laser scattering (DLS) were obtained in hydrated state in solution, while those obtained by TEM had been dried after being dropped on the carbon-coated copper meshes.
Morphology of NPs and IDA/mPEG-PLGA NPs
The surface morphology of mPEG-PLGA1 NPs and IDA/mPEG-PLGA1 NPs was characterized using TEM. Results showed that all mPEG-PLGA1 NPs and IDA/mPEG-PLGA1 had exhibited spherical morphology, which were about 50 nm in diameter (Figure 2A and B). These results demonstrated the successful formation of NPs and IDA/mPEG-PLGA NPs. However, the diameter evaluated by TEM was smaller than that evaluated by DLS analysis. We speculate that dry NPs was inclined to shrinking and collapsing.20,21
 | Figure 2 TEM images of mPEG-PLGA1 NPs (A) and IDA/mPEG-PLGA NPs (B). |
IDA EE and drug release in vitro
UV–vis spectrophotometer was used to determine the DL and EE of IDA/mPEG-PLGA1 NPs at 485 nm. The standard curve of IDA indicated that the absorbance was increased in a liner relationship with the increasing concentration of IDA (Figure 3A). After centrifuging using the ultrafiltration centrifuge tube, pictures were taken by camera (inserted) and free IDA was separated and collected to measure the concentration via the standard curve of IDA. Compared with the IDA, IDA/mPEG-PLGA1 NPs after centrifuging exhibited slight red. These results reveal that most of the IDA is encapsulated into IDA/mPEG-PLGA1 NPs.
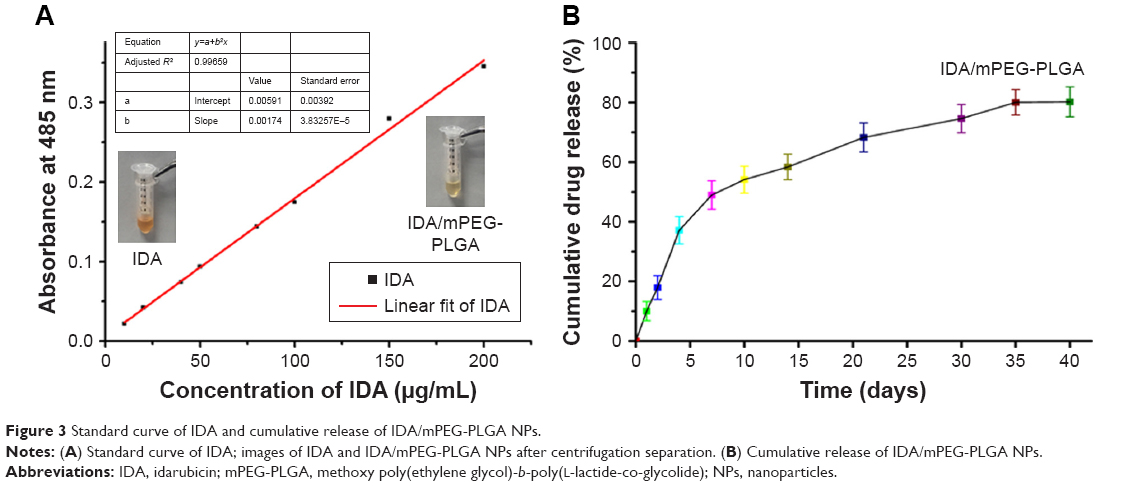 | Figure 3 Standard curve of IDA and cumulative release of IDA/mPEG-PLGA NPs. |
Results from quantitative analysis showed that DL and EE of IDA/mPEG-PLGA1 NPs were 8.59±0.96 and 93.95%±3.2%, respectively. Therefore, IDA/mPEG-PLGA1 NPs had showed excellent DL efficiency in the present investigation. High values of DL and EE could be attributed to the hydrophobic segments of PLGA. Suitable PLGA segments provided enough hydrophobic force to encapsulate plenty of IDA in their cores of mPEG-PLGA1 NPs. Herein, mPEG-PLGA1 NPs are the potential and efficient drug delivery system and might possess superb effect against leukemia.
To investigate the release of IDA, IDA/mPEG-PLGA1 NPs were incubated in PBS for 40 days in vitro. About 37.13±0.9 wt% of IDA was released during the first 4 days (Figure 3B). At day 7, 48.97±0.2 wt% of the loaded IDA was released (Figure 3B). After 40 days, the drug release from IDA/mPEG-PLGA1 NPs was to be 80.2 wt% (Figure 3B). These results indicate that the reduced and steady levels of IDA prolong the release time. Sustained release behavior of IDA/mPEG-PLGA1 NPs was mainly ascribed to the degradation of mPEG-PLGA1 diblock copolymers and drug diffusion from the hydrophobic cores. Sustained release of IDA might benefit leukemia treatment with suitable drug concentration and avoid drug invalid loss at the same time. Therefore, mPEG-PLGA1 NPs act as biocompatible carriers for IDA, and IDA/mPEG-PLGA1 NPs were used for the following experiments.
Finally, the release rate of IDA from IDA/mPEG-PLGA1 NPs in complete medium (10% fetal calf serum, shaking in water bath, 120 rpm, 37°C) was detected. The release rate in complete medium was compared with that in PBS. As shown in Table 2, no significant difference of IDA release rate was found in complete medium compared with that in PBS.
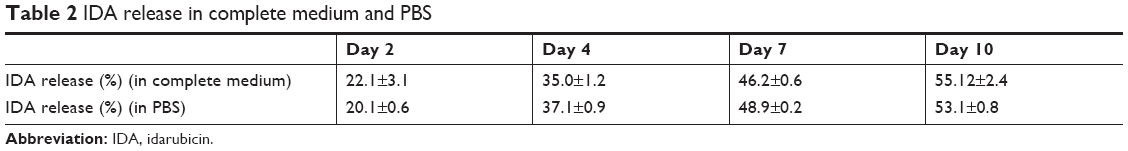 | Table 2 IDA release in complete medium and PBS |
Cytotoxicity of IDA/mPEG-PLGA1 NPs in mouse fibroblast cells and human normal CD34+ HSPCs in vitro
To investigate the cytotoxicity of IDA/mPEG-PLGA1 NPs in normal cells, L929 cell line (mouse fibroblast) and human CD34+ HSPCs were utilized to characterize the biocompatibility of IDA/mPEG-PLGA1 NPs and mPEG-PLGA1 NPs. The cell viability was first measured in L929 cells treated with different concentrations of mPEG-PLGA1 NPs, IDA, and IDA/mPEG-PLGA1 NPs for 24 hours. As shown in Figure 4A, the relative cell activity values of IDA/mPEG-PLGA1 NPs and mPEG-PLGA1 NPs in L929 cells were more than 90% at three different concentrations (0.1–10 μM). However, the relative activity value of pure IDA was decreased with the increase in concentration. When the concentration of IDA reached to 10 μM, the value declined to 43.9%. Furthermore, cell viability was detected in CD34+ HSPCs treated with different concentrations of mPEG-PLGA1 NPs, IDA, and IDA/mPEG-PLGA1 NPs for 24 hours. Low concentrations of IDA/mPEG-PLGA1 NPs (0.1 and 1 μM) present no cytotoxicity, but high concentration of IDA/mPEG-PLGA1 NPs (10 μM) presents moderate cytotoxicity in normal CD34+ HSPCs (Figure 4B). However, different concentrations of IDA/mPEG-PLGA1 NPs present less cytotoxicity than pure IDA (Figure 4B). All these results demonstrate that mPEG-PLGA1 NPs have no cytotoxicity for normal cells and IDA/mPEG-PLGA1 NPs have less cytotoxicity than pure IDA in normal mouse fibroblast cells and human HSPCs.
 | Figure 4 Cytotoxicity of mPEG-PLGA1 NPs, IDA/mPEG-PLGA NPs, and IDA in L929 cells and normal human CD34+ HSPCs. |
Biodistribution and pharmacokinetics of IDA/mPEG-PLGA1 NPs
To investigate the in vivo biodistribution of IDA/mPEG-PLGA1 NPs, liver, spleen, heart, lung, kidney, and intestine were dissected out after the mice were injected with IDA/mPEG-PLGA1 NPs for 6 hours. An ex vivo imaging analysis was performed to measure the biodistribution of IDA. As shown in Figure 5A, IDA from IDA/mPEG-PLGA1 NPs was mainly distributed in liver, spleen, and intestine but not in heart, lung, and kidney. As expected, IDA/mPEG-PLGA1 NPs were distributed in BM (Figure 5B), suggesting that IDA/mPEG-PLGA1 NPs can deliver to BM.
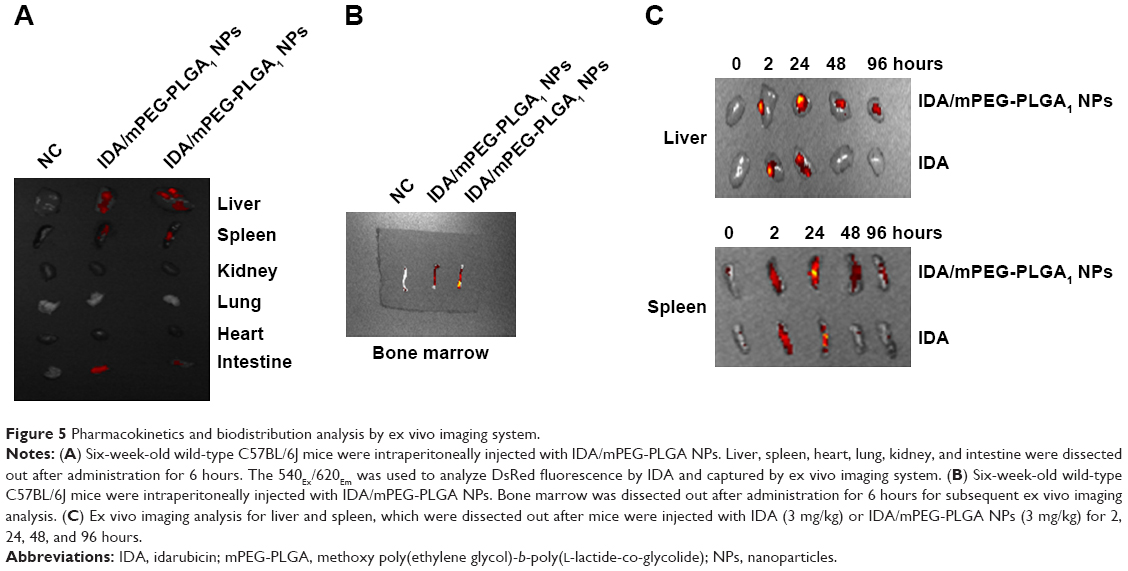 | Figure 5 Pharmacokinetics and biodistribution analysis by ex vivo imaging system. |
To further determine whether IDA/mPEG-PLGA1 NPs have elongated circulation time than free IDA, we further analyzed the pharmacokinetics of IDA/mPEG-PLGA1 NPs and IDA in liver and spleen, which were dissected out after the mice were injected with IDA/mPEG-PLGA1 NPs or IDA for 2, 24, 48, and 96 hours. In IDA/mPEG-PLGA1 NPs injected mice, the fluorescence levels analyzed by ex vivo imaging system in the liver and spleen appeared at 2 hours, reached the highest concentration 24 hours later, and decreased after 48 and 96 hours (Figure 5C). However, in IDA-injected mice, the fluorescence levels in the liver and spleen appeared at 2 hours, reached the highest concentration 24 hours later, and almost completely cleared after 48 and 96 hours (Figure 5C).
mPEG-PLGA enhances the cellular uptake of IDA
To determine whether mPEG-PLGA can enhance the cellular uptake of IDA, red fluorescence representing excitation light by IDA was detected by flow cytometry in HL-60 cells, which were incubated with IDA/mPEG-PLGA1 NPs or IDA. The shift in fluorescence peak from the untreated group (negative control [NC]) to the treated samples in the flow cytometry plot confirms the binding and uptake of IDA/mPEG-PLGA1 NPs and IDA to leukemia cells (Figure 6A). Most importantly, PE+ cells in IDA/mPEG-PLGA1 NPs treated cells were higher than that in IDA-treated cells (Figure 6A), suggesting that mPEG-PLGA enhances the uptake of IDA in leukemia cells.
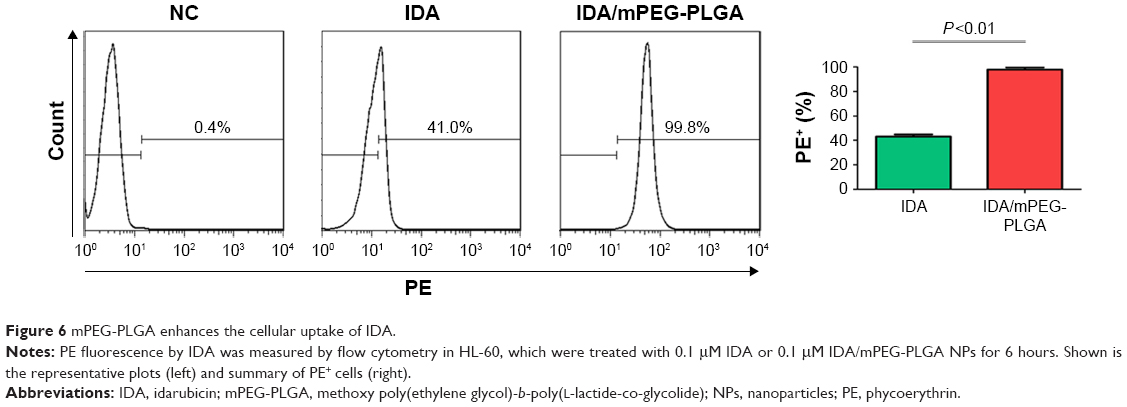 | Figure 6 mPEG-PLGA enhances the cellular uptake of IDA. |
Antileukemia activity of mPEG-PLGA NPs and IDA/mPEG-PLGA NPs in leukemia cell lines
To assess the antileukemia activity of mPEG-PLGA NPs in leukemia, we measured the IC50 of IDA/mPEG-PLGA NPs and pure IDA in two leukemia cell lines. HL-60 and U937 were treated with different concentrations of IDA/mPEG-PLGA NPs and IDA for 4 days, and cell proliferation was measured. IDA/mPEG-PLGA NPs exhibited the lower IC50 (Figure 7A; 0.014 μM in HL-60 and 0.0084 μM in U937) than IDA (Figure 7A; 0.048 μM in HL-60 and 0.013 μM in U937), suggesting that IDA/mPEG-PLGA NPs have approximately two- to fourfold higher effect on inhibiting cell growth than IDA.
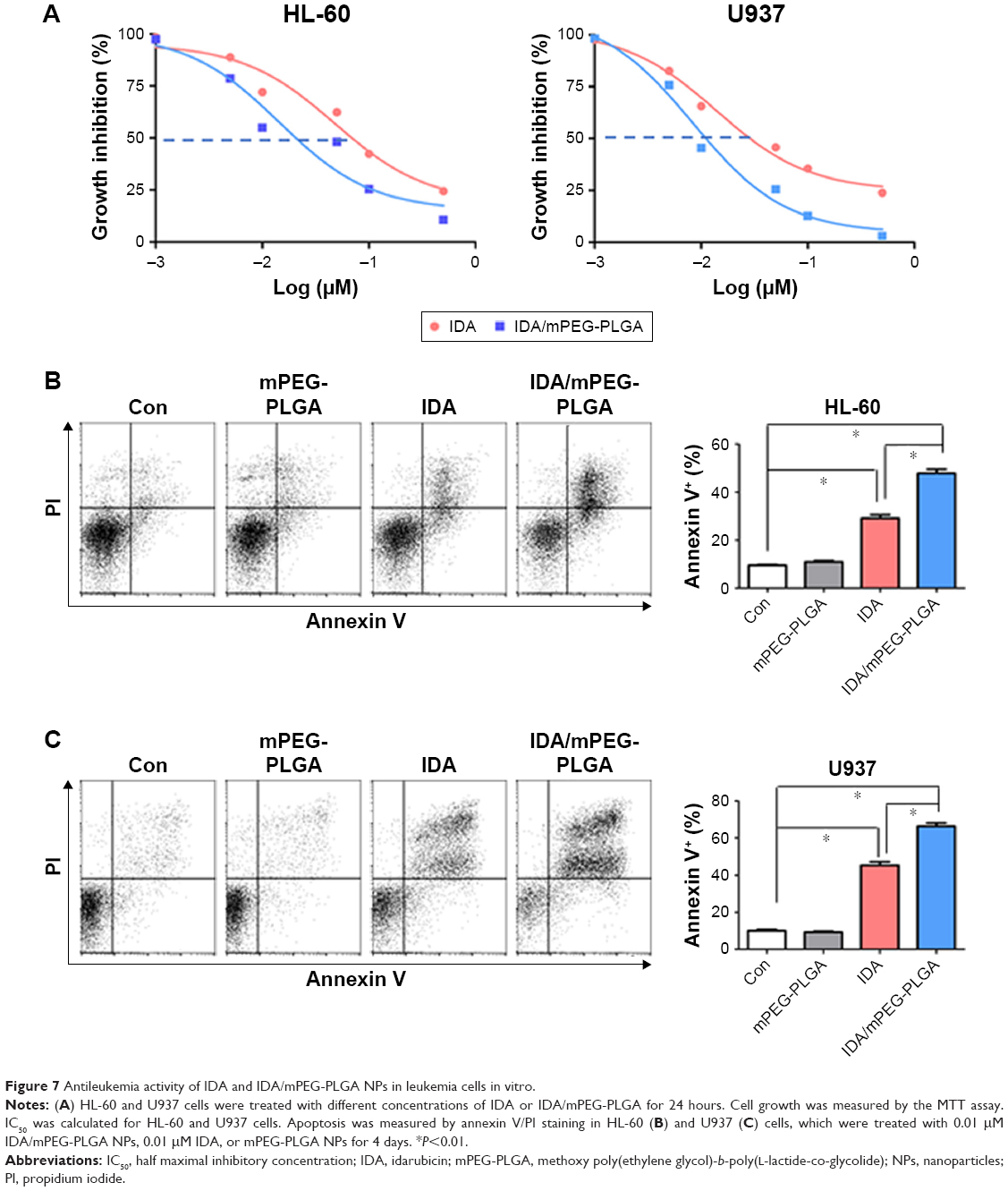 | Figure 7 Antileukemia activity of IDA and IDA/mPEG-PLGA NPs in leukemia cells in vitro. |
As chemotherapy drugs present antileukemia activity mainly by inducing apoptosis, we then measured the apoptosis in HL-60 and U937 cells incubated with 0.01 μM IDA/mPEG-PLGA NPs and 0.01 μM IDA for 4 days. IDA/mPEG-PLGA NPs and IDA substantially induced apoptosis in leukemia cells than negative control (Figure 7B and C). However, IDA/mPEG-PLGA NPs are more effective in inducing apoptosis in leukemia cells than IDA (Figure 7B and C).
Antileukemia activity of IDA/mPEG-PLGA NPs in MLL-AF9-induced murine leukemia
As reported, MLL-rearranged leukemia has been used as a classical model to assess the antileukemia activity of chemotherapy drugs, because MLL fusion proteins can efficiently transform HSPC into leukemia stem cell.22 Therefore, MLL-AF9-induced mice leukemia model was used to further investigate the antileukemia activity by IDA/mPEG-PLGA NPs and IDA. GFP+ cells were isolated from MLL-AF9-transduced mice and transplanted into irradiated mice, which were divided into four groups (IDA/mPEG-PLGA NPs, IDA, mPEG-PLGA, and vehicle). Three vehicle mice and three drug-treated mice were sacrificed when vehicle mice developed full-blown leukemia. GFP-positive cells were measured in BM mononuclear cells. GFP-positive cells were significantly decreased in IDA-treated and IDA/mPEG-PLGA NPs treated mice than in vehicle mice (Figure 8A). However, IDA/mPEG-PLGA NPs are more effective in reducing GFP-positive cells than did IDA (Figure 8A). Moreover, blood, BM, and spleen smears indicated an obvious decrease in leukemia blasts in IDA-treated and IDA/mPEG-PLGA1 NPs treated mice than in vehicle mice (Figure 8B). Also, leukemia blasts in IDA/mPEG-PLGA NPs treated mice were less than those in IDA-treated mice (Figure 8B). Accordingly, although IDA/mPEG-PLGA NPs and IDA reduced spleen weight, IDA/mPEG-PLGA NPs induced more decrease in spleen weight than IDA did (Figure 8C). Finally, IDA/mPEG-PLGA NPs and IDA treatment markedly prolonged the survival of MLL-AF9-transduced mice than vehicle mice (Figure 8D). The OS in IDA/mPEG-PLGA1 NPs treated mice was longer than that in IDA-treated mice (Figure 8D).
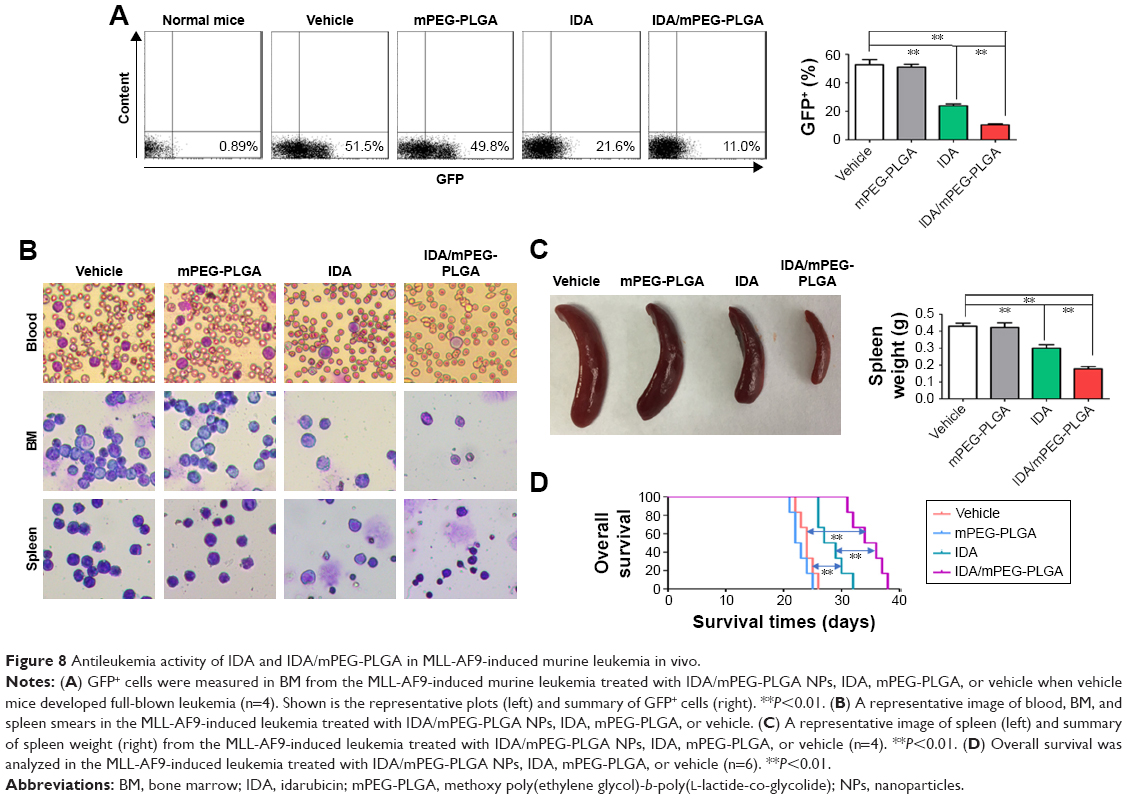 | Figure 8 Antileukemia activity of IDA and IDA/mPEG-PLGA in MLL-AF9-induced murine leukemia in vivo. |
Discussion
Chemotherapy is one of the main strategies for AML patients. However, high doses of chemotherapy drugs cause a series of severe side effects, including BM suppression, gastrointestinal reactions, kidney damage, and hair loss. These side effects severely limit its further application. NP drug delivery systems have a potential to sustain the release of drugs and enhance the accumulation of drugs in cancer cells.23 Optimization of these carriers extends the release property and reduces the dose of cytotoxic drugs, thus reducing the associated side effects.
IDA is the first-line drug in the treatment of AML.6 To reduce the dose of IDA, solid lipid NPs,24 propyl starch NPs,25 and folic acid conjugated magnetic NPs have been developed as carriers to conjugate IDA.26 However, the potential use of mPEG-PLGA to deliver IDA has not been reported. Here, we successfully developed mPEG-PLGA-loaded NPs as a modified dialysis method. Our results indicate that mPEG-PLGA is little toxic in normal mouse fibroblast. IDA/mPEG-PLGA NPs act as a controlled release formulation for IDA and maintains a longer release time than pure IDA. We confirm the efficiency of IDA/mPEG-PLGA1 NPs in inducing cytotoxicity by both in vitro studies with leukemia cell lines and in vivo study in MLL-AF9-induced mice model. IDA/mPEG-PLGA1 NPs are substantially more toxic than IDA in vitro and in vivo, suggesting that mPEG-PLGA is suitable carrier to deliver IDA.
MLL-AF9-induced murine leukemia is a widely used model to assess the antileukemia activity of chemotherapy drugs.27,28 However, few reports use this model to assess NP-coated drugs. Preclinical models using subcutaneous xenografts have often been used to nanomedicine.29,30 Subcutaneous models are easy to create and to evaluate tumor progression in response to treatment, but this approach does not reflect the in vivo situation because all leukemia cells are not subcutaneous. MLL-AF9 model accurately mimics the in vivo disease condition where the leukemia blasts are in the blood and BM. Furthermore, therapeutic efficiencies of IDA/mPEG-PLGA1 NPs and pure IDA were assessed by direct IP injection. This situation closely resembles the treatment of AML patients in the clinic, suggesting that this model will be a valuable tool in the evaluation of nanotherapeutics for hematological malignancies. Our data indicate that IDA/mPEG-PLGA1 NPs decrease leukemia blasts and prolong the OS in MLL-AF9-induced leukemia mice. Moreover, IDA/mPEG-PLGA1 NPs present stronger antileukemia ability than IDA in the same concentration in leukemia mice. We speculate that IDA/mPEG-PLGA1 NPs sustain a longer release than pure IDA, resulting in a longer duration of action. Our results are consistent with the report by Krishnan et al,31 who encapsulated dexamethasone in polymeric NPs and found that dexamethasone NPs significantly improved the quality of life and survival of leukemia mice than the free drug.
One of the AML patients’ characteristics is associated with splenomegaly and infiltration of leukemia blasts in spleen.32 The normal structure of spleen is destroyed, and the spleen is infiltrated by single leukemia cells. Furthermore, leukemia blasts in spleen often escape from the treatment of chemical drugs and become the origin of relapse. Therefore, reducing the weight of spleen and decreasing the infiltration of leukemia blasts in spleen have been considered as successful treatments of AML patients. Consistent with AML patients’ characteristic, splenomegaly and leukemia cell infiltration always occur in MLL-AF9-induced murine leukemia.33 Reducing spleen weight and decreasing leukemia cell infiltration by chemical drugs such as IDA suggest the efficient treatment in murine leukemia. Therefore, in our study, we measured the spleen weight and leukemia cell infiltration in spleen and found that IDA/mPEG-PLGA1 NPs reduced the spleen weight and decreased leukemia blasts’ infiltration than pure IDA did. Our data demonstrate that the spleen weight is a suitable indicator to reflect the antileukemia activity during IDA treatment.
Because of biocompatibility and biodegradability, mPEG-PLGA-based polymeric NPs are the suitable carriers for the delivery and controlled release of anticancer drugs. The blank NPs are nontoxic to cultured normal cells in vitro, suggesting that mPEG-PLGA-based polymeric NPs might be safe for AML patients. Also, PLGA is approved by the US FDA for biomedical usage, because it can create NPs like nanocarriers displaying an internal hydrophobic PLGA portion and it is suitable for entrapping lipophilic molecules to form a stabilizing shell.34,35 To investigate the potential usage of PLGA for the carrier of chemotherapy drugs, Tamborini et al reported that PLGA NPs (PNPs) are conjugated to chlorotoxin and silver NPs are entrapped inside the functionalized NPs. These functionalized NPs can efficiently target the tumor.36 Also, Duo et al37 use core/shell gene carriers with different lengths of PLGA chains to promote the transfection of endothelial cells. These results with our study confirm that PLGA is a suitable carrier to encapsulate drugs.
mPEG is a neutral polymer and PLGA presents negative charges. However, the mPEG-PLGA NPs present negative charges but not neutral charges. We speculate that the presence of PEG on the surface of PLGA cores only covers the part of PLGA-induced negative charges, resulting in the negative NPs. Our results are consistent with the previous reports.38,39
We detected the biodistribution of IDA/mPEG-PLGA NPs by ex vivo imaging system in murine organs and found that IDA was mainly distributed in liver, spleen, and intestine but not in lung, heart, and kidney. Although no accumulation of IDA was detected in lung, heart, and kidney, it is possible that the ex vivo imaging analysis is not sensitive enough to detect the very low accumulation of IDA/mPEG-PLGA in these organs. We speculate that IDA/mPEG-PLGA NPs have low toxicity for these organs.
Conclusion
Our study demonstrates that the IDA/mPEG-PLGA NPs’ formulation has high DL capacity, good EE, and good aqueous dispersion. Thus, mPEG-PLGA NPs have the properties of sustained drug release. IDA/mPEG-PLGA NPs have significant antileukemia activity than pure IDA. IDA/mPEG-PLGA should be a promising systemic chemotherapy for leukemia and can be expanded for use on other types of cancers. IDA encapsulated in NPs may enable the use of reduced doses of IDA to reduce side effects and improve survival. The next step is to establish targeted drug delivery platforms. For example, CD33 antibody can be covalently linked with IDA/mPEG-PLGA1 NPs to selectively kill leukemia blasts.40 Translation of this technology into the clinic can reduce the severe side effects and improve the survival and quality of AML patients.
Acknowledgment
The authors are thankful for the financial support from the National Natural Science Foundation (81672087) and the Natural Science Foundation of Zhejiang Province (LY12H08002 and Y19H080019).
Disclosure
The authors report no conflicts of interest in this work.
References
Wouters BJ, Delwel R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood. 2016;127(1):42–52. | ||
Dombret H, Gardin C. An update of current treatments for adult acute myeloid leukemia. Blood. 2016;127(1):53–61. | ||
Oun R, Moussa YE, Wheate NJ. The side effects of platinum-based chemotherapy drugs: a review for chemists. Dalton Trans. 2018;47:6645–6653. | ||
Berman E, Heller G, Santorsa J, et al. Results of a randomized trial comparing idarubicin and cytosine arabinoside with daunorubicin and cytosine arabinoside in adult patients with newly diagnosed acute myelogenous leukemia. Blood. 1991;77(8):1666–1674. | ||
Gallois L, Fiallo M, Garnier-Suillerot A. Comparison of the interaction of doxorubicin, daunorubicin, idarubicin and idarubicinol with large unilamellar vesicles. Circular dichroism study. Biochim Biophys Acta. 1998;1370(1):31–40. | ||
Ohtake S, Miyawaki S, Fujita H, et al. Randomized study of induction therapy comparing standard-dose idarubicin with high-dose daunorubicin in adult patients with previously untreated acute myeloid leukemia: the JALSG AML201 Study. Blood. 2011;117(8):2358–2365. | ||
Liu H, Fu R, Li L, et al. Comparison of Reduced-Intensity Idarubicin and Daunorubicin Plus Cytarabine as Induction Chemotherapy for Elderly Patients with Newly Diagnosed Acute Myeloid Leukemia. Clin Drug Investig. 2017;37(2):167–174. | ||
Dutta RC. Drug carriers in pharmaceutical design: promises and progress. Curr Pharm Des. 2007;13(7):761–769. | ||
Yuan YY, Mao CQ, Du XJ, Du JZ, Wang F, Wang J. Surface charge switchable nanoparticles based on zwitterionic polymer for enhanced drug delivery to tumor. Adv Mater. 2012;24(40):5476–5480. | ||
Misra R, Sahoo SK. Coformulation of doxorubicin and curcumin in poly(D,L-lactide-co-glycolide) nanoparticles suppresses the development of multidrug resistance in K562 cells. Mol Pharm. 2011;8(3):852–866. | ||
Yadav KS, Sawant KK. Modified nanoprecipitation method for preparation of cytarabine-loaded PLGA nanoparticles. AAPS PharmSciTech. 2010;11(3):1456–1465. | ||
An X, Zhu A, Luo H, Ke H, Chen H, Zhao Y. Rational Design of Multi-Stimuli-Responsive Nanoparticles for Precise Cancer Therapy. ACS Nano. 2016;10(6):5947–5958. | ||
Mohanty C, Sahoo SK. The in vitro stability and in vivo pharmacokinetics of curcumin prepared as an aqueous nanoparticulate formulation. Biomaterials. 2010;31(25):6597–6611. | ||
Dilnawaz F, Singh A, Mohanty C, Sahoo SK. Dual drug loaded superparamagnetic iron oxide nanoparticles for targeted cancer therapy. Biomaterials. 2010;31(13):3694–3706. | ||
Jiang X, Huang H, Li Z, et al. Blockade of miR-150 maturation by MLL-fusion/MYC/LIN-28 is required for MLL-associated leukemia. Cancer Cell. 2012;22(4):524–535. | ||
Li Z, Chen P, Su R, et al. Overexpression and knockout of miR-126 both promote leukemogenesis. Blood. 2015;126(17):2005–2015. | ||
Xiong W, Peng L, Chen H, Li Q. Surface modification of MPEG-b-PCL-based nanoparticles via oxidative self-polymerization of dopamine for malignant melanoma therapy. Int J Nanomedicine. 2015;10:2985–2996. | ||
Rakhmatullin RM, Kurkin IN, Pavlov VV, Semashko VV. EPR, optical, and dielectric spectroscopy of Er-doped cerium dioxide nanoparticles. Physica Status Solidi. 2014;251(8):1545–1551. | ||
Acharya S, Sahoo SK. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv Drug Deliv Rev. 2011;63(3):170–183. | ||
Zeng X, Tao W, Mei L, Huang L, Tan C, Feng SS. Cholic acid-functionalized nanoparticles of star-shaped PLGA-vitamin E TPGS copolymer for docetaxel delivery to cervical cancer. Biomaterials. 2013;34(25):6058–6067. | ||
Liu T, Seiffert S, Thiele J, Abate AR, Weitz DA, Richtering W. Non-coalescence of oppositely charged droplets in pH-sensitive emulsions. Proc Natl Acad Sci U S A. 2012;109(2):384–389. | ||
Krivtsov AV, Twomey D, Feng Z, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442(7104):818–822. | ||
Zhang Y, Sun T, Jiang C. Biomacromolecules as carriers in drug delivery and tissue engineering. Acta Pharm Sin B. 2018;8(1):34–50. | ||
Ma P, Dong X, Swadley CL, et al. Development of idarubicin and doxorubicin solid lipid nanoparticles to overcome Pgp-mediated multiple drug resistance in leukemia. J Biomed Nanotechnol. 2009;5(2):151–161. | ||
Jain R, Dandekar P, Loretz B, et al. Enhanced cellular delivery of idarubicin by surface modification of propyl starch nanoparticles employing pteroic acid conjugated polyvinyl alcohol. Int J Pharm. 2011;420(1):147–155. | ||
Gunduz U, Keskin T, Tansık G, et al. Idarubicin-loaded folic acid conjugated magnetic nanoparticles as a targetable drug delivery system for breast cancer. Biomed Pharmacother. 2014;68(6):729–736. | ||
Maifrede S, Martinez E, Nieborowska-Skorska M, et al. MLL-AF9 leukemias are sensitive to PARP1 inhibitors combined with cytotoxic drugs. Blood Adv. 2017;1(19):1467–1472. | ||
Pikman Y, Puissant A, Alexe G, et al. Targeting MTHFD2 in acute myeloid leukemia. J Exp Med. 2016;213(7):1285–1306. | ||
Dhar S, Kolishetti N, Lippard SJ, Farokhzad OC. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc Natl Acad Sci U S A. 2011;108(5):1850–1855. | ||
Wang X, Li J, Wang Y, et al. A folate receptor-targeting nanoparticle minimizes drug resistance in a human cancer model. ACS Nano. 2011;5(8):6184–6194. | ||
Krishnan V, Xu X, Barwe SP, et al. Dexamethasone-loaded block copolymer nanoparticles induce leukemia cell death and enhance therapeutic efficacy: a novel application in pediatric nanomedicine. Mol Pharm. 2013;10(6):2199–2210. | ||
Fujishima M, Fujishima N, Kitadate A, et al. Successful management of splenomegaly with ruxolitinib prior to allogeneic hematopoietic stem cell transplantation in acute myeloid leukemia transformed from post-polycythemia vera myelofibrosis. Rinsho Ketsueki. 2017;58(7):743–748. | ||
Nguyen AT, Taranova O, He J, Zhang Y. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood. 2011;117(25):6912–6922. | ||
Locatelli E, Gil L, Israel LL, et al. Biocompatible nanocomposite for PET/MRI hybrid imaging. Int J Nanomedicine. 2012;7:6021–6033. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Tamborini M, Locatelli E, Rasile M, et al. A Combined Approach Employing Chlorotoxin-Nanovectors and Low Dose Radiation To Reach Infiltrating Tumor Niches in Glioblastoma. ACS Nano. 2016;10(2):2509–2520. | ||
Duo X, Li Q, Wang J, et al. Core/Shell Gene Carriers with Different Lengths of PLGA Chains to Transfect Endothelial Cells. Langmuir. 2017;33(46):13315–13325. | ||
Avgoustakis K, Beletsi A, Panagi Z, et al. Effect of copolymer composition on the physicochemical characteristics, in vitro stability, and biodistribution of PLGA-mPEG nanoparticles. Int J Pharm. 2003;259(1–2):115–127. | ||
Gryparis EC, Hatziapostolou M, Papadimitriou E, Avgoustakis K. Anticancer activity of cisplatin-loaded PLGA-mPEG nanoparticles on LNCaP prostate cancer cells. Eur J Pharm Biopharm. 2007;67(1):1–8. | ||
Walter RB, Appelbaum FR, Estey EH, Bernstein ID. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood. 2012;119(26):6198–6208. |
Supplementary material
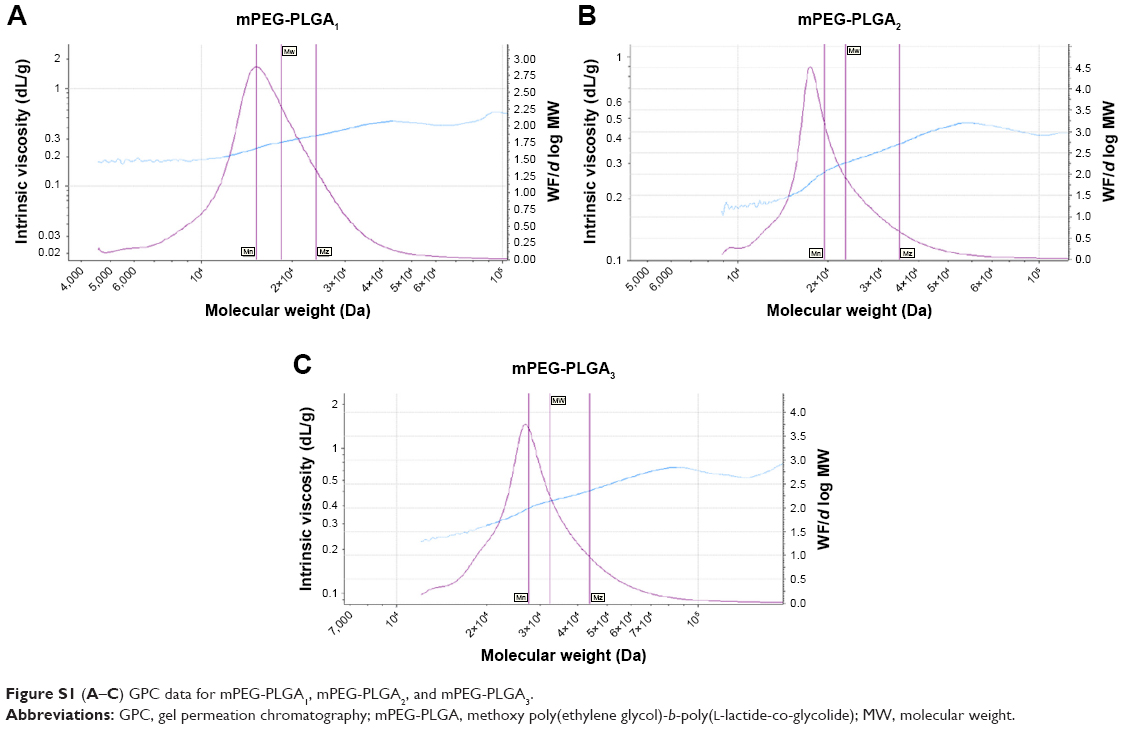 | Figure S1 (A–C) GPC data for mPEG-PLGA1, mPEG-PLGA2, and mPEG-PLGA3. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.