Back to Journals » Drug Design, Development and Therapy » Volume 9
How we make cell therapy in Italy
Authors Montemurro T, Viganò M, Budelli S, Montelatici E, Lavazza C, Marino L, Parazzi V, Lazzari L, Giordano R
Received 6 January 2015
Accepted for publication 25 March 2015
Published 20 August 2015 Volume 2015:9 Pages 4825—4834
DOI https://doi.org/10.2147/DDDT.S80403
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Tiziana Montemurro, Mariele Viganò, Silvia Budelli, Elisa Montelatici, Cristiana Lavazza, Luigi Marino, Valentina Parazzi, Lorenza Lazzari, Rosaria Giordano
Cell Factory, Unit of Cell Therapy and Cryobiology, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milano, Italy
Abstract: In the 21st century scenario, new therapeutic tools are needed to take up the social and medical challenge posed by the more and more frequent degenerative disorders and by the aging of population. The recent category of advanced therapy medicinal products has been created to comprise cellular, gene therapy, and tissue engineered products, as a new class of drugs. Their manufacture requires the same pharmaceutical framework as for conventional drugs and this means that industrial, large-scale manufacturing process has to be adapted to the peculiar characteristics of cell-containing products. Our hospital took up the challenge of this new path in the early 2000s; and herein we describe the approach we followed to set up a pharmaceutical-grade facility in a public hospital context, with the aim to share the solutions we found to make cell therapy compliant with the requirements for the production and the quality control of a high-standard medicinal product.
Keywords: advanced therapy medicinal product, good manufacturing practices, stem cells
Introduction
Cell therapy is the administration of autologous, allogeneic or xenogeneic living cells to humans, which have been manipulated or processed ex vivo. When cell therapy is applied to restore the function of cells, tissues and organs using healthy cells (ie, mesenchymal stem or stromal cells, MSC), we enter in the field of regenerative medicine. To make regenerative medicine as a real therapeutic tool, it is necessary to manipulate cells and tissues as for any other sterile medicinal products in absolute microbiological safety with special equipment and complex procedures, in compliance with pharmaceutical quality standards in order to obtain cell products to be transplanted in humans. These structures, in Italy commonly called “Cell Factories”, are therefore pharmaceutical factories and their products have been defined in the European regulatory framework as advanced therapy medicinal products (ATMPs), together with gene therapy products and tissue engineered products, the latter being essentially cell therapy product used alone or with biomaterial with tissue replacing purposes. Since ATMPs are drugs, we need precise and defined standards and guidelines not only as regards the manufacturing and quality control (QC) but also for everything that surrounds the cell therapy product including facility (how to build the production site and the QC area and how to maintain them), staff (how to train and to keep them updated) and to periodic QCs of the environment (where the operators make the production process).
All the facilities for the production of cell therapy, even those located within public hospitals, must meet good manufacturing practice (GMP) standards.1
The aim of this article is to describe our GMP experience from the beginning to today and to give an overview of all the programs where our Cell Factory is involved. Our goal is to openly share our experience in this novel fascinating therapeutic scenario.
Cell Factory authorization
In 2000, a project to develop a GMP facility for ATMP production has been started by the Fondazione IRCCs Ca’ Granda Ospedale Maggiore Policlinico in Milan and supported by other co-fundings.
In 2007, the Cell Factory was certified by the Italian Drug Agency (Agenzia Italiana del Farmaco – AIFA) in compliance with European GMP regulations (authorization number 120/2007 of July 5, 2007). The following were obtained in 2011 (authorization number 22/2011 of February 2, 2011) and in 2013 (authorization number 98/2013 of July 23, 2013, the last at the time when this manuscript was written).
GMP facility production site
When we decided to build a GMP facility in a public hospital, the layout of the system was carried out in order to have restricted access of personnel to the areas of production, changing rooms with all the appropriate and approved clothing and accessories for cleanrooms, as easy as possible cleaning together with minimizing the introduction of airborne contaminants into the production area through appropriate standard operating procedures (SOPs), an orderly placement of equipment and materials in order to avoid cross-contamination or cross-path, organization of space and separation processes sufficient to maintain a clean working environment and a proper flow of operations, personnel, and materials.
The GMP facility is located in a controlled area with limited access: the production area includes laboratories with class-A biosafety cabinet for the production in a Class B-GMP surround, with the aseptic gowning rooms and corridors of access in increasing value of air cleanliness from D to B, a specific area for the storage of materials (NC, not classified) and a class D room dedicated to the preparation and packaging of materials for the passage in Class B with ventilated pass-boxes (Figure 1). The classification of different areas has been carried out in accordance with EudraLex – Vol 4, Annex 1 of GMP,1 both at resting and in operation conditions; the maximum permitted number of particles per grade is reported in Table 1. The classified areas are maintained at positive pressure relative to the surrounding areas, with a Δp value between contiguous rooms (with different classification) of at least 10 Pa. Clearly, the higher number of air changes is at the production area. In addition, the temperature and humidity conditions are maintained within 18°C–25°C with 60% relative humidity.
 | Figure 1 Representative pictures of the classified areas inside the Cell Factory. |
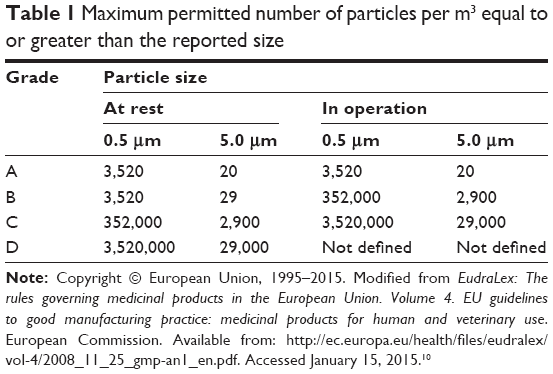 | Table 1 Maximum permitted number of particles per m3 equal to or greater than the reported size |
Description of the air-conditioning system
In our facility there is a single conditioning system with no re-circulation (100% expulsion of air), protected by absolute filters both inbound and outbound. The production site and the non-sterile changing rooms are completely isolated from each other and the critical areas are protected by appropriate pressure gradients. The pressures are articulated in order to separate the laboratories of production maintained at positive pressure relative to the surrounding areas with respect to the rest of the structure, and to protect the manufacturing processing. This system is constituted by the following elements: the air treatment unit, two groups of air expulsion “high-efficiency particulate air” (HEPA) filter, terminal filters installed in laboratories, and a temperature control system with specific sensors. Importantly, critical systems (“heating, ventilation and air conditioning” (HVAC) system, process systems, and equipment for the storage of materials for the production) are equipped with a 24 hour alarm system phone availability.
In compliance with the GMP standards, the production site is constantly monitored on the basis of specific procedures with planned maintenance programs. In addition to the viable and non-viable particle counts, the pressures, the efficiency of HEPA filters, and other critical components of the air conditioning system are maintained under control according to the internal procedures and according to the GMP rules.1
Maintenance of the system
The ordinary maintenance of the HVAC system is followed by specific technical services of our hospital: the specifications of acceptability of the system have been fixed in advance in the validation phase. All the checks, calibrations, and maintenance are carried out by qualified personnel with a good knowledge of air conditioning system, and in particular of mechanical, electrical, and instrumental components. In case of anomalies on the state of efficiency of the components, it is necessary to notify the qualified person (QP) to define the specific actions to be taken. The maintenance of the entire system in our facility is scheduled through controls at different frequencies: fortnightly (such as the replacement of wavy pre-filters), monthly (check of ventilation group, check of valves for heat transfer fluids), half-yearly (replacement of the HEPA filters, calibration of flow rates, control and calibration of differential pressure for environmental pressures, etc), while requalification of the HVAC system is annual, in order to confirm that the facility is under control.
Cleaning validation and procedures
In our GMP facility, all the equipment, tools, and facilities must be properly cleaned, sanitized, and maintained under an efficient condition for appropriate intervals to prevent malfunctions or contamination that could affect the quality of the final product. In fact, a suitable cleaning process of the production site is a requirement to avoid the risk of contamination. For that reason a cleaning validation is designed with the objective to confirm the effectiveness of any cleaning procedure. Therefore, our cleaning validation was focused on the areas to be cleaned, the equipment for cleaning, the detergents to be used, their concentrations of use and the contact time, the method of removal of detergents, and also the proper qualification and training of operators. In this context, a critical aspect has also been the validation of microbicide efficacy of the disinfectants, also evaluated towards autochthon strains eventually isolated in the production site during the routine microbial monitoring.
Personnel: training and microbiological monitoring
The GMP staff is the crucial point of any good facility. Staff must have the necessary skills, be properly trained and always updated both in theory and in practice on the concept of quality assurance and standards of GMP. We planned an initial training and an ongoing training, as requested by the GMP guidelines (EudraLex – Vol 4, chapter 2)1 with frequent staff performance and training courses. At our facility, the training plan follows an annual schedule and it is differentiated by various topics for both production and QC staff: an example of training program is reported in Figure 2. In addition, special training schedules are set up for the technical, maintenance, and cleaning personnel. All the GMP staff duties, including the management staff and the QP, responsible for implementation and enforcement of GMP are specified in our job descriptions. Among the various activities of the initial training, microbiological validation of the operators is also crucial: only after successfully overcoming the microbial validation including aseptic gowning and operations also with media fill (as thereafter illustrated) the production staff is authorized to work in aseptic procedures. Moreover, the single operator involved in the production process is constantly monitored at the end of each work session. The personnel microbiological monitoring consists in testing the forearms as well as the five fingers. All the plates are incubated at 20°C–25°C for 5 days and then at 30°C–35°C for 5 days. The results are expressed as TAMC (total aerobic microbial count) and as UFC/plate (unit-forming colony) as requested by EuGMP/Annex1. Autochthon strains are tested in the validation of final product sterility testing. Every 3 months environmental microbiological, particulate, and staff data are collected in specific forms (Figure 3) in order to prepare our periodic reports (Trend Analysis). These trend analyses are built according to the theory of control of Shewhart charts, to fit the warning and alarm limits to the real situation of the production site. Adequate alarm and action limits have been established and are continuously updated to determine the immediate investigation and appropriate corrective action in case of deviations.
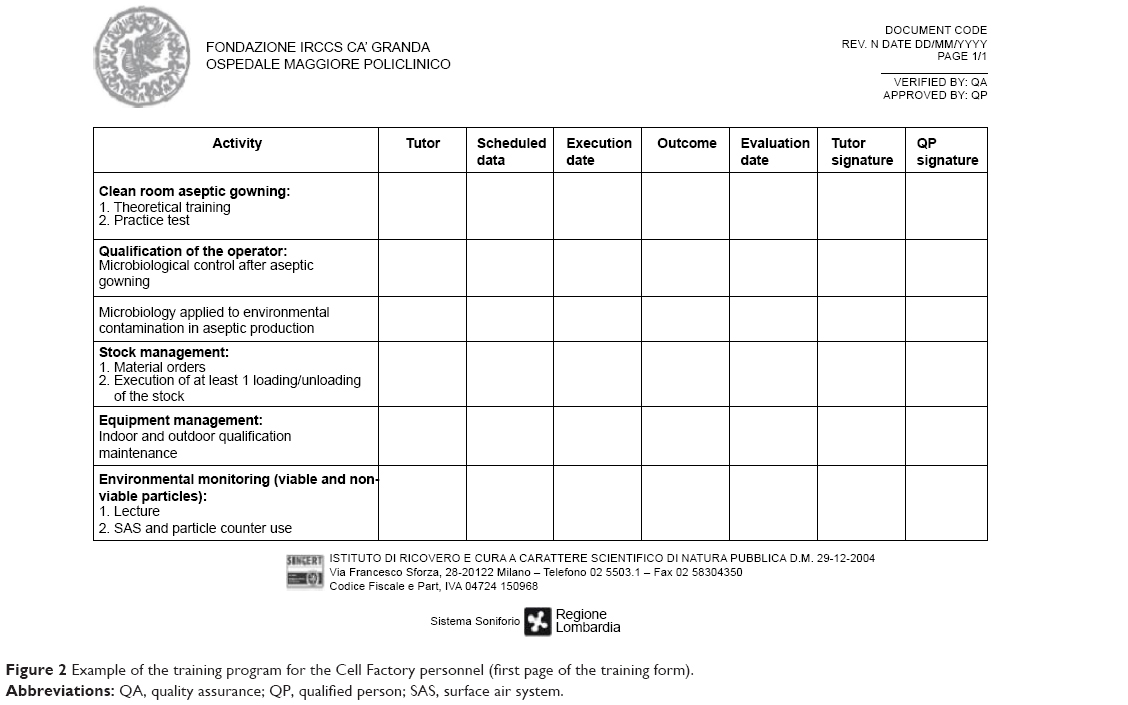 | Figure 2 Example of the training program for the Cell Factory personnel (first page of the training form). |
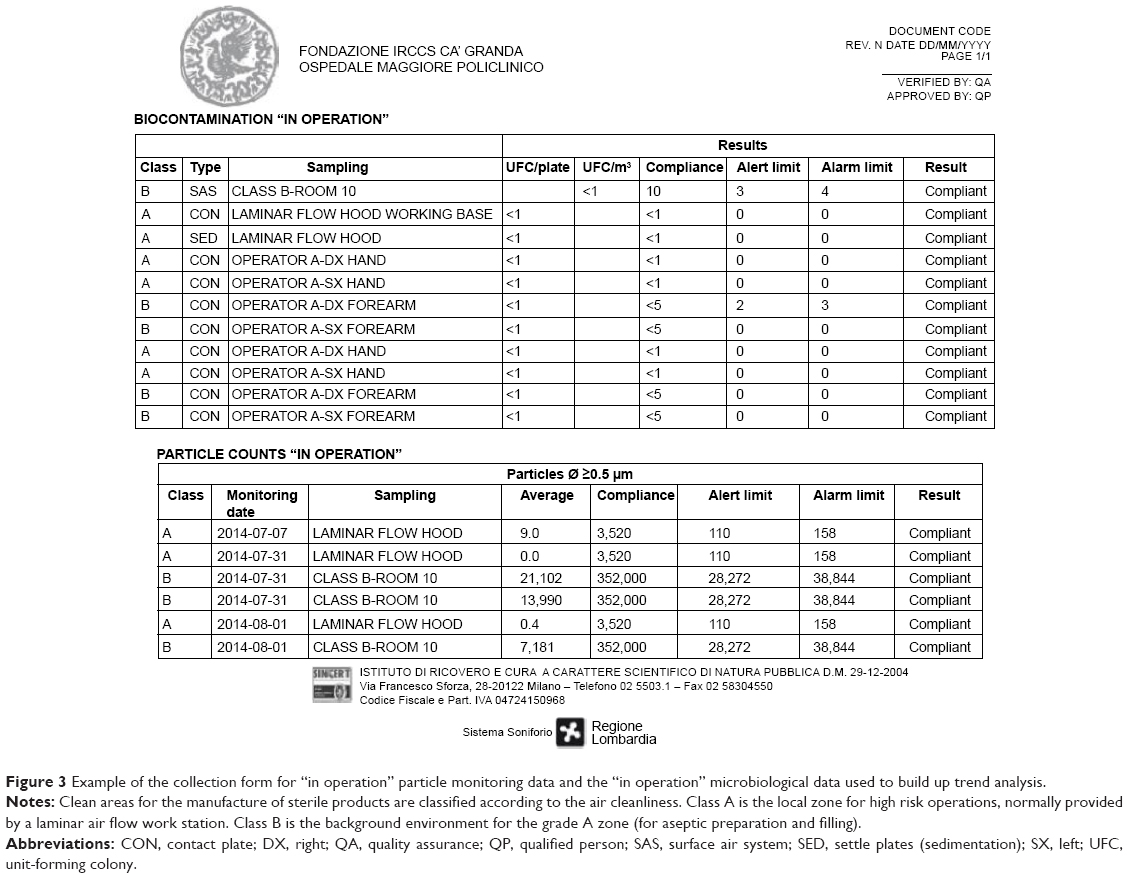 | Figure 3 Example of the collection form for “in operation” particle monitoring data and the “in operation” microbiological data used to build up trend analysis. |
To control the risks for the process and the products, in our Cell Factory there are specific SOPs to manage unexpected or unusual events, especially for out of specifications in continuous environmental monitoring. Briefly, in case of an exceeding of the alert (or action) limits for the air particle count during cell manipulation, the operators are trained to stop their activities, to close the containers (in case of open procedures), and to wait until the reestablishment of the normal conditions. Additional in process controls may be performed upon indication of the responsible for QC and QP authorization. In case of deviations in particle counts or microbial environmental controls, the Director of Production may decide to cancel all the planned manufacturing processes, to perform additional sanitization of both equipments and clean rooms, and to repeat environmental controls. The QP authorizes to resume the production only when the results of repeated environmental controls are within the limits.
The corrective actions are defined after the analysis of the causes and may include also additional operations on the HVAC system and staff re-training.
Environmental control monitoring
As requested by EuGMP/Annex1, several environmental monitoring are performed during cell manipulations at our facility. These include the microbial air (surface air system – SAS) and surface controls of the clean rooms (Classes A and B) and the particle count of laminar flow hoods (Class A) that must be monitored continuously. The personnel and the environmental monitoring are conducted during the manufacturing process in working areas, based on the results obtained during the media fill process in which laminar flow hoods and ventilated pass-box are tested with contact plates and settle plates (TSA, tryptic soy agar, contact plates with LTHTh-ICR, Heipha Dr Muller GmbH, Heidelberg, Germany). During the entire production process, only 1.06% out of specification result values for microbiological environmental and personnel monitoring have been detected at our GMP site from 2007 to the date when this manuscript has been written.
There have been no over-specifications in the averages of the daily values under the laminar flow hood (Class A) within the clean room processing (Class B), for the microbial air and surface controls of the clean rooms (Classes A and B) and the particle count of laminar flow hoods (Class A). All the requested trainings and staff performance evaluations have been performed following the internal SOPs.
Instruments and supplies
According to the GMP guidelines (Eudralex – Vol 4, chapter 6)1 specific procedures for each type of equipment, instrument, and other devices have be established, with regard to the extent of which they are used, verified, and calibrated at regular intervals according to SOPs.
Therefore, all the instruments available and used in our GMP facility have been subject to installation qualification, in accordance with the manufacturer’s specifications and to operational qualification, demonstrating the instrument functions according to the operational procedures. The maintenance schedule of the instruments is strictly monitored based on a GANTT chart.
The choice of systems and equipments is crucial for a GMP facility: whenever possible a closed system with advanced integrated solutions for streamlining cell-processing workflows, with disposable sterile and non-pyrogenic sets for single use only, should always be preferred. As an example, for cell sorting we choose closed and automated systems based on immunomagnetic selection processes. Moreover, for cell culture we use large-scale multiple layer closed plastic systems designed for scale-up applications and finally to preserve the sterility during cell manipulation we use specific equipment for automatic sealing of different types/sizes of tubings (sterile heating connectors).
Main aspects of a production process
The translation from the bench to the bedside takes place according to precise planning and following some steps clearly defined in order to reach the clinical phase.
A kind of roadmap is summarized in Figure 4. Briefly, there is an important and initial phase of planning where the GMP staff has to carefully analyze preclinical data before moving into a GMP process. Soon after, crucial is the identification of GMP reagents and the development of appropriate SOPs. At the QC level it is mandatory to establish precise and well-defined assays and parameters, providing that any method and results consistently fulfil particular quality requirements. Next, there is the step of validation that must normally be completed for the authorization of the cell therapy production by the QP.
 | Figure 4 Summary of all the steps to be followed for the cell therapy translational process from the bench to the bedside. |
During this validation at least three consecutive batches of the final cell therapy product must be run to confirm the reproducibility, efficacy, and robustness of the entire production and QC process. In this context, a very important step is the validation of the aseptic process (media fill), which must be performed on the most risky production process (worst case).
Media fill
At our facility and in our experience, the media fill applied to the ATMP production, as for any other aseptic drug manufacturing processes, represents a very important issue that brought us to devote to it a specific chapter. First of all, the media fill must be carried out for asepsis validation in order to ensure that even the most critical production activities do not create events that could affect the sterility of the final product, using a specific microbiological culture medium instead of the medicinal product. We select the tryptic soy broth because it is a non-selective broad-spectrum microbiological medium. We simulate all the steps of production that are considered critical using the same procedures and the same equipment as during the real manufacturing process. Obviously to avoid any risk of contamination at the end of the test, the surfaces are thoroughly cleaned, sterilized, and disinfected. During the media fill the microbial sampling of the air and the surface and the particle count of the clean rooms and laminar flow hoods are performed extensively, in order to define the environmental monitoring plan to be carried out during each production. The environment is monitored through microbiological monitoring and particle count for each type of actions. Taking the example of the production of adherent cells, such as the MSCs, the actions are represented by seeding, trypsinization, freezing, and thawing. Before the beginning of a specific production process, three runs of media fill have to be carried out in order to generate a statistically significant amount of data.
In our facility these are the monitored points: bench top, laminar flow hoods, ventilated pass-box, water bath, handle door, basket centrifuge, work surface, and external wall of incubator with contact plates, while laminar flow cabin with settle plates exposing for 4 hours. In addition, the operators are extensively monitored through contact plates: right forearm and left, right five fingers and left, thorax and mask. Periodically, and at least once/twice a year, the process is reviewed and if no critical change is requested, one run of media fill is performed. Herein as an example we briefly summarize the results of the media fill that we performed for our workhorse process: the MSC production. At the end of three runs of media fill (30 days of total time for MSC) the tryptic soy broth medium was always sterile at all stages of the process and conformed to the “growth promotion” test. Two hundred and three plates were used for the test: 18 SAS controls, 3 settle plates, 62 contact plates, and 120 contact plates for the personnel. No out-of-specification value for environmental and personnel microbiological monitoring was detected, as well as for microbiological monitoring on the personnel. All the representative samples of QCs and final products were sterile. There was no over-specification in the averages of the daily values under the laminar flow hood (Class A) within the clean room processing (Class B). All the results were collected within an official report.
Planning the resources and the budget for a cell factory
An important issue in planning a GMP-hospital-based facility is represented by its economical aspects. ATMP production has indeed, a significant economic impact even when manufactured inside a public hospital. All the public GMP facilities are still far away from defining an appropriate business model that starts from the definition of a correct plan but several interesting approaches are recently emerging. What we can definitely assume is that one of the main obstacles towards the appropriate use of ATMPs is the final costs of these products. A big challenge is to define a model to analyze and control the costs. The manufacturing centers have the terrific potential to answer the fundamental question: when promising therapies reach the later stage of clinical trial, what type of process issues may be encountered and how best can we address it in a cost-effective manner? Optimization of the manufacturing process and times, automation, application of a quality-by design approaches are the main tools that should be used.
The role of the Cell Factory in Italy and in Europe
Our Cell Factory, in addition to producing cell therapy products and perform QCs according to GMP in experimental clinical protocols, may take part to authorized multicenter studies for institutions that do not have GMP facilities. In particular, based on our previous experiences, the Cell Factory can support studies where a cell process is involved, as an example we can work with hematopoietic progenitor cells from cord blood expanded in vitro, dendritic cells with the aim of realizing anti-tumor vaccination, any kind of regulatory cell population isolation and expansion with immune suppress activity, or various lymphocyte populations (CD3, CD19, CD4, CD8, and CD25) for protocols supporting bone marrow transplantation; and endothelial progenitors from bone marrow for any type of tissue revascularization. In this regard, in Europe GMP is mandatory for cell manipulation that, even though “minimal” (eg, immunomagnetic cell selection) is used to provide cells that are not intended for the same essential function or functions in the recipient as in the donor (the so-called “non-homologous” use). An example is CD133+ cells for vascular heart coronary repair. In any case, as a service to accreditated cell processing laboratories inside bone marrow transplantation program not under GMP rules, the Cell Factory can also provide hematopoietic stem/progenitor cells (CD34+ and CD133+ cells) to treat leukemia, lymphoma, myeloma, and solid tumors.
One of the skills in which the Cell Factory is much stronger is the GMP production of MSCs from bone marrow, adipose tissue, and human cord blood, for various applications in hematology and regenerative medicine (running applications in neurology, nephrology, pneumology, dermatology, orthopedics, and hematology). In particular, we underline the peculiarity of being able to produce under GMP the cord blood mesenchymal stem cells that have already been the subject of several preclinical studies2,3 and that are now used as ATMP in innovative experimental clinical trials (ie, Eudract number 2011-001387-21). All the information regarding our activities are available at our website www.cellfactorymilano.com.
Our hospital has recently decided to further support its program in regenerative medicine and a new building was built to host the Cell Factory in new and larger areas including three class-B production rooms, new QC laboratories, and ancillary areas (storage room, archives, and offices). This decision was taken to satisfy the increasing number of requests due to the involvement of the Cell Factory into national and international multicentre studies. This is perfectly in compliance with the mission of the hospital that focuses specifically on regenerative medicine.
Past clinical programs
Table 2 lists the Phase I/II clinical protocols conducted to date by the Cell Factory in cooperation with clinical groups belonging to different specialties within or outside our hospital on a national basis.
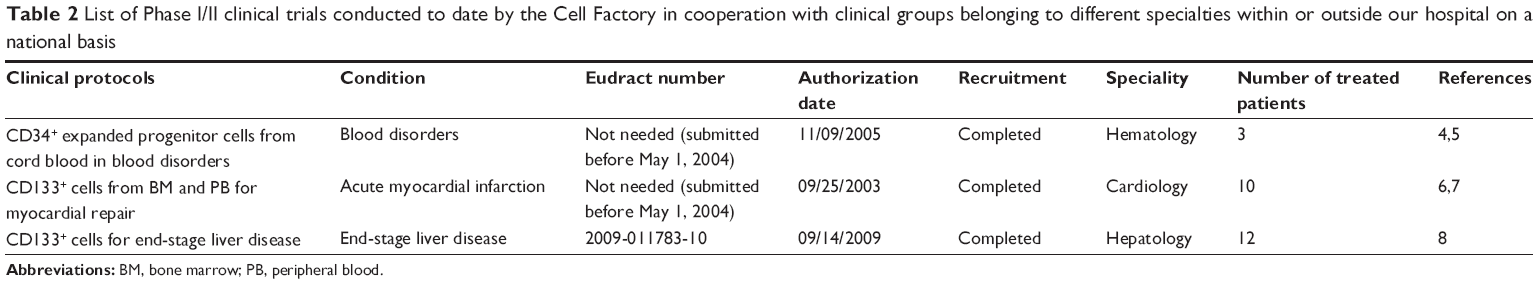 | Table 2 List of Phase I/II clinical trials conducted to date by the Cell Factory in cooperation with clinical groups belonging to different specialties within or outside our hospital on a national basis |
Clinical ongoing programs
Table 3 lists the Phase I/II clinical protocols conducted to date by the Cell Factory in cooperation with clinical groups belonging to different specialties within our hospital, or outside on a national and European basis.
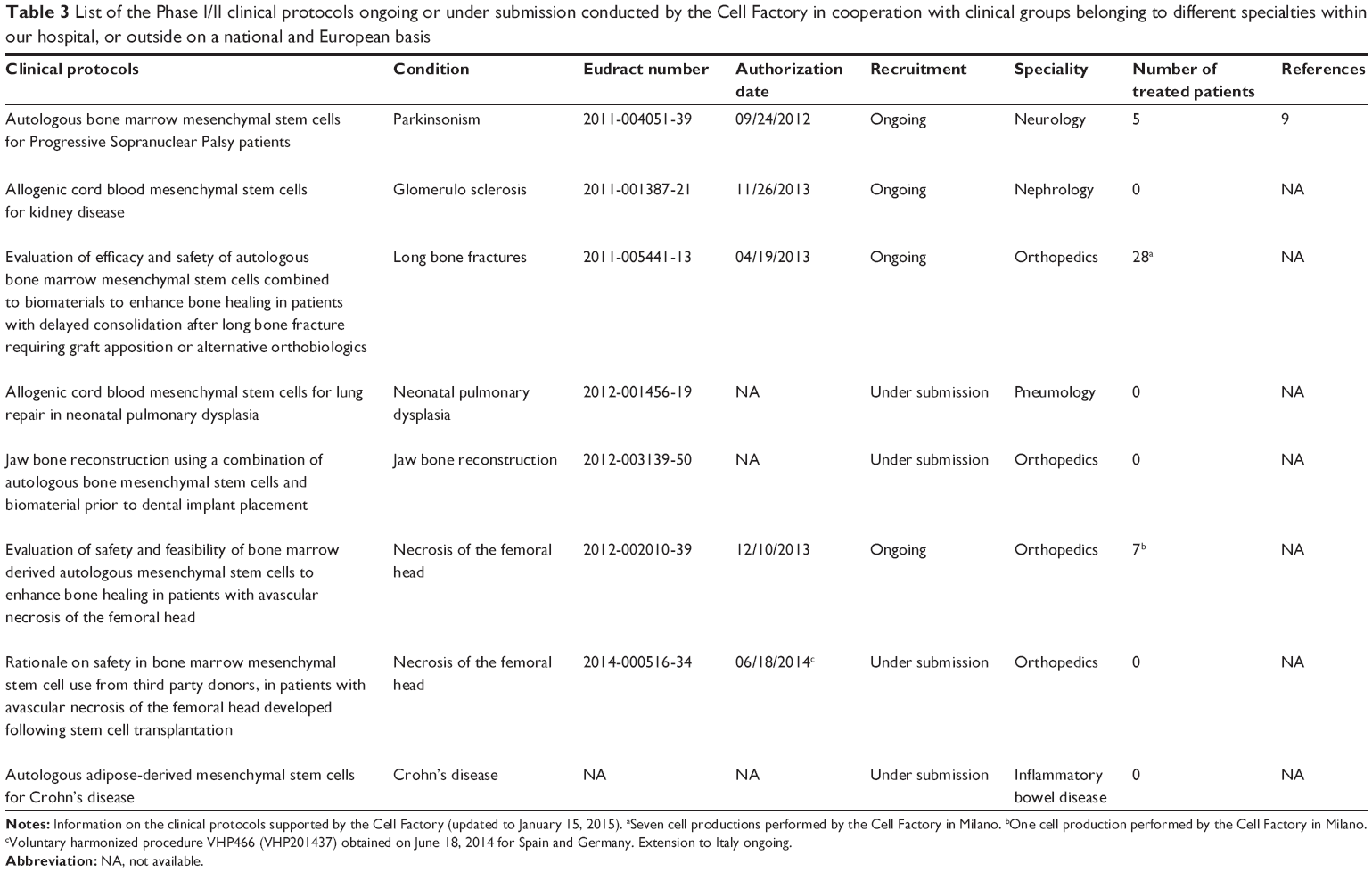 | Table 3 List of the Phase I/II clinical protocols ongoing or under submission conducted by the Cell Factory in cooperation with clinical groups belonging to different specialties within our hospital, or outside on a national and European basis |
Considerations and conclusions
First of all with this report we want to prove that making cell therapy in Italy in compliance with national and European rules, for the protection of the subjects treated with ATMPs, is feasible. All the herein reported steps are needed primarily to protect the cell product that has to be given to the patient who needs cell therapy. The safety of the patient is the first goal of a GMP project and this is why we follow these strict regulations.
As a final statement, we would like to underline that producing ATMP following GMP rules and using them in the context of Good Clinical Practice-compliant clinical trials is the only way to prove the safety and efficacy of novel cell-therapy based approaches. Results from Phase I to III studies obtained using reliable products and methodologies are urgently needed in order to understand both the limits and the advantages of these new therapies. Anyway, if you want to move translational research into the GMP model, we strongly believe that the concepts described in this paper are the first brick to set up a GMP facility for ATMP production. Sharing this kind of vision and the strategy to realize it may be an important help for those who want to start this GMP “journey”.
Acknowledgments
The authors would like to thank the patients and their families who continuously support our work, all the researchers working till now at our facility, the Cell Factory, for their support to this project. In particular, special mention must be made to all the colleagues who have contributed over the years to create this facility: Simona Lucchi, Viviana Lo Cicero, Gabriella Andriolo, Gabriella Spaltro, Francesca Chelli, Alessandra Giorgetti, Stefano Zangrossi, Barbara Baluce, Paolo Rebulla, and Girolamo Sirchia. In addition, authors would like to thank the management of Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico for their support, with a special regard to the administrative officers. This project was partially supported by funds from Fondazione Il Sangue, from Regione Lombardia, from Ministero della Salute Italiano and from European Union’s Sixth and Seventh Programme.
Disclosure
The authors report no conflicts of interest in this work.
References
European Commission. EudraLex – Volume 4 Good Manufacturing Practice (GMP) Guidelines. Brussels: European Commission. Available from: http://ec.europa.eu/health/documents/eudralex/vol-4/index_en.htm. Accessed January 15, 2015. | ||
Morigi M, Rota C, Montemurro T, et al. Life-sparing effect of human cord-blood mesenchymal stem cells in experimental acute kidney injury. Stem Cells. 2010;28:513–522. | ||
Zanier ER, Montinaro M, Vigano M, et al. Human umbilical cord blood mesenchymal stem cells protect mice after brain trauma. Crit Care Med. 2011;39:2501–2510. | ||
Lazzari L, Giordano R, Giorgiani G, et al. Transplantation of ex vivo expanded cord blood progenitor cells: first experience in two children affected by hemoglobinopathies. Blood. 2005;106:2187. | ||
Giordano R, Lazzari L, Giorgiani G, et al. Ex vivo expansion of cord-blood derived progenitor cells for allogeneic transplantation: a phase I–II clinical study. Bone Marrow Transp. 2006;37(S1):S246. | ||
Colombo A, Castellani M, Piccaluga E, et al. Myocardial blood flow and infarct size after CD133+ cell injection in large myocardial infarction with good recanalization and poor reperfusion: results from a randomized controlled trial. J Cardiovasc Med. 2011;12:239–248. | ||
Castellani M, Colombo A, Giordano R, et al. The role of PET with 13N-ammonia and 18F-FDG in the assessment of myocardial perfusion and metabolism in patients with recent AMI and intracoronary stem cell injection. J Nucl Med. 2010;51:1908–1916. | ||
Catani L, Lorenzini S, Giordano R, et al. CD133+ pluripotent stem cells for the treatment of chronic liver failure. Blood. 2010;116:1185. | ||
Giordano R, Canesi M, Isalberti M, et al. Autologous mesenchymal stem cell therapy for progressive supranuclear palsy: translation into a phase I controlled, randomized clinical study. J Transl Med. 2014;12:14. | ||
EudraLex: The rules governing medicinal products in the European Union. Volume 4. EU guidelines to good manufacturing practice: medicinal products for human and veterinary use. European Commission. Available from: http://ec.europa.eu/health/files/eudralex/vol-4/2008_11_25_gmp-an1_en.pdf. Accessed January 15, 2015. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.