Back to Journals » International Medical Case Reports Journal » Volume 15
Homozygous Insulin Promotor Gene Mutation Causing Permanent Neonatal Diabetes Mellitus and Childhood Onset Autoantibody Negative Diabetes in the Same Family
Authors Haris B, Mohammed I, Al-Khawaga S, Hussain K ![]()
Received 14 November 2021
Accepted for publication 12 January 2022
Published 1 February 2022 Volume 2022:15 Pages 35—41
DOI https://doi.org/10.2147/IMCRJ.S349424
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ronald Prineas
Basma Haris,1 Idris Mohammed,1 Sara Al-Khawaga,2 Khalid Hussain1
1Department of Pediatric Endocrinology, Sidra Medicine, Education City, Doha, Qatar; 2Department of Dermatology, Hamad General Hospital, Doha, Qatar
Correspondence: Khalid Hussain
Department of Pediatric Medicine, Sidra Medicine, Education City, OPC, C6-340 |PO Box 26999, Al Luqta Street, North Campus, Doha, Qatar
, Tel +974-4003-7608
, Email [email protected]
Purpose: To report a family with a homozygous INS promotor gene mutation causing permanent neonatal diabetes mellitus (PNDM) in one sibling and autoantibody negative childhood onset diabetes in another sibling.
Case Presentation: Patient 1 is a 12-year-old girl born at term with low birth weight to a consanguineous family, diagnosed with PNDM at 26 days of life. She presented with ketoacidosis and has a severe course of disease with high insulin requirement. Patient 2 is a 9-year-old girl born at term with normal weight, who presented with ketoacidosis at 2 years of age. Both subjects have negative type 1 autoantibodies. On genetic testing, a mutation in the promoter region of INS gene c.-331 C>G was found in homozygous state in both subjects and in a heterozygous state in parents.
Conclusion: Homozygous INS gene promotor mutations may present with either PNDM or later onset autoantibody negative diabetes in childhood. This suggests that homozygous INS gene promotor mutations show marked heterogeneity in clinical presentation within individuals in the same family. The pathophysiology of this is not well known but could be related to a number of factors, including the position of the variant, penetrance, other associated genetic defects, HLA etc. Premarital screening and genetic counselling is recommended for highly consanguineous families to reduce occurrence of such conditions.
Keywords: pediatric diabetes, neonatal diabetes mellitus, type 1b diabetes, INS mutation
Introduction
Neonatal diabetes mellitus is defined as hyperglycemia occurring in the first 6 months of life. It is a rare condition with a minimal incidence of 1 in 90,000 live births; however, variations have been reported in various ethnic groups.1 There are two main types of neonatal diabetes: permanent neonatal diabetes mellitus (PNDM) and transient neonatal diabetes mellitus (TNDM). The causes of PNDM vary around the world but the most common cause in the Western world is due to mutations in the genes (ABCC8/KCNJ11) coding for the SUR1 and KIR6.2 subunits of the potassium ATP channels, respectively.2 Insulin promotor gene (INS) mutations are a rare cause of PNDM.3 Both heterozygous and homozygous INS promotor gene mutations may lead to PNDM.
Heterozygous INS promotor gene mutations were first reported to cause neonatal diabetes in 2007 where 10 mutations in 16 probands with neonatal diabetes were reported.3 Subsequently, many more variants have been reported. The mechanism of diabetes in heterozygous mutations is due to improper folding of proinsulin, which leads to endoplasmic reticulum stress and beta cell apoptosis.4 In addition, homozygous INS promotor gene mutations were first reported in 2010 in 15 probands with neonatal diabetes.5 The mechanism of diabetes in homozygous INS mutations is thought to be due to the disruption of polyadenylation signal causing disturbed mRNA stability and lack of translation initiation signaling.5
Maturity-onset diabetes of the young (MODY) is an autosomal dominant disorder characterized by insufficient insulin secretion with autoantibody negativity. The MODY subtype 10 is caused by INS gene mutations and the mechanism of the diabetes is also due to improper folding of the proinsulin with intracellular accumulation of proinsulin and defective insulin biosynthesis.4
In this study, we report a family with two siblings having the same homozygous INS promotor gene mutation causing PNDM in one subject and autoantibody negative childhood onset diabetes in the other subject with different severity of phenotype.
Case Presentation
We present a consanguineous family of Qatari ethnicity. Patient 1 is now a 12-year-old girl born at term by caesarian section with a birth weight of 1.6kg. There was a history of gestational diabetes during the pregnancy but it was otherwise uneventful. At the age of 26 days she presented to the clinic with fatigue and investigations confirmed hyperglycemia with a blood glucose level of 33mmol/L and c-peptide of 0.29ng/mL. Based on ADA guidelines, she was classified as having neonatal diabetes mellitus since the age of onset of diabetes was less than 6 months. Treatment with insulin was started immediately and HbA1c improved. Thyroid peroxidase and celiac autoantibodies were negative. She is being treated with insulin Glargine 22 units daily and insulin Lispro 10 divided units daily. She has been on insulin treatment continuously for 12 years; however, she has had episodes of diabetic ketoacidosis needing hospitalization, which are mainly due to non-compliance to insulin dosage and diet restrictions as the child is an adolescent now. Current Hba1c is 12.1%.
Patient 2 is a 9-year-old girl born at term with birth weight of 2kg. She first presented to the clinic with dysglycaemia with blood glucose level of 17mmol/L at 2 years of age. She reported a history of polydipsia, polyuria, weight loss and fatigue. She is physically and developmentally within normal limits. Treatment with insulin was started immediately and she has been on insulin therapy for 7 years now. Autoantibody testing was performed to look for Glutamic Acid Decarboxylase 65 (GAD65), Insulin Autoantibody (IAA), Islet Antigen-2 Auto Antibody (IA-2A), and Zinc Transporter 8 (ZnT8A) and these were found to be negative; c-peptide was undetectable <0.08 ng/mL. Thyroid peroxidase as well as celiac autoantibody screening was negative. She is being treated with insulin Glargine 8 units daily and insulin Lispro 5 divided units daily. Current Hba1c is 8.2%. Liver function test and other metabolites were within normal limits. As per ADA guidelines, idiopathic type 1 diabetes is a rare form of type 1 diabetes with permanent insulinopenia without autoimmunity. MODY is classified as onset of DM at an early age due to mutations in 13 genes inherited in autosomal dominant pattern. Since these 2 entities are clinically not distinguishable, genetic testing was performed to accurately classify the patient.
Genetic Analysis
Genomic DNA was extracted from peripheral blood of all individuals and whole exome sequencing (WES) was performed on Illumina HiSeq platform using a 150-base paired-end single-index-read format. Genomic variants in genes already known to be implicated in MODY and NDM (Table 1) were extracted. The analysis revealed a homozygous c.-331C > G in INS gene (NM_001185098) in both patients whereas the parents were heterozygous carriers of this mutation. To confirm the variant identified by WES, the genomic DNA was used for polymerase chain reaction (PCR using Sanger sequencing. Specific Primers (forward: 5’ GCTGACGACCAAGGAGATCT 3’ and reverse: 5’ TGCTTGATGGCCTCTTCTGA 3’, product size: 226bp) were designed using primer3 software (http://primer3.ut.ee/). For PCR, a total of 25ul reaction (12.5uL of Amplitaq Gold master mix, 2uL of DNA, 2uL primer mix and 8.5uL of nuclease free water) was amplified with PCR conditions; 5 min, 95°C initial denaturation, 35 cycles of 30 sec, 95°C denaturation, 30 sec, 55°C annealing, 30 sec, 72°C elongation and 10 min, 72°C for the final elongation. The product was purified with PCR purification kit (Qiagen), followed by cycle sequencing PCR using a BigDye Terminator Sequencing v.3.1 (Thermo Fisher Scientific, Inc.,) and sequenced using a 96-capillary ABI 3500xl automated genetic analyzer. The results were analyzed using SeqScape (Applied Biosystems; Thermo Fisher Scientific, USA). Sanger sequencing verified the c.-331C>G transversion homozygous point mutation in both patients and heterozygous state in both parents. The pedigree and Sanger sequencing electropherogram results of the whole family are shown in Figures 1 and 2, respectively.
 |
Table 1 Salient Clinical Features Observed |
 |
Figure 1 Pictorial representation of the pedigree of the family. |
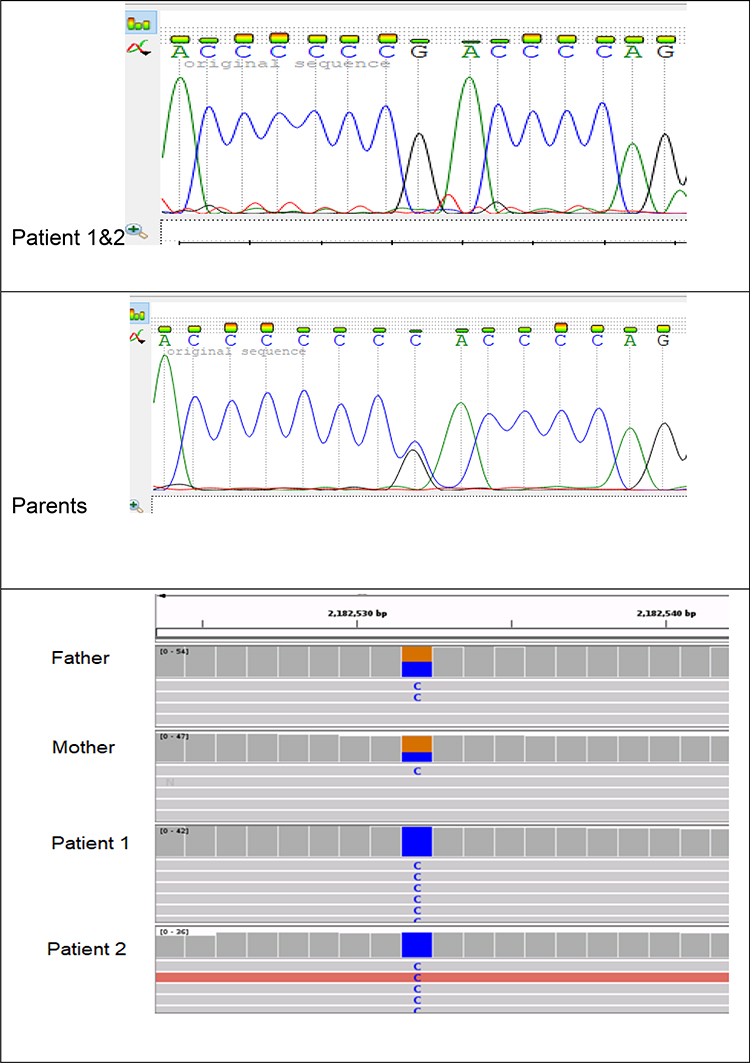 |
Figure 2 Sanger sequencing and IGV analysis of the family. |
Discussion
Mutations in the INS gene promotor can result in defective production of insulin and present as DM. Both homozygous and heterozygous mutations have been described in INS gene with different pathophysiology.6 Homozygous mutations are primarily loss of function mutations resulting in absent insulin bio-synthesis by affecting the transcription or translation of the protein.6 The commonly observed phenotype in such individuals is early onset PNMD in the neonatal period, presenting with severely low birth weight, insulin deficiency and diabetic ketoacidosis soon after birth.7 Homozygous mutations are reported to account for about 20% of INS mutations.6
Heterozygous mutations in INS gene are reported to cause autosomal dominant insulin dependent diabetes in older children and adults.7 Although there is 80% reduction in insulin secretion, the remaining is enough to maintain blood glucose levels initially and over the course of time they develop cell stress and beta cell dysfunction which may be the cause for later age of diagnosis in heterozygous mutation carriers.6
Here, we present a family with a complex pedigree. The parents of the subjects are heterozygous for the variant; however, they have not developed diabetes yet. Patient 1 presented with severely low birthweight and all features consistent with PNDM. However, patient 2 presented at 2 years of age with a relatively milder version of the disease even though she has the same homozygous variant. This is evident from the course of the disease observed in both subjects, including serial HBA1c measurements and daily insulin requirement. The trigger for the different ages of onset of diabetes is not well understood. However there have been previous reports that there is great variability in the phenotype observed in INS mutations and variable periods of remission have also been observed.8 Table 2 describes all the previous reports with the homozygous INS promoter mutation and their phenotype. These studies confirm that the severity of the phenotype observed in homozygous INS mutations varies from severe intrauterine insulin deficiency to milder forms that resemble type 2 diabetes or MODY.13
 |
Table 2 Describes All Previous Reports with the Homozygous INS Promoter Mutation and Their Phenotype |
Our patient 2 is the first patient to be reported with promoter INS mutation causing diabetes with onset after the neonatal period and all 4 autoantibodies negative. The father and mother aged 45 years and 42 years, respectively, are heterozygous for the variant but have not developed diabetes yet. Our study supports the idea that there is clinical heterogeneity in the severity and expression of INS mutation even within families with all the similar characteristics. Although consanguinity is an important factor, the exact mechanism of this variability is not clearly understood. HLA loci reported to increase susceptibility to diabetes in Qatari children such as HLA class II DQB1 and DQA1 genes14 could be playing a role in the pathophysiology in addition to the INS mutation. Another reason could be the difference in penetrance of mutation affecting each individuals. Motzo et al15 reported that INS predisposing genotype was less frequent in high-risk HLA genotype positive patients than intermediate risk and low risk groups and proposed an interaction between the proteins of the HLA and INS alleles. However, this has not been studied extensively.
Genetic testing should be considered in all children with onset of diabetes before 6 months of age and in all siblings of such subjects, especially if the parents are of a consanguineous marriage. Genetic testing is also useful in all cases where all autoantibodies are negative and do not display features of type 2 diabetes to accurately classify into different types of monogenic diabetes or idiopathic DM. Accurate genetic classification can help to change the management of diabetes in some subjects. Mutations in certain genes, for example NDM due to ABCC8/KCNJ11, can be treated with oral sulfonylureas only.16 MODY due to GCK mutations sometimes do not need any treatment at all.17
As a preventative measure in countries with high degree of consanguinity like Qatar, premarital screening can be offered to all individuals marrying within their family to reduce the occurrence of such debilitating conditions. Genetic screening can be offered to all siblings of subjects with homozygous mutations as followed over time.
Ethics Statement
The protocol for this research project has been approved by the ‘Institutional Review Board (IRB) for the protection of human subjects’ in Sidra Medicine, Qatar and it conforms to the provisions of the Declaration of Helsinki. Informed consent to take part in study and publish findings was obtained from the subject(s) and/or guardian(s) prior to study commencement.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Beltrand J, Busiah K, Vaivre-Douret L, et al. Neonatal diabetes mellitus. Front Pediatr. 2020;8:540718. doi:10.3389/fped.2020.540718
2. Gloyn AL, Siddiqui J, Ellard S. Mutations in the genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) in diabetes mellitus and hyperinsulinism. Hum Mutat. 2006;27(3):220–231. doi:10.1002/humu.20292
3. Støy J, Edghill EL, Flanagan SE, et al.; Neonatal Diabetes International Collaborative Group. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci U S A. 2007;104(38):15040–15044. doi:10.1073/pnas.0707291104
4. Polak M, Dechaume A, Cavé H, et al.; (Neonatal Diabetes) Study Group. Heterozygous missense mutations in the insulin gene are linked to permanent diabetes appearing in the neonatal period or in early infancy: a report from the French ND (Neonatal Diabetes) Study Group. Diabetes. 2008;57(4):1115–1119. doi:10.2337/db07-1358
5. Garin I, Edghill EL, Akerman I, et al. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc Natl Acad Sci U S A. 2010;107(7):3105–3110. doi:10.1073/pnas.0910533107
6. Liu M, Sun J, Cui J, et al. INS-gene mutations: from genetics and beta cell biology to clinical disease. Mol Aspects Med. 2015;42:3–18. doi:10.1016/j.mam.2014.12.001
7. Raile K, O’Connell M, Galler A, et al. Diabetes caused by insulin gene (INS) deletion: clinical characteristics of homozygous and heterozygous individuals. Eur J Endocrinol. 2011;165(2):255–260. doi:10.1530/EJE-11-0208
8. Demiral M, Demirbilek H, Çelik K, Okur N, Hussain K, Ozbek MN. Neonatal diabetes due to homozygous INS gene promoter mutations: highly variable phenotype, remission and early relapse during the first 3 years of life. Pediatr Diabetes. 2020;21(7):1169–1175. doi:10.1111/pedi.13079
9. Shaikh AA, Shirah B, Alzelaye S. A homozygous mutation in the insulin gene (INS) causing autosomal recessive neonatal diabetes in Saudi families. Ann Pediatr Endocrinol Metab. 2020;25(1):42–45. doi:10.6065/apem.2020.25.1.42
10. Deeb A, Habeb A, Kaplan W, et al. Genetic characteristics, clinical spectrum, and incidence of neonatal diabetes in the Emirate of Abu Dhabi, United Arab Emirates. Am J Med Genet A. 2016;170(3):602–609. doi:10.1002/ajmg.a.37419
11. Demirbilek H, Arya VB, Ozbek MN, et al. Clinical characteristics and molecular genetic analysis of 22 patients with neonatal diabetes from the South-Eastern region of Turkey: predominance of non-KATP channel mutations. Eur J Endocrinol. 2015;172(6):697–705. doi:10.1530/EJE-14-0852
12. Yildiz M, Akcay T, Aydin B, et al. Emergence of insulin resistance following empirical glibenclamide therapy: a case report of neonatal diabetes with a recessive INS gene mutation. J Pediatr Endocrinol Metab. 2018;31(3):345–348. doi:10.1515/jpem-2017-0325
13. Glaser B. Insulin mutations in diabetes: the clinical spectrum. Diabetes. 2008;57(4):799–800. doi:10.2337/db08-0116
14. Haris B, Ahmed I, Syed N, et al. Clinical features, epidemiology, autoantibody status, HLA haplotypes and genetic mechanisms of type 1 diabetes mellitus among children in Qatar. Sci Rep. 2021;11(1):18887. doi:10.1038/s41598-021-98460-4
15. Motzo C, Contu D, Cordell HJ, et al. Heterogeneity in the magnitude of the insulin gene effect on HLA risk in type 1 diabetes. Diabetes. 2004;53(12):3286–3291. doi:10.2337/diabetes.53.12.3286
16. Codner E, Flanagan SE, Ugarte F, et al. Sulfonylurea treatment in young children with neonatal diabetes: dealing with hyperglycemia, hypoglycemia, and sick days. Diabetes Care. 2007;30(5):e28–9. doi:10.2337/dc06-2134
17. Urakami T. Maturity-onset diabetes of the young (MODY): current perspectives on diagnosis and treatment. Diabetes Metab Syndr Obes. 2019;12:1047–1056. doi:10.2147/DMSO.S179793
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.