Back to Journals » International Medical Case Reports Journal » Volume 18
Homozygous α-Spectrin (SPTA1) Variant Causing Persistent Hereditary Pyropoikilocytosis in a Newborn: A Case Report and Literature Review
Authors Sayed J ![]() , Alabdulhadi AS
, Alabdulhadi AS ![]() , Alzahrani WA, Joueidi F
, Alzahrani WA, Joueidi F ![]() , Alzahrani GA
, Alzahrani GA ![]() , Sayed AG, Ebid GT
, Sayed AG, Ebid GT ![]()
Received 19 May 2025
Accepted for publication 27 September 2025
Published 10 October 2025 Volume 2025:18 Pages 1303—1309
DOI https://doi.org/10.2147/IMCRJ.S483359
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Thomas E Hutson
Jamal Sayed,1 Alanoud Sulaiman Alabdulhadi,2 Waheed Abdullah Alzahrani,1 Faisal Joueidi,2 Ghaida Ali Alzahrani,3 Ahmed Gamal Sayed,2 Gamal T Ebid1
1Department of Pediatrics, Security Forces Hospital Makkah, (SFHM), Makkah, Saudi Arabia; 2College of Medicine, Alfaisal University, Riyadh, 11533, Saudi Arabia; 3Ibn Sina National College for Medical Studies, Jeddah, Saudi Arabia
Correspondence: Jamal Sayed, Email [email protected]
Abstract: Hereditary pyropoikilocytosis is an inherited, rare form of severe hemolytic anemia that is associated with a disordered erythrocyte membrane. Such a disordered membrane is due to the quantitative and qualitative α-spectrin defects that associate with homozygous or doubly heterozygous mutations in the SPTA1 gene. Characterized by marked poikilocytosis, anisocytosis, and thermal sensitivity of erythrocytes, often leading to severe hemolytic anemia and neonatal jaundice. We report the case of a full-term newborn admitted in the first day of life to the neonatal intensive care unit with significant jaundice and anemia. Peripheral blood smear revealed severe anemia with pronounced anisopoikilocytosis and moderate elliptocytosis, suggestive of a hereditary RBC membrane disorder. The whole exome sequencing (WES) did identify a SPTA1 gene homozygous likely pathogenic missense variant, p. (Leu260Pro) confirming the diagnosis of hereditary pyropoikilocytosis. The patient received intensive phototherapy and a red blood cell transfusion during hospitalization. Over an 18-month follow-up period, the infant remained clinically stable with no further transfusion requirements. However, the features consistent with HPP tended to be persistent during the follow‑up, highlighting the chronic nature of the disorder in this case. Furthermore, it underscores the importance of considering rare hereditary causes of hemolytic anemia in neonates presenting with early-onset jaundice and anemia. It highlights the diagnostic value of genetic testing in confirming SPTA1-related disorders.
Keywords: hereditary anemia, neonatal pyropoikilocytosis, neonatal red cell membrane disorder
Introduction
Hereditary pyropoikilocytosis (HPP) is a severe red blood cell (RBC) membrane disorder that is classified as a subset of hereditary elliptocytosis. It is characterized by striking morphological abnormalities, including marked anisopoikilocytosis, microspherocytosis, red cell fragmentation, and thermal sensitivity, often resulting in chronic hemolytic anemia.1 In neonates, it may present with severe anemia and jaundice within the first days of life. This early presentation often necessitates prompt intervention, but distinguishing HPP from other hemolytic conditions can be difficult given the rarity and the overlapping features of this disease. As a condition, inheritance is mainly in an autosomal recessive pattern primarily resulting from SPTA1 gene mutations, which encodes α-spectrin. That is crucial for the structural component of the RBC cytoskeleton. These mutations do impair the self-association and tetramer formation of spectrin, thus weakening the RBC membrane and increasing the susceptibility to hemolysis. The manifestation of the disorder can be either homozygous or compound heterozygous forms, thus both the qualitative and quantitative defects of spectrin can be involved, playing a significant role in the physiopathology of HPP.2 Although most cases present during infancy or early childhood, delayed diagnosis into adolescence or adulthood is not uncommon due to phenotypic variability. Up to one-third of the patients have been found to have a family member with similar erythrocyte morphology or a known diagnosis.3 Diagnosing HPP can be challenging, especially in a resource-limited setting, due to the rarity and the overlapping features with other hemolytic anemias. Conventional diagnostic approaches often fall short in identifying the underlying genetic etiology. Although confirming HPP requires ektacytometry and/or genetic analysis, initial clinical suspicion is often based on characteristic peripheral blood smear findings in the setting of hemolytic anemia. Molecular diagnostics advances, particularly whole-exome sequencing, have significantly improved the detection of mutations, allowing for better diagnostic accuracy. Expanded genetic screening is increasingly recognized as essential for guiding early diagnosis and tailored management.4 The burden of rare hereditary anemias is especially pronounced in developing regions, where diagnostic limitations and lack of awareness may lead to underdiagnosis and suboptimal care.5 Early recognition of characteristic clinical and hematological findings—such as fragmented RBCs and significant anisopoikilocytosis on peripheral smear—is critical for timely intervention.6
Our case, together with a focused review of the literature on the identified SPTA1 gene mutation, contributes to the understanding of hereditary pyropoikilocytosis (HPP) by presenting a rare neonatal presentation confirmed through genetic analysis. This highlights the importance of maintaining a high index of clinical suspicion and utilizing advanced diagnostic approaches in vulnerable pediatric populations. Given the rarity of persistent forms of HPP, this report further underscores the value of integrating morphological, clinical, and genetic data to achieve an accurate and timely diagnosis.
Case Report
We present the case of a female neonate born at 37 weeks’ gestation via vacuum-assisted delivery due to fetal heart rate decelerations. She was vigorous at birth, with Apgar scores of 7 at one minute and 8 at five minutes.
At birth, her weight was 2.3 kg, which placed her below the 10th percentile for her gestational age on standard newborn growth charts. Her head circumference measured 34 cm, which is at the 50th percentile, and her length was 48 cm, also at the 50th percentile. These findings are consistent with asymmetrical intrauterine growth restriction. She is the first child from a consanguineous marriage, and the pregnancy was uneventful. The mother has a history of hereditary anemia, which has been present since she was two months old. This condition has not been thoroughly investigated, and a definitive diagnosis has not been established. The infant was admitted to the neonatal intensive care unit at 8 hours of age due to neonatal jaundice and anemia. Her initial hemoglobin level at birth was 8.9 g/dL (reference range 14–20 g/dL), indicating significant anemia. The reticulocyte count was 6.8% (reference range 2.0–6.0%), consistent with a compensatory marrow response. At 8 hours of age, her total serum bilirubin was 8.4 mg/dL (143.6 µmol/L), exceeding the expected physiologic range <5 mg/dL (85.5 µmol/L) in the first 24 hours of life and suggestive of early pathological hyperbilirubinemia. Both the mother and the baby had blood type O positive, making hemolysis due to ABO incompatibility unlikely. Upon physical examination, the patient appeared alert, was active, and showed mild signs of pallor and jaundice. Her vital signs were normal, and the systemic examination revealed no significant abnormalities.
Blood tests indicated high levels of hemolysis markers, while the direct antiglobulin test result was negative. Enzymatic assays for glucose-6-phosphate dehydrogenase and pyruvate kinase were normal. The analysis of the blood film and peripheral blood smear indicates severe anemia, characterized by unusual morphological changes in the red blood cells (RBCs). The findings include abnormal red cell morphology, microspherocytosis, anisopoikilocytosis, schistocytosis, and a moderate degree of elliptocytosis. Additionally, polychromasia is present. In addition to the characteristic morphological abnormalities observed in the peripheral blood smear [Figure 1], the complete blood count (CBC) showed findings consistent with chronic hemolysis. The patient exhibited significant anemia along with an elevated reticulocyte count, indicating a robust bone marrow response. Notably, there was microcytosis, as reflected by a low mean corpuscular volume (MCV). This level of microcytosis is commonly seen in hereditary pyropoikilocytosis (HPP) and indicates the pronounced red blood cell fragmentation and membrane instability characteristic of the disorder. These hematological abnormalities, summarized in [Table 1], support the diagnosis of HPP and correlate with the clinical severity observed in this case. Further evidence supporting the diagnosis was obtained from the red blood cell (RBC) histogram generated by the automated hematology analyzer. The histogram displayed a bimodal distribution, with one peak representing a population of microcytic cells and the other indicating fragmented or abnormally shaped erythrocytes. This pattern is highly indicative of hereditary pyropoikilocytosis (HPP) and is consistent with the significant anisopoikilocytosis and microcytosis observed in the peripheral blood smear.
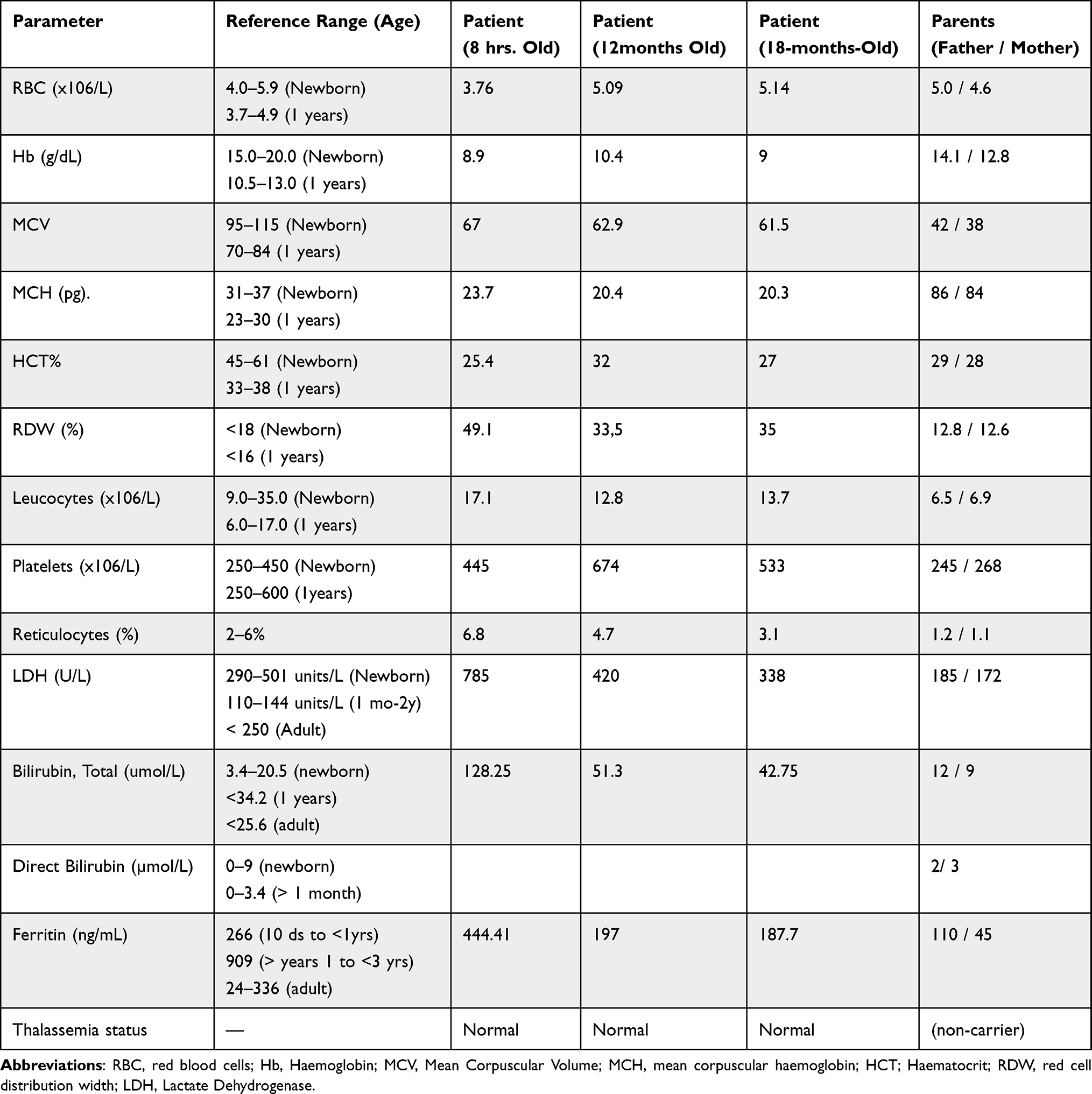 |
Table 1 Hematological and Biochemical Findings of the Index Patient and Parents |
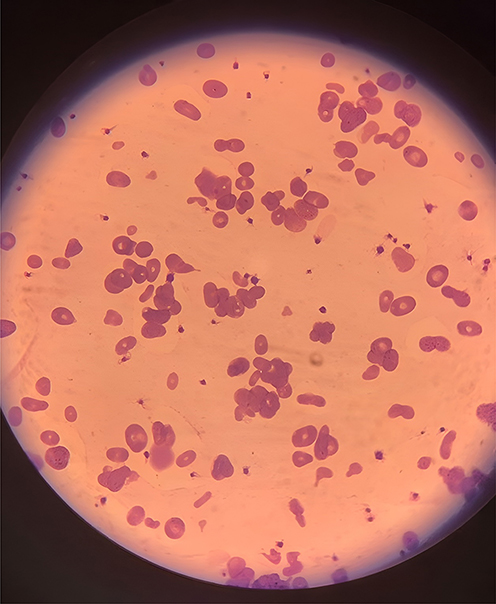 |
Figure 1 Peripheral blood smear, Peripheral blood film demonstrated marked red blood cell morphological abnormalities, including microspherocytosis, anisopoikilocytosis, schistocytosis, and a moderate degree of elliptocytosis. |
Haemoglobin Electrophoresis Was Performed Using Capillarys (Sebia)
A moving band (FM) in the zone of Haemoglobin Bart is present at a level of 1.2%. The clinical significance of this fraction is currently uncertain [Figure 2].
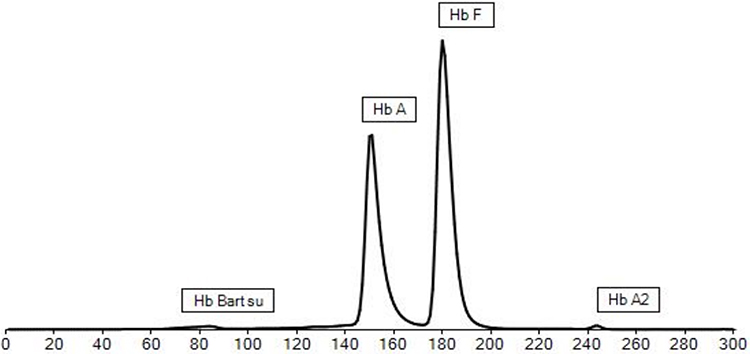 |
Figure 2 Hemoglobin Electrophoresis (Capillary), A moving band (FM) in the zone of Haemoglobin Bart is present at a level of 1.2%. The clinical significance of this fraction is currently uncertain. |
The patient was treated in the neonatal intensive care unit, where he received intensive phototherapy and a blood transfusion. After being discharged with timely follow-up, his haemoglobin level remained stable He did not need further management over the next 18 months of follow-up. [Table 1]. Whole exome sequencing was performed using CentoXome® MOx Solo at CENTOGENE Laboratory, a commercial laboratory. Sequencing was conducted on an Illumina platform with at least 20× coverage for >98% of targeted bases. Analysis followed the laboratory’s validated bioinformatics pipeline, and relevant variants were confirmed by Sanger sequencing. Molecular analysis identified a homozygous missense variant in the SPTA1 gene: c.779T>C (p. Leu260Pro), which has been previously reported in individuals with hereditary elliptocytosis (HE) and hereditary pyropoikilocytosis (HPP). According to HGMD Professional 2022., this variant has previously been described as disease-causing for Elliptocytosis by Sahr et al, 1989 (PMID: 2794061), Glele-Kakai et al, 1996 (PMID: 8857939), Harper et al, 2013 (PMID: 23974198). ClinVar reports this variant with an interpretation of pathogenic or likely pathogenic (Variation ID: 12844). It is classified as likely pathogenic (class 2) according to ACMG guidelines. The variant is considered rare and very low frequency in the population (0.02% in gnomAD). Both parents were found to be heterozygous carriers of this variant. The pattern is consistent with autosomal recessive inheritance, indicating a hereditary pyropoikilocytosis. The patient was treated in the neonatal intensive care unit, where he received intensive phototherapy and a blood transfusion. After being discharged with timely follow-up, his hemoglobin level remained stable He did not need further management over the next 18 months of follow-up.
Discussion
HPP is a clinical and genetic red cell membranopathy that was previously described by Zarkowsky et al in 1975 in two patients as a severely rare form of congenital hemolytic anemia characterized typically by a striking morphological abnormality in red blood cells (micro-spherocytosis, poikilocytosis), a moderate degree of elliptocytosis, and a low threshold for heat-induced red cell lysis and fragmentation at a temperature between 46–49c.7 The pathophysiology of HPP is now well established and is primarily related to mutations in SPTA1 and SPTB, which impair spectrin self-association and membrane stability.2 In human disease, we have found that such mutations have been associated with membranopathies, HPP in particular.2 α-spectrin forms heterodimers with β-spectrin and protein 4.1R, maintaining RBC stability.2 In the red blood cell (RBC), membrane cytoskeleton a key component is the spectrin, which is composed of α-spectrin (SPTA1) and β-spectrin (SPTB) subunits. In erythrocytes, spectrin forms tetramers through the association of α- and β-spectrin heterodimers, creating the principal structural unit that preserves cell shape and deformability. Mutations in these genes weaken the membrane stability, leading to poikilocytosis, fragmentation, and shortened RBC survival.8,9 Frequently, HPP is a transient condition, presenting with hemolytic anemia in the first months of life and often evolving into asymptomatic elliptocytosis by the end of the first year. The development of chronic persistent hemolytic anemia has been rarely reported in a few instances, as described in this manuscript. HPP typically follows an autosomal recessive pattern, unlike hereditary elliptocytosis (HE), which is usually inherited in a dominant pattern.10 Villalobos et al reported a 13-day-old newborn who presented with severe hemolytic anemia and neonatal jaundice. That needed a DNA sequence test that showed a double heterozygous mutation for mutant α-spectrin SPTA1 involving the Arg28 His variant and homozygous αLELY polymorphism, which was suggestive of HPP. The patient accordingly did require a blood transfusion and subsequently improved after two years.2 It has been reported that HPP and HE phenotypes can coexist, thus suggesting variability in the clinical expression of spectrin defects.11 Several families with spectrin mutations have shown both HPP and HE phenotypes, supporting this overlap.11 Five infants were reported by Prchal et al, who were affected by pyropoikilocytosis based on the morphological criteria and the heat-induced fragmentation.10 In two of these cases, features of elliptocytosis became evident by one year of age, highlighting the evolving nature of red cell morphology in infancy and the diagnostic overlap between HPP and HE. In this case, the diagnostic approach followed a structured hierarchy guided by the clinical presentation, laboratory findings, and availability of diagnostic tests. The Initial evaluation with routine hematological parameters and peripheral smear morphology suggested a red blood cell membrane disorder. Although osmotic gradient ektacytometry is the gold standard for confirming hereditary elliptocytosis and related conditions, it was not available in our setting. Given the severity of presentation, the inconclusive nature of preliminary findings, and the importance of establishing a definitive diagnosis for management and family counseling, we proceeded directly to whole exome sequencing (WES), particularly in our inconclusive case. Such a genetic approach does provide a comprehensive tool for clinical assessment that enables a simultaneous evaluation of multiple candidate genes in one single analysis. Moreover, given the complexity of the presentation and the practical limitations of available resources, the WES was selected over other molecular methods, allowing for a thorough and targeted genetic evaluation. A similar variant has been reported in a heterozygous state in Case 39 of the study by Mansour-Hendili et al, where the individual also exhibited an HPP phenotype. Compared with our patient, who is homozygous for this variant, the previously described heterozygous case demonstrated a milder clinical course. This observation supports the hypothesis that zygosity may influence phenotypic severity, although additional studies are needed to confirm this relationship.12 In instances where the genetic findings may help predict the persistence of anemia beyond infancy, it is essential to have an ongoing clinical follow-up with complete blood count, reticulocyte count, and hemolysis parameters for assessing disease severity and progression. Hence genetic approaches alone cannot determine the hematological progression, as this is implied by given hematological investigations. As they do offer clinical insights into the condition of the patient overall. Hence HPP management can be complex as HPP can strictly affect the focus. In the clinical care there are multiple points that should be relied on. This ranges from the supportive care of symptoms and the implicated hemolytic complications. Hence includes blood transfusion, exchange transfusion, phototherapy, and splenectomy.6 The Biallelic mutations may imply the need for a long-term transfusion support as it is a cause of chronic anemia. Splenectomy is partially effective in reducing hemolysis in some cases of severe HPP, though responses vary and anticoagulation may be required postoperatively.9,11 Previous case reports describe variable outcomes after splenectomy.11 Our case highlights the importance of early detection of SPTA1 and correlating genetic results with the clinical course to better predict prognosis in HPP
Conclusion
As a rare red cell membranopathy, hereditary pyropoikilocytosis may present in the neonatal period with severe hemolysis. Our case expands the spectrum of reported SPTA1 variants associated with HPP and emphasizes the molecular testing role when such conventional diagnostic tools are unavailable or inconclusive. For a proper diagnosis to be reached, genetic testing can be of aid in clarifying the diagnosis and supporting the development of an individualized management plan based on the diagnosis. Thus, families affected can have their counseling addressed appropriately, as a continued follow-up is essential in certain cases. The clinical course may evolve, ranging from a transient hemolysis in infancy to persistent anemia requiring long-term support.
Ethics Approval
This case report received approval from the Institutional Review Board (IRB) at Security Forces Hospital in Makkah. The research was conducted in line with the principles outlined in the Declaration of Helsinki and its subsequent amendments.
Informed Consent Statement
Written informed consent for publication was obtained from the patient’s parent for publication of the case details, including the history, physical findings, laboratory reports, and images.
Acknowledgment
There was no funding source for this paper.
Funding
This research received no external funding.
Disclosure
The author(s) report no conflicts of interest in this work.
References
1. Floyd PB, Gallagher PG, Valentino LA, Davis M, Marchesi SL, Forget BG. Heterogeneity of the molecular basis of hereditary pyropoikilocytosis and hereditary elliptocytosis associated with increased levels of the spectrin alpha I/74-kilodalton tryptic peptide. Blood. 1991;78(5):1364–1372. doi:10.1182/blood.V78.5.1364.1364
2. Sánchez Villalobos M, Salido Fiérrez E, Martínez Nieto J, et al. Case report: α-spectrin mutation associated with αLELY polymorphism responsible for hereditary pyropoikilocytosis. Hematol Rep. 2022;14(4):300–304. doi:10.3390/hematolrep14040043
3. Kim SJ, Song J, Reading NS, et al. Novel mechanism of hereditary pyropoikilocytosis phenotype due to co-inheritance of β globin and α spectrin mutations. Am J Hematol. 2021;96(5):E150–E154. doi:10.1002/ajh.26121
4. Costa DB, Lozovatsky L, Gallagher PG, Forget BG. A novel splicing mutation of the alpha-spectrin gene in the original hereditary pyropoikilocytosis kindred. Blood. 2005;106(13):4367–4369. doi:10.1182/blood-2005-05-1813
5. Kalfa TA. Diagnosis and clinical management of red cell membrane disorders. Hematology Am Soc Hematol Educ Program. 2021;2021(1):331–340. doi:10.1182/hematology.2021000265
6. Mallouh A, Sa’di AR, Ahmad MS, Salamah M. Hereditary pyropoikilocytosis: report of two cases from Saudi Arabia. Am J Med Genet. 1984;18(3):413–417. doi:10.1002/ajmg.1320180309
7. Wang X, Liu A, Lu Y, Hu Q. Novel compound heterozygous mutations in the SPTA1 gene, causing hereditary spherocytosis in a neonate with Coombs-negative hemolytic jaundice. Mol Med Rep. 2019;19(4):2801–2807. doi:10.3892/mmr.2019.9947
8. Anil More T, Kedar P. Unravelling the genetic and phenotypic heterogeneity of SPTA1 gene variants in hereditary elliptocytosis and hereditary pyropoikilocytosis patients using next-generation sequencing. Gene. 2022;843:146796. doi:10.1016/j.gene.2022.146796
9. Prchal JT, Castleberry RP, Parmley RT, Crist WM, Malluh A. Hereditary pyropoikilocytosis and elliptocytosis: clinical, laboratory, and ultrastructural features in infants and children. Pediatr Res. 1982;16(6):484–489. doi:10.1203/00006450-198206000-00017
10. Brancamp R, Hughes CE, Dar A, Polic A, Zuckerwise LC, Booth GS. Homozygous SPTA1-associated hereditary pyropoikilocytosis presenting as hydrops fetalis. Transfusion. 2024;64(1):189–193. doi:10.1111/trf.17617
11. Schuster F, Graubner U, Schön C, et al. Hereditary pyropoikilocytosis in a German patient. Pediatr Res. 1999;45:761. doi:10.1203/00006450-199905010-00140
12. Mansour-Hendili L, Aissat A, Badaoui B, et al. Exome sequencing for diagnosis of congenital hemolytic anemia. Orphanet J Rare Dis. 2020;15(1):180. doi:10.1186/s13023-020-01425-5
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.