Back to Archived Journals » Neurobehavioral HIV Medicine » Volume 7
HIV-associated progressive multifocal leukoencephalopathy: current perspectives
Authors Iannetta M, Zingaropoli MA, D'Abramo A, Oliva A, Mastroianni CM, Vullo V, Ciardi MR
Received 7 March 2016
Accepted for publication 2 June 2016
Published 8 August 2016 Volume 2016:7 Pages 43—52
DOI https://doi.org/10.2147/NBHIV.S107941
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Stephen Ferrando
Marco Iannetta, Maria Antonella Zingaropoli, Alessandra D’Abramo, Alessandra Oliva, Claudio Maria Mastroianni, Vincenzo Vullo, Maria Rosa Ciardi
Department of Public Health and Infectious Diseases, Sapienza University, Rome, Italy
Abstract: Progressive multifocal leukoencephalopathy (PML) is a demyelinating disease of the central nervous system, caused by the polyomavirus JC and occurring almost exclusively in the context of severe immune depression. AIDS represents the most common predisposing condition for PML development. Antiretroviral treatment has reduced PML incidence in HIV-infected subjects, but the disease remains a severe and life-threatening complication of AIDS, considering thus far the lack of an effective anti-JC virus (JCV) direct-acting antiviral drug. In the last decade, the use of monoclonal antibodies for treating immune-based diseases evidenced new predisposing conditions for PML development, promoting a renewed interest in PML pathogenesis. In this article, we review the current knowledge on JCV epidemiology and AIDS-associated PML incidence, JCV viral cycle, pathogenesis, and the interplay with HIV infection. We give an updated overview of diagnostic and prognostic tools available for PML diagnosis and describe past and current therapeutic approaches, including new strategies for PML cure.
Keywords: PML, JCV, NeuroAIDS, IRIS, diagnosis, therapy
Introduction
Progressive multifocal leukoencephalopathy (PML) is a debilitating and demyelinating disease of the central nervous system (CNS) which occurs almost exclusively in the context of severe impairment of the immune system, like AIDS, immunomodulatory therapy with monoclonal antibodies (mAbs), immune suppression after solid organ or hematopoietic stem cell transplants, and cancer.1–3
PML is caused by a polyomavirus first identified from brain lesions of a patient with PML, and named JC virus (JCV) from the patient’s initials (John Cunningham).4-6
Although JCV infects the worldwide population at a very high rate, PML remains a rare disorder. The introduction of combination antiretroviral therapies (cART) improved recovery of HIV-treated patients from severe immunosuppression, thus reducing the PML risk.7
In this review, we will summarize the epidemiology, the biology, and clinical presentation of HIV-related PML. We will also show what is known about the delicate interplay between JCV and the host immune system during HIV infection and discuss future perspectives.
PML history and epidemiology
The first case of PML was described in 1958 by Astrom et al,4 who showed the presence of inclusion bodies in the nuclei of damaged oligodendrocytes with electron microscopy, suggesting a possible viral cause. It was only in 1971 that Padgett et al characterized the etiologic agent of PML, by culturing JCV from PML brain lesions in human fetal spongioblasts.6
Before the AIDS pandemic, PML has been a rare and obscure disorder affecting old patients with predisposing conditions.4 From 1958 to 1984, Brooks and Walker identified 230 cases of PML, 40 of which were pathologically and virologically confirmed and 69 only pathologically confirmed.8 The most common PML-associated predisposing illnesses were lymphoproliferative diseases (62.2%), myeloproliferative diseases (6.5%), carcinoma (2.2%), and granulomatous and inflammatory diseases (7.4%). In 5.6% of PML patients, no immunosuppressive predisposing disorders were identified.9
After the spread of HIV in the early 1980s, the PML incidence has increased 50-fold compared to previous years10 and occurred in 1%–10% of AIDS cases, with AIDS accounting for 55%–85% of PML cases.11 With the introduction of cART, the PML incidence in HIV-infected patients has decreased because of the restoration of anti-JCV T-cell immunity.7 For this reason, the history of HIV-associated PML is characterized by two distinct phases: before and after cART introduction. According to the data from the EuroSIDA cohort, in the pre-cART era, PML incidence in HIV-infected subjects was 2–10 cases for 1,000 person-years and dropped down to one case for 1,000 person-years in the post-cART era.12,13 Furthermore, cART improved 1-year survival of HIV-infected patients with PML, ranging from 9% in the pre-cART era7 to 30% in the post-cART era. Today the 1-year survival rate is 50% or higher in patients under effective antiretroviral treatment, compared to 5% or lower in patients not receiving cART.14 According to the Italian NeuroAIDS study 2000–2002 (IRINA), PML is the third most common cause of encephalopathy in HIV-infected patients, after Toxoplasma encephalitis and HIV encephalitis.15
More recently, PML has become a growing concern in patients under immunosuppressive therapies and mAbs treatment (such as natalizumab).7 PML cases are also associated with organ transplantation, solid malignancies, sarcoidosis, autoimmune disorders, and congenital immune deficiencies, even though these populations represent less than 10% of all reported PML cases.15 Some cases of PML have been described in patients affected by idiopathic CD4+ lymphocytopenia.16
JCV structure and replication
The JC polyomavirus was the first polyomavirus to be discovered and, similar to other members of the polyomavirus family, has a double-stranded circular DNA genome of approximately 5 kb in size, enclosed in an icosahedral capsid.17 The JCV genome consists of three parts: a hypervariable noncoding control region (NCCR), a clockwise early coding region, and an anticlockwise late coding region.18 The early JCV transcript is responsible for encoding the majority of regulatory proteins: large and small tumor antigen proteins (T-antigen and t-antigen, respectively). The T-antigen is the key viral regulatory protein and acts as a transcriptional factor to direct DNA replication and to drive expression of the late viral transcript.19,20
The late JCV transcript encodes the capsid viral protein VP1, VP2, VP3, and a small regulatory phosphoprotein called agnoprotein, accounting for 71 amino acids.20,21 The capsid proteins (resulting from an alternative splicing of the late transcript) are essential for viral particles’ formation.17 Agnoprotein forms highly stable dimers and oligomers and plays an important role in viral DNA replication by enhancing T-antigen binding to the origin of replication.21
The early and late genes are characterized by highly conserved sequences, while NCCR is the most variable portion of the viral genome within a single viral population, as well as across different species and genera.17,19 For this reason, NCCR is considered to be the main determinant of viral tropism, and changes in the NCCR region are associated with an increase in viral transcription and replication in patients with PML.22 The NCCR acts as a bidirectional viral promoter, separating the viral genome into early (clockwise) and late (anticlockwise) genes and each NCCR form always contains the highly conserved origin of DNA replication (ori), one or more TATA boxes, T-antigen binding sites, multiple cellular transcription factor binding sites, a promoter, and an enhancer for transcription of early and late genes.17
Alignments of JCV NCCR sequences from several different patients and tissues allowed the identification of six blocks of sequences, named from A to F.17,19 The original isolate of JCV, known as MAD-1, contained an enhancer element constituted by a 98 bp tandem repeat (A-C-E-A-C-E-F) due to the duplication of the TATA box. Several NCCR variants containing tandem repeat-like structures have been isolated from tissue of PML patients and all of these NCCR variants are named “prototype” sequences.22 Later, a non-pathogenic variant of the NCCR region was found in the kidney and urine of healthy subjects and this strain was named “archetype”. Archetype-associated NCCR was composed of a single A-C-E unit that contained the 23 bp B block and the 66 bp D block resulting in an A-B-C-D-E-F NCCR region. Notably, insertion of the B and D block sequences in the NCCR region disrupts the integrity of several transcriptional binding factor sites, including YB-1/Purα, Oct-6, and NF-1. In conclusion, JCV exists in at least two different forms: a neurotropic form with rearranged NCCR (prototype) and a nonpathogenic form (archetype).22
Figure 1 shows JCV viral cycle in target cells.
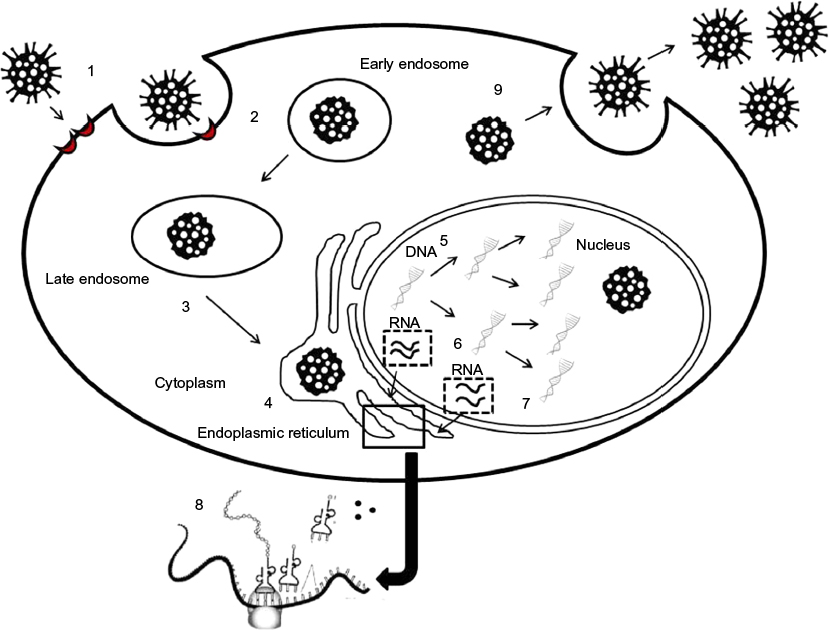 | Figure 1 JC virus (JCV) life cycle in target cells. Notes: 1) JCV adhesion to receptors in target cells. 2) JCV internalization through clathrin mediated endocytosis and transport to the early endosomes. 3) Transport to either recycling or late endosomes. 4) Virus transport in the endoplasmic reticulum up to the nucleus. 5) DNA replication in the nucleus. 6) Early gene transcription (regulatory genes). 7) Late gene transcription (structural genes). 8) Translation of the transcripts from either early or late genes. 9) Viral particles’ assembly and virus release from target cells with either lytic (oligodendrocytes) or nonlytic mechanisms (tubular epithelial cells). |
Mechanism of cell invasion
JCV first attaches to the cell surface via an interaction between the JCV capsid protein VP1 and the pentasaccharide NeuNAc-α2,6-Gal-β1,4-GlcNAc-β1,3-Gal-β1,4-Glc (LSTc).23 Viral particles then bind to receptors containing either α 2,3- or α 2,6-linked sialic acids.24 It has been shown that JCV can infect glial cells after binding the 2A form of the 5HT2AR,24 but the presence of this receptor seems to be sufficient but not necessary, as evidenced by infecting cell types lacking 5HT2AR (such as human brain microvascular endothelial cells) with JCV.25,26 As a consequence of viral interaction with target receptors, the virus enters into the cells by a clathrin-dependent endocytosis mechanism,27,28 as demonstrated by JCV infection inhibition by molecules able to block the clathrin-mediated pathway.28,29
Following clathrin-dependent endocytosis, ligands and viruses are usually carried to early endosomes and then shunted to recycling endosomes or to a late-endosomal/lysosomal compartment.30,31 The proper trafficking of cargos is regulated by early endosome membrane domains32 and by different Rab-family GTPases.33,34 Inside endosomal vesicles, viral particles are transported through the endoplasmic reticulum to the nucleus.
The first step of viral replication is the induction of early gene transcription by JCV T-antigen. This step could represent a potential key target for blocking JCV reactivation. JCV exploits several pathways to maintain cells in an immature stem cell-like phenotype. Expression of T-antigen, as well as the JCV late protein, agnoprotein, can also inhibit the differentiation of glial cells into a mature form.35,36 T-antigen can also allow infected cells to evade apoptosis. Through its interactions with the IGF-1 receptor, T-antigen enhances mTOR signaling and antiapoptotic protein expression.37 A strategy for arresting JCV replication could be represented by the induction of autophagy with the molecular chaperone regulator-3 (BAG3), facilitating T-antigen degradation.38
Once JCV infects permissive cells, the viral genome is readily transcribed, due to several binding sites for cellular transcription factors. After encoding for capsid proteins, the transcribed viral DNA is encapsulated and virions are released in a lytic process with the host cell death.39
T-antigen proteins are able to sequester key cell-cycle regulators, such as p53 and Rb, “hijacking” cell cycle machinery into an S-phase like state to maintain active viral replication.40 JCV T-antigen selectively binds to wild type p53, a potent inducer of G1-phase cell cycle arrest, apoptosis, and DNA repair.41 This protein complex also enhances the expression of IGF-1, the receptor of which is another T-antigen binding partner, to further stimulate cell growth.42 When JCV infects tubular epithelial cells in the kidney, it usually establishes a nonlytic cycle, characterized by persistent urinary shedding of viral particles.9
JCV/HIV interaction
The high incidence of PML in HIV-positive patients seems to be due to several factors: 1) CD4+ T-cell loss, with impairment of immune surveillance of the CNS,43 2) the activation of CD8+ CTL responses with destruction of infected oligodendrocytes,39 and 3) the presence of HIV in the brain, which may directly or indirectly participate in the pathogenesis of PML.43
In HIV-infected subjects with PML, NCCR is characterized by multiple duplications of the upstream Tat-responsive element (up-TAR), which is also constantly found at the 5′-end of HIV-1 mRNAs. Up-TAR is involved in HIV Tat induction of the JCV late promoter.44 Considering that Tat can induce its own expression and translation, establishing a positive feedback, JCV viral expression and viral protein production can be massively induced in PML HIV-infected subjects, by Tat transactivation of JCV viral genome transcription.45–47 Notably, Tat enhances the ability of Pur-α48 to bind the up-TAR element in the JCV-NCCR region, thus synergistically activating transcription49 and contributing to PML pathogenesis during HIV infection. Despite HIV and JCV infecting different cell types, Tat protein is secreted by HIV-infected cells, such as microglial cells, astrocytes, and monocytes, and can be internalized by JCV-infected oligodendrocytes,50 contributing to the enhancement of viral replication and transcription.49
Concerning the different HIV-1 clades and JCV interactions, HIV-1 clade B-associated Tat protein was found to be more effective in activating JCV early and late gene transcription in glial cells, than Tat from other clades, in vitro.50
Immune deficiency and JCV reactivation: immune pathogenesis of PML
The PML pathogenesis is still not completely clarified, since the source of the primary infection, the site of viral latency, and the route by which JCV enters into the brain have not been exactly understood. However, the conditions facilitating JCV-induced PML onset are well known: 1) changes in the NCCR with enhancement of viral transcription and replication, 2) availability of transcription factors binding the rearranged NCCR, 3) immunodeficiency, and 4) individual genetic predisposition.
JCV infection is highly prevalent in the worldwide population and after primary infection, which usually occurs during childhood, the virus can establish a latent infection with lifelong persistence.39 Considering anti-JCV antibody titers, infection rate increases with age, ranging from 10% of children aged 1–5 years and 40%–60% of children aged 10 years, up to 50% and 68% of adults aged 20–29 and 60–100 years, respectively.51
JCV infection is restricted to kidney epithelial cells, tonsillar stromal cells, bone marrow-derived cell lineages, oligodendrocytes, and astrocytes.17 The virus seems to replicate efficiently in kidneys and bone marrow-derived cells, establishing a low-level persistent or latent infection. In the CNS, the virus replicates vigorously in oligodendrocytes causing the appearance of demyelinating lesions.17 The immune system plays a critical role in clearing JCV, through the action of specific B, CD4, and CD8 T-cell responses.52
Initial infection results in a subclinical viral disease and generally remains clinically silent, despite active virus replication in the kidneys with urinary JCV-shedding in a large number of individuals.17 Upper respiratory airway may represent a feasible JCV entry site in humans, since JCV-DNA was detected in both stromal cells and lymphocytes of children’s and adults’ tonsils. Contrasting evidences showed the presence of both prototype and archetype JCV in tonsillar tissue,53,54 so it is difficult to define the JCV circulating form causing primary infection. Moreover, JCV can infect splenic and bone marrow mononuclear cells.55 Notably, bone marrow has been considered one of the most important latency and neurotropism acquiring sites for JCV.56 Specifically, bone marrow B-lymphocytes and CD34+ hematopoietic precursor cells are susceptible to JCV infection and are supposed to be a viral reservoir involved in NCCR rearrangement and viral carriage to the brain, especially in those conditions in which CD34+ progenitors and B-cell precursors are increased in peripheral blood (natalizumab treatment).17,52
Nevertheless, studies in hematopoietic stem cell transplant recipients showed that JCV can reactivate from kidney tubular cells independently from bone marrow latency sites, as demonstrated by the persistent JCV-urinary shedding in these patients before and after hematopoietic stem cell transplantation.57
Classical PML in AIDS
PML is an AIDS defining disease, occurring almost exclusively in the context of severe immune depression consequent to HIV infection.58 In this setting, we define the disease as AIDS-associated classical PML.
The majority of HIV patients who develop PML have a CD4 cell count <200 cells/μL, although there are some reports of PML cases in HIV-infected subjects with higher CD4 cell count. The incidence of PML in HIV-infected subjects has decreased after the introduction of cART, but it remains a major concern because of the lack of an effective direct acting anti-JCV drug.58
PML lesions are histopathologically characterized by demyelination, astrocyte modifications, and oligodendroglial nuclei changes, while edema, lymphocyte infiltration, and blood -brain barrier (BBB) damage are usually absent.59,60
In AIDS-presenter therapy-naïve HIV-infected subjects with PML, the only therapeutic intervention is cART initiation, with the aim of controlling HIV replication and consequently improving the host immune system.61 If PML occurs in a cART-treated patient, who is experiencing either virological or immunological failure, a therapeutic optimization (switch or intensification) should be considered in order to better control HIV replication and improve CD4 cell count. 62
PML-immune reconstitution inflammatory syndrome
The immune reconstitution after cART initiation has been associated with the paradoxical worsening or the unmasking of an opportunistic infection, asymptomatic or clinically controlled before ART initiation. Immune reconstitution inflammatory syndrome (IRIS) represents an exuberant inflammatory response to a dead, dying, or viable pathogen from a persistent infection.60,63,64
PML-IRIS manifests as a neurologic deterioration with evidences of CNS JCV infection after cART initiation.63 PML-IRIS lesions are pathologically different from classical PML, being characterized by lymphocyte perivascular infiltrates, BBB damage with brain edema, and consequent mass effect, which are lacking in classical PML lesions. Interestingly, PML-IRIS brain lesions are infiltrated by cytotoxic CD8+ T lymphocytes that recognize and kill JCV-infected oligodendrocytes, controlling JCV dissemination but causing oligodendrocyte cell death and demyelination.65 Neuroimaging of PML-IRIS lesions is characterized by contrast enhancement as well as mass effect and interstitial edema.60
Considering the lack of an effective anti-JCV treatment, PML-IRIS therapy is essentially based on corticosteroids administration, with the aim of reducing brain inflammation. However, a case series of 54 patients with AIDS-associated PML-IRIS treated with corticosteroids showed contrasting results.66
PML diagnosis
PML clinical manifestations are heterogeneous and reflect the brain areas affected by the lesions. Most common clinical findings are motor weakness, gait abnormalities, speech and language disturbances, incoordination, visual field deficit (due to involvement of the optic radiations, whereas the optic nerve is usually spared). Less common clinical findings are sensory impairment, seizures, headache, and diplopia.59
Definitive PML diagnosis relies on the presence of the classic histopathologic triad: demyelination, bizarre astrocytes, and enlarged oligodendroglial nuclei in the brain lesion biopsies, associated with the demonstration of the presence of JCV by either electron microscopy/immunohistochemistry or tissue polymerase chain reaction (PCR) positivity. The presence of the classic triad without evidence of JCV CNS infection by microscopy or molecular biology makes the diagnosis probable, whereas evidence of JCV through electron microscopy/immunohistochemistry or tissue PCR without evidence of the typical histopathologic brain lesions makes the diagnosis possible.59,60
Considering the difficulties in obtaining CNS biopsies, the diagnosis of PML can be achieved on the basis of clinical features, neuroimaging, and cerebrospinal fluid (CSF) molecular biology testing. Notably, the presence of consistent clinical signs and symptoms, with positive neuroimaging findings and CSF JCV detection by PCR, make the PML diagnosis definite. The CSF JCV-PCR positivity in addition to either consistent clinical features or radiological alterations, suggest a diagnosis of probable PML, while the CSF JCV-PCR positivity alone or merely the presence of clinical and neuroradiological findings consistent with PML, makes PML diagnosis possible. PML should be excluded when either compatible clinical features or compatible neuroimaging is associated with negative JCV-PCR in CSF.59
Beyond the classical diagnostic tools, several new diagnostic approaches have been evaluated for PML diagnosis in AIDS patients.
CSF PCR viral load associated with NCCR sequencing is a useful tool for the diagnostic and prognostic evaluation of HIV-infected subjects affected by PML.67 JCV-DNA detection and quantification in plasma have been evaluated for their predictive value in terms of PML onset, but lacked a definitive demonstration of any association.68
NCCR deep sequencing is providing new insights into PML pathogenesis. It has been shown that archetype-like JCV is the predominant population in urine, in the absence of minor variants. In CSF, JCV viral population is a mixture of several recombined forms, contributing to the constitution of a “quasispecies”. Moreover, viral quasispecies in CSF and blood seem to be strongly correlated in PML-affected patients, suggesting the main role of the hematogenous route in JCV dissemination from latency sites to the CNS.69,70
JCV serology has been a very useful tool in identifying JCV-infected multiple sclerosis patients, candidates for natalizumab treatment, and those at higher risk of JCV reactivation. However, JCV serologic screening alone does not identify all JCV-infected individuals, given the presence of seronegative subjects with PCR detectable JCV-DNA in blood or urine.71 Accordingly, JCV-DNA urine testing is complementary to anti-JCV serostatus assessment in identifying patients at risk for PML (at least in natalizumab-treated patients).72 Furthermore, no evidence of correlations between serum anti-JCV-specific IgG titers and PML onset was found.68
JCV urinary shedding may also occur in healthy subjects73 and JCV viral load in urine seems to be unchanged when comparing HIV-positive subjects with and without PML.74 A new diagnostic tool could be represented by JCV-associated microRNAs’ detection and quantification in human blood, urine, and CSF.75 Specifically, Jcv-miR-J1 can be used in order to identify the presence of a latent JCV infection, with a higher detection rate than JCV-specific serology.76 Further studies are needed to understand the predictive value of microRNAs for PML diagnosis.
Magnetic resonance imaging (MRI) represents a paramount noninvasive tool for PML diagnosis. Classically, PML lesions appear as single or multiple hyperintense areas in T2-weighted images, slightly hypointense in T1-weighted images, mainly subcortical, involving the white matter (although the gray matter can be involved), without mass effect. Typically, lesions do not appear enhanced after contrast administration. Diffusion-weighted images with apparent diffusion coefficient maps, diffusion tensor imaging (which analyzes the degree of fractional anisotropy), white matter magnetic resonance spectroscopy, and magnetization transfer imaging represent magnetic resonance techniques which can improve differential diagnosis and prognostic assessment of PML.77 PML-IRIS-associated lesions may show contrast enhancement on MRI examination, because of inflammation and BBB disruption. Brain edema, swelling, and mass effect can also be found with MRI in this setting.68
PML therapy
The introduction of cART improved the prognosis of patients with AIDS-related PML.61 In particular, protease inhibitor based ART seems to be associated with better survival, regardless of the CNS penetration score.62
Several drugs targeting serotonin receptors and inhibiting viral entry have shown an antiviral effect in vitro, such as chlorpromazine, citalopram, risperidone, ziprasidone, and mirtazapine. Risperidone’s efficacy was challenged by negative results in inhibiting JCV infection of glial cells in vitro.78 Furthermore, a definitive demonstration of the in vivo efficacy of these drugs is still lacking and for some of them is weakly supported by some case reports.79 Retro-2cycl and brefeldin A are able to interfere with viral replication by inhibiting retrograde transport of JCV to the endoplasmic reticulum in vitro, while no in vivo data are available so far. Nucleoside analog cytarabine; nucleotide analogs cidofovir and brincidofovir; small molecules like ganciclovir, topotecan, and leflunomide; the antimalarial drug mefloquine; Poly (ADP-ribose) polymerase inhibitors; the protein kinase inhibitor imatinib; and siRNA Ag122 (which targets JCV agnoprotein) have shown anti-JCV activity in vitro, by inhibiting viral DNA replication. Cytarabine, cidofovir, and brincidofovir activity also affects host cell polymerase causing toxicity, which limits their utilization in clinical settings. The in vivo efficacy of ganciclovir and leflunomide was supported by some case reports. siRNA Ag122 seemed to be effective in inhibiting JCV replication in a PML mouse model, while no human data are available so far. 7 A recent paper evidenced the potential antiviral effect of the antimalarial drug artesunate, which showed the ability to inhibit JCV viral cycle in vitro in the monkey kidney cell line COS-7. In vitro, artesunate’s half maximal effective concentration can be achieved in human plasma without toxic effects and its active metabolite dihydroartemisinin accumulates in the CNS. Clinical studies are needed to demonstrate the in vivo antiviral efficacy of artesunate.79
Furthermore, several drugs have been tested in clinical studies, for their anti-JCV potential activity during AIDS-related PML. In one Phase II clinical trial, HIV-positive patients with PML were randomly assigned to three different arms: treatment with cART alone, cART in combination with either intravenous or intrathecal cytarabine. The overall survival in the three arms was similar and cytarabine did not improve the prognosis of HIV-infected patients with PML.7 A pilot study and analysis of data from one prospective study and five cohort studies on cidofovir administration to HIV-positive patients with PML did not show any benefit in reducing mortality and residual disability.7 A Phase II clinical uncontrolled trial failed to demonstrate topotecan’s efficacy in HIV-related PML, because of the small number of enrolled subjects and the high rate of adverse events directly related to this drug.7 Retrospective analyses of observational studies demonstrated the inefficacy of IFN-alpha, while for IFN-beta only anecdotal reports are available. Both IFN-alpha and -beta are no longer considered as potential treatments for PML. For IL-2 and IL-7, only anecdotal data are available.7,58 More recently, treatment of AIDS-related PML with mefloquine and mirtazapine has been described in several case reports, with contrasting outcomes.80–82 Limited evidences of mefloquine efficacy in non-AIDS-related PML and AIDS PML-IRIS have been described. In both cases, the demonstration of mefloquine efficacy was not definitive, considering that in non-AIDS-related PML the immune suppressive condition is often removed (drug suspension and plasmapheresis),83 while in AIDS PML-IRIS the immune reconstitution due to cART could play a major role.84 Larger studies did not confirm the efficacy of mefloquine for PML treatment.85 The CCR5 blocker maraviroc was associated with clinical improvement in three non-HIV subjects with PML (patients had immunological disorders: two patients had sarcoidosis, the third had idiopathic CD4+ lymphocytopenia). Maraviroc can act indirectly, by positively influencing CD4+ T lymphocyte reconstitution and modulating immune activation, and directly by interfering with the levels of nuclear transcription factor which enhance JCV replication in oligodendrocytes. Maraviroc seems to increase the survival of HIV-positive and natalizumab-treated patients affected by PML-IRIS. Further studies are needed to assess maraviroc’s efficacy in the treatment of HIV-associated classical PML.86
Passive immunization strategies (recombinant JCV VP1-specific mAbs and JCV-specific cytotoxic T lymphocyte) and active immunization approaches (by using VP1 epitopes with and without IL-7 boosting) are currently under development.7 Recently, a fluorescent JCV viral construct was obtained in laboratory, allowing for the possibility of rapid screening of several molecules with the capacity of inhibiting JCV replication in cell cultures.87 A new and promising approach for PML treatment could be the gene editing procedures based on the clustered regularly interspaced short palindromic repeats (CRISPR)/CAS9 technology.88,89 Table 1 summarizes molecules with potential anti-PML activity with in vitro or in vivo efficacy in inhibiting JCV replication.
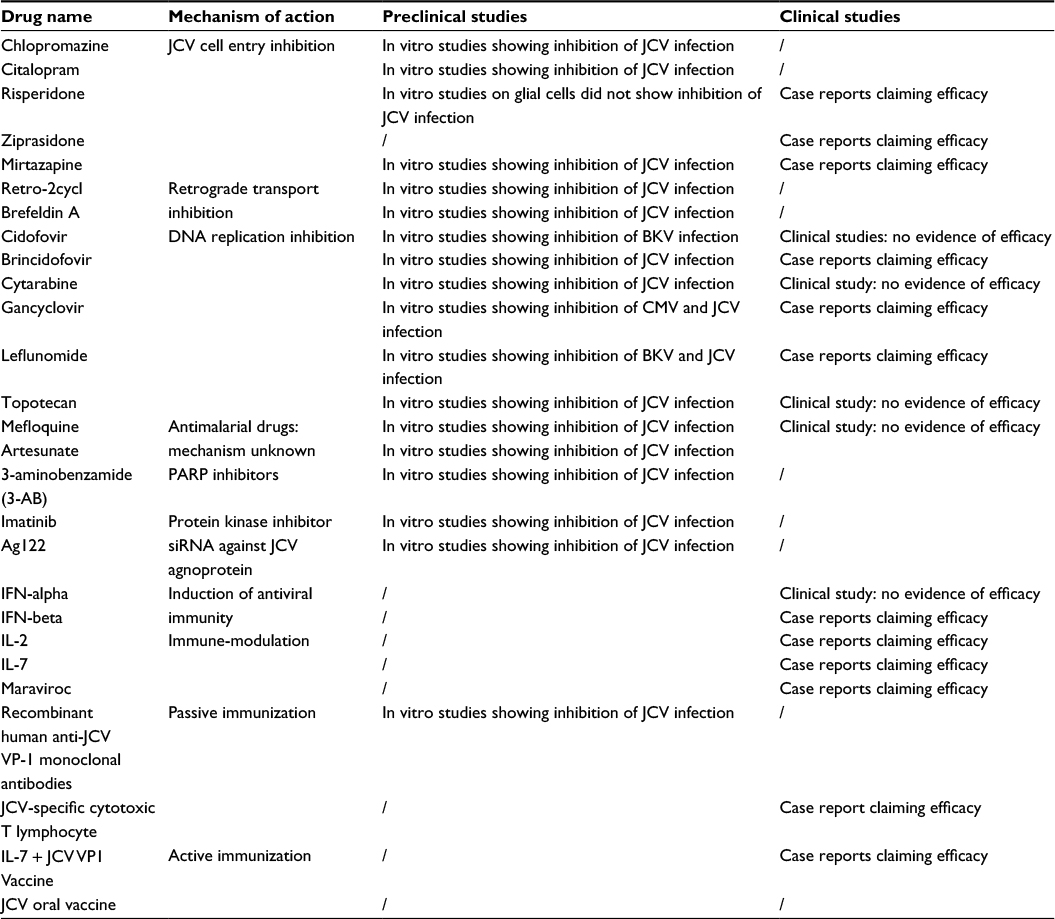 | Table 1 Potential candidates for PML treatment with anti-JCV activity Note: “/”, no data. Abbreviations: JCV, JC virus; PML, progressive multifocal leukoencephalopathy; IFN, interferon; IL, interleukin; VP, viral protein; PARP, Poly (ADP-ribose) polymerase; BKV, BK virus; CMV, cytomegalovirus. |
Acknowledgments
The authors thank Società Italiana di Malattie Infettive e Tropicali (SIMIT) for the fellowship awarded to MI. Thanks to the RaP group for fruitful discussions.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Berger JR. Progressive multifocal leukoencephalopathy and newer biological agents. Drug Saf. 2010;33(11):969–983. | ||
White MK, Khalili K. Pathogenesis of progressive multifocal leukoencephalopathy--revisited. J Infect Dis. 2011;203(5):578–586. | ||
Major EO. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annu Rev Med. 2010;61:35–47. | ||
Astrom KE, Mancall EL, Richardson EP Jr. Progressive multifocal leuko-encephalopathy: a hitherto unrecognized complication of chronic lymphatic leukaemia and Hodgkin’s disease. Brain. 1958;81(1):93–111. | ||
Zurhein G, Chou SM. Particles resembling papova viruses in human cerebral demyelinating disease. Science. 1965;148(3676):1477–1479. | ||
Padgett BL, Walker DL, ZuRhein GM, Eckroade RJ, Dessel BH. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. Lancet. 1971;1(7712):1257–1260. | ||
Pavlovic D, Patera AC, Nyberg F, Gerber M, Liu M; Progressive Multifocal Leukeoncephalopathy Consortium. Progressive multifocal leukoencephalopathy: current treatment options and future perspectives. Ther Adv Neurol Disord. 2015;8(6):255–273. | ||
Brooks BR, Walker DL. Progressive multifocal leukoencephalopathy. Neurol Clin. 1984;2(2):299–313. | ||
Berger JR. Progressive multifocal leukoencephalopathy in acquired immunodeficiency syndrome: explaining the high incidence and disproportionate frequency of the illness relative to other immunosuppressive conditions. J Neurovirol. 2003;9(Suppl 1):38–41. | ||
Berger JR, Major EO. Progressive multifocal leukoencephalopathy. Semin Neurol. 1999;19(2):193–200. | ||
Berger JR, Pall L, Lanska D, Whiteman M. Progressive multifocal leukoencephalopathy in patients with HIV infection. J Neurovirol. 1998;4(1):59–68. | ||
d’ Arminio Monforte A, Cinque P, Mocroft A, et al. Changing incidence of central nervous system diseases in the EuroSIDA cohort. Ann Neurol. 2004;55(3):320–328. | ||
Engsig FN, Hansen AB, Omland LH, et al. Incidence, clinical presentation, and outcome of progressive multifocal leukoencephalopathy in HIV-infected patients during the highly active antiretroviral therapy era: a nationwide cohort study. J Infect Dis. 2009;199(1):77–83. | ||
Khanna N, Elzi L, Mueller NJ, et al. Incidence and outcome of progressive multifocal leukoencephalopathy over 20 years of the Swiss HIV Cohort Study. Clin Infect Dis. 2009;48(10):1459–1566. | ||
Antinori A, Cingolani A, Lorenzini P, et al. Clinical epidemiology and survival of progressive multifocal leukoencephalopathy in the era of highly active antiretroviral therapy: data from the Italian Registry Investigative Neuro AIDS (IRINA). J Neurovirol. 2003;9(Suppl 1):47–53. | ||
Haider S, Nafziger D, Gutierrez JA, Brar I, Mateo N, Fogle J. Progressive multifocal leukoencephalopathy and idiopathic CD41 lymphocytopenia: a case report and review of reported cases. Clin Infect Dis. 2000;31(4):E20–22. | ||
Ferenczy MW, Marshall LJ, Nelson CD, et al. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev. 2012;25(3):471–506. | ||
Bellizzi A, Anzivino E, Rodio DM, Palamara AT, Nencioni L, Pietropaolo V. New insights on human polyomavirus JC and pathogenesis of progressive multifocal leukoencephalopathy. Clin Dev Immunol. 2013;2013:839719. | ||
Major EO, Amemiya K, Tornatore CS, Houff SA, Berger JR. Pathogenesis and molecular biology of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev. 1992;5(1):49–73. | ||
Lashgari MS, Tada H, Amini S, Khalili K. Regulation of JCVL promoter function: transactivation of JCVL promoter by JCV and SV40 early proteins. Virology. 1989;170(1):292–295. | ||
Khalili K, White MK, Sawa H, Nagashima K, Safak M. The agnoprotein of polyomaviruses: a multifunctional auxiliary protein. J Cell Physiol. 2005;204(1):1–7. | ||
Martin JD, King DM, Slauch JM, Frisque RJ. Differences in regulatory sequences of naturally occurring JC virus variants. J Virol. 1985;53(1):306–311. | ||
Neu U, Maginnis MS, Palma AS, et al. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host Microbe. 2010;8(4):309–319. | ||
Dugan AS, Gasparovic ML, Atwood WJ. Direct correlation between sialic acid binding and infection of cells by two human polyomaviruses (JC virus and BK virus). J Virol. 2008;82(5):2560–2564. | ||
Neu U, Stehle T, Atwood WJ. The Polyomaviridae: contributions of virus structure to our understanding of virus receptors and infectious entry. Virology. 2009;384(2):389–399. | ||
Chapagain ML, Verma S, Mercier F, Yanagihara R, Nerurkar VR. Polyomavirus JC infects human brain microvascular endothelial cells independent of serotonin receptor 2A. Virology. 2007;364(1):55–63. | ||
Pho MT, Ashok A, Atwood WJ. JC virus enters human glial cells by clathrin-dependent receptor-mediated endocytosis. J Virol. 2000;74(5):2288–2292. | ||
Querbes W, Benmerah A, Tosoni D, Di Fiore PP, Atwood WJ. A JC virus-induced signal is required for infection of glial cells by a clathrin- and eps15-dependent pathway. J Virol. 2004;78(1):250–256. | ||
Benmerah A, Bayrou M, Cerf-Bensussan N, Dautry-Varsat A. Inhibition of clathrin-coated pit assembly by an Eps15 mutant. J Cell Sci. 1999;112(Pt 9):1303–1311. | ||
Bantel-Schaal U, Hub B, Kartenbeck J. Endocytosis of adeno-associated virus type 5 leads to accumulation of virus particles in the Golgi compartment. J Virol. 2002;76(5):2340–2349. | ||
Bousarghin L, Touzé A, Sizaret PY, Coursaget P. Human papillomavirus types 16, 31, and 58 use different endocytosis pathways to enter cells. J Virol. 2003;77(6):3846–3850. | ||
Pfeffer S. Membrane domains in the secretory and endocytic pathways. Cell. 2003;112(4):507–517. | ||
Nielsen E, Severin F, Backer JM, Hyman AA, Zerial M. Rab5 regulates motility of early endosomes on microtubules. Nat Cell Biol. 1999;1(6):376–382. | ||
Sönnichsen B, De Renzis S, Nielsen E, Rietdorf J, Zerial M. Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J Cell Biol. 2000;149(4):901–914. | ||
Merabova N, Kaniowska D, Kaminski R, et al. JC virus agnoprotein inhibits in vitro differentiation of oligodendrocytes and promotes apoptosis. J Virol. 2008;82(3):1558–1569. | ||
Tretiakova A, Krynska B, Gordon J, Khalili K. Human neurotropic JC virus early protein deregulates glial cell cycle pathway and impairs cell differentiation. J Neurosci Res. 1999;55(5):588–599. | ||
Gualco E, Urbanska K, Perez-Liz G, et al. IGF-IR-dependent expression of Survivin is required for T-antigen-mediated protection from apoptosis and proliferation of neural progenitors. Cell Death Differ. 2010;17(3):439–451. | ||
Sariyer IK, Merabova N, Patel PK, et al. Bag3-induced autophagy is associated with degradation of JCV oncoprotein, T-Ag. PloS One. 2012;7(9):e45000. | ||
Beltrami S, Gordon J. Immune surveillance and response to JC virus infection and PML. J Neurovirol. 2014;20(2):137–149. | ||
Caracciolo V, Reiss K, Khalili K, De Falco G, Giordano A. Role of the interaction between large T antigen and Rb family members in the oncogenicity of JC virus. Oncogene. 2006;25(38):5294–5301. | ||
Krynska B, Gordon J, Otte J, et al. Role of cell cycle regulators in tumor formation in transgenic mice expressing the human neurotropic virus, JCV, early protein. J Cell Biochem. 1997;67(2):223–230. | ||
Del Valle L, Wang JY, Lassak A, et al. Insulin-like growth factor I receptor signaling system in JC virus T antigen-induced primitive neuroectodermal tumors--medulloblastomas. J Neurovirol. 2002;8(Suppl 2):138–147. | ||
Chun TW, Fauci AS. HIV reservoirs: pathogenesis and obstacles to viral eradication and cure. AIDS. 2012;26(10):1261–1268. | ||
Wortman MJ, Krachmarov CP, Kim JH, et al. Interaction of HIV-1 Tat with Puralpha in nuclei of human glial cells: characterization of RNA-mediated protein-protein binding. J Cell Biochem. 2000;77(1):65–74. | ||
Berkhout B, Silverman RH, Jeang KT. Tat trans-activates the human immunodeficiency virus through a nascent RNA target. Cell. 1989;59(2):273–282. | ||
Berkhout B, Gatignol A, Rabson AB, Jeang KT. TAR-independent activation of the HIV-1 LTR: evidence that tat requires specific regions of the promoter. Cell. 1990;62(4):757–767. | ||
Berkhout B, Jeang KT. Functional roles for the TATA promoter and enhancers in basal and Tat-induced expression of the human immunodeficiency virus type 1 long terminal repeat. J Virol. 1992;66(1):139–149. | ||
Chepenik LG, Tretiakova AP, Krachmarov CP, Johnson EM, Khalili K. The single-stranded DNA binding protein, Pur-alpha, binds HIV-1 TAR RNA and activates HIV-1 transcription. Gene. 1998;210(1):37–44. | ||
Daniel DC, Wortman MJ, Schiller RJ, et al. Coordinate effects of human immunodeficiency virus type 1 protein Tat and cellular protein Puralpha on DNA replication initiated at the JC virus origin. J Gen Virol. 2001;82(Pt 7):1543–1553. | ||
Wright CA, Nance JA, Johnson EM. Effects of Tat proteins and Tat mutants of different human immunodeficiency virus type 1 clades on glial JC virus early and late gene transcription. J Gen Virol. 2013;94(Pt 3):514–523. | ||
Borie D, Kremer JM. Considerations on the appropriateness of the John Cunningham virus antibody assay use in patients with rheumatoid arthritis. Semin Arthritis Rheum. 2015;45(2):163–166. | ||
Durali D, de Goër de Herve MG, Gasnault J, Taoufik Y. B cells and progressive multifocal leukoencephalopathy: search for the missing link. Front Immunol. 2015;6:241. | ||
Monaco MC, Jensen PN, Hou J, Durham LC, Major EO. Detection of JC virus DNA in human tonsil tissue: evidence for site of initial viral infection. J Virol. 1998;72(12):9918–9923. | ||
Kato A, Kitamura T, Takasaka T, et al. Detection of the archetypal regulatory region of JC virus from the tonsil tissue of patients with tonsillitis and tonsilar hypertrophy. J Neurovirol. 2004;10(4):244–249. | ||
Houff SA, Major EO, Katz DA, et al. Involvement of JC virus-infected mononuclear cells from the bone marrow and spleen in the pathogenesis of progressive multifocal leukoencephalopathy. N Engl J Med. 1988;318(5):301–305. | ||
Tan CS, Dezube BJ, Bhargava P, et al. Detection of JC virus DNA and proteins in the bone marrow of HIV-positive and HIV-negative patients: implications for viral latency and neurotropic transformation. J Infect Dis. 2009;199(6):881–888. | ||
Tan CS, Broge TA Jr, Ngo L, et al. Immune reconstitution after allogeneic hematopoietic stem cell transplantation is associated with selective control of JC virus reactivation. Biol Blood Marrow Transplant. 2014;20(7):992–999. | ||
Cinque P, Koralnik IJ, Gerevini S, Miro JM, Price RW. Progressive multifocal leukoencephalopathy complicating HIV-1 infection. Lancet Infect Dis. 2009;9(10):625–636. | ||
Berger JR, Aksamit AJ, Clifford DB, et al. PML diagnostic criteria: consensus statement from the AAN Neuroinfectious Disease Section. Neurology. 2013;80(15):1430–1438. | ||
Post MJ, Thurnher MM, Clifford DB, et al. CNS-immune reconstitution inflammatory syndrome in the setting of HIV infection, part 1: overview and discussion of progressive multifocal leukoencephalopathy-immune reconstitution inflammatory syndrome and cryptococcal-immune reconstitution inflammatory syndrome. AJNR Am J Neuroradiol. 2013;34(7):1297–1307. | ||
Mikita K, Maeda T, Fujikura Y, et al. Does anti-JCV therapy improve the prognosis of AIDS-related PML? Clin Neurol Neurosurg. 2013;115(9):1853–1854. | ||
Fanjul F, Riveiro-Barciela M, Gonzalez J, et al. Evaluation of progressive multifocal leukoencephalopathy treatments in a Spanish cohort of HIV-infected patients: do protease inhibitors improve survival regardless of central nervous system penetration-effectiveness (CPE) score? HIV Med. 2013;14(5):321–325. | ||
Sidhu N, McCutchan JA. Unmasking of PML by HAART: unusual clinical features and the role of IRIS. J Neuroimmunol. 2010;219(1-2):100–104. | ||
Shelburne SA 3rd, Hamill RJ. The immune reconstitution inflammatory syndrome. AIDS Rev. 2003;5(2):67–79. | ||
Martin-Blondel G, Bauer J, Cuvinciuc V, et al. In situ evidence of JC virus control by CD8+ T cells in PML-IRIS during HIV infection. Neurology. 2013;81(11):964–970. | ||
Tan K, Roda R, Ostrow L, McArthur J, Nath A. PML-IRIS in patients with HIV infection: clinical manifestations and treatment with steroids. Neurology. 2009;72(17):1458–1464. | ||
Delbue S, Elia F, Carloni C, et al. JC virus load in cerebrospinal fluid and transcriptional control region rearrangements may predict the clinical course of progressive multifocal leukoencephalopathy. J Cell Physiol. 2012;227(10):3511–3517. | ||
Viscidi RP, Khanna N, Tan CS, et al. JC virus antibody and viremia as predictors of progressive multifocal leukoencephalopathy in human immunodeficiency virus-1-infected individuals. Clin Infect Dis. 2011;53(7):711–715. | ||
Van Loy T, Thys K, Ryschkewitsch C, et al. JC virus quasispecies analysis reveals a complex viral population underlying progressive multifocal leukoencephalopathy and supports viral dissemination via the hematogenous route. J Virol. 2015;89(2):1340–1347. | ||
Van Loy T, Thys K, Tritsmans L, Stuyver LJ. Quasispecies analysis of JC virus DNA present in urine of healthy subjects. PloS One. 2013;8(8):e70950. | ||
Berger JR, Houff SA, Gurwell J, Vega N, Miller CS, Danaher RJ. JC virus antibody status underestimates infection rates. Ann Neurol. 2013;74(1):84–90. | ||
Laroni A, Giacomazzi CG, Grimaldi L, et al. Urinary JCV-DNA testing during natalizumab treatment may increase accuracy of PML risk stratification. J Neuroimmune Pharmacol. 2012;7(3):665–672. | ||
Kling CL, Wright AT, Katz SE, et al. Dynamics of urinary polyomavirus shedding in healthy adult women. J Med Virol. 2012;84(9):1459–1463. | ||
Behzad-Behbahani A, Klapper PE, Vallely PJ, Cleator GM, Khoo SH. Detection of BK virus and JC virus DNA in urine samples from immunocompromised (HIV-infected) and immunocompetent (HIV-non-infected) patients using polymerase chain reaction and microplate hybridisation. J Clin Virol. 2004;29(4):224–229. | ||
Pietilä T, Nummi M, Auvinen P, Mannonen L, Auvinen E. Expression of BKV and JCV encoded microRNA in human cerebrospinal fluid, plasma and urine. J Clin Virol. 2015;65:1–5. | ||
Lagatie O, Van Loy T, Tritsmans L, Stuyver LJ. Circulating human microRNAs are not linked to JC polyomavirus serology or urinary viral load in healthy subjects. Virology Journal. 2014;11:41. | ||
Sahraian MA, Radue EW, Eshaghi A, Besliu S, Minagar A. Progressive multifocal leukoencephalopathy: a review of the neuroimaging features and differential diagnosis. Eur J Neurol. 2012;19(8):1060–1069. | ||
Chapagain ML, Sumibcay L, Gurjav U, Kaufusi PH, Kast RE, Nerurkar VR. Serotonin receptor 2A blocker (risperidone) has no effect on human polyomavirus JC infection of primary human fetal glial cells. J Neurovirol. 2008;14(5):448-454. | ||
Sharma BN, Marschall M, Rinaldo CH. Antiviral effects of artesunate on JC polyomavirus replication in COS-7 cells. Antimicrob Agents Chemother. 2014;58(11):6724–6734. | ||
Iannetta M, Bellizzi A, Lo Menzo S, et al. HIV-associated progressive multifocal leukoencephalopathy: longitudinal study of JC virus non-coding control region rearrangements and host immunity. J Neurovirol. 2013;19(3):274–279. | ||
Moenster RP, Jett RA. Mirtazapine and mefloquine therapy for progressive multifocal leukoencephalopathy in a patient infected with human immunodeficiency virus. Am J Health Syst Pharm. 2012;69(6):496–498. | ||
Cettomai D, McArthur JC. Mirtazapine use in human immunodeficiency virus-infected patients with progressive multifocal leukoencephalopathy. Arch Neurol. 2009;66(2):255–258. | ||
Beppu M, Kawamoto M, Nukuzuma S, Kohara N. Mefloquine improved progressive multifocal leukoencephalopathy in a patient with systemic lupus erythematosus. Intern Med. 2012;51(10):1245–1247. | ||
Adachi E, Koibuchi T, Imai K, et al. Favourable outcome of progressive multifocal leukoencephalopathy with mefloquine treatment in combination with antiretroviral therapy in an HIV-infected patient. Int J STD AIDS. 2012;23(8):603–605. | ||
Clifford DB, Nath A, Cinque P, et al. A study of mefloquine treatment for progressive multifocal leukoencephalopathy: results and exploration of predictors of PML outcomes. J Neurovirol. 2013;19(4):351–358. | ||
Middel A, Arends JE, van Lelyveld SF, et al. Clinical and immunologic effects of maraviroc in progressive multifocal leukoencephalopathy. Neurology. 2015;85(1):104-106. | ||
Dang X, Chalkias S, Koralnik IJ. JC virus-iLOV fluorescent strains enable the detection of early and late viral protein expression. J Virol Methods. 2015;223:25–29. | ||
Loignon M, Toma E. Treatment options for progressive multifocal leukoencephalopathy in HIV-infected persons: current status and future directions. Expert Rev Anti Infect Ther. 2016;14(2):177–191. | ||
Wollebo HS, Woldemichaele B, Khalili K, Safak M, White MK. Epigenetic regulation of polyomavirus JC. Virol J. 2013;10:264. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.